

Abstract
In photosynthetic organisms, sudden changes in light intensity perturb the photosynthetic electron flow and lead to an increased production of reactive oxygen species. At the same time, thioredoxins can sense the redox state of the chloroplast. According to our hypothesis, thioredoxins and related thiol reactive molecules downregulate the activity of H2O2-detoxifying enzymes, and thereby allow a transient oxidative burst that triggers the expression of H2O2 responsive genes. It has been shown recently that upon light stress, catalase activity was reversibly inhibited in Chlamydomonas reinhardtii in correlation with a transient increase in the level of H2O2. Here, it is shown that Arabidopsis thaliana mutants lacking the NADP–malate dehydrogenase have lost the reversible inactivation of catalase activity and the increase in H2O2 levels when exposed to high light. The mutants were slightly affected in growth and accumulated higher levels of NADPH in the chloroplast than the wild-type. We propose that the malate valve plays an essential role in the regulation of catalase activity and the accumulation of a H2O2 signal by transmitting the redox state of the chloroplast to other cell compartments.
Keywords: malate valve, catalase, reactive oxygen species, light stress
1. Introduction
Life in an oxygenic atmosphere unavoidably leads to the production of toxic reactive oxygen species (ROS) during normal cell metabolism and increasingly under stress [1]. Plants are especially susceptible to oxidative stress, because ROS are formed during photosynthetic electron transport in the light. When plants are exposed to higher light intensities than that needed for saturation of photosynthetic electron transport, the yield of light-induced ROS formation increases. Plants have different strategies to cope with high light intensities such as non-photochemical quenching [2] and downregulation of photosynthetic electron transport by photosynthetic control [3], both activated by a low pH value in the thylakoid lumen. In the presence of a proton gradient that is larger than needed for the generation of ATP, the yield of singlet oxygen (1O2) generation is decreased, thanks to the pH-dependent component of non-photochemical quenching [4]. The yield of superoxide (O2·−) production at photosystem I (PSI), the so-called Mehler reaction, is also lowered, because the linear electron transport is slowed down. Furthermore, plants have a set of defence mechanisms involving antioxidant molecules such as carotenoids, tocopherols, glutathione, ascorbate and enzymes such as superoxide dismutase, peroxiredoxins, glutathione peroxidase, ascorbate peroxidase and catalase to combat ROS. Despite these protection mechanisms, a certain amount of ROS is generated in the light by photosynthetic electron transfer reactions and plays an important physiological role that allows plants to adapt to changes in their environment.
It is recognized that ROS act as signalling molecules that can trigger cell responses [5–13]. Specific and distinct signalling pathways are activated by 1O2 and its products such as lipid peroxides and carotenoid radicals and by O2− and hydrogen peroxide (H2O2) [9,14,15]. Here, we want to focus on H2O2, the most stable of the ROS. H2O2 is generated in the light when O2 is reduced to O2·− at the acceptor side of PSI and then disproportionated by superoxide dismutase to H2O2 and O2 and during photorespiration. H2O2 generated in chloroplasts during the light phase is able to diffuse out into the cytosol [16] where it can function as a signalling agent. To act in this function, the H2O2 concentration must rise rapidly to a threshold concentration and remain high enough (10–100 µM) for a sufficient time so that it can oxidize the molecules involved in the cell signalling events [17]. Therefore, enzymes that metabolize ROS must play a dual role: in an active state, they keep ROS concentrations at safe levels, and in an inactivated state, they can allow internal ROS concentrations to reach critical levels for activation of signalling components. We have shown recently that in the unicellular green alga Chlamydomonas reinhardtii, H2O2 accumulated transiently during the first 10 min of exposure of the cultures to high light [18]. This transient accumulation of H2O2 correlated with a reversible inactivation of catalase activity. The activity of other antioxidant enzymes such as superoxide dismutase and ascorbate peroxidase did not follow the same trend. Catalase activity has been shown to be inactivated in vitro by low concentrations of dithiothreitol (DTT) and by reduced thioredoxin (TRX) [18–20]. The midpoint potential of the inactivation was determined to be at −330 mV at pH 8.0, which makes it possible that catalase is inactivated in vivo by reduced glutaredoxin (GRX), TRX or glutathione [18]. In the C. reinhardtii genome, only one catalase gene (CAT1, A8J537, UNIPROT) is present, whereas higher plants contain three catalase genes (CAT1–3 in Arabidopsis). In higher plants, catalases are localized in peroxisomes, whereas in C. reinhardtii, which lacks leaf-like peroxisomes, the enzyme is most likely to be located in cytosolic microbodies that were visualized by subcellular localization of green fluorescent proteins fused to several putative Chlamydomonas peroxisomal targeting sequences [21].
According to our hypothesis (figure 1a), light stress increases the production of ROS (1O2, O2·−, H2O2) and modifies the reduction state of TRX. TRX could regulate the catalase activity either directly or via other redox mediators. Reversible inactivation of catalase induces a transient H2O2 burst that acts as a signal and could alter the expression level of nuclear genes encoding stress-related proteins, such as catalases, ascorbate peroxidases, glutathione peroxidases, heat shock proteins and peroxiredoxins. The upregulation of these genes would help the plant to cope with oxidative stress, and thereby to adapt to changes in its environment. We hypothesize that the following scenario takes place: under high light, NADP+ and other electron acceptors, such as TRX, become more reduced. O2 acts as an alternative electron sink, and O2− is generated and disproportionates into H2O2. H2O2 diffuses out of the chloroplast while at the same time the peroxisomal catalase activity is transiently downregulated via a yet unknown reducing mechanism. Hence, the concentration of H2O2 is expected to increase in all cell compartments. As a consequence, the expression levels of nuclear H2O2-responsive genes increase. During prolonged high light stress, downregulation mechanisms of the electron transport chain (such as non-photochemical quenching) take place, and the reduction pressure is lowered. Therefore, the cellular redox state becomes less reducing, and the catalase is reactivated. The upregulation of the expression level of stress-induced genes results in an increase in the content of antioxidant enzymes. The plant/alga thus adapts to the changes in the environment.
Figure 1.
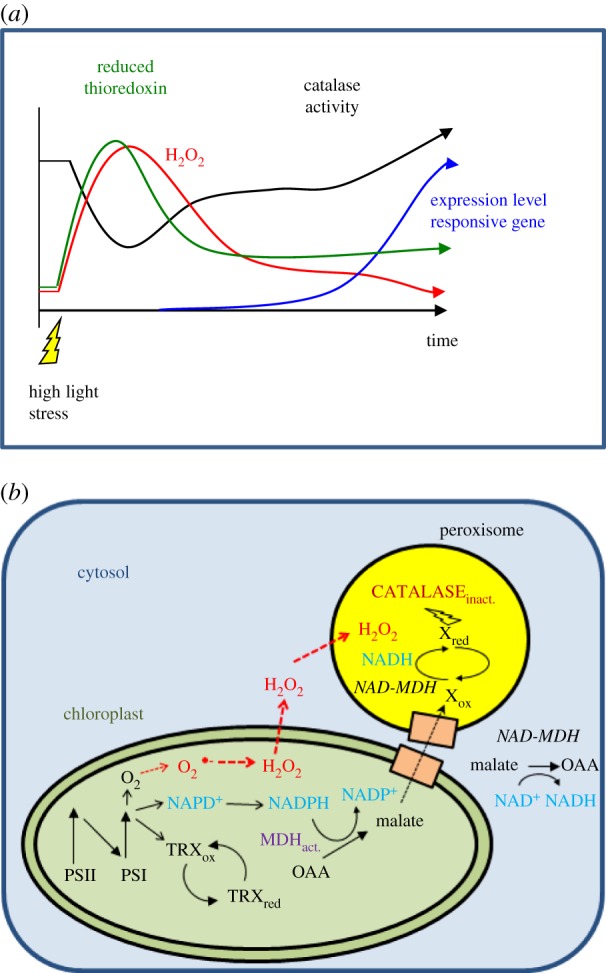
(a) Scheme describing signalling by a H2O2 pulse according to our hypothesis. When a plant is suddenly exposed to a change in the light intensity from low to high light, the pool of NADP+ in the chloroplast becomes quickly reduced. In parallel, the reduction state of the thioredoxin pool increases and O2 acts as final electron acceptor leading to the generation of O2.− and H2O2. H2O2 is partially detoxified in the chloroplast and partially diffuses into the cytosol and other cell compartments leading to an increase in the overall intracellular H2O2 concentration. Because the activity of catalase is partially inhibited by reducing equivalents originating from the chloroplast (see (b) for further explanation), the H2O2 concentration reaches its maximum. After a given time, the photosynthetic electron transfer chain is downregulated by different mechanisms, such as photosynthetic control, non-photochemical quenching and changes in the cellular metabolism. These phenomena increase the availability of NADP+ and as the reduction pressure in the chloroplast declines, TRX get more oxidized and the catalase is re-activated and decreases the intracellular H2O2 concentration. According to our hypothesis, this H2O2 burst is sufficient to induce the expression of H2O2-responsive nuclear genes. (b) Reactions occurring in the chloroplast and peroxisomes leading to a reversible inactivation of the catalase activity via the malate valve. At the acceptor side of photosystem I, NADP+, TRXs and O2 are reduced. Reduced TRXs activate the NADP-dependent malate dehydrogenase (MDH indicated in purple in the chloroplast). NADPH is oxidized and oxalacetate is reduced to malate by NADP–MDH. Malate is exported from the chloroplast via dicarboxylate transporters (orange boxes). NAD-dependent MDH isoforms present in the cytosol and in the peroxisomes convert malate into oxaloacetate and reduce NAD+ to NADH. NADH in the peroxisomes reduces a compound called X in the scheme that inactivates the catalase activity partially and reversibly by a still unknown reduction mechanism. H2O2 is generated in the chloroplasts and diffuses into the cytosol and other cell compartments. (Online version in colour.)
In mammalian cells, the ‘floodgate hypothesis’ [22] posits that an overoxidation of the peroxidatic cysteine of 2-Cys peroxiredoxin (Prx) to the sulfinate form, and its subsequent slow reduction by sulfiredoxin [23,24], leads to a peak in H2O2 that allows this ROS to take on a signalling role. In addition to the ‘floodgate hypothesis’, or alternatively, the hyperoxidation of Prx that inactivates its TRX-dependent peroxidase activity was proposed to be important to maintain a high level of reduced TRXs under oxidative stress conditions. This would allow to keep a number of other TRX-dependent targets reduced when H2O2 levels are high and to thereby maintain cell viability [25]. In plants, the role of a transient accumulation of ROS during light stress may be achieved by the reversible inactivation of catalase activity instead of inactivation of Prx.
Changes in the chloroplastic redox state have to be sensed, transmitted to the other parts of the cell and reach the unknown reductant that inactivates catalase in peroxisomes. One proposed partner in this signalling pathway is chloroplastic NADP-dependent malate dehydrogenase (NADP–MDH), which catalyses the reduction of oxaloacetate to malate using NADPH. When the NADPH pool becomes over-reduced, NADP–MDH is involved in the export of reducing power from the chloroplast to the cytosol, feeding malate to the chloroplast envelope dicarboxylate transporter (the so-called malate valve) [13,26]. Furthermore, NADP–MDH is activated by reduced TRX, making the link between the reduction state of the electron transport chain, the redox state of the chloroplast and the redox state of the cytosol and other cell compartments such as peroxisomes that host catalase (figure 1b). NAD-dependent isoforms of MDHs are present in cytosol, peroxisomes, mitochondria and plastids. These NAD–MDHs are not redox-regulated and they are involved in generating NADH through malate oxidation. Thus, the reducing power of malate could reduce the putative redox regulator of catalase in peroxisomes.
Here, we tested whether the malate valve plays a crucial role in transmitting the redox state of the chloroplast to other cell compartments. To do so, we measured catalase activity and H2O2 levels under high light stress in Arabidopsis thaliana knockout mutants deficient for chloroplastic NADP–MDH.
2. Material and methods
(a). Material
Arabidopsis thaliana (ecotype Columbia) plants were grown for five to seven weeks in soil under short-day conditions (8 h white light, 120 µmol quanta m−2 s−1, 20°C/16 h dark, 18°C).
The Arabidopsis nadp–mdh mutant lines named ‘M44’ (Salk_063444) and ‘M81’ (Salk_156181) are T-DNA mutant lines created at the Salk Institute (Salk Institute Genomic Analysis Laboratory, San Diego, USA; http://signal.salk.edu/) [27] and were obtained from the Nottingham Stock Centre (http://arabidopsis.info/).
(b). Screen for NADP–MDH mutants
Initial screening of nadp–mdh mutant lines was made by genotyping using PCR to track the presence of T-DNA in the gene region of At5g58330 (the single gene encoding plastidial NADP–MDH in the Arabidopis genome). PCR screening was performed as described previously [28] to amplify the T-DNA left border/gene-specific junction using the following oligonucleotide couples: newLB2 (5′-GGTGATGGTTCACGTAGTGGGCCATCG-3′)/mdhsens2 (5′-CCCTCACGAGGTTAGACGAAAATCG-3′) for M81 line and newLB2/mdhExtNrev (5′-GATCATAGGTGAGGCAGAACACTCC-3′) for M44 line. A second PCR using the following oligonucleotide couples: mdhsens2/mdhrev4 (5′-GGAAACAGCAGTAGAAGCAGCAGAAGATCG-3′) for M81 line and mdhsens1 (5′-TAATGGCCATGGCAGAGCTCTCAAC-3′)/mdhExtNrev for M44 line, was performed to amplify a wild-type, gene-specific fragment encompassing the site of T-DNA insertion. Sequencing of PCR products allowed the confirmation of PCRs specificity and positioning of T-DNA insertion at the locus of NADP–MDH.
(c). Extraction of soluble proteins from crude extracts
After the light treatment, leaves were frozen immediately in liquid nitrogen, homogenized and the homogenate was suspended in 100 mM Tris–HCl pH 8.0 or in 100 mM sodium phosphate buffer supplemented with a plant-specific protease inhibitor cocktail obtained from Sigma. The homogenate was centrifuged at 20 800g at 4°C for 5 min, and the clear supernatant was recovered.
(d). Protein determination, SDS–PAGE and immunoblot analysis
The protein content of soluble leaf crude extracts was determined using the bicinchoninic acid assay (Sigma). For western blot analysis, equal amounts of soluble proteins (50 µg per lane) were electrophoresed on SDS–PAGE (10%) gels and electrotransferred onto nitrocellulose membrane as described by Keryer et al. [28]. For detection of NADP–MDH, polyclonal antibodies raised against NADP–MDH from sorghum [29] were used at a 1 : 2000 dilution.
(e). Protoplast preparation
Cell wall digestion buffer was prepared by adding 0.75% cellulase YC and 0.03% pectolyase Y-23 (MP Biomedicals) in the protoplast buffer (0.5 M sorbitol, 1 mM CaCl2, 10 mM MES–KOH, pH 5.6). To allow complete dissolving of the enzymes, the solution was heated to 37°C while stirring for 5 min. Two grams of rosette leaves were cut with a scalpel in approximately 2 mm stripes and placed under vacuum for 2 × 5 min in 30 ml of digestion buffer. The material was gently shaken at 70 rpm for 4–6 h at room temperature. When most of the leaf material was digested, it was filtered through a nylon net (mesh of 60 μm) and centrifuged for 10 min at 10g, 20°C. The protoplast pellet was gently washed twice with the protoplast buffer (centrifugation 10 min, 10g, 20°C), and resuspended in a small volume of the same buffer. The intactness and the number of protoplasts were monitored microscopically.
(f). Photoinhibition
Photoinhibition of photosystem II (PSII) was carried out on detached leaves at 500 µmol quanta m−2 s−1 at room temperature. To block chloroplast-encoded protein synthesis, detached leaves were vacuum-infiltrated with lincomycin (1 g l−1) and floated on the lincomycin solution for 3 h under dim light prior to the photoinhibitory treatment. During photoinhibition, the leaves were kept hydrated on wet filter paper.
(g). Chlorophyll fluorescence
Room temperature chlorophyll fluorescence was measured on attached leaves, except during photoinhibition assays, using a pulse-amplitude modulation fluorometer (DUAL-PAM, Walz, Effeltrich, Germany). As actinic light, red light at 635 nm was used. The maximum quantum yield of PSII was calculated as the ratio of the variable fluorescence, Fv (Fm − F0), to maximal fluorescence, Fm, (Fv/Fm) with F0 being the fluorescence level obtained by the measuring light in dark-adapted leaves. The quantum yield of PSII (ΦPSII) was assayed after 10 min of actinic light (600 µmol quanta m−2 s−1) as (Fm′ − F)/Fm′ with Fm′ being the maximal fluorescence in the actinic light and F the steady-state fluorescence. qN was calculated as (Fm − Fm′)/(Fm − F0) and qP as (Fm′ − F)/(Fm′ − F0′) with F0′ = F0/Fv/(Fm + F0/Fm′), with F0′ being the dark fluorescence level after illumination. Plants were taken from the growth chamber and dark-adapted for 10–15 min prior to the measurement to allow most of the reversible quenching to relax.
(h). Electron paramagnetic resonance spectroscopy
Spin-trapping assays were performed with 4-pyridyl-1-oxide-N-tert-butylnitrone (4-POBN) using protoplasts. Samples were collected at given time points and incubated for 5 min in the presence of 50 mM 4-POBN, 4% ethanol, 50 μM Fe–EDTA. The Fe–EDTA is used as a Fenton reagent catalysing the reduction of H2O2 into −OH and •OH, the latter being detected in the spin-trapping assay. Electron paramagnetic resonance (EPR) spectra were recorded at room temperature in a standard quartz flat cell using an ESP-300 X-band (9.7 GHz) spectrometer (Bruker, Rheinstetten, Germany). The following parameters were used: microwave frequency, 9.7 GHz; modulation frequency, 100 kHz; modulation amplitude, 1G; microwave power, 6.3 milliwatt; receiver gain, 2 × 104; time constant, 40.96 ms; number of scans, 4.
(i). Measurements of enzyme activities
NAD(P)-dependent MDH activity of Arabidopsis leaf crude extracts was assayed spectrophotometrically as in Keryer et al. [28]. Activities were measured without (extractable, i.e. initial activity of the enzyme) or after pre-treatment (reductive activation giving the maximal activity, i.e. the enzyme capacity) of extracts with 25 mM DTT and 10 µM TRX f1 from Arabidopsis [30] for 20 min at room temperature.
Catalase activity was measured as O2-evolution in the soluble protein fraction of crude extracts with a Clark electrode at 20°C. 1 mM H2O2 was added as substrate. To determine the loss of activity by reduction, the sample was incubated in the presence of 5 mM DTT for 5 min prior to the measurement. The reduction-sensitive fraction of catalase activity was determined as the ratio between the activity measured in the absence of DTT and the activity measured in the presence of DTT. This ratio was set to 100% at time point zero.
3. Results
The two Arabidopsis nadp–mdh T-DNA insertion mutants M44 and M81 were compared with the isogenic wild-type S44 segregated from the parent heterozygous plant of M44 initially identified by PCR genotyping (figure 2a). Protein levels of NADP–MDH in leaf extracts from both mutant lines were strongly decreased down to the limit of detection (figure 2b). Consistently, leaf NADP–MDH capacity was found to be drastically decreased in both mutant lines to a level corresponding to less than 10% of that measured for the wild-type S44 line (figure 2c). NAD–MDH activity corresponding to non-redox-regulated plastidial isoforms and isoforms in other cell compartments (cytosol, mitochondria and microbodies) [31] remained unaffected by the mutation (figure 2c). The strong decrease in NADP–MDH protein level and activity had a slight effect on the phenotype of the M44 and M81 mutant lines. After six weeks growth in short-day conditions, the leaves of the mutant lines M44 and M81 were smaller, and the rosettes were lighter than those of the segregation line S44 (see electronic supplementary material, figure S1). Arabidopsis mutants of NADPH–MDH have been described previously and, in contrast to this study, showed no growth restriction phenotype [32]. At the present state, we do not have an explanation for this difference. To show whether the difference in weight under our conditions was caused by an alteration of the photosynthetic electron transport chain, chlorophyll fluorescence curves were measured. As shown in the electronic supplementary material, table S1, there were no significant differences in chlorophyll fluorescence parameters in the different lines. The loss of the NADP–MDH did not affect the maximum quantum yield of PSII (Fv/Fm). The quantum yield of PSII (Fv/Fm), non-photochemical quenching (NPQ) and photochemical quenching (qP) showed small differences that were not statistically significant but may indicate a trend that the loss of the NADP–MDH affects photosynthesis mildly. The efficiency of PSII photochemistry (Φ(II)) after 5 min illumination was also almost unchanged. However, these effects were so small that an impact on the overall growth seems to be very unlikely. In accordance with these observations, the mutant lines showed no higher susceptibility to photoinhibition compared with S44 (see electronic supplementary material, figure S2). These results are in accordance with a previous publication on Arabidopsis mutants lacking NADP–MDH [32]. However, we observed a significant difference in the post-illumination F0′ rise (figure 3). This rise in the dark level fluorescence reflects the reduction of the plastoquinone (PQ) pool by NADPH [33]. NADPH is oxidized by the chloroplastic NADPH dehydrogenase (NDH) complex and PQ is reduced. The chlorophyll fluorescence level increases, because plastoquinol is in equilibrium with the PQ molecules QB and QA at the acceptor side of PSII. It is the reduction of QA that leads to an increase in F0′. In the two mutant lines the transient F0′ rise was much higher than in S44, indicating a higher NADPH/NADP+ ratio that can possibly be a consequence of the lack of the export of reducing equivalents via the malate valve in the absence of NADP–MDH.
Figure 2.

Molecular characterization of the NADP–MDH knockout lines. (a) PCR screening with genomic DNA by amplification of either the T-DNA left border/nadp–mdh gene junction (m) or a wild-type nadp–mdh gene-specific fragment encompassing the site of T-DNA insertion (wt). Expected sizes (in base pairs, bp) of PCR products are shown as well as molecular weight (MW) markers. (b) Detection of NADP–MDH protein in wild-type (S44) and in mutant plants (M44 and S81). Western blot analysis of Arabidopsis leaf proteins along with varying amounts of sorghum NADP–MDH as control was made using antibodies raised against sorghum NADP–MDH. (c) NAD(P)–MDH leaf activity. Total leaf NADP–MDH capacity (black columns) was measured after a reducing (enzyme activation) treatment of soluble crude leaf extracts. For comparison, NAD–MDH activity (grey columns) was also measured.
Figure 3.
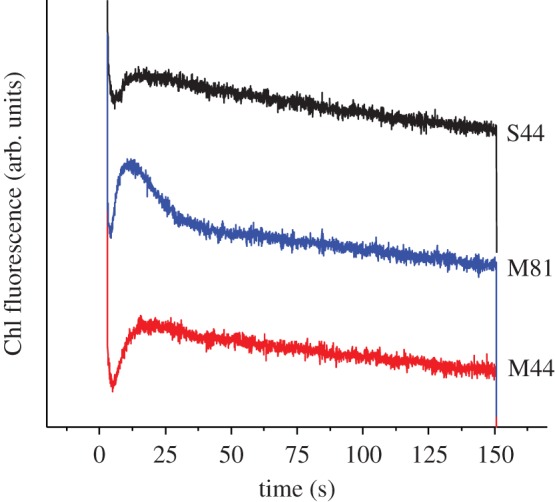
F0′ rise after illumination with actinic light in leaves from S44 (black, top), M44 (red, bottom) and M81 (blue, middle). Leaves were pre-illuminated with actinic light (I = 500 µmol quanta m−2 s−1) for 5 min. At time 0, the actinic light was switched off and the F0′ rise was followed. For clarity, the curves were displaced vertically. (Online version in colour.)
According to the current hypothesis, the redox state of the chloroplast is communicated to other cell compartments via the malate valve. We have shown previously that the catalase activity of C. reinhardtii and Arabidopsis is inhibited under reducing conditions [18]. Higher plant catalases are localized in peroxisomes and are therefore ideal candidates to investigate the role of the malate valve in the communication between chloroplasts and peroxisomes. We first tested whether the catalase activity was differently affected in S44 and in the two nadp–mdh mutant lines under reducing conditions. To maintain the maximum reduction state that is not damaging for the plant, we illuminated the leaves with 500 µmol quanta m−2 s−1 at 10°C, which minimizes the activity of the Calvin cycle and of the photorespiratory pathway, so that a high reduction state of the chloroplast was maintained during the illumination. The catalase activity was measured in crude leaf extracts containing the soluble proteins, prepared immediately after the illumination. Figure 4 shows that in S44 the catalase activity was decreased by about 20% during the first 30 min of illumination and recovered its initial activity at the end of the illumination time. In contrast, the two mutant lines showed no decrease of the catalase activity but rather an increase during the first 10 min of illumination. Inactivation of the catalase should allow a higher accumulation of H2O2 according to our previous data on C. reinhardtii. H2O2 was measured using a spin-trapping assay in which the size of the EPR signal reflects the amount of H2O2 generated in the light [16,18]. As we were using leaf material in this study, the spin-trapping assay was performed with protoplasts, because reliable measurements of early time points of high light stress (10 min) cannot be assured by infiltrating leaves with the probe; the spin trap will not be equilibrated between a leaf disc and the medium that is measured. A transient increase in the H2O2 level was indeed seen in protoplasts isolated from S44 but not in those from M44 and M81 (figure 5). A 40% increase in the H2O2 level was seen in the first 10 min in protoplasts from S44 exposed to high light, whereas the H2O2 level in protoplasts from the two mutant lines increased only slightly (maximum 10%) upon the onset of high light. The amount of detectable H2O2 decreased in S44 after 60 min of illumination, the time point at which the transient loss in the catalase activity was reverted (figure 5). In S44, in parallel to the decrease in catalase activity and the increase in H2O2 levels, the activation state of the NADP–MDH increased during the first 30 min of high light and decreased slightly towards the end of the light treatment (figure 6).
Figure 4.
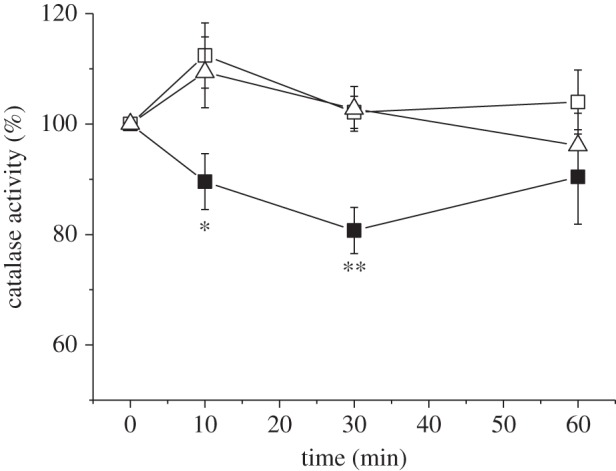
Extractable catalase activity from leaves. Leaves were exposed to 500 µmol quanta m−2 s−1 at 10°C for the indicated time. 100% activity corresponds to the activity measured in the absence of 10 mM DTT divided by the activity measured in the presence of DTT. The samples were treated with DTT to determine the proportion of catalase activity that is sensitive to reduction. Closed squares, S44; open squares, M44; open triangles, M81. Data are mean ± s.e. of seven to eight measurements. The significance levels (*p < 0.05 and **p < 0.005) are indicated.
Figure 5.
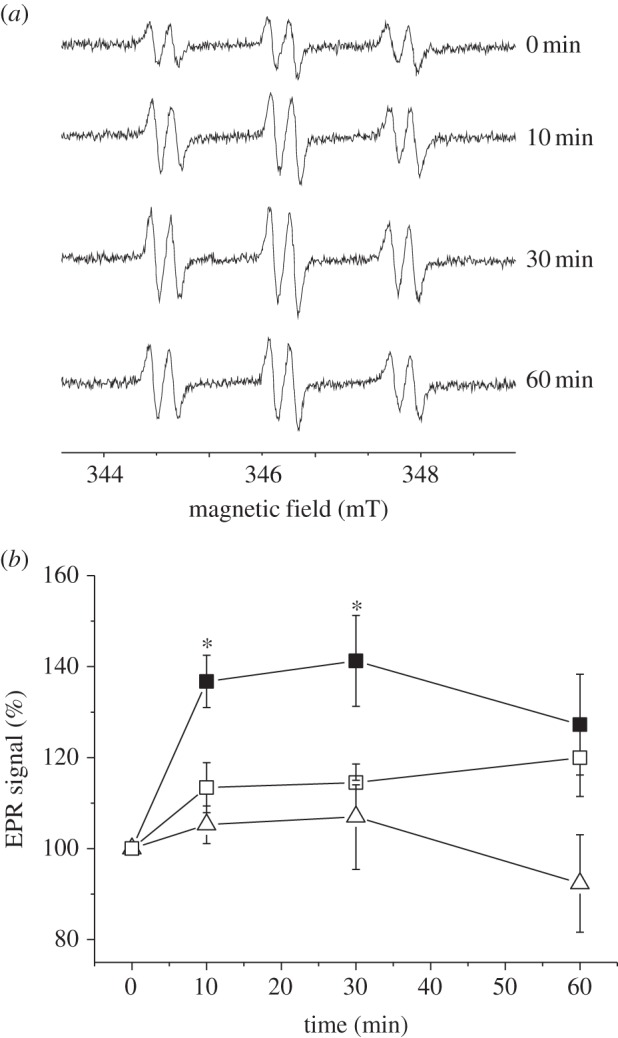
Transient accumulation of H2O2 in isolated protoplasts from S44, M44 and M81 after exposure to 500 µmol quanta m−2 s−1 at 10°C for a given time. (a) Typical EPR spectra of the 4-POBN/α-hydroxyethyl adduct recorded at 0, 10, 30, 60 min after illumination obtained in protoplasts of S44. The size of the signal indicates the concentration of H2O2 indirectly by the trapping of hydroxyl radicals (•OH). •OH radicals were generated from H2O2 in a Fe2+-catalysed Fenton reaction. (b) Average amplitude of the EPR signals ± s.e. (closed squares, S44; open squares, M44; open triangles, M81). Data are mean ± s.e. of four to five measurements. The significance level (*p < 0.05) is indicated.
Figure 6.
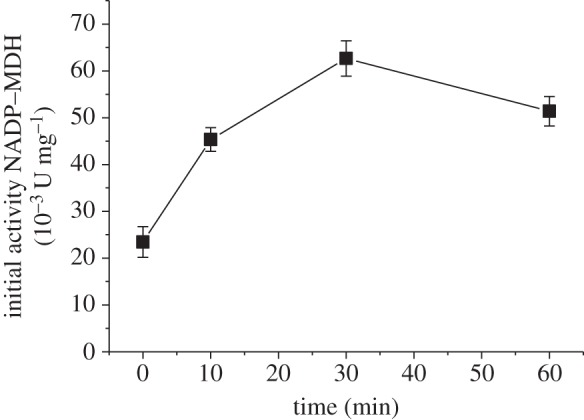
Leaf-extractable NADP–MDH activity. Activities correspond to the initial NADP–MDH activity measured from wild-type (S44) leaves exposed to 500 µmol quanta m−2 s−1 at 10°C for the indicated time. Data are mean ± s.e. of three measurements.
4. Discussion
It has been previously suggested that the malate valve is essential for counteracting the over-reduction of the photosynthetic electron transport chain [26]. According to our hypothesis (figure 1), the malate valve plays, in addition, an important role in H2O2 signalling by permitting the transport of reducing equivalents from the chloroplast to other cell compartments such as the peroxisomes, and could thereby regulate catalase activity. We investigated the effect of NADP–MDH deficiency on both the redox state of the chloroplast and H2O2 signalling by measuring the catalase activity and H2O2 accumulation. Here, we show that the chlorophyll fluorescence (F0′) after illumination increases to a much higher level in NADP–MDH mutants than in the wild-type S44 (figure 3). The rise in the F0′ level indicates that the NADP pool in the stroma is more reduced and that it is oxidized by the NDH complex leading to a higher reduction state of the PQ pool. A loss of the NADP–MDH activity not only leads to a higher reduction state of the chloroplast but also interrupts the transmission of the redox state of the chloroplast to the peroxisomes. Figures 4 and 5 show that the transient decrease in catalase activity is accompanied by a transient increase in the H2O2 level in S44. In these experiments, leaves were illuminated at 10°C to keep the photorespiratory activity low and to slow down biochemical reactions in general. Thereby, a high reduction state of the chloroplast was maintained. It is known that at low temperatures (10°C) Rubisco has a lower Km value for CO2 than at higher temperatures (room temperature), whereas the affinity for O2 remains unchanged [34]. At lower temperatures, a greater proportion of the ribulose-1,5-bisphosphate is carboxylated per absorbed quantum and the CO2 fixation is preferred to the oxygenation reaction compared with elevated temperatures [35]. At 20°C, the reversible inactivation of catalase activity after short-term exposure to high light was less pronounced than at 10°C (data not shown). In our experimental conditions (high light at 10°C), NADP–MDH showed a transient activation mirroring the transient inactivation of catalase and matching the transient H2O2 increase (figure 6). As knockout of NADP–MDH suppresses both the transient inactivation of catalase activity and the concomitant increase in H2O2 levels, it is indeed the malate valve that seems to play a crucial role for shuttling redox equivalents and for communicating the chloroplastic redox state to the cytosol and further on to other cell compartments.
Previous in vitro studies have shown that catalase from C. reinhardtii was inactivated by reduced TRX [19] and that both C. reinhardtii and Arabidopsis catalases were inactivated by DTT [18,20]. We have proposed that the catalase from C. reinhardtii was inactivated through the reduction of a cysteine residue [18]. In Arabidopsis, three isoforms of catalase have been found (for a recent review, see [36]). CAT2 is the most abundant catalase in mesophyll cells, it is involved in photorespiration and it exhibits a circadian rhythm in mRNA abundance with a peak during the early morning [37]. In this study, all measurements of catalase activity were performed in the morning, 1 h after the onset of light. The sequence comparison of CAT1 from C. reinhardtii with CAT1, CAT2 and CAT3 from Arabidopsis reveals that Cys230 is conserved among all isoforms and that Cys325 is conserved between CAT1 from C. reinhardtii and CAT1 and CAT2 from Arabidopsis. Thus, redox modification of at least one of these two conserved cysteine residues is very likely responsible for the redox-dependent inactivation of catalase activity in the peroxisomes of mesophyll cells. With the exception of GSH and glutathione reductase (GR1) [38], up to now no classical components of thiol redox control such as TRX, TRX reductases or GRXs have been identified in peroxisomes. Compared with other cell compartments, the knowledge on the peroxisomal proteome is lagging behind because of the difficulty of purifying peroxisomes free of contaminations from chloroplasts or mitochondria [39]. However, in proteome studies, TRX H3 and 5 have been found [40], but their presence in peroxisomes has yet not been independently verified. Proteins that lack the target sequence for peroxisomes may nevertheless be transported into peroxisomes. It has been shown recently that protein heteromers of plastid-destined glucose-6-phosphate dehydrogenases 1 and 4 can alternatively be targeted to peroxisomes after undergoing the cytosol oxidation of cysteine residues mediated by thioredoxin [41]. Further studies are needed to confirm the presence of the thiol-modulating compounds in peroxisomes. Furthermore, the glutathione level has been recently shown to increase in peroxisomes of mesophyll cells when Arabidopsis plants are exposed to high light [42]. Inactivation of catalase strongly impacts the leaf glutathione status, because photorespiratory H2O2 production increases the total glutathione content and leads to a dramatic oxidation of the glutathione pool in the Arabidopsis cat2 mutant [12]. Changes in the glutathione content and in the ratio of reduced to oxidized glutathione clearly modify redox- and H2O2-dependent gene regulation [43]. Further work is needed to decipher whether glutathione alone or in concert with TRX, and/or GRX, is directly or indirectly responsible for the reversible reductive inactivation of catalase in peroxisomes.
Acknowledgement
This work was supported by the Agence Nationale de Recherche, ANR-09-BLAN-0005-01.
References
- 1.Apel K, Hirt H. 2004. Reactive oxygen species: metabolism, oxidative stress, and signal transduction. Annu. Rev. Plant Biol. 55, 373–399. ( 10.1146/annurev.arplant.55.031903.141701) [DOI] [PubMed] [Google Scholar]
- 2.Müller P, Li XP, Niyogi KK. 2001. Non-photochemical quenching. A response to excess light energy. Plant Physiol. 125, 1558–1566. ( 10.1104/pp.125.4.1558) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Foyer CH, Neukermans J, Queval G, Noctor G, Harbinson J. 2012. Photosynthetic control of electron transport and the regulation of gene expression. J. Exp. Bot. 63, 1637–1661. ( 10.1093/jxb/ers013) [DOI] [PubMed] [Google Scholar]
- 4.Roach T, Krieger-Liszkay A. 2012. The role of the PsbS protein in the protection of photosystems I and II against high light in Arabidopsis thaliana. Biochim. Biophys. Acta 1817, 2158–2165. ( 10.1016/j.bbabio.2012.09.011) [DOI] [PubMed] [Google Scholar]
- 5.Bechtold U, Richard O, Zamboni A, Gapper C, Geisler M, Pogson B, Karpinski S, Mullineaux PM. 2008. Impact of chloroplastic- and extracellular-sourced ROS on high light-responsive gene expression in Arabidopsis. J. Exp. Bot. 59, 121–133. ( 10.1093/jxb/erm289) [DOI] [PubMed] [Google Scholar]
- 6.Foyer CH, Noctor G. 2011. Ascorbate and glutathione: the heart of the redox hub. Plant Physiol. 155, 2–18. ( 10.1104/pp.110.167569) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gadjev I, et al. 2006. Transcriptomic footprints disclose specificity of reactive oxygen species signaling in Arabidopsis. Plant Physiol. 141, 436–445. ( 10.1104/pp.106.078717) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gough DR, Cotter TG. 2011. Hydrogen peroxide: a Jekyll and Hyde signalling molecule. Cell Death Dis. 2, e213 ( 10.1038/cddis.2011.96) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Laloi C, Stachowiak M, Pers-Kamczyc E, Warzych E, Murgia I, Apel K. 2007. Cross-talk between singlet oxygen- and hydrogen peroxide-dependent signaling of stress responses in Arabidopsis thaliana. Proc. Natl Acad. Sci. USA 104, 672–677. ( 10.1073/pnas.0609063103) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mittler R, Vanderauwera S, Suzuki N, Miller G, Tognetti VB, Vandepoele K, Gollery M, Shulaev V, Van Breusegem F. 2011. ROS signaling: the new wave? Trends Plant Sci. 16, 300–309. ( 10.1016/j.tplants.2011.03.007) [DOI] [PubMed] [Google Scholar]
- 11.op den Camp RG, et al. 2003. Rapid induction of distinct stress responses after the release of singlet oxygen in Arabidopsis. Plant Cell 15, 2320–2332. ( 10.1105/tpc.014662) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Queval G, et al. 2007. Conditional oxidative stress responses in the Arabidopsis photorespiratory mutant cat2 demonstrate that redox state is a key modulator of daylength-dependent gene expression, and define photoperiod as a crucial factor in the regulation of H2O2-induced cell death. Plant J. 52, 640–657. ( 10.1111/j.1365-313X.2007.03263.x) [DOI] [PubMed] [Google Scholar]
- 13.Scheibe R, Dietz KJ. 2012. Reduction-oxidation network for flexible adjustment of cellular metabolism in photoautotrophic cells. Plant Cell Environ. 35, 202–216. ( 10.1111/j.1365-3040.2011.02319.x) [DOI] [PubMed] [Google Scholar]
- 14.Galvez-Valdivieso G, Mullineaux PM. 2010. The role of reactive oxygen species in signalling from chloroplasts to the nucleus. Physiol. Plant. 138, 430–439. ( 10.1111/j.1399-3054.2009.01331.x) [DOI] [PubMed] [Google Scholar]
- 15.Fischer BB, Hideg É, Krieger-Liszkay A. 2013. Production, detection, and signaling of singlet oxygen in photosynthetic organisms. Antioxid. Redox Signal. 18, 2145–2162. ( 10.1089/ars.2012.5124) [DOI] [PubMed] [Google Scholar]
- 16.Mubarakshina MM, Ivanov BN, Naydov IA, Hillier W, Badger MR, Krieger-Liszkay A. 2010. Production and diffusion of chloroplastic H2O2 and its implication to signalling. J. Exp. Bot. 61, 3577–3587. ( 10.1093/jxb/erq171) [DOI] [PubMed] [Google Scholar]
- 17.Stone JR, Yang S. 2006. Hydrogen peroxide: a signaling messenger. Antioxid. Redox Signal. 8, 243–270. ( 10.1089/ars.2006.8.243) [DOI] [PubMed] [Google Scholar]
- 18.Michelet L, Roach T, Fischer BB, Bedhomme M, Lemaire SD, Krieger-Liszkay A. 2013. Down-regulation of catalase activity allows transient accumulation of a hydrogen peroxide signal in Chlamydomonas reinhardtii. Plant Cell Environ. 36, 1204–1213. ( 10.1111/pce.12053) [DOI] [PubMed] [Google Scholar]
- 19.Lemaire SD, Guillon B, Le Maréchal P, Keryer E, Miginiac-Maslow M, Decottignies P. 2004. New thioredoxin targets in the unicellular photosynthetic eukaryote Chlamydomonas reinhardtii. Proc. Natl Acad. Sci. USA 101, 7475–7480. ( 10.1073/pnas.0402221101) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shao N, Krieger-Liszkay A, Schroda M, Beck CF. 2007. A reporter system for the individual detection of hydrogen peroxide and singlet oxygen: its use for the assay of reactive oxygen species produced in vivo. Plant J. 50, 475–487. ( 10.1111/j.1365-313X.2007.03065.x) [DOI] [PubMed] [Google Scholar]
- 21.Hayashi Y, Shinozaki A. 2011. Visualization of microbodies in Chlamydomonas reinhardtii. J. Plant Res. 125, 579–586. ( 10.1007/s10265-011-0469-z) [DOI] [PubMed] [Google Scholar]
- 22.Wood ZA, Poole LB, Karplus PA. 2003. Peroxiredoxin evolution and the regulation of hydrogen peroxide signaling. Science 300, 650–653. ( 10.1126/science.1080405) [DOI] [PubMed] [Google Scholar]
- 23.Biteau B, Labarre J, Toledano MB. 2003. ATP-dependent reduction of cysteine-sulphinic acid by S. cerevisiae sulphiredoxin. Nature 425, 980–984. ( 10.1038/nature02075) [DOI] [PubMed] [Google Scholar]
- 24.Woo HA, Yim SH, Shin DH, Kang D, Yu DY, Rhee SG. 2010. Inactivation of peroxiredoxin I by phosphorylation allows localized H2O2 accumulation for cell signaling. Cell 140, 517–528. ( 10.1016/j.cell.2010.01.009) [DOI] [PubMed] [Google Scholar]
- 25.Day AM, Brown JD, Taylor SR, Rand JD, Morgan BA, Veal EA. 2012. Inactivation of a peroxiredoxin by hydrogen peroxide is critical for thioredoxin-mediated repair of oxidized proteins and cell survival. Mol. Cell 45, 1–11. ( 10.1016/j.molcel.2011.11.027) [DOI] [PubMed] [Google Scholar]
- 26.Scheibe R. 2004. Malate valves to balance cellular energy supply. Physiol. Plant. 120, 21–26. ( 10.1111/j.0031-9317.2004.0222.x) [DOI] [PubMed] [Google Scholar]
- 27.Alonso JM, et al. 2003. Genome-wide insertional mutagenesis of Arabidopsis thaliana. Science 301, 653–657. ( 10.1126/science.1086391) [DOI] [PubMed] [Google Scholar]
- 28.Keryer E, Collin V, Lavergne D, Lemaire SD, Issakidis-Bourguet E. 2004. Characterization of Arabidopsis mutants for the variable subunit of ferredoxin:thioredoxin reductase. Photosynth. Res. 79, 265–274. ( 10.1023/B:PRES.0000017173.46185.3e) [DOI] [PubMed] [Google Scholar]
- 29.Jacquot JP, Keryer E, Issakidis E, Decottignies P, Miginiac-Maslow M, Schmitter JM, Crétin C. 1991. Properties of recombinant NADP-malate dehydrogenase from Sorghum vulgare leaves expressed in Escherichia coli. Eur. J. Biochem. 199, 47–51. ( 10.1111/j.1432-1033.1991.tb16090.x) [DOI] [PubMed] [Google Scholar]
- 30.Collin V, Issakidis-Bourguet E, Marchand C, Hirasawa M, Lancelin JM, Knaff DB, Miginiac-Maslow M. 2003. The Arabidopsis plastidial thioredoxins: new functions and new insights into specificity. J. Biol. Chem. 278, 23 747–23 752. ( 10.1074/jbc.M302077200) [DOI] [PubMed] [Google Scholar]
- 31.Berkemeyer M, Scheibe R, Ocheretina O. 1998. A novel non-redox-regulated NAD-dependent malate dehydrogenase from chloroplasts of Arabidopsis thaliana. J. Biol. Chem. 273, 27 927–27 933. ( 10.1074/jbc.273.43.27927) [DOI] [PubMed] [Google Scholar]
- 32.Hebbelmann I, et al. 2012. Multiple strategies to prevent oxidative stress in Arabidopsis plants lacking the malate valve enzyme NADP-malate dehydrogenase. J. Exp. Bot. 63, 1445–1459. ( 10.1093/jxb/err386) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rumeau D, Bécuwe-Linka N, Beyly A, Louwagie M, Garin J, Peltier G. 2005. New subunits NDH-M, -N, and -O, encoded by nuclear genes, are essential for plastid Ndh complex functioning in higher plants. Plant Cell 17, 219–232. ( 10.1105/tpc.104.028282) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Servaites JC, Ogren WL. 1978. Oxygen inhibition of photosynthesis and stimulation of photorespiration in leaf cells. Plant Physiol. 61, 62–67. ( 10.1104/pp.61.1.62) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ehleringer J, Björkman O. 1977. Quantum yields for CO2 uptake in C3 and C4 plants. Plant Physiol. 59, 86–90. ( 10.1104/pp.59.1.86) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mhamdi A, Noctor G, Baker A. 2012. Plant catalases: peroxisomal redox guardians. Arch. Biochem. Biophys. 525, 181–194. ( 10.1016/j.abb.2012.04.015) [DOI] [PubMed] [Google Scholar]
- 37.Zhong HH, Young JC, Pease EA, Hangarter RP, McClung CR. 1994. Interactions between light and the circadian clock in the regulation of C A R expression in Arabidopsis . Plant Physiol. 104, 889–898. ( 10.1104/pp104.3.889) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kataya AR, Reumann S. 2010. Arabidopsis glutathione reductase 1 is dually targeted to peroxisomes and the cytosol. Plant Signal. Behav. 5, 171–175. ( 10.4161/psb.5.2.10527) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bussel JD, Behrens C, Ecke W, Eubel H. 2013. Arabidopsis peroxisome proteomics. Front. Plant Sci. 4, 101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Reumann S, et al. 2009. In-depth proteome analysis of Arabidopsis leaf peroxisomes combined with in vivo subcellular targeting verification indicates novel metabolic and regulatory functions of peroxisomes. Plant Physiol. 150, 125–143. ( 10.1104/pp.109.137703) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Meyer T, Hölscher C, Schwöppe C, von Schaewen A. 2011. Alternative targeting of Arabidopsis plastidic glucose-6-phosphate dehydrogenase G6PD1 involves cysteine-dependent interaction with G6PD4 in the cytosol. Plant J. 66, 745–758. ( 10.1111/j.1365-313X.2011.04535.x) [DOI] [PubMed] [Google Scholar]
- 42.Heyneke E, Luschin-Ebengreuth N, Krajcer I, Wolkinger V, Müller M, Zechmann B. 2013. Dynamic compartment specific changes in glutathione and ascorbate levels in Arabidopsis plants exposed to different light intensities. BMC Plant Biol. 13, 104 ( 10.1186/1471-2229-13-104) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Han Y, Chaouch S, Mhamdi A, Queval G, Zechmann B, Noctor G. 2013. Functional analysis of Arabidopsis mutants points to novel roles for glutathione in coupling H2O2 to activation of salicylic acid accumulation and signaling. Antioxid. Redox Signal. 18, 2106–2121. ( 10.1089/ars.2012.5052) [DOI] [PMC free article] [PubMed] [Google Scholar]