

Abstract
AIM
To investigate the effect of 5-Aza-2′-deoxycytidine (5-Aza-CdR), a DNA methyltransferase (DNMT) inhibitor, on the growth and survival of the Chinese retinoblastoma (RB) cell line HXO-RB44.
METHODS
The DNA methylation status of the Ras association domain family (RASSF1A) promoter in the presence of 5-Aza-CdR at different concentrations was analyzed by methylation-specific polymerase chain reaction (MSP). RASSF1A mRNA and protein levels were measured by semiquantitative RT-PCR and immunohistochemistry staining, respectively, when cells were treated with 5.0µmol/L of 5-Aza-CdR. The effect of 5.0µmol/L 5-Aza-CdR on the proliferation and viability of HXO-RB44 cells was examined using 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay and flow cytometry.
RESULTS
5-Aza-CdR efficiently induced cell cycle arrest at G0/G1 and apoptotic death in HXO-RB44 cells. MSP analysis showed that unmethylated RASSF1A DNA increased and methylated RASSF1A decreased in a dose-dependent manner in a range of 0.5-5.0µmol/L 5-Aza-CdR. Accordingly, RASSF1A expression was reactivated at both mRNA and protein levels. Incubation time of 5-Aza-CdR treatment also functioned as a factor for the demethylation status of RASSF1A promoter DNA, with a plateau on day four. 5-Aza-CdR at 5.0µmol/L completely demethylated the RASSF1A promoter in HXO-RB44 cells on day four, and as a result, RASSF1A expression increased significantly from day 4 to day 7.
CONCLUSION
5-Aza-CdR inhibits the growth of the HXO-RB44 RB cell line and induces apoptosis by demethylating the RASSF1A gene.
Keywords: 5-Aza-2′-deoxycytidine, retinoblastoma, methylation, apoptosis, Ras association domain family
INTRODUCTION
Long-term survival rates of retinoblastoma (RB) patients are low in some developing and undeveloped countries, and diagnostic delay or lack of access to multidisciplinary coordinated therapies account for the low rates[1],[2]. Intracranial invasion, metastasis or second nonocular tumors lead to death even after enucleation or orbital exenteration[3].And aggressive systemic approaches might improve the survival rate of high-risk cases.However, antineoplastic drugs can adversely affect the physiological function and development of normal cells, especially in children. Moreover, some RB cases are resistant to these drugs. Thus, there is a need for a new drug with lower toxicity or synergistic effects.
Recent studies have focused on the epigenetic changes of-tumorigenesis, and the silencing of multiple tumor suppressor genes (TSGs) by aberrant methylation of CpG islands in promoter regions, is now widely recognized as an important contributing factor[4]-[8]. In contrast to genetic mutation, epigenetic silencing of gene expression can be effectively reversed by DNA methylation inhibitors in many tumor types, rendering the inhibitors as attractive tools for therapeutic interventions. 5-Aza-2′-deoxycytidine (5-Aza-CdR) is a cytotoxic drug with severe side effects when used at maximum tolerance doses and it can trap and rapidly deplete cellular DNA methyltransferase (DNMT) through a stable covalent DNA-5-Aza-CdR-DNMT reaction[9],[10]. Therefore, selective degradation of DNMT1 through a proteasomal pathway may be a new alternative mechanism for 5-Aza-CdR activity[11]-[13]. Hypermethylation occurs frequently in a variety of human cancers, but rarely in normal tissues, suggesting that methylation inhibitors could be more specific to tumors and less toxic to normal cells[14]. Based on the results of previous studies, 5.0µmol/L 5-Aza-CdR chemical intervention were the median lethal dose concentration[15].
Moreover, the promoter hypermethylation of RASSF1A gene is one of the most frequent epigenetic inactivation events, observed in at least 37 human tumor types[16]. Meanwhile, some studies report that RASSF1A also has a high methylation rate in RB patients and it can be reversed by 5-Aza-CdR in RB WERI-RB1 and Y79 cell lines[17],[18]. Futhernore, a recent study show that 5-Aza-CdR has a new function in activating the expression of silenced RASSF1A at a much lower concentration[19].
However, few data are available regarding RASSF1A gene inactivation and reactivation in Chinese, especially in the HXO-RB44 cell line To find the optimal concentration of 5-Aza-CdR with the least toxicity and the greatest effect on RB cells, we investigated the effect of different concentrations or different treatment times of 5-Aza-CdR on the demethylation of the RASSF1A promoter and reestablishment of RASSF1A expression that consequently inhibits the growth of HXO-RB44 cells. Our data might provide a new strategy for the treatment of advanced RB.
MATERIALS AND METHODS
Cell Culture
The HXO-RB44 human RB cell line was obtained from the Center of Cell Biology, Xiangya Medical College, Central South University, China. HXO-RB44 cells were cultured in RPMI 1640 with 10% fetal bovine serum (FBS ) in a humidified incubator at 37°C in 5% CO2.
5-Aza-CdR Treatment
HXO-RB44 cells (1×104) in six-well plates were treated with various concentrations of 5-Aza-CdR (0.1, 0.5, 1.0, 5.0, and 10.0µmol/L) in DMSO (Sigma Chemical Co., St. Louis, MO, USA). Concentrations used in this study were based on previous findings[20]. The medium containing 5-Aza-CdR was replaced every 24h for the indicated period.
Methylation-Specific PCR Assay
Genomic DNA was extracted from HXO-RB44 cells treated with different doses of 5-Aza-CdR for four days by the proteinase K digestion procedure, according to the manufacturer's instructions (Sigma-Aldrich, USA). To evaluate demethylation levels of the RASSF1A promoter, DNA extracted from HXO-RB44 cells was analyzed by methylation-specific PCR (MSP), based on the sequence differences between methylated and unmethylated DNA after sodium bisulfite modification (EZ DNA Methylation-Gold Kit™, Zymo Research, USA)[21]. Unmethylated cytosine was converted to uracil, which is recognized as thymine by Taq polymerase. The primer pairs specific for methylated (M) and unmethylated (U) RASSF1A DNA were as follows: U-sense primer, 5′-GGTTTTGTGAGAGTGTGTTTAG-3′; U-antisense, 3′-CACTAACAAACACAAACCAAAC-5′; M-sense primer, 5′-GGGTTTTGCGAGAGCGCG-3′, and M-antisense, 3′-GCTAACAAACGCGAACCG-5′. DNA from normal lymphocytes with or without SssI methyltransferase treatment (New England Biolabs, Boston, MA, USA) was used as the positive control for methylated and unmethylated alleles, respectively. PCR products were separated on a 3% agarose gel containing ethidium bromide, and visualized under ultraviolet illumination. Photodensity (OD) of the methylated and unmethylated products of RASSF1A was measured by quantitation software (Quantity One; Bio-Rad, Hercules, CA, USA). The methylation status of the RASSF1A promoter represented the average OD value of methylated or unmethylated products from triplicate samples. The experiment was repeated once.
Semi-Quantitative RT-PCR
HXO-RB44 cells (1×107) were harvested at different time points before (D0) or after (days 1-7) treatment with 5.0µmol/L 5-Aza-CdR. Total RNA was extracted using Trizol reagent (Sigma-Aldrich), dissolved in RNase-free water and quantified with a UV spectrometer. cDNA was synthesized from 2µg total RNA as described previously[22]. ß-actin mRNA was used as an internal control. Primers for RT-PCR were as follows: RASSF1A sense, 5′-GGCGTCGTGCGCAAAGGCC-3′, antisense, 3′-GGGTGGCTTCTTGCTGGAGGG-5′, Tm 56°C, 35 cycles for a 329 bp PCR product; ß-actin sense 5′-CGCACCACTGGCATTGTCAT-3′, and antisense 3′-TTCTCCTTGATGTCACGCAC-5′. Blank controls containing all PCR components except for sample cDNA were included in all PCRs. RASSF1A and ß-actin band density was measured from three independent experiments with Quantity One software.
Immunohistochemistry
Expression of RASSF1A in HXO-RB44 cells with or without 5.0µmol/L 5-Aza-CdR treatment for five days was analyzed by immunohistochemical staining using purified anti-human RASSF1A antibody (14-6888-80, Clone eB114-10H1, dilution 1:200; eBioscience, USA) according to the manufacturer's protocol.
MTT Assay MTT
assay was performed according to the method of Carmichael using 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT). HXO-RB44 cells were seeded in 96-well plates, and treated the next day with 5-Aza-CdR at the indicated concentrations. Medium containing 5-Aza-CdR was replaced every 24h. Untreated tumor cells (no 5-Aza-CdR and no DMSO) were used for a control (untreated control), and cells with only complete medium were used as a blank control. Cells incubated only with 0.0125% DMSO, the solvent concentration in the highest concentration of 5-Aza-CdR (10.0µmol/L), were used as a vehicle control to exclude the cytotoxic effect of DMSO on cells. At the indicated time points (24, 48, 72, 96, 120, and 144h) after incubation, 20µL MTT solution was added to each well and cells were incubated at 37°C for 4h. The supernatant medium containing drugs and unconverted MTT was removed, followed by the addition of 150µL DMSO per well to dissolve the produced formazan. Absorbance at 492nm was read using a spectrophotometric microplate reader (Bio-Rad). Triplicate samples from four separate experiments were performed for each concentration/exposure time combination.
Cell Cycle Analysis
HXO-RB44 cells were plated and treated with different dosages of 5-Aza-CdR or 0.0125% DMSO. Cells (5×106) were harvested and washed with Hank's balanced salt solution (HBSS; Sigma-Aldrich), fixed in 70% ethanol at 4°C for 2h, and then treated with RNasin (20µg/mL) and stained with propidium iodide (PI, 50µg/mL). DNA contents were determined by flow cytometry (BD Company, USA).
Apoptotic Assay
The Annexin V staining kit (Roche, USA) and flow cytometry analysis were used for apoptotic assays, according to the manufacturer's instructions.
Statistical Analysis
All data were tested for non-normal distribution and heterogeneity of variance. Overall, Kruskal-Wallis H analysis was performed to test the differences among the treatment groups. The Wilcoxon method was used for pair-wise comparison in contexts. Results were considered statistically significant when P<0.05. The SPS software package (Brothersoft) was used for all statistical analyses.
RESULTS
5-Aza-CdR Induces HXO-RB44 Cell Cycle Arrest and Apoptosis
We tested the effect of 5-Aza-CdR on the viability of HXO-RB44 cells using MTT assays. As shown in Figure 1, 5-Aza-CdR effectively suppressed HXO-RB44 cell growth and proliferation in a dose-dependent manner. Just like conventional cytostatic agents, prolonged incubation further enhanced the cytotoxicity of 5-Aza-CdR in HXO-RB44 cells (Figure 1).
Figure 1. Inhibition of HXO-RB44 cell growth by 5-Aza-CdR.
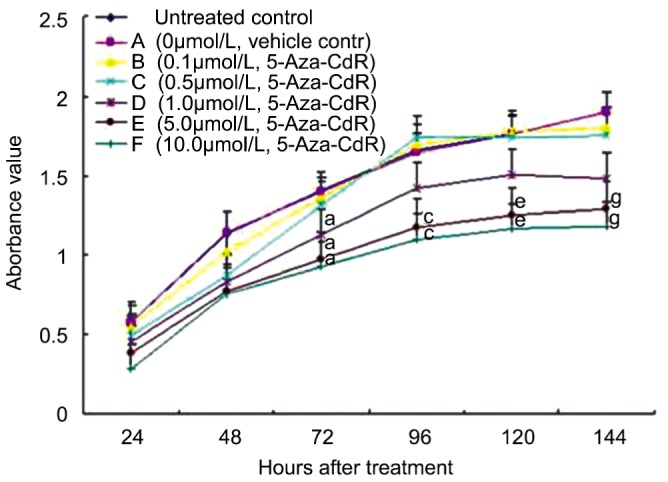
HXO-RB44 cells were treated with 0.0125% DMSO or 5-Aza-CdR at indicated concentrations. Cell viability was measured using MTT assay at indicated time points. Values represent the mean±S.D. from four independent experiments. P<0.05 was compared with the control (a, c, e, g).
To more closely examine the cellular events induced by 5-Aza-CdR, we evaluated the cell cycle distribution of HXO-RB44 cells treated with 5-Aza-CdR at different concentrations. In comparison with the DMSO control cells, 5-Aza-CdR induced a significant cell cycle arrest in S phase (G2/M phase blockage) in a dose-dependent manner (Table 1). Interestingly, the cell cycle arrest induced by 5-Aza-CdR was also time dependent, indicating that the cell cycle arrest induced by 5-Aza-CdR may not be ascribed to its direct cytotoxicity (Table 1). At the end point of 144h, the percentages of HXO-RB44 cells blocked in G0/G1 phase in response to 5.0 and 10.0µmoL 5-Aza-CdR treatment were 45.1% and 43.3%, respectively.
Table 1. Effect of 5-Aza-CdR on cell cycle distribution (%).
| 48h | 96h | 144h | ||||||||||||||||
| A | 0.1 | 0.5 | 1.0 | 5.0 | 10 | A | 0.1 | 0.5 | 1.0 | 5.0 | 10 | A | 0.1 | 0.5 | 1.0 | 5.0 | 10 | |
| PI | 1.04 | 1.05 | 1.23 | 1.29 | 1.79 | 1.34 | 1.72 | 2.42 | 2.33 | 2.54 | 6.62 | 6.88 | 2.00 | 2.60 | 3.31 | 6.71 | 18.5 | 17.6 |
| G0/G1 | 18.1 | 23.1 | 25.6 | 29.9 | 32.5 | 33.3 | 19.6 | 23.7 | 29.2 | 30.5 | 33.5 | 34.0 | 20.4 | 27.0 | 31.8 | 32.1 | 45.1 | 43.3 |
| S | 57.3 | 52.0 | 43.4 | 50.1 | 52.0 | 49.3 | 59.5 | 63.5 | 54.3 | 53.0 | 47.3 | 51.5 | 64.0 | 50.5 | 56.0 | 50.4 | 37.5 | 42.6 |
| G2 | 24.6 | 24.9 | 31.0 | 20.0 | 15.5 | 17.4 | 21.9 | 10.8 | 16.5 | 16.5 | 19.2 | 14.5 | 11.6 | 22.5 | 12.2 | 17.5 | 17.4 | 18.1 |
A indicates DMSO (0.0125%) controls. 5-Aza-CdR was used at indicated concentrations (0-10.0µmol/L).
5-Aza-CdR also induced apoptosis of HXO-RB44 cells, and this activity was also dose and time dependent. After 48h treatment with 5.0 or 10.0µmol/L of 5-Aza-CdR, apoptosis of HXO-RB44 cells increased slightly compared with the DMSO control, but the apoptotic cell death was enhanced to 6% at 96h and 18% at 144h, levels that were significantly higher than that of the vehicle control group.
5-Aza-CdR Inhibits RASSF1A Methylation
To elucidate the mechanism of cell cycle arrest and apoptosis induced by 5-Aza-CdR, we examined the demethylation of the RASSF1A promoter using MSP in HXO-RB44 cells after a four-day exposure. Our results showed that 5-Aza-CdR at 0.1µmol/L did not affect the RASSF1A promoter methylation status compared with the control, but partial methylation and demethylation was observed at 0.5-5.0µmol/L, demonstrating a concentration-dependent effect (Figure 2). At 5.0µmol/L of 5-Aza-CdR, the RASSF1A promoter was completely demethylated. This concentration was used for the studies below.
Figure 2. Inhibition of RASSF1A promoter methylation by 5-Aza-CdR.

Methylation status of RASSF1Apromoter in HXO-RB44 retinoblastoma cells was analyzed by MSP. A: MSP products. The PCR products showed the presence of unmethylated templates (U) or methylated templates (M). Controls were DNA from normal lymphocytes treated or untreated with SssI methyltransferase; B: Photodensity of PCR products. Values denote mean MSP products measured from triplicate samples of two independent experiments. All P<0.05 was compared with the control.
5-Aza-CdR Reactivates RASSF1A Expression
We further determined whether 5-Aza-CdR could re-establish RASSF1A expression in HXO-RB44 cells. Using semi-quantitative reverse-transcription polymerase chain reaction (RT-PCR), we measured RASSF1A mRNA levels in HXO-RB44 cells. We found that 5-Aza-CdR at 5.0µmol/L significantly reactivated RASSF1A mRNA expression in a time-dependent manner, peaking on the fifth day and lasting for at least three days (Figure 3).
Figure 3. Reactivation of silenced RASSF1A mRNA expression by 5.0µmol/L 5-Aza-CdR.
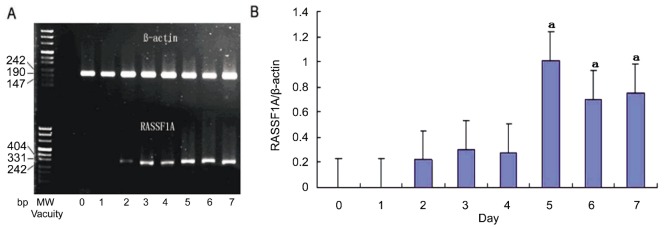
HXO-RB44 cells were treated with 5.0µmol/L 5-Aza-CdR for the indicated days. Prominent RASSF1A mRNA was detected by semi-quantitative RT-PCR. A: RT-PCR products, RASSF1A (bottom) and ß-actin (top). MW: molecular weight marker; B: Ratios of RASSF1A/ß-actin mRNA levels. Photodensity values of RASSF1A mRNA were normalized to ß-actin for each sample. Values represent the ratios of RASSF1A/ß-actin from three independent experiments. aP <0.05 was compared to the control.
RASSF1A gene reactivation also resulted in enhanced protein levels, as demonstrated by immunocytochemistry staining. Control HXO-RB44 cells showed undetectable RASSF1A protein levels, but cells treated with 5.0µmol/L 5-Aza-CdR for five days exhibited strong RASSF1A protein staining (Figure 4). Taken together, our data showed that silenced RASSF1A gene expression was reactivated by 5-Aza-CdR at 5.0µmol/L both at the mRNA and protein level, which may trigger cell cycle arrest and apoptosis.
Figure 4. Immunohistochemistry of RASSF1A protein.

A: DMSO (0.0125%) vehicle treatment of control HXO-RB44 cells. RASSF1A protein was undetectable; B: 5-Aza-CdR at 5.0µmol/L for five days. RASSF1A protein was re-expressed in HXO-RB44 cells. Magnification, ×200.
DISCUSSION
In this study, we investigated the activity of 5-Aza-CdR in inhibiting the growth and proliferation of HXO-RB44 RB cells. Our data revealed that 5-Aza-CdR significantly inhibited HXO-RB44 cell growth at a low concentration of 1.0µmol/L and led to a dramatic growth suppression and apoptosis at 5.0µmol/L on the 4th day. This study also evaluated the effects of 5-Aza-CdR on the demethylation and reactivation of expression of the tumor suppressor gene RASSF1A. 5-Aza-CdR at a low concentration of 0.5µmol/L was sufficient to inhibit the methylation of the RASSF1A promoter. With increasing concentrations from 0.5µmol/L to 5.0µmol/L, demethylation of RASSF1A promoter DNA significantly increased and peaked at 5.0µmol/L treatment, while methylated DNA decreased down to zero at 5.0µmol/L. Length of 5-Aza-CdR treatment was also a factor that influenced demethylation of the RASSF1A promoter. At 5.0µmol/L 5-Aza-CdR for four days, the RASSF1A promoter was completely demethylated, and silenced RASSF1A was strongly reexpressed at the mRNA level on the 4th day and at the protein level on the 5th day, indicating a time dependency in the demethylation of RASSF1A promoter by 5-Aza-CdR. These data suggested that RASSF1A demethylation and reactivation may be responsible for the cell growth inhibition and apoptosis induced by 5-Aza-CdR. These results also supported the observation that RASSF1A is epigenetically silenced via promoter hypermethylation in the HXO-RB44 RB cell line. 5-Aza-CdR induced cell cycle arrest and apoptosis of HXO-RB44 cells. With increasing concentration or incubation time of 5-Aza-CdR, the percentage of cells in G0/G1 gradually increased to approximately 45% at 5µmol/L after exposure for 144h. Meanwhile, the percentage of apoptotic cells increased markedly from 1% at 48h to approximately 18% at 144h. In parallel, RASSF1A mRNA and protein levels significantly increased. These findings displayed that RASSF1A reexpression could arrest the cell cycle at G1/G0 and promote cell apoptosis in vitro. These biological consequences may be ascribed to the function of RASSF1A in repressing cyclin A and D1, stabilizing microtubules, activating other tumor suppressor genes (including p14ARF, RB, P53) through p120E4F, revitalizing the RAS-regulated pro-apoptotic pathway, or interacting with CNK1 or other factors[23],[24]. Our study further supported the existing report that RASSF1A induces G0/G1 cell cycle arrest in some human solid tumor cell lines[25]. However, Choy describe negligible alterations of cell cycle or apoptosis in two other RB cell lines, WER1-Rb1 and Y79, after exposure to 5.0µmol/L 5-Aza-CdR for four days[17]. The most likely explanation for the different effects is the disparity in RASSF1A transcription level. Even at the same concentration of 5.0µmol/L, 5-Aza-CdR triggered only partial demethylation and reactivation of RASSF1A transcription in WER1-Rb1 and Y79 cells, and thus had limited effects on cell cycle profile or apoptosis. However, complete demethylation of the RASSF1A promoter may completely restore RASSF1A expression in HXO-RB44 cells and induce cell cycle arrest and apoptosis.
Two major obstacles limiting clinical implementation of 5-Aza-CdR in solid tumor therapies are toxicity and remethylation. Clinically, 5-Aza-CdR at the cytotoxic dose or maximum tolerated dose is active against leukemia and myelodysplastic syndrome, but leads to disappointing therapeutic response and severe side effects in the initial phase I/II trials in solid tumors[26],[27]. Researchers have refocused on lower concentrations to explore the best balance between the highest demethylation activity and minimal cytotoxicity. Bender[28] showed that 0.1µmol/L or 0.5µmol/L 5-Aza-CdR demethylates T24 bladder carcinoma-derived cells with very low cytotoxicity. Demethylation activity, such as re-expression of TSGs, cellular growth suppression and G1/G0 arrest, could be transmitted to cells for at least the ninth passage, unlike cytotoxicity of Ara-C, which incorporates into replicating DNA but does not inhibit DNA methylation. Although remethylation may occur in several days, scientists believe that 5-Aza-CdR may potentiate the anti-tumor effects of other drugs by epigenetic reactivation of pro-apoptotic genes during the window of demethylation. The secondary agents would target the reexpressed gene and/or reconstituted signal transduction pathways[29]. Zebularine, a stable DNMT inhibitor with lower cytotoxicity, is an alternative approach to solve the remethylation problems[30]. In our study, 5-Aza-CdR at 0.5µmol/L led to demethylation of the RASSF1A gene promoter and completely reversed the hypermethylation at 5.0µmol/L. The reactivation of RASSF1A appeared to last for more than five days, indicating the therapeutic potential of this agent in RB.
To date, an increasing number of RBs exhibit RASSF1A promoter hypermethylation, especially in unilateral RB, indicating the role of epigenetic alterations in this disease. Our study showed the capability of 5-Aza-CdR to effectively reverse the hypermethylation of RASSF1A, reestablish RASSF1A expression, and suppress the growth and proliferation of HXO-RB44 RB cells by restraining cells at G0/G1 and triggering apoptosis. These results suggested that methylation inhibition may be a potential approach in the prevention and treatment of RB in vitro. Further studies are merited to explore the possible application of demethylating agents, such as 5-Aza-CdR, in the treatment of RB.
Acknowledgments
The authors thank Dr. De-Liang Cao in Department of Microbiology, Immunology and Cell Biology, Simmons Cancer Institute, Southern Illinois University School of Medicine for proofreading this article. The authors thank Guang-Xiu Lu for approval of the project, which was undertaken in the Institute of Reproduction and Stem Cell Engineering, and all the researchers in the Institute for technical support.
Foundation: Supported by the National Natural Science Foundation of China (No.3087282; No.81072221).
Conflicts of Interest: Liu R, None; Zhang XH, None; Zhang K, None; Li W, None; Wang WJ, None; Luo DX, None; Gao L, None.
REFERENCES
- 1.Swaminathan R, Rama R, Shanta V. Childhood cancers in Chennai, India, 1990-2001: incidence and survival. Int J Cancer. 2008;122(11):2607–2611. doi: 10.1002/ijc.23428. [DOI] [PubMed] [Google Scholar]
- 2.Bowman RJ, Mafwiri M, Luthert P, Luande J, Wood M. Outcome of retinoblastoma in east Africa. Pediatr Blood Cancer. 2008;50(1):160–162. doi: 10.1002/pbc.21080. [DOI] [PubMed] [Google Scholar]
- 3.Kim JW, Kathpalia V, Dunkel IJ, Wong RK, Riedel E, Abramson DH. Orbital recurrence of retinoblastoma following enucleation. Br J Ophthalmol. 2009;93(4):463–467. doi: 10.1136/bjo.2008.138453. [DOI] [PubMed] [Google Scholar]
- 4.Brown R, Strathdee G. Epigenomics and epigenetic therapy of cancer. Trends Mol Med. 2002;8(4 Suppl):S43–48. doi: 10.1016/s1471-4914(02)02314-6. [DOI] [PubMed] [Google Scholar]
- 5.Bjornsson HT, Cui H, Gius D, Fallin MD, Feinberg AP. The new field of epigenomics: implications for cancer and other common disease research. Cold Spring Harb Symp Quant Biol. 2004;69:447–456. doi: 10.1101/sqb.2004.69.447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jones PA, Baylin SB. The Epigenomics of Cancer. Cell. 2007;128(4):683–692. doi: 10.1016/j.cell.2007.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Toyota M, Suzuki H, Yamashita T, Hirata K, Imai K, Tokino T, Shinomura Y. Cancer epigenomics: implications of DNA methylation in personalized cancer therapy. Cancer Sci. 2009;100(5):787–791. doi: 10.1111/j.1349-7006.2009.01095.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shenker N, Flanagan JM. Intragenic DNA methylation: implications of this epigenetic mechanism for cancer research. Br J Cancer. 2012;106(2):248–253. doi: 10.1038/bjc.2011.550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ding YB, Long CL, Liu XQ, Chen XM, Guo LR, Xia YY, He JL, Wang YX. 5-aza-2′-deoxycytidine leads to reduced embryo implantation and reduced expression of DNA methyltransferases and essential endometrial genes. PLoS One. 2012;7(9):e45364. doi: 10.1371/journal.pone.0045364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lyko F, Brown R. DNA methyltransferase inhibitors and the development of epigenetic cancer therapies. J Natl Cancer Inst. 2005;97(20):1498–1506. doi: 10.1093/jnci/dji311. [DOI] [PubMed] [Google Scholar]
- 11.Ghoshal K, Datta J, Majumder S, Bai S, Kutay H, Motiwala T, Jacob ST. 5-Aza-deoxycytidine induces selective degradation of DNA methyltransferase 1 by a proteasomal pathway that requires the KEN box, bromo-adjacent homology domain, and nuclear localization signal. Mol Cell Biol. 2005;25(11):4727–4741. doi: 10.1128/MCB.25.11.4727-4741.2005. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 12.Karahoca M, Momparler RL. Pharmacokinetic and pharmacodynamic analysis of 5-aza-2′-deoxycytidine (decitabine) in the design of its dose-schedule for cancer therapy. Clin Epigenetics. 2013;5(1):3. doi: 10.1186/1868-7083-5-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Müller-Thomas C, Schuster T, Peschel C, Götze KS. A limited number of 5-azacitidine cycles can be effective treatment in MDS. Ann Hematol. 2009;88(3):213–219. doi: 10.1007/s00277-008-0583-8. [DOI] [PubMed] [Google Scholar]
- 14.Kawaguchi K, Oda Y, Saito T, Yamamoto H, Takahira T, Kobayashi C, Tamiya S, Tateishi N, Iwamoto Y, Tsuneyoshi M. DNA hypermethylation status of multiple genes in soft tissue sarcomas. Mod Pathol. 2006;19(1):106–114. doi: 10.1038/modpathol.3800502. [DOI] [PubMed] [Google Scholar]
- 15.Tang QB, Sun HW, Zou SQ. Effects of 5-aza-2-deoxycytidine on the growth and apoptosis of bile duct cancer cell line. J Huazhong Univ Sci Tech (Health Sci) 2004;33(1):34–36. [Google Scholar]
- 16.Agathanggelou A, Cooper WN, Latif F. Role of the Ras-association domain family 1 tumor suppressor gene in human cancers. Cancer Res. 2005;65(9):3497–3508. doi: 10.1158/0008-5472.CAN-04-4088. [DOI] [PubMed] [Google Scholar]
- 17.Choy KW, Lee TC, Cheung KF, Fan DS, Lo KW, Beaverson KL, Abramson DH, Lam DS, Yu CB, Pang CP. Clinical implications of promoter hypermethylation in RASSF1A and MGMT in retinoblastoma. Neoplasia. 2005;7(3):200–206. doi: 10.1593/neo.04565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cohen Y, Merhavi-Shoham E, Avraham RB, Frenkel S, Pe'er J, Goldenberg-Cohen N. Hypermethylation of CpG island loci of multiple tumor suppressor genes in retinoblastoma. Exp Eye Res. 2008;86(2):201–206. doi: 10.1016/j.exer.2007.10.010. [DOI] [PubMed] [Google Scholar]
- 19.Issa JP, Garcia-Manero G, Giles FJ, Mannari R, Thomas D, Faderl S, Bayar E, Lyons J, Rosenfeld CS, Cortes J, Kantarjian HM. Phase 1 study of low-dose prolonged exposure schedules of the hypomethylating agent 5-aza-20-deoxycytidine (decitabine) in hematopoietic malignancies. Blood. 2004;1103(5):1635–1640. doi: 10.1182/blood-2003-03-0687. [DOI] [PubMed] [Google Scholar]
- 20.Liu X, Dai X, Wu B. Study of 5-Aza-CdR on transcription regulation of RASSF1A gene in the BIU87 cell line. Urol Int. 2009;82(1):108–112. doi: 10.1159/000176036. [DOI] [PubMed] [Google Scholar]
- 21.Herman JG, Graff JR, Myohanen S, Nelkin BD, Baylin SB. Methylation-specific PCR: a novel PCR assay for methylation status of CpG islands. Proc Natl Acad Sci USA. 1996;93(18):9821–9826. doi: 10.1073/pnas.93.18.9821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Miyoshi N, Ishii H, Mimori K, Nishida N, Tokuoka M, Akita H, Sekimoto M, Doki Y, Mori M. Abnormal expression of PFDN4 in colorectal cancer: a novel marker for prognosis. Ann Surg Oncol. 2010;17(11):3030–3036. doi: 10.1245/s10434-010-1138-5. [DOI] [PubMed] [Google Scholar]
- 23.Khokhlatchev A, Rabizadeh S, Xavier R, Nedwidek M, Chen T, Zhang XF, Seed B, Avruch J. Identification of a novel Ras-regulated proapoptotic pathway. Curr Biol. 2002;12(4):253–265. doi: 10.1016/s0960-9822(02)00683-8. [DOI] [PubMed] [Google Scholar]
- 24.Rabizadeh S, Xavier RJ, Ishiguro K, Bernabeortiz J, Lopez-Ilasaca M, Khokhlatchev A, Mollahan P, Pfeifer GP, Avruch J, Seed B. The scaffold protein CNK1 interacts with the tumor suppressor RASSF1A and augments RASSF1A-induced cell deaths. J Biol Chem. 2004;279(28):29247–29254. doi: 10.1074/jbc.M401699200. [DOI] [PubMed] [Google Scholar]
- 25.Xu J, Zhou JY, Tainsky MA, Wu GS. Evidence that tumor necrosis factor-related apoptosis-inducing ligand induction by 5-Aza-2-deoxycytidine sensitizes human breast cancer cells to adriamycin. Cancer Res. 2007;67(3):1203–1211. doi: 10.1158/0008-5472.CAN-06-2310. [DOI] [PubMed] [Google Scholar]
- 26.Takahashi Y, Kimura S, Okano M. Pharmacological profiles and clinical roles of 5-azacitidine (Vidaza®) for injection 100 mg) for the treatment of myelodysplastic syndrome (MDS) Nihon Yakurigaku Zasshi. 2012;140(5):235–243. doi: 10.1254/fpj.140.235. [DOI] [PubMed] [Google Scholar]
- 27.Aparicio A, Weber JS. Review of the clinical experience with 5-azacytine and 5-aza-2′-deoxycytidine in solid tumors. Curr Opin Investig Drugs. 2002;3(4):627–633. [PubMed] [Google Scholar]
- 28.Bender CM, Pao MM, Jones PA. Inhibition of DNA methylation by 5-Aza-2-deoxycytidine suppresses the growth of human tumor cell lines. Cancer Res. 1998;58(1):95–101. [PubMed] [Google Scholar]
- 29.Shakya R, Gonda T, Quante M, Salas M, Kim S, Brooks J, Hirsch S, Davies J, Cullo A, Olive K, Wang TC, Szabolcs M, Tycko B, Ludwig T. Hypomethylating therapy in an aggressive stroma-rich model of pancreatic carcinoma. Cancer Res. 2013;73(2):885–896. doi: 10.1158/0008-5472.CAN-12-1880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Soengas MS, Capodieci P, Polsky D, Mora J, Esteller M, Opitz-Araya X, McCombie R, Herman JG, Gerald WL, Lazebnik YA, Cordón-Cardó C, Lowe SW. Inactivation of the apoptosis effector Apaf-1 in malignant melanoma. Nature. 2001;409(6817):207–211. doi: 10.1038/35051606. [DOI] [PubMed] [Google Scholar]