

Abstract
Small fast folding subdomains with low contact order have been postulated to facilitate the folding of larger proteins. We have tested this idea by determining how the fastest folding linear β-hairpin, CLN025, which folds on the nanosecond time scale, folds within the context of a two-hairpin WW domain system, which folds on the microsecond time scale. The folding of the wild type FBP28 WW domain was compared to constructs in which each of the loops was replaced by CLN025. A combination of FTIR spectroscopy and laser-induced temperature-jump coupled with infrared spectroscopy was used to probe changes in the peptide backbone. The relaxation dynamics of the β-sheets and β-turn were measured independently by probing the corresponding bands assigned in the amide I region. The folding rate of the CLN025 β-hairpin is unchanged within the larger protein. Insertion of the β-hairpin into the second loop results in an overall stabilization of the WW domain and a relaxation lifetime five times faster than the parent WW domain. In both mutants, folding is initiated in the turns and the β-sheets form last. These results demonstrate that fast folding subdomains can be used to speed the folding of more complex proteins, and that the folding dynamics of the subdomain is unchanged within the context of the larger protein.
INTRODUCTION
Protein structures are shaped by the demands of folding, stability, and function in their native environment. Evolution of ultrafast folding sequences is one way nature may have balanced the need for stability of a structure against the need to maintain the flexibility necessary for it to function, or the need for fast unfolding rates for degradation and regulation.1,2 Exactly how nature achieves ultrafast folding is not well understood, however, at least not well enough for the rational design of fast folding sequences. Many of the fastest folding proteins that have been studied as model systems are α-helical subdomains of larger, naturally occurring proteins.3–6 Small α-helical structures like these have a low contact order, native interactions between residues that are close in sequence, which has been correlated with fast folding.7 These fast folding subdomains may facilitate folding of the larger proteins by acting as nucleation sites, but this has not yet been demonstrated. In contrast to helical structures, higher contact orders are found in β-sheet proteins. They exhibit slower folding rates that span a wide range.8–10 The turns that connect multiple strands within a β-sheet fold have low contact order, however, and are thought to act as nucleation sites of folding.11 If turn formation is rate limiting, it should be possible to speed folding by increasing the rate of turn formation. For example, optimization of the turns of Pin1 WW domain, a two-turn structure, resulted in a 2 orders of magnitude increase in folding of the WW domain.12
Here, we have explored an alternative way to increase the folding rate of a β-sheet protein, by replacing a native turn with a faster folding turn sequence. The fastest folding linear β-hairpin, the model peptide CLN025, folds within 100 ns, a similar time scale to folding of α-helical peptides.13 It remains to be seen how this ultrafast folding β-hairpin folds in the context of a larger protein, and in turn how it impacts folding of a host system. We have addressed this question by incorporating CLN025 into WW domains, multistranded β-sheet structures, that comprise two β-hairpins.14 WW domains are good hosts for the CLN025 β-hairpin, because their small size (28–37 residues) is accessible by standard solid phase peptide synthesis and they are resistant to mutation, folding into a WW domain after modification at nearly any position.15
The WW domain family consists of an antiparallel and highly twisted three-stranded β-sheet structure with a small hydrophobic core and two highly conserved tryptophan residues.16–19 Members of this family, including Pin1, FBP28, and hYAP, have been found to fold in less than 100 µs.15,20,21 The fastest folding WW domain mutants are Fip35 and GTT35, derivatives of the Pin1 WW domain, which have relaxation lifetimes as fast as 14.5 and 3.7 µs, respectively.12,22 There have been extensive computational and experimental studies of WW domains, which predict folding by both two-state and three-state models.12,15,20,23–31 Formin binding protein 28 (FBP28) was selected as the host WW domain for our studies, because previous work postulated that it folds through an intermediate state in which the first hairpin is highly structured.15,30,32 Since FBP28 folds through such a polarized intermediate, it is an ideal system for exploring the effect of turn stability and folding rate on the overall mechanism. Therefore we determined the effect on the folding dynamics of inserting the optimized, fast-folding CLN025 construct into loop 1 (residue 11–20), FBP28 1L, and loop 2 (residue 20–29), FBP28 2L, of the FBP28 structure.
We compared the folding dynamics of the mutated WW domains to the parent CLN025 and wild type FBP28 WW domain. The dynamics of WW domain formation were studied using temperature-jump (T-jump), time-resolved IR spectroscopy. Pulsed laser excitation was used to rapidly initiate a shift in the folding equilibrium. The relaxation of the WW domain was measured by independently probing the components of the IR amide I band assigned to the β-sheet and β-turn. We found that insertion of CLN025 into the first loop has little affect on folding kinetics and stability. This is expected as previous studies report the first loop of FBP28 is structured in the intermediate state, and thus its formation is not rate-limiting. Insertion of CLN025 into the second loop stabilizes loop 2, resulting in a global stabilization of the peptide. This mutant folds five times faster than the parent WW domain.
The folding mechanisms of both the WW domain and β-hairpin are robust. CLN025 folds at nearly the same rate inside the larger protein as it does independently, indicating that the folding mechanism is not disrupted by insertion into a larger system. Although large regional mutations were made to the native WW domain sequence, the folding mechanism that we observe is consistent with previous studies. We propose a multistep mechanism of WW domain formation initiated in the turns. Analysis of the frequency dependence of the relaxation dynamics reveals that the turn of loop 1 forms on the nanosecond time scale. The turn of loop 2 forms second, followed by the sheet of loop 1 on the microsecond time scale. The final step in WW domain folding is formation of the second sheet. When the ultrafast folder was introduced into the second loop this mechanism collapsed to a simple three-state model with the turns forming before the sheets. Frequency-dependent IR measurements allow resolution of the folding mechanism with greater structural detail than has previously been reported. These experiments provide direct evidence for turn formation as the earliest step in the folding of a β-sheet protein, and further, that fast folding subdomains can speed formation of larger proteins.
EXPERIMENTAL SECTION
Protein Structure Modeling
The web-service QUARK33 was used for the prediction of the structure of a nonaggregating FBP28 WW domain mutant34 with CLN025 β-hairpin, YYDPETGTWY,35 inserted into one of the turns for each of the two mutants (Figure 1). The program used an estimated template modeling score (TM-score) to predict the quality of the protein structure prediction. A score below 0.2 corresponds to a random selection, and a score above 0.5 corresponds to a high confidence in the fold.36 The sequence with the highest TM-score when CLN025 is inserted into each loop of the WW domain was selected for peptide synthesis. The estimated TM-score of the best of the top 10 models with CLN025 inserted into loop 1, FBP28 1L, was 0.3408 ± 0.0764. The estimated TM-score of the best of the top 10 models with CLN025 inserted into loop 2, FBP28 2L, was 0.5594 ± 0.0764.
Figure 1.

Prediction of FBP28 1L and FBP28 2L structures. (A) Sequence alignment of wild type FBP28 protein with synthetic constructs containing the CLN025 sequence (in blue). The three β-strands are labeled above and colored red in the FBP28 sequence. (B) The three-dimensional models constructed using QUARK coordinates aligned with the FBP28 structure (PDB entry: 1E0L). The wild type FBP28 is shown in gray, FBP28 1L is shown in red, and FBP28 2L is shown in blue. The figure was prepared using PyMOL (www.pymol.org).
Protein Synthesis and Purification
FBP28 1L, FBP28 2L, and wild type FBP28 were synthesized via standard 9-fluornylmethoxycarbonyl (Fmoc)-based solid-phase chemistry on a Liberty1 microwave peptide-synthesizer (CEM, Matthews, NC). Fmoc-PAL-PS resin (Applied Biosystems, Foster City, CA) was used to form a peptide amide. The peptide was purified by reverse-phase chromatography (C18 column) using a water/acetonitrile gradient with 0.1% trifluoracetate (TFA) as the counterion. TFA interferes in the amide I IR measurements at 1672 cm−1, so we remove it by anion exchange. The peptide was lyophilized and dissolved in a 2 mM HCl solution to allow exchange of the TFA counterion for chloride.37 The identity of the peptide was confirmed by matrix-assisted laser desorption ionization time-of-flight mass spectrometry. The peptide was lyophilized and dissolved in D2O to allow deuterium−hydrogen exchange of the amide protons. The peptide was lyophilized a second time and resuspended in D2O buffer with 20 mM potassium phosphate buffer at pD* 7.0 (pD* refers to the uncorrected pH meter reading). Sample concentrations of 0.5–1.0 mM were prepared for both IR and fluorescence experiments.
CD Spectroscopy
CD spectra and CD melting curves were recorded on a Jasco J-810 spectropolarimeter equipped with a PFD-425S Jasco temperature controller module (Jasco, Inc., Easton, MD). Peptides were dissolved at 50 µM in 20 mM potassium phosphate buffer at pH 7.0. All measurements were obtained using a 1-mm path length cell. Wavelength scans were recorded over the range of 260 to 190 nm with an average of 3 repeats. A bandwidth of 2 nm and scan rate of 50 nm/min were used for spectral acquisition. Thermal unfolding experiments were performed by monitoring the signal at 226 nm from 5 to 90 °C using a 0.1 °C interval and scan rate of 30 °C/h. During the thermal unfolding experiment a full wavelength scan was obtained every 5 °C after a 60 s delay. The buffer and protein concentrations were the same as used in the wavelength scan experiment.
FTIR Spectroscopy
The equilibrium melting behavior was monitored on a Varian Excalibur 3100 FTIR spectrometer (Varian Inc., Palo Alto, USA) using a temperature-controlled IR cell. The IR cell consists of two CaF2 windows stacked and separated by a 100 µm Teflon spacer split into two compartments, a sample and a reference. The same cells are used for equilibrium FTIR and T-jump experiments. No aggregation was observed in the infrared at reported concentrations. All spectra shown at a specific temperature are constructed by subtracting the spectrum of reference buffer solution without protein from sample solution with protein. The temperature-dependent difference spectra were then generated by subtracting the spectrum at the lowest temperature from the spectra at higher temperatures. The second derivative spectra were computed in IGOR PRO after smoothing the data with a sixth order binomial algorithm to remove any residual water vapor (WaveMetrics, Lake Oswego, OR).
Time-Resolved Temperature-Jump (T-Jump) Relaxation Measurements
The IR T-jump apparatus has been described previously.38 Pulsed laser excitation is used to rapidly perturb the folding equilibrium on a time scale faster than the molecular dynamics of interest. Time-resolved infrared is then use to probe the reaction. A Q-switched DCR-4 Nd:YAG laser (Spectra Physics, Mountainview, CA) fundamental at 1064 nm is Raman shifted (one Stokes shift in 200 psi H2 gas) to produce a 10 ns pulse at 2 µm. The magnitude of the T-jump is calculated using the change in reference absorbance with temperature. The T-jump reference is taken from D2O buffer with 20 mM potassium phosphate buffer at pD* 7.0 at the same temperature and frequency as the sample. Absorbance changes at the reference frequency are due only to changes in D2O absorbance, which is used as an internal thermometer.38
The change in signal induced by the T-jump is probed in real time by a continuous laser with a frequency in the amide I′ band of the IR. The mid-IR probe beam is generated by a continuous wave quantum cascade laser (Daylight Solutions Inc., San Diego, CA) with a tunable output range of 1570–1730 cm−1. The transient transmission of the probe beam through the sample is measured using a fast, 100 MHz, photovoltaic MCT IR detector/preamplifier (Kolmar Technologies, Newburyport, MA). Transient signals are digitized and signal averaged (1000 shots) using a Tektronics digitizer (7612D, Beaverton, OR). Instrument control and data collection are controlled using a LabVIEW computer program.
Analysis of Kinetics Data
The peptide relaxation kinetics must be deconvolved from the observed kinetics. Accurate deconvolution is possible as the instrument response is determined from the reference measurement under the exact conditions of the sample measurements. In order to minimize detector artifacts, the reference is scaled prior to subtraction from the sample. The decay function is a triple exponential decay with the following formula:
| (1) |
where A0 is an offset, n is the number of exponentials to fit, An is a preexponential factor, τn is the relaxation lifetime of the sample and x0 is the time offset. In order to best fit the data, the minimum number of exponentials with unique relaxation lifetimes was selected. The data are fit over the interval from 90 ns to an order of magnitude outside the slowest exponential. In the cases where a fast phase is not reported the fit starts at 400 ns. The data analysis was performed in IGOR PRO (WaveMetrics, Lake Oswego, OR).
RESULTS AND DISCUSSION
QUARK Prediction of WW Domain Structure
The web-service QUARK33 was used to predict the Formin binding protein 28 (FBP28) WW domain structure with the fast-folding CLN025 β-hairpin inserted into each of the loops (Figure 1). Glutamine and asparagine mutations were made to the wild type FBP28 sequence to eliminate aggregation.34 There is good agreement between the tertiary structure prediction of the mutated sequences and the wild type WW domain (Figure 1B). The locations of each of the sheets, β1, β2, and β3, align well with the NMR structure of the FBP28 WW domain (PDB ID 1E0L). In both models QUARK predicts two additional secondary structure elements, an N-terminal β-sheet and a C-terminal α-helix. These small differences at the termini of the peptide do not affect the arrangement of the core structure. There is no experimental evidence by either CD or IR spectroscopy that these additional structural elements are formed, meaning they are likely artifacts of the QUARK prediction.
Far-UV CD Spectroscopy
The FBP28 1L and FBP28 2L peptides were constructed to test the affect of CLN025 insertion into each loop of FBP28 on WW domain folding. Far-UV CD is a good indicator of peptide secondary structure. The typical β-sheet signal has a negative peak at ~218 nm and a positive peak at ~195 nm.39 Folded WW domains do not exhibit this behavior; instead they have a negative peak at ~202 nm and a positive peak at ~230 nm.40,41 Small variations in the position and intensities of these peaks have been observed across the WW domain family.41 The peak at ~202 nm resembles the random coil peak usually found at 200 nm. Disorder in the N- and C- termini of the folded structure of WW domains is thought to contribute to the negative peak in the CD spectrum.40,42–44 The peak at ~230 nm arises from the presence of ordered aromatic amino acids.20,39,40 Analysis of the CD spectrum of FBP28 1L and 2L reveals both a positive peak at 226 nm and a negative peak at 197 nm (Figure 2A), consistent with a WW domain topology and the structure predicted by QUARK.
Figure 2.

(A) Far-UV CD spectra of 50 µM solutions of the FBP28 1L (red) and FBP28 2L (blue) WW domains in 20 mM potassium phosphate buffer (pH 7.0) acquired at 2 °C (solid line) and 95 °C (dashed line) during the course of a thermal denaturation in a 0.1 cm path length cell. (B) Thermal denaturation of the WW domains monitored by CD at 226 nm. The continuous line represents the best fit to the data to eq 2.
Thermal denaturation was monitored by recording the absorbance change at 226 nm with temperature (Figure 2B). The WW domains exhibit the typical heat-induced unfolding behavior with a loss of intensity at 226 nm and a shift of the minimum at 197 to 200 nm, corresponding to a change in the secondary structure from WW domain to random coil (Figure 2A).40 The melting curves (Figure 2B) were fit to an apparent two-state equilibrium model:
| (2) |
where Ai and Af are the extrapolated absorbance values at the two end points of the transition taking into account a linear baseline m, Tm is the transition midpoint and ΔT is the overall temperature range of the transition. The transition for FBP28 1L was too broad to obtain a reliable fit. FBP28 2L has a greater thermodynamic stability than FBP28 1L; at the highest temperature (Figure 2A) of the melt of FBP28 2L, there is still some intensity in the positive band at 230 nm, indicating that the peptide has not completely unfolded. The melting temperature of FBP28 2L is 74.4 ± 0.4 °C. This is 23 °C higher than the melting temperature of FBP28, and closer to the 69.9 °C melting temperature of the CLN025 β-hairpin.34,35 The transition of FBP28 2L is also much sharper (smaller ΔT) than the transition of FBP28 1L, revealing an increased cooperativity in the melt of FBP28 2L.45 This is evidence that insertion of CLN0225 into the second loop of FBP28 stabilizes the WW domain.
FTIR Spectroscopy
The temperature-induced unfolding of the two mutants was studied over the range from 5 to 95 °C in 5 °C intervals using FTIR spectroscopy to monitor the amide I′ region. An example of the temperature-dependent absorption spectra of the amide I′ spectral region (amide I region of peptides in D2O) of FBP28 2L is shown in Figure 3A (similar data for FBP28 1L is shown in Supporting Information, Figure S1). The amide I′ absorbance arises from C=O stretching vibrations of the polypeptide backbone carbonyls and is an established indicator of secondary structure.46–48 This relatively broad band contains contributions from the entire polypeptide backbone, which in the case of the WW domain includes β-sheet, β-turn, and random coil structure. The changes with temperature are highlighted by the difference spectra for each peptide (Figure 3B). The difference spectra are generated by subtracting the lowest temperature spectrum from each absorbance spectrum at higher temperature. Negative peaks correspond to specific structures or interactions present in the folded state, and positive peaks correspond to new interactions with solvent in the unfolded state. The individual peaks are more easily distinguished in the second derivative of the FTIR spectra at the lowest temperature (Supporting Information, Figure S2).
Figure 3.

Temperature-dependent FTIR spectra of 1 mM FBP28 2L in 20 mM potassium phosphate buffer (pH 7). (A) Absorbance spectra in the amide I′ region; the temperature of the individual traces varies from 5 to 85 °C in 5 °C intervals. (B) Difference spectra obtained by subtracting the spectrum at 5 °C from the spectra at higher temperatures. (C) Normalized second derivative of FTIR difference spectrum at 33 (blue) and 67 °C (red).
At low temperature there are three major peaks that are characteristic of specific secondary structures, centered at 1614, 1634, and 1679 cm−1. In the difference spectra the peak at 1614 appears as a shoulder on the dominant negative component at 1634 cm−1. The negative peak at 1679 cm−1 overlaps with the broad positive spectral feature at 1660 cm−1, a characteristic peak due to the disordered polypeptide at high temperatures.38,49,50 The Gai group previously observed these peaks in the Pin1 WW domain.51 A peak at 1614 cm−1 was observed in CLN025 and attributed to a tertiary amide interaction between the glutamic acid backbone in the turn and the aspartic acid side chain.13,52 In the FBP28 WW domain there is a tertiary amide interaction between the backbone of lysine and side chain of threonine in the first turn, and there are no native tertiary amide interactions in the second turn.17,53 We assign the peak at 1614 cm−1 to tertiary amide interactions in the turns of the WW domain. In FBP28 1L the peak at 1614 cm−1 is assigned to tertiary amide interactions in the first loop, found in the turn of CLN025. In FBP28 2L the peak is assigned to tertiary amide interactions in the turns of both loops, stemming from the FBP28 WW domain in loop 1 and CLN025 in loop 2. IR bands at 1634 and 1681 cm−1 are well-established markers of antiparallel β-sheets.45 The peak at 1634 cm−1 arises from inward directed carbonyl groups in the sheet, and the peak at 1681 cm−1 arises from solvent exposed carbonyl bonds in the sheet. The FBP28 amide I′ bands at 1634 and 1679 cm−1 are consistent with these characteristic β-sheet markers.
Analysis of the second derivative of the difference spectra with temperature (Figure 3C) highlights the temperature-dependent contributions to the 1634 cm−1 difference peak. Below 46 °C the second derivative peak maximum is at lower frequency, 1629 cm−1. As the temperature increases, this peak shifts to a higher frequency, 1635 cm−1 (slightly higher than the peak observed in the difference spectrum, because the second derivative represents the actual peak maximum). A similar phenomenon was observed by the Keiderling group in their extensive studies of multistranded β-sheet structures.54 They assign the main maximum to out-of-phase amide I vibrations of neighboring amides in the β-sheet. As the number of strands was increased, the peak maximum decreased in frequency. This is due to interstrand coupling as the number of β-sheets increases. The efficiency of the interstrand coupling depends on the orientation of the dipole moments of the individual carbonyl oscillators. The splitting is less in WW domains than other extended β-sheet structures, because the twist of the sheets misaligns the carbonyl oscillators so that they do not couple as efficiently as in more planar structures, causing only a small change in frequency with strand number. We attribute the second derivative 1635 cm−1 feature to C=O bonds in the β-sheet of a single hairpin. As the extended three β-strand WW domain structure forms, there is a shift of the peak maximum to lower frequency, 1629 cm−1. In the difference spectra (Figure 3B), as the peptide is melted we observe the melt of the first strand of the WW domain, loss of the 1629 cm−1 band, followed by loss of the band at 1635 cm−1 due to the second strand. We conclude that the second loop melts first, since the first loop has been previously identified as the more stable of the two loops.
The normalized melting curves for both proteins derived from the temperature-dependent IR absorbance at 1634 cm−1 are shown in Figure 4. The data were normalized after being fit to an apparent two-state equilibrium model using eq 1. The melting temperature of FBP28 2L measured at 1634 cm−1 is 65.3 ± 0.8 °C. The melting temperature derived from fits to other IR frequencies agreed within the error of the fit. The melting temperature derived from the IR melt of FBP28 2L is similar to that reported for CLN025, 69.9 °C.35 Thermodynamic parameters were derived from a van’t Hoff analysis (inset of figure 4) of the FTIR data at 1634 cm−1. The free energy of unfolding of these peptides over the temperature range studied is linear. The thermodynamic parameters observed in FBP28 2L ΔHf, −73 ± 4 kJ/mol, and ΔSf, −216 ± 11 J/mol·K, are similar to the ΔHf, −62.8 kJ/mol, and ΔSf, −182.8 J/mol·K reported for the CLN025 hairpin.13 Since the thermodynamic parameters of FBP28 2L mimic those of the isolated hairpin, we conclude that insertion of CLN025 into the second turn of FBP28 stabilizes the WW domain.
Figure 4.

FTIR melt curves for the FBP28 1L (red) and FBP28 2L (blue) WW domains obtained by plotting the change in IR difference spectra at 1634 cm−1 versus temperature. The data are fit to an apparent 2-state model (eq 2) and then normalized. Inset: van’t Hoff analyses using equilibrium constants generated from the melt curves.
Temperature-Jump Relaxation Kinetics
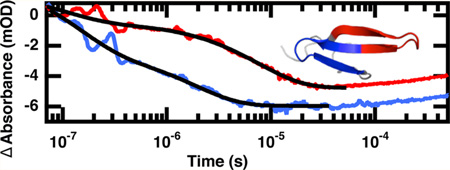
The relaxation kinetics of the folding/unfolding transition following a laser-induced temperature jump were probed using time-resolved infrared spectroscopy of the amide I′ frequency of the tertiary amide linkage of the turns (1619 cm−1) and the β-sheet of the first loop (1633 cm−1) and β-sheets of the fully formed WW domain (1629 cm−1). Jumps were performed slightly off peak center to maximize the signal. Figure 5 displays the relaxation kinetics for the peptides following a jump from 30 to 45 °C (Figure 5A) and from 50 to 65 °C (Figure 5B). These jumps are to the midpoint of the transition of FBP28 1L and FBP28 2L, respectively. The relaxation transient is fit by a triple exponential at low temperature and a single or double exponential at high temperature. The complete relaxation parameters observed for the peptides are reported in the Supporting Information (Tables S1–S3). Both peptides exhibit three relaxation lifetimes (Figure 6). The first two, a fast 100 ns phase (τ1) and a µs (τ2) phase, are relatively temperature independent. The third relaxation lifetime (τ3) is highly temperature dependent. The observed relaxation of τ3 spans 100s of microseconds through a few µs. As the temperature increases, τ2 becomes indistinguishable from τ3.
Figure 5.

Representative IR T-jump relaxation kinetics of FBP28 1L (red), FBP28 2L (blue) and wild type FBP28 (gray) monitored in the amide I′ spectral region at 1633 cm−1 following a T-jump from (A) 30 to 45 °C and (B) 50 to 65 °C. A triple exponential fit, eq 1, is overlaid on each kinetic trace on top and a double exponential fit is overlaid on each kinetic trace on the bottom (black solid line). The data are normalized at 1 × 10−4 for comparison.
Figure 6.

Arrhenius plot showing the temperature dependence of the folding region at 1633 cm−1. The values of T used for the (1/T) axis are the final temperatures reached during the jump. k is the value obtained from a fit (see text) of the T-jump transient of FBP28 1L (▲) and FBP28 2L (●). Lines are a result of fitting τ1 (solid line), τ2 (dashed line), and τ3 (dotted line) of FBP28 2L.
At jumps to low final temperatures there is little difference between the observed kinetics and relative intensities of the relaxation lifetimes of FBP28 1L and 2L (Figure 5A). The absorbance change associated with each of the three phases is of approximately equal intensity (Supporting Information, Tables S1–S3). At jumps to higher temperatures FBP28 1L and FBP28 2L exhibit different behaviors (Figure 5B). Most of the absorbance change in FBP28 1L is observed in the slow phase, whereas most of the absorbance change in FBP28 2L is in the fast phase, τ1. An Arrhenius plot (Figure 6) of the observed kinetics reveals that at low temperature there is little difference between the kinetics of the three phases of FBP28 1L and FBP28 2L. At higher final jump temperatures, although not all the phases are observed, the kinetics that are observed follow the same trend.
Frequency-dependent T-jump measurements (Figure 7) reveal differences in the kinetics of the turns (1619 cm−1), the β-sheet of a single hairpin (1633 cm−1) and β-sheet of the complete WW domain (1629 cm−1). There are multiple exponentials observed at each of these probe frequencies due to overlap between the bands. It is also likely that the spectral response is coupled such that formation of one structure affects the others. The following discussion focuses on FBP28 1L, which has the ultrafast folding CLN025 sequence in the first loop. The 1619 cm−1 band in this case arises exclusively from the first turn, as there are no tertiary amide interactions in the second turn. The kinetics monitored at 1619 cm−1 exhibits the largest amplitude of the fast (~100 ns) phase (Table 1). On the basis of this observation we assign the fast phase to turn formation in loop 1 and the intermediate (8 µs) phase to sheet formation. This supports the hypothesis that folding is initiated in the turn, where there is lower contact order.7 The kinetics at 1633 cm−1 is sensitive to the β-sheet of a single hairpin. The observed kinetics is similar to that at 1619 cm−1, which probes the turn of the first loop, but with more intensity in the intermediate phase than the fast phase. The 1633 cm−1 band in FBP28 1L was assigned to the β-sheet of the first loop on the basis of its higher stability in the temperature-dependent FTIR measurements. Thus we assign the fast (~100 ns) phase to the formation of the turn in loop 1 and the intermediate (8 µs) phase to sheet formation in loop 1. This conclusion is consistent with previous studies that postulated FBP28 folds through an intermediate in which the first loop is fully formed. Replacing the first loop with the ultrafast folding CLN025 hairpin should not perturb this mechanism.
Figure 7.

Representative IR T-jump relaxation kinetics of FBP28 1L (A) and FBP28 2L (B) monitored in the amide I′ spectral region at 1619, 1629, and 1633 cm−1 following a T-jump from 50 to 65 °C. A double exponential fit is overlaid on each kinetic trace (black solid line).
Table 1.
Relaxation Kinetics Following a Jump from 50 to 65 °C
| FBP28 1L | FBP28 2L | |||||||
|---|---|---|---|---|---|---|---|---|
| A1 (%) | τ1 (ns) | A2 (%) | τ2 (µs) | A1 (%) | τ1 (ns) | A2 (%) | τ2 (µs) | |
| 1619 cm−1 | 65 ± 2 | 105 ± 6 | 35 ± 1 | 8.6 ± 0.5 | 45 ± 3 | 185 ± 15 | 55 ± 2 | 1.7 ± 0.8 |
| 1629 cm−1 | 28 ± 8 | 2300 ± 400 | 72 ± 1 | 17.2 ± 0.4 | 67 ± 2 | 213 ± 10 | 33 ± 1 | 3.1 ± 0.2 |
| 1633 cm−1 | 20 ± 1 | 151 ± 14 | 80 ± 1 | 7.9 ± 0.1 | 42 ± 2 | 154 ± 11 | 58 ± 1 | 2.1 ± 0.1 |
The probe at 1629 cm−1 is sensitive to interstrand coupling across all three sheets of the WW domain. Since formation of the second loop is thought to be the rate-limiting step for WW domain formation, the kinetics at 1629 cm−1 should probe this final folding step. As expected, most of the 1629 cm−1 intensity is in the slowest (17 µs) phase, which we assign to β-sheet formation in loop 2. We postulate that the 2.3 µs phase observed at 1629 cm−1 is the turn of loop 2; however, we do not have a direct probe of the second turn of FBP28 1L. Similar to previously studied WW domains, we propose a mechanism of FBP28 1L WW domain formation initiated in the turn of loop 1 (Figure 8). The turn of loop 2 forms second, followed by the sheet of loop 1. The final step in WW domain formation is the completion of the second loop.
Figure 8.

Proposed mechanism of WW domain folding.
Using this model we can interpret the three phases observed in the Arrhenius analysis (Figure 6). The folding mechanism of FBP28 1L includes four steps (Figure 8), one more than was observed in the Arrhenius analysis at 1633 cm−1, a probe of loop 1. Because the IR band at 1633 cm−1 is relatively broad and overlaps with the band at 1619 and 1629 cm−1, there may be a relatively smaller contribution from kinetics associated with each of these wavelengths. The ns and µs temperature-independent fast phases are assigned to the formation of the turn and sheet of loop 1, respectively. The third phase that is highly temperature-dependent is assigned to formation of the β-sheet of loop 2. At high temperatures, the dynamics of the sheet of loop 2 and sheet of loop 1 become indistinguishable. Analysis of frequency-dependent measurements and relative intensities of the absorbance changes give strong evidence for a fourth phase, formation of turn 2, in between the formation of the turn and sheet of loop 1 (Table 1). This turn forms on a similar time scale to the sheet of loop 1, the dominant contribution monitored at 1633 cm−1, so it is not possible to distinguish a fourth phase in the Arrhenius analysis (Figure 6).
The following discussion focuses on the kinetics of FBP28 2L, which has the ultrafast folding turn inserted in the second loop. Whereas speeding the formation of the first loop of FBP28 has little affect on the observed dynamics and folding mechanism, speeding the formation of the second loop speeds the overall folding dynamics and simplifies the folding mechanism (Table 1). In FBP28 2L, measurements at 1619 cm−1 include contributions from both turns, because there are tertiary amide interactions found in both of the turns. The fast kinetics is again assigned to the turns. It is not possible to determine which turn is forming first as it is not possible to independently probe each turn. The slower phases are assigned to β-sheet formation. When the kinetics is probed at either the frequency associated with the β-sheet of a single hairpin or that of the complete WW domain (1633 and 1629 cm−1, respectively), there is only a small difference between the observed slow rates. Speeding the folding of the second loop greatly speeds up the rate-limiting step, formation of the β-sheet of the second loop, simplifying the folding model. The folding of FBP28 2L WW domain is initiated in the turns followed by zipping of the β-sheets.
Previous fluorescence studies of FBP28 revealed biexponential kinetics below 65 °C with relaxation lifetimes of 15 µs and >900 µs.28 Above 65 °C single exponential kinetics were reported, with a relaxation lifetime of <15 µs.15 Infrared measurements offer a level of detail not available from the fluorescence experiments. IR measurements of the wild type FBP28 (Figure 5) and mutants reveal fast nanosecond and microsecond phases that have not been previously observed in this sequence. A jump to a final temperature of 65 °C shows that mutation of the second loop results in an order of magnitude increase in the observed relaxation lifetimes (Table 1). The same experiment with the mutation to the first loop results in no change in the folding dynamics. Insertion of the CLN025 hairpin into loop 2 speeds its folding, allowing the WW domain to form faster. The relaxation lifetime of the fastest folding WW domain, GTT35, is 3.7 µs at a temperature for which the equilibrium constant for folding is ~2.12 Comparing the relaxation lifetime of the FBP28 2L mutant at the same stability, a final jump temperature of 60 °C, the relaxation lifetime is <5 µs, comparable to the GTT35 WW domain. GTT35 is a mutant of the Pin1 WW domain, which resulted from optimization of both of the turns in the WW domain. Our experiments show that large regional mutations is an effective approach to increase the folding rate of a larger system.
Protein mutations have mixed affects on the mechanism of protein folding. SH3 domains were found to have a discrete order to folding. Computational studies of src, Fyn and α-spectrin SH3 domains showed that while there is local variation in the transition state ensembles of each, they share a topology characteristic of the SH3 domain.55 Experiments on SH3 domain where mutants included lengthening of the n-src by 10 residues and an introduction of disulfide cross-links all exclusively affected the folding rates.56 Similarly the folding mechanism after mutation of the first loop of the FBP28 WW domain is consistent with previous studies. CLN025 folds at nearly the same rate inside the larger protein as it does independently, indicating that its folding mechanism is not disrupted by insertion into a larger system. Studies of protein G and protein L, which share a similar topology, show different transition state ensembles where the hairpin with the lowest free energy forms first.57 The overall order of folding was switched in protein G, by changing the stability of each of the turns.58 The folding of the slower turn was sped by selecting a low energy sequence with termini that superimposed on the terminal residues of the hairpin. However, in order to change the folding mechanism a hydrogen bond in the faster turn was disrupted. The folding of the protein G mutant without this point mutation in the turn was five times faster. While this shows that it is possible to change the order of folding, it came at the cost of the folding rate. We observe that the folding rate is limited by the folding of the slower turn. Our measurements of FBP28 2L (Table 1) show that the insertion of the fast folding β-hairpin into the second turn speeds not only the formation of the second turn, but also of the sheets of both loop 1 and loop 2. Increasing the rate of folding of the turns is one way that nature may create faster folding β-sheet proteins. We have shown that small fast folding single domain β-hairpin models can be used to speed the folding of a larger protein without exhaustively searching for low energy sequences.
CONCLUSION
Insertion of the CLN025 β-hairpin into each loop of the FBP28 WW domain reveals that the folding dynamics of the ultrafast folding hairpin is the same within the context of a larger protein. CLN025 folds in ~100 ns both independently13 and inside the WW domain. Furthermore, the ultrafast folding hairpin accelerates the overall folding of the WW domain by an order of magnitude when placed in loop 2, since the formation of this loop clearly limits the overall rate of folding. Previous studies have focused on speeding the folding of larger systems by point mutations or sequence homology based on the energy of the sequence.12,58 While these methods have proven effective, we have demonstrated an alternative method that introduces a fast folding subdomain to nucleate the larger structure. Since the folding of the CLN025 domain is unchanged in the context of the larger WW domain, it is a robust nucleation structure that may be generally applicable to speeding the folding of other β-sheet systems.
We propose a model to describe WW domain folding initiated in the turns, similar to that proposed for other WW domains.15,30,32 Previous studies using fluorescence T-jump have been unable to observe the level of detail available from infrared spectroscopy. We are able to observe subtle differences in the time scale of formation of each of the turns and sheets of the loops in the WW domain. We observe that the turn of both loop 1 and loop 2 form prior to the β-sheets. This highlights the importance of the turn in folding of β-proteins. The folding mechanism of the WW domain is robust and is not disrupted by mutation of the loops. We observe that the speed of folding is limited by formation of the second turn. When the fast folding β-hairpin is inserted into the second turn, the folding of the sheets of both loops is increased. Thus, by applying our understanding of the basic principles of protein folding and small subdomains and models like the CLN025 β-hairpin, we can speed formation of larger multidomain systems.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by a grant from the National Institutes of Health (NIH R01 GM53640) to R.B.D.
Footnotes
ASSOCIATED CONTENT
Supporting Information
Temperature-dependent FTIR spectra of FBP28 1L, second derivative spectra of FBP28 1L and 2L absorbance data at 5 °C, FBP28 1L difference data at 33 and 67 °C, and a complete table of relaxation kinetics for FBP28 1L and FBP28 2L at 1619, 1629, and 1633 cm−1. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
REFERENCES
- 1.Dyer RB. Curr. Opin. Struct. Biol. 2007;17:38. doi: 10.1016/j.sbi.2007.01.001. [DOI] [PubMed] [Google Scholar]
- 2.Eaton WA, Munoz V, Hagen SJ, Jas GS, Lapidus LJ, Henry ER, Hofrichter J. Annu. Rev. Biophys. Biomol. Struct. 2000;29:327. doi: 10.1146/annurev.biophys.29.1.327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Takada S. Proteins: Struct. Funct. Genet. 2001;42:85. [Google Scholar]
- 4.Mayor U, Guydosh NR, Johnson CM, Grossmann JG, Sato S, Jas GS, Freund SMV, Alonso DOV, Daggett V, Fersht AR. Nature. 2003;421:863. doi: 10.1038/nature01428. [DOI] [PubMed] [Google Scholar]
- 5.Bai YW, Karimi A, Dyson HJ, Wright PE. Protein Sci. 1997;6:1449. doi: 10.1002/pro.5560060709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zagrovic B, Snow CD, Shirts MR, Pande VS. J. Mol. Biol. 2002;324:1051. doi: 10.1016/s0022-2836(02)00997-x. [DOI] [PubMed] [Google Scholar]
- 7.Plaxco KW, Simons KT, Baker D. J. Mol. Biol. 1998;277:985. doi: 10.1006/jmbi.1998.1645. [DOI] [PubMed] [Google Scholar]
- 8.Dinner AR, Lazaridis T, Karplus M. Proc. Natl. Acad. Sci. U. S. A. 1999;96:9068. doi: 10.1073/pnas.96.16.9068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Klimov DK, Thirumalai D. Proc. Natl. Acad. Sci. U. S. A. 2000;97:2544. doi: 10.1073/pnas.97.6.2544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pande VS, Rokhsar DS. Proc. Natl. Acad. Sci. U. S. A. 1999;96:9062. doi: 10.1073/pnas.96.16.9062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vu DM, Brewer SH, Dyer RB. Biochemistry. 2012;51:9104. doi: 10.1021/bi301296y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Piana S, Sarkar K, Lindorff-Larsen K, Guo MH, Gruebele M, Shaw DE. J. Mol. Biol. 2011;405:43. doi: 10.1016/j.jmb.2010.10.023. [DOI] [PubMed] [Google Scholar]
- 13.Davis CM, Xiao S, Raleigh DP, Dyer RB. J. Am. Chem. Soc. 2012;134:14476. doi: 10.1021/ja3046734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kubelka J, Hofrichter J, Eaton WA. Curr. Opin. Struct. Biol. 2004;14:76. doi: 10.1016/j.sbi.2004.01.013. [DOI] [PubMed] [Google Scholar]
- 15.Jager M, Nguyen H, Crane JC, Kelly JW, Gruebele MJ. Mol. Biol. 2001;311:373. doi: 10.1006/jmbi.2001.4873. [DOI] [PubMed] [Google Scholar]
- 16.Macias MJ, Hyvonen M, Baraldi E, Schultz J, Sudol M, Saraste M, Oschkinat H. Nature. 1996;382:646. doi: 10.1038/382646a0. [DOI] [PubMed] [Google Scholar]
- 17.Macias MJ, Gervais V, Civera C, Oschkinat H. Nat. Struct. Biol. 2000;7:375. doi: 10.1038/75144. [DOI] [PubMed] [Google Scholar]
- 18.Verdecia MA, Bowman ME, Lu KP, Hunter T, Noel JP. Nat. Struct. Biol. 2000;7:639. doi: 10.1038/77929. [DOI] [PubMed] [Google Scholar]
- 19.Wiesner S, Stier G, Sattler M, Macias MJ. J. Mol. Biol. 2002;324:807. doi: 10.1016/s0022-2836(02)01145-2. [DOI] [PubMed] [Google Scholar]
- 20.Ferguson N, Johnson CM, Macias M, Oschkinat H, Fersht A. Proc. Natl. Acad. Sci. U. S. A. 2001;98:13002. doi: 10.1073/pnas.221467198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ferguson N, Pires JR, Toepert F, Johnson CM, Pan YP, Volkmer-Engert R, Schneider-Mergener J, Daggett V, Oschkinat H, Fersht A. Proc. Natl. Acad. Sci. U. S. A. 2001;98:13008. doi: 10.1073/pnas.221467398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jager M, Zhang Y, Bieschke J, Nguyen H, Dendle M, Bowman ME, Noel JP, Gruebele M, Kelly JW. Proc. Natl. Acad. Sci. U. S. A. 2006;103:10648. doi: 10.1073/pnas.0600511103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Karanicolas J, Brooks CL. 3rd Proc. Natl. Acad. Sci. U. S. A. 2003;100:3954. doi: 10.1073/pnas.0731771100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Karanicolas J, Brooks CL. 3rd Proc. Natl. Acad. Sci. U. S. A. 2004;101:3432. doi: 10.1073/pnas.0304825101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mu Y, Nordenskiold L, Tam JP. Biophys. J. 2006;90:3983. doi: 10.1529/biophysj.105.076406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Noe F, Schutte C, Vanden-Eijnden E, Reich L, Weikl TR. Proc. Natl. Acad. Sci. U. S. A. 2009;106:19011. doi: 10.1073/pnas.0905466106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.a Beccara S, Skrbic T, Covino R, Faccioli P. Proc. Natl. Acad. Sci. U. S. A. 2012;109:2330. doi: 10.1073/pnas.1111796109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nguyen H, Jager M, Moretto A, Gruebele M, Kelly JW. Proc. Natl. Acad. Sci. U. S. A. 2003;100:3948. doi: 10.1073/pnas.0538054100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ferguson N, Berriman J, Petrovich M, Sharpe TD, Finch JT, Fersht AR. Proc. Natl. Acad. Sci. U. S. A. 2003;100:9814. doi: 10.1073/pnas.1333907100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Petrovich M, Jonsson AL, Ferguson N, Daggett V, Fersht AR. J. Mol. Biol. 2006;360:865. doi: 10.1016/j.jmb.2006.05.050. [DOI] [PubMed] [Google Scholar]
- 31.Liu F, Du D, Fuller AA, Davoren JE, Wipf P, Kelly JW, Gruebele M. Proc. Natl. Acad. Sci. U. S. A. 2008;105:2369. doi: 10.1073/pnas.0711908105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Deechongkit S, Nguyen H, Powers ET, Dawson PE, Gruebele M, Kelly JW. Nature. 2004;430:101. doi: 10.1038/nature02611. [DOI] [PubMed] [Google Scholar]
- 33.Xu D, Zhang Y. Proteins: Struct. Funct. Bioinf. 2012;80:1715. doi: 10.1002/prot.24065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tremmel S, Beyermann M, Oschkinat H, Bienert M, Naumann D, Fabian H. Angew. Chem. Int. Ed. 2005;44:4631. doi: 10.1002/anie.200500547. [DOI] [PubMed] [Google Scholar]
- 35.Honda S, Akiba T, Kato YS, Sawada Y, Sekijima M, Ishimura M, Ooishi A, Watanabe H, Odahara T, Harata K. J. Am. Chem. Soc. 2008;130:15327. doi: 10.1021/ja8030533. [DOI] [PubMed] [Google Scholar]
- 36.Zhang Y, Skolnick J. Nucleic Acids Res. 2005;33:2302. doi: 10.1093/nar/gki524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Andrushchenko VV, Vogel HJ, Prenner EJ. J. Pept. Sci. 2007;13:37. doi: 10.1002/psc.793. [DOI] [PubMed] [Google Scholar]
- 38.Williams S, Causgrove TP, Gilmanshin R, Fang KS, Callender RH, Woodruff WH, Dyer RB. Biochemistry. 1996;35:691. doi: 10.1021/bi952217p. [DOI] [PubMed] [Google Scholar]
- 39.Manning MC, Woody RW. Biopolymers. 1987;26:1731. doi: 10.1002/bip.360261007. [DOI] [PubMed] [Google Scholar]
- 40.Koepf EK, Petrassi HM, Sudol M, Kelly JW. Protein Sci. 1999;8:841. doi: 10.1110/ps.8.4.841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Koepf EK, Petrassi HM, Ratnaswamy G, Huff ME, Sudol M, Kelly JW. Biochemistry. 1999;38:14338. doi: 10.1021/bi991105l. [DOI] [PubMed] [Google Scholar]
- 42.Viguera AR, Arrondo JL, Musacchio A, Saraste M, Serrano L. Biochemistry. 1994;33:10925. doi: 10.1021/bi00202a011. [DOI] [PubMed] [Google Scholar]
- 43.Knapp S, Mattson PT, Christova P, Berndt KD, Karshikoff A, Vihinen M, Smith CI, Ladenstein R. Proteins. 1998;31:309. doi: 10.1002/(sici)1097-0134(19980515)31:3<309::aid-prot7>3.0.co;2-d. [DOI] [PubMed] [Google Scholar]
- 44.Reid KL, Rodriguez HM, Hillier BJ, Gregoret LM. Protein Sci. 1998;7:470. doi: 10.1002/pro.5560070228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Maness SJ, Franzen S, Gibbs AC, Causgrove TP, Dyer RB. Biophys. J. 2003;84:3874. doi: 10.1016/S0006-3495(03)75115-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang-Jih Yang PRG, Michael Byler D, Heino Susi. Appl. Spectrosc. 1985;39:282. [Google Scholar]
- 47.Arrondo JLR, Blanco FJ, Serrano L, Goni FM. FEBS Lett. 1996;384:35. doi: 10.1016/0014-5793(96)00261-x. [DOI] [PubMed] [Google Scholar]
- 48.Susi H, Byler DM. Methods Enzymol. 1986;130:290. doi: 10.1016/0076-6879(86)30015-6. [DOI] [PubMed] [Google Scholar]
- 49.Werner JH, Dyer RB, Fesinmeyer RM, Andersen NH. J. Phys. Chem. B. 2002;106:487. [Google Scholar]
- 50.Dyer RB, Gai F, Woodruff WH. Acc. Chem. Res. 1998;31:709. [Google Scholar]
- 51.Wang T, Xu Y, Du DG, Gai F. Biopolymers. 2004;75:163. doi: 10.1002/bip.20101. [DOI] [PubMed] [Google Scholar]
- 52.Hilario J, Kubelka J, Keiderling TA. J. Am. Chem. Soc. 2003;125:7562. doi: 10.1021/ja030039e. [DOI] [PubMed] [Google Scholar]
- 53.Chan DC, Bedford MT, Leder P. EMBO J. 1996;15:1045. [PMC free article] [PubMed] [Google Scholar]
- 54.Kubelka J, Keiderling TA. J. Am. Chem. Soc. 2001;123:12048. doi: 10.1021/ja0116627. [DOI] [PubMed] [Google Scholar]
- 55.Lindorff-Larsen K, Vendruscolo M, Paci E, Dobson CM. Nat. Struct. Mol. Biol. 2004;11:443. doi: 10.1038/nsmb765. [DOI] [PubMed] [Google Scholar]
- 56.Grantcharova VP, Riddle DS, Baker D. Proc. Natl. Acad. Sci. U. S. A. 2000;97:7084. doi: 10.1073/pnas.97.13.7084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.McCallister EL, Alm E, Baker D. Nat. Struct. Biol. 2000;7:669. doi: 10.1038/77971. [DOI] [PubMed] [Google Scholar]
- 58.Nauli S, Kuhlman B, Baker D. Nat. Struct. Biol. 2001;8:602. doi: 10.1038/89638. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.