

Abstract
Background: The BRAFV600E mutation is the most common genetic alteration in papillary thyroid carcinomas (PTCs). Transgenic mice overexpressing BRAFV600E in their thyroids under control of the thyroglobulin promoter (Tg-BRAF2 mice) developed invasive PTCs with high penetrance. However, these mice showed elevated thyrotropin (TSH) levels, which also stimulate the proliferation of thyrocytes and tumorigenesis. The purpose of the present study was to investigate how TSH signaling cooperates with BRAFV600E in the process of thyroid carcinogenesis.
Methods: We crossed Tg-BRAF2 mice with TSH receptor knockout (TshR−/−) mice. Four genetically distinct mice groups—Brafwt/TshR+/− (group 1), Brafwt/TshR−/− (group 2), Tg-BRAF2/TshR+/− (group 3), and Tg-BRAF2/TshR−/− (group 4)—were sacrificed at 12 and 24 weeks of age. We performed histopathological analysis. Genomic instability was evaluated by immunofluorescence for p53-binding protein 1 (53BP1) and γH2AX. Invasiveness and genomic instability were also evaluated using thyroid PCCL3 cells expressing BRAFV600E.
Results: Groups 3 and 4 developed distinct neoplasias comparable to human PTCs. Group 3 developed typically larger, more aggressive, invasive tumors compared to group 4. The frequency of 53BP1 and γH2AX foci—indicators of genomic instability—in group 3 was higher than that in group 4. TSH also enhanced invasiveness and genomic instability in PCCL3 cells with BRAFV600E expression.
Conclusions: These data demonstrate that the TSH signaling confers more aggressive features in BRAFV600E-induced thyroid tumors in mice. This might be due, in part, to accelerated genomic instability.
Introduction
Papillary thyroid cancer (PTC) represents the most frequently occurring type of differentiated thyroid cancer (1). PTCs are associated with characteristic genetic alterations including RET/PTC rearrangements and activating point mutations involving BRAF and the RAS family genes. These mutually exclusive gene mutations typically result in activation of the MAPK signaling pathway, thus providing strong genetic evidence that constitutive activation of this cascade is critical to the transformation of thyrocytes to PTC (2–4).
The BRAF activating mutation is the most common genetic aberration in PTCs (29–83%) (5). It is most commonly due to a thymine-to-adenine transversion at nucleotide1799 (c.1799T>A), resulting in a valine-to-glutamic acid substitution at amino acid 600 (p.V600E). It is believed that this mutation destabilizes the interactions that maintain the DFG motif in an inactive conformation, and that the mutation flips the activation segment into the active position (6). Therefore, BRAFV600E constitutively activates the MAPK pathway and is thought to play a major role in PTC tumorigenesis (7,8).
Although the majority of PTCs have an excellent outcome with treatment (9), several studies have shown that the BRAFV600E mutation in PTC is associated with aggressive clinicopathological features such as advanced clinical stage, extrathyroidal extension, and lymph-node metastasis (7). These tumors also have a high risk of recurrence (10). Transgenic mice overexpressing BRAFV600E in thyrocytes under control of the thyroglobulin (Tg) gene promoter developed PTC with distinct tall cells and poorly differentiated areas (11), which are aggressive features of human PTCs. However, these mice were severely hypothyroid with consequently high thyrotropin (TSH) levels because the function of all thyrocytes was impaired by the expression of the oncoprotein. In rodents, it is known that increasing serum TSH by prolonged administration of an antithyroid drug leads to development of thyroid cancer, albeit at low penetrance (12). Most thyroid cancer mouse models similarly show elevated TSH levels because of this hormonal feedback system. However, the role of the high TSH levels in these mouse models remains less clear.
So far, two mouse models have been developed to study the role of TSH in thyroid carcinogenesis. Lu et al. crossed TRβPV/PV mice—a model of follicular thyroid cancer—with TSH receptor (TSHR) knockout mice (13). Surprisingly, the tumorigenic effect of TRβPV/PV was totally abolished in the TRβPV/PV/TshR−/− mice. Only a few, small-sized follicles (no tumors) were observed in their thyroids. Recently, Franco et al. established another mouse model (14). They crossed mice with a thyroid-specific knock-in of BrafV600E (LSL-BrafV600E/TPO-Cre) with TshR−/− mice. By three weeks (at this time point, thyroid cancer was seen in LSL-BrafV600E/TPO-Cre mice), they developed benign tumors without characteristic nuclear features of human PTCs. However, by nine weeks, they progressed to low-grade PTCs.
Genomic instability (GIN) is a hallmark of cancers and has been suggested to play a crucial role in the progression of thyroid tumors (15,16). p53-binding protein 1 (53BP1) is a DNA damage response protein that rapidly localizes at the site of DNA double-strand breaks (DSB) together with phosphorylated ATM and many other related proteins such as γH2AX (17–20). One of the manifestations of GIN is the induction of DSB, and it has been demonstrated that 53BP1 focus formation can be used as a marker of GIN (16,21). Advanced and more malignant thyroid cancer tissues showed intense and abnormal nuclear 53BP1 staining patterns compared with low-grade cancers, suggesting increased GIN in advanced cancer cells.
To examine the role of TSH signaling in thyroid carcinogenesis in vivo, we developed mice with thyrocyte-targeted BRAFV600E expression crossed with the TshR gene knockout mice. We also evaluated cell proliferation and GIN status in their thyroids, and demonstrate that TSH signaling may be related to the induction of GIN and may then enhance the malignant phenotype.
Materials and Methods
Cell lines
The PC-BRAFV600E-6 line was derived from PCCL3 cells, a clonal rat thyroid cell line, to obtain doxycycline-inducible BRAFV600E expression as previously described (22). The cells were maintained in H4 complete medium consisting of Coon's F-12 medium (Sigma) supplemented with 5% tetracycline screened fetal bovine serum (FBS; Hyclone), 0.3 mg/mL l-glutamine, 1 mIU/mL TSH, 10 μg/mL insulin, 5 μg/mL apo-transferrin, 10 nM hydrocortisone, and penicillin/streptomycin. H3 medium contained the same constituents except for TSH. To induce BRAFV600E expression, the cells were treated with 1 μg/mL of doxycycline.
Mice
FVB/N, bovine Tg promoter-driven BRAFV600E transgenic mice—Tg-BRAF2 mice (11)—were obtained from Dr. James Fagin (University of Cincinnati; currently Memorial Sloan-Kettering Cancer Center). C57/Bl6J-129 hybrids—thyrotropin receptor null mice (TshR−/−) (23)—were obtained from Jackson Laboratory Inc. Both strains were crossed, and offspring were genotyped using tail DNA as described previously (11,23). All the mice were bred in a specific pathogen-free facility. Mice with TshR−/− were fed with a desiccated thyroid powder (100 ppm)-supplemented diet (23). Animal care and all experimental procedures were performed in accordance with the Guidelines for Animal Experimentation of Nagasaki University with the approval of the Institutional Animal Care and Use Committee.
Thyroid weight and histology
Mice were sacrificed at 12 and 24 weeks of age. Thyroid glands were removed, weighed, and frozen in liquid nitrogen for real-time reverse-transcription PCR (RT-PCR) described below. Formalin-fixed and paraffin-embedded (FFPE) thyroid tissue sections were stained with hematoxylin and eosin (H&E) for histology and pathological scoring of lesions. Aggressiveness of the tumor tissues was scored based on the sum of the following parameters: invasion, yes=1 or no=0; solid pattern, ≥40%=1 or <40%=0.
Real-time quantitative RT-PCR
Total RNA was isolated from homogenized thyroid tissues using ISOGEN reagent (Nippon Gene). Two hundred nanograms of the total RNA was reverse transcribed with High Capacity RNA-to-cDNA Kit (Applied Biosystems) in the presence of random hexamers to generate cDNA. The following quantitative PCR was carried out in a Thermal Cycler Dice Real-time system (TaKaRa Bio) using QuantiTect SYBR Green PCR Kit (Qiagen). The sequences of used primer pairs were described previously (11). The cycle threshold value, which was determined using second derivative, was used to calculate the normalized expression of the indicated mRNAs with Q-Gene software (24) using β-actin for normalization.
Immunohistochemistry
For cleaved caspase-3 detection, FFPE sections were deparaffinized and subjected to antigen retrieval by microwave treatment in citrate buffer. After blocking, the sections were incubated with anticleaved caspase-3 antibody #9661 (1:250 dilution; Cell Signaling Technology) overnight at 4°C. Staining was performed using the Vectastain Elite ABC Kit (Vector Laboratories) and 3,3′-diaminobenzidine (DAB). For F4/80 staining, FFPE sections were deparaffinized and treated with proteinase K. After blocking, the sections were incubated with anti-mouse F4/80 antibody Cl:A3-1 (1:50 dilution; AbD Serotec) overnight at 4°C followed by incubation with HRP-conjugated anti-rat IgG secondary antibody (Dako). Visualization was done with DAB.
Immunofluorescence
FFPE sections were deparaffinized and subjected to antigen retrieval by microwave treatment in citrate buffer. For 53BP1/Ki67 co-staining, the tissues were then incubated with a mixture of rabbit anti-53BP1 antibody A300-272A (1:200 dilution; Bethyl Laboratories Inc.) and rat monoclonal anti-mouse Ki67 antibody Clone TEC 3 (1:50 dilution; Dako) in 5% milk in TBS-T buffer. After two hours of incubation at room temperature, the tissues were incubated with another mixture of Alexa Fluor 532-conjugated goat anti-rabbit and Alexa Fluor 647-conjugated goat anti-rat antibodies (Invitrogen). For γH2AX staining, anti-phosphorylated H2AX (Ser139) Clone 2F3 (1:500 dilution; BioLegend) and Alexa Fluor 488-conjugated anti-mouse antibody (Invitrogen) were used. After counterstaining with 4′,6-diamidino-2-phenylindole (DAPI), images were visualized and captured using a fluorescence microscope (Leica DM 6000B). Immunoreactivity signals were analyzed at 400× magnification in four to six fields of view per sample, and analyzed with the NIH Image J toolkit (http://rsbweb.nih.gov/ij/index.html).
For detection of γH2AX in PC-BRAFV600E cells, the cells were grown on coverslips and pretreated with H3 medium for seven days. The medium was then replaced by H3 or H4 medium with or without doxycycline. After incubation for 24 hours, the cells were fixed with 10% formalin and permeabilized with 0.5% Triton-X in PBS. Anti-phosphorylated H2AX (Ser139) antibody Clone JBW301 (1:1000 dilution, Millipore) was used as a primary antibody. After labeling with a secondary antibody conjugated with Alexa Fluor 488, images were acquired using a fluorescence microscope.
Matrigel invasion assay
The Matrigel invasion assay was performed in triplicate using the PC-BRAFV600E-6 cells. Matrigel (BD Biosciences; 60 μg/transwell) diluted with serum-free medium was placed on 24-well format transwell cell culture inserts (Corning; 8-μm pore size). After seven days of TSH starvation in H3 medium, the cells were resuspended in 200 μL serum-free medium with and without doxycycline in the presence or absence of TSH and added to the upper chamber at 8×104 cells per insert. Next, 750 μL complete medium with 10% FBS was added to the lower chamber of each transwell. After 24 hours' incubation, nonmigrated cells at the upper side of the membrane were mechanically removed with cotton swabs, and the transwells were washed with PBS, fixed with 100% methanol, and stained with Giemsa Stain solution (Sigma). Cells that invaded the Matrigel and migrated to the lower side of the membrane were quantified in 10 random images per transwell taken at 10× magnification in a bright field microscope. The number of migrated cells were counted using the NIH Image J toolkit.
Statistical analysis
Differences between groups were examined for statistical significance by unpaired t-test or analysis of variance (ANOVA) followed by Tukey's post-hoc test. A p-value <0.05 was considered statistically significant.
Results
BRAFV600E generates tumors in mice, and TSH increases tumor size
We first crossed Tg-BRAF2 mice with TshR−/− mice to obtain Tg-BRAF2/TshR+/− mice, and then the Tg-BRAF2/TshR+/− mice were further crossed with TshR−/− mice. Offspring were genotyped and divided into four groups: Brafwt/TshR+/− (group 1), Brafwt/TshR−/− (group 2), Tg-BRAF2/TshR+/− (group 3), and Tg-BRAF2/TshR−/− (group 4). Although previous reports demonstrated Tg expression in the thyroids of TshR−/− mice (23,25), we assessed BRAFV600E expression by real-time RT-PCR because the Tg promoter activity is dependent on TSH stimulation in vitro, and the bovine Tg promoter drove the expression of BRAFV600E in our model. In the thyroid lobes collected at 12 weeks, the BRAFV600E expression levels in the group 3 mice were comparable to those in group 4 (Fig. 1), indicating that BRAFV600E was properly transcribed regardless of TSH signaling. We also examined other thyroid-specific genes, the sodium-iodide symporter (Nis) and Tg. Nis expression was remarkably downregulated in TshR−/− mice (groups 2 and 4; Fig. 1). Regarding Tg, its expression in group 3 was also reduced, as well as in groups 2 and 4 (Fig. 1), suggesting that the dedifferentiating effect of BRAFV600E on the expression of Tg was strong.
FIG. 1.
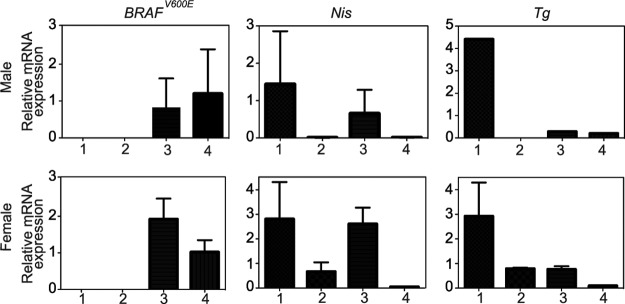
Effect of TshR knockout on mRNA levels of transgene and thyroid-specific genes. Total RNA was extracted from the thyroids at 12 weeks and subjected to quantitative reverse-transcription polymerase chain reastion (qRT-PCR) for the expression of the indicated genes after normalization to β-actin. Columns represent means and standard error of the mean (SEM) of values obtained from three independent biological replicates. 1, group 1 (Brafwt/TshR+/−); 2, group 2 (Brafwt/TshR−/−); 3, group 3 (Tg-BRAF2/TshR+/−); 4, group 4 (Tg-BRAF2/TshR−/−).
The ratio of thyroid weight to body weight was compared among the four groups at both 12 and 24 weeks. Note that body weights of group 2 and group 4 mice (TshR−/−) at five weeks were smaller than those of group 1 and group 3 mice (TshR+/−), presumably due to hypothyroidism. However, these differences were diminished until 12 weeks (following a thyroid powder-supplemented diet beginning at four weeks (Supplementary Fig. S1; Supplementary Data are available online at www.liebertpub.com/thy). The ratio of thyroid weight to body weight was the highest in group 3 (Fig. 2A). However, there was no significant difference between groups 1, 2, and 4 (Fig. 2A). These data demonstrate the obvious influence of TSH signaling on BRAFV600E-induced goiter formation. Representative low-power images of thyroid sections in each group are shown in Figure 2B. Group 1 showed a normal structure (Fig. 2B). The thyroids in group 2 had fewer follicles and some adipose tissue (Fig. 2B). In group 3, the thyroid glands were markedly enlarged, and no normal follicles were observed (Fig. 2B). These data are consistent with previous reports (11,23). In group 4 mice with BRAFV600E without TSH signaling, the normal follicular architecture was also completely disrupted, and their morphology was similar to that in group 3 (Fig. 2B). However, the size of the glands was much smaller (Fig. 2B), which is in accordance with their lower weights (Fig. 2A). No metastasis was found in either group 3 or group 4.
FIG. 2.

Effect of TSH signaling on BRAFV600E-induced thyroid tumor formation. (A) Dot plots represent individual values and means (bars) of ratio of thyroid lobe weights (mg) to body weights (g) at sacrifice. (B) Representative hematoxylin and eosin (H&E) stained sections of thyroid lobes at 24 weeks. *p<0.05.
TSH signaling confers more aggressive features in BRAFV600E-induced thyroid tumors
Next, we performed microscopic analyses of groups 3 and 4 using H&E stained sections. Figure 3 shows representative findings of BRAFV600E-induced tumors in the group 3 mice. The mice developed neoplastic lesions distorting the entire thyroid glands (Fig. 3A). Solid pattern (Fig. 3C), capsular invasion (Fig. 3D), and tall cells lining papillary structures (Fig. 3E) were observed, and these features were similar to human aggressive PTCs. Papillary projection (Fig. 3B) and nuclear clearing (Fig. 3F) were also observed.
FIG. 3.

Representative images of H&E stained sections from thyroid tissues of group 3 (Tg-BRAF2/TshR+/−) mice: (A) low power view (×100) of the thyroid neoplasm; (B) intrafollicular papillary projection (×200); (C) solid pattern replacing normal thyroid architecture (×100); (D) extracapsular extension (arrows depict remnant of the capsule [R; ×200]); (E) tall cells (T; ×400); (F) nuclear clearing (×400).
Most of the mice in both groups developed neoplasias with features characteristic of human PTC, including nuclear changes such as nuclear clearing, grooves, and inclusions (Table 1). Tall cells, which are thought to be an aggressive subtype, were also seen in 7/20 and 8/19 mice in group 3 and group 4 respectively (Table 1). By contrast, a solid pattern, which is another aggressive feature in PTC, was observed more frequently in group 3 (6/20) than in group 4 (3/19; Table 1). Capsular invasion was only observed in group 3 (5/20), while group 4 showed no features suggestive of capsular invasion or extrathyroidal extension (Table 1). A papillary pattern was more common in group 4 (data not shown). A time-dependent trend was observed, particularly in group 4 where the papillary pattern at 12 weeks was replaced by solid features at 24 weeks (Table 1; data not shown). We carried out histopathological scoring of the neoplastic lesions in groups 3 and 4. The aggressiveness score for group 3 (0.550±0.170) was higher than that of group 4 (0.158±0.086), which was statistically significant (p=0.0498, unpaired t-test). These data suggest that TSH signaling results in a more aggressive phenotype in the BRAFV600E-induced thyroid tumors.
Table 1.
Histopathological Scoring of Thyroid Lesions in BRAFV600E/TshR+/− and BRAFV600E/TshR−/− Mice
| Overall process | Cellular features | |||||
|---|---|---|---|---|---|---|
| Genotype | Age (weeks) | Neoplastic | Nuclear changes | Tall cells | Solid pattern | Capsular invasion |
| BRAFV600E/TshR+/− | 12 | 9/10 | 9/10 | 3/10 | 2/10 | 2/10 |
| 24 | 10/10 | 9/10 | 4/10 | 4/10 | 3/10 | |
| BRAFV600E/TshR−/− | 12 | 8/9 | 7/9 | 3/9 | 0/9 | 0/9 |
| 24 | 9/10 | 8/10 | 5/10 | 3/10 | 0/10 | |
TSH prevents apoptosis but not macrophage infiltration
To examine the influence of apoptosis on the difference between groups 3 and 4, we performed immunohistochemistry for cleaved caspase-3. The percentage of positively stained cells was significantly higher in group 4 than in group 3 at 12 weeks (Fig. 4A). There was also a similar tendency at 24 weeks, but no statistical significance was observed (Fig. 4A). These results suggest that the TSH signal has a role in preventing apoptosis, and this may cause, at least in part, the difference in thyroid size between groups 3 and 4.
FIG. 4.

Evaluation of apoptosis and macrophage infiltration. Immunostaining was performed using cleaved caspase-3 (A) and F4/80 (B) antibodies. Each dot represents the percentage of indicated positive cells relative to ∼500 cells in four to six fields of view counted under bright field microscopy. Horizontal bars indicate means. *p<0.05.
Recently, it has been demonstrated that tumor-associated macrophages have a role in the progression of BRAFV600E-induced thyroid cancer, particularly PTC, to poorly differentiated thyroid carcinoma (26). To test this possibility, we also performed immunohistochemistry for F4/80. As shown in Fig. 4B, however, there was no difference between groups 3 and 4.
TSH enhances BRAFV600E-induced cell invasiveness in rat thyroid cells
To support the in vivo observation, we utilized a doxycycline-inducible BRAFV600E rat thyroid cell line—PC-BRAFV600E-6 cells (22)—and performed a cell invasion assay in vitro. As shown in Fig. 5, BRAFV600E expression (treatment with doxycycline) robustly enhanced cell invasiveness in the presence of TSH (4H). However, deprivation of TSH (3H) significantly suppressed the invasive ability. These data also demonstrate that BRAFV600E and TSH cooperatively induce invasiveness in thyroid cells.
FIG. 5.
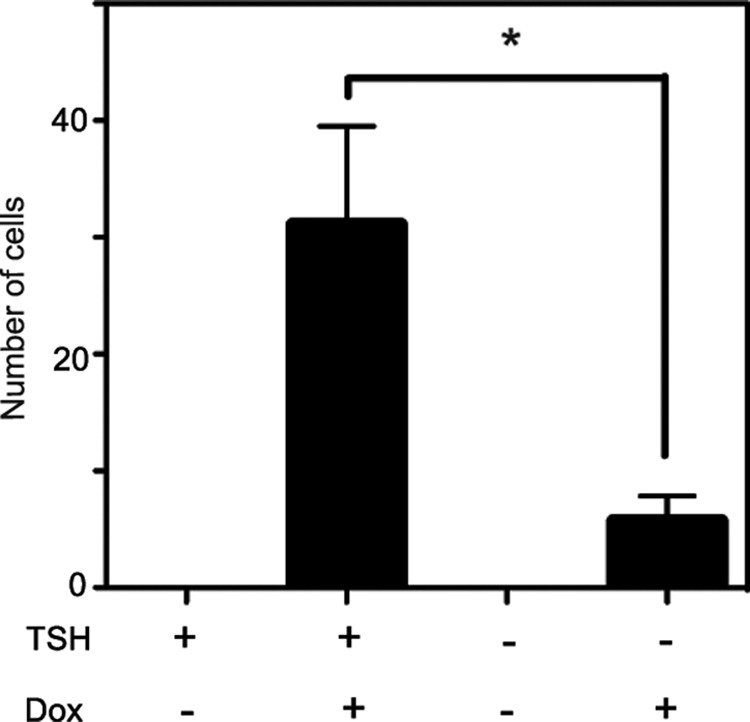
Effect of TSH and/or BRAFV600E on cell invasion. Invasion was examined using Matrigel and Transwell chambers. After TSH starvation for seven days, PC-BRAFV600E-6 cells were treated with or without doxycycline in the presence or absence of thyrotropin (TSH) for 24 hours, and subjected to Matrigel invasion assay. Each bar indicates mean and SEM of three wells. *p<0.05. Similar results were obtained in two independent experiments.
TSH signaling induces GIN
To explore the influence of TSH signaling and BRAFV600E on tumor growth and induction of aggressive features, we performed dual-label immunofluorescence for 53BP1 and Ki67. Representative merged images of each group are presented in Fig. 6A. The percentage of 53BP1 focus-positive and Ki67-positive cells was calculated. Regarding the comparison between group 3 and 4, the frequency of 53BP1 focus-positive cells in group 3 was significantly higher than that in group 4 in 24-week male and 12-week male mice (Fig. 6B). The frequency in 24-week female mice in group 3 also showed a higher trend compared to group 4. However, it did not reach statistical significance (Fig. 6B). Notably, the group 1 mice also exhibited higher percentages at 24 weeks, and the frequencies in female mice were generally higher than those in male mice (Fig. 6B). Regarding the proliferative index, Ki67 positivity in the group 1 and 2 mice was very low (Fig. 6B). By contrast, in groups 3 and 4, BRAFV600E robustly increased the Ki67-positive cells (Fig. 6B). However, there was no statistical difference between groups 3 and 4 (Fig. 6B). To confirm the above observations regarding GIN, we also used another DNA damage marker—γH2AX (phosphorylated H2AX at Ser-139). The frequency of γH2AX foci in group 3 was significantly higher than that in group 4 in all age/sex pairs (Fig. 6C). Similar findings were also obtained in vitro using the PC-BRAFV600E-6 cells (Fig. 6D).
FIG. 6.

Evaluation of genomic instability and cell proliferative activity. (A) Representative images of immunofluorescence of the thyroid tissues obtained from indicated groups. White and yellow arrowheads indicate Ki67 positive cells (green) and 53BP1 foci (red) respectively. Nuclei were stained with DAPI (blue). (B) Dot plot analysis of 53BP1 foci and Ki67 in thyrocytes. Each data point represents a single subject and depicts the percentage of cells having 53BP1 foci or Ki67 at 12 and 24 weeks. In each point, at least 2000 cells were analyzed. Horizontal bars indicate means; p-values generated by analysis of variance (ANOVA) and post-test values are also provided. (C) Dot plot analysis of γH2AX foci. In each point, at least 2000 cells were analyzed. Horizontal bars indicate means. *p<0.05 by unpaired t-test. (D) γH2AX staining in PC-BRAFV600E-6 cells. After TSH starvation for seven days, the cells were treated with or without doxycycline in the presence or absence of TSH for 24 hours, and subjected to immunofluorescence for γH2AX. Each bar indicates mean and SEM of three wells. *p<0.05 by ANOVA and post-test. Color images available online at www.liebertpub.com/thy
Discussion
In the present study, we show that TSH signaling confers more aggressive features in BRAFV600E-induced thyroid tumors in vivo. We utilized the Tg-BRAF2 mice in which the Tg gene promoter drives BRAFV600E expression in thyroid follicular cells (11). Even though Tg expression was highly suppressed in group 3 and group 4 mice thyroids, BRAFV600E expression was still maintained in both groups. This might be due to a lack of distant regulatory element(s) or particularities of the integration site of the transgene in this animal model. Compared with the first report of the Tg-BRAF2 mice (11), our group 3 mice (Tg-BRAF2/TshR+/−) showed weaker phenotypes: a lower percentage of tall cells (79–87% in (11) vs. 30–40% in group 3) and capsular invasion (73–93% in (11) vs. 20–30% in group 3). This is presumably due to hemizygosity of the TshR and consistent with the report by Franco et al. demonstrating that thyroid weights in LSL-BrafV600E/TPO-Cre/TshR+/− mice were approximately half of those in LSL-BrafV600E/TPO-Cre mice (14). Although growth, histology, and serum T3/T4/TSH levels were all normal in the TshR+/− mice (23), the influence of difference in TSH signal between wild type and TshR+/− on thyroid tumor formation is probably significant, especially with elevated serum TSH levels. These findings also suggest that TSH signaling plays an important role in BRAFV600E-induced thyroid tumorigenesis.
Although our current study and the report by Franco et al. (14) used similar mouse models (Tg-BRAF2 and LSL-BrafV600E/TPO-Cre respectively), there are some differences. First, Ii our model, BRAFV600E expression was supposedly induced from embryonic day 15, while in the model by Franco et al., the recombination of LSL-BrafV600E was gradually induced and completed until 7–10 days postnatally. Second, although our group 4 (Tg-BRAF2/TshR−/−) mice had some characteristics of human PTC such as tall cells and nuclear features at both 12 and 24 weeks, the LSL-BrafV600E/TPO-Cre/TshR−/− mice by Franco et al. did not show any nuclear features of human PTC at three weeks and no tall cells even at nine weeks. This difference might be due to the time of onset of the expression of the oncoprotein. Prolonged expression beginning at the embryonic stage may induce characteristic features of human PTC even in the absence of TSH signaling. Despite some differences, however, our overall results are quite consistent with the study by Franco et al. (14).
We analyzed GIN using immunofluorescence for 53BP1. Interestingly, 53BP1 foci formation in the group 1 mice was increased in a time-dependent manner (12 and 24 weeks) even though proliferation of thyrocytes (Ki67) was absent, suggesting that the foci were not caused by TSH-mediated DNA replication. Since Nis and thyroid peroxidase (Tpo) expression (but not Tg) was highly suppressed [(25) and our data] in mice with TshR−/−, oxidative stress by iodide and/or Tpo during thyroid hormonogenesis may be responsible for DSB formation. However, further research is needed to confirm this connection. On the other hand, in BRAFV600E-induced thyroid tumor cells, the frequency of 53BP1 foci in group 3 was also higher than that in group 4. Since the 53BP1 frequency in group 1 was lower than that in group 3 in 12-week-old mice, BRAFV600E (22) and TSH signaling may cooperatively induce GIN in thyrocytes. Our experiments showing an increased number of γH2AX foci strongly support this notion.
Our data in the present study suggest that the activity of the TSH signaling pathway confers more aggressive features in BRAFV600E-induced thyroid tumors in vivo. This might be due, in part, to accelerated genomic instability. Generally, these results suggest that TSH signaling plays an important role in tumor progression rather than tumor initiation in our model. Since several epidemiological studies have accumulated evidence of an association between serum TSH levels and risk of malignancy in patients with thyroid nodules (27–30), the current study may provide additional insight in terms of prevention of this type of cancer.
Supplementary Material
Acknowledgments
We thank Dr. James Fagin (Memorial Sloan-Kettering Cancer Center) for kindly providing the Tg-BRAF2 mice and PC-BRAFV600E-6 cells. This work was supported in part by Grants-in-Aid for Scientific Research (NM #23591357, SY #22390189) from the Japan Society for the Promotion of Science.
Author Disclosure Statement
No competing financial interests exist.
References
- 1.Hundahl SA, Cady B, Cunningham MP, Mazzaferri E, McKee RF, Rosai J, Shah JP, Fremgen AM, Stewart AK, Holzer S.2000Initial results from a prospective cohort study of 5583 cases of thyroid carcinoma treated in the united states during 1996. U.S. and German Thyroid Cancer Study Group. An American College of Surgeons Commission on Cancer Patient Care Evaluation study. Cancer 89:202–217 [DOI] [PubMed] [Google Scholar]
- 2.Kimura ET, Nikiforova MN, Zhu Z, Knauf JA, Nikiforov YE, Fagin JA.2003High prevalence of BRAF mutations in thyroid cancer: genetic evidence for constitutive activation of the RET/PTC-RAS-BRAF signaling pathway in papillary thyroid carcinoma. Cancer Res 63:1454–1457 [PubMed] [Google Scholar]
- 3.Soares P, Trovisco V, Rocha AS, Lima J, Castro P, Preto A, Maximo V, Botelho T, Seruca R, Sobrinho-Simoes M.2003BRAF mutations and RET/PTC rearrangements are alternative events in the etiopathogenesis of PTC. Oncogene 22:4578–4580 [DOI] [PubMed] [Google Scholar]
- 4.Frattini M, Ferrario C, Bressan P, Balestra D, De Cecco L, Mondellini P, Bongarzone I, Collini P, Gariboldi M, Pilotti S, Pierotti MA, Greco A.2004Alternative mutations of BRAF, RET and NTRK1 are associated with similar but distinct gene expression patterns in papillary thyroid cancer. Oncogene 23:7436–7440 [DOI] [PubMed] [Google Scholar]
- 5.Xing M.2005BRAF mutation in thyroid cancer. Endocr Relat Cancer 12:245–262 [DOI] [PubMed] [Google Scholar]
- 6.Wan PT, Garnett MJ, Roe SM, Lee S, Niculescu-Duvaz D, Good VM, Jones CM, Marshall CJ, Springer CJ, Barford D, Marais R.2004Mechanism of activation of the RAF-ERK signaling pathway by oncogenic mutations of B-RAF. Cell 116:855–867 [DOI] [PubMed] [Google Scholar]
- 7.Xing M.2007BRAF mutation in papillary thyroid cancer: pathogenic role, molecular bases, and clinical implications. Endocr Rev 28:742–762 [DOI] [PubMed] [Google Scholar]
- 8.Knauf JA, Fagin JA.2009Role of MAPK pathway oncoproteins in thyroid cancer pathogenesis and as drug targets. Curr Opin Cell Biol 21:296–303 [DOI] [PubMed] [Google Scholar]
- 9.Hundahl SA, Fleming ID, Fremgen AM, Menck HR.1998A National Cancer Data Base report on 53,856 cases of thyroid carcinoma treated in the U.S., 1985–1995 [see comments]. Cancer 83:2638–2648 [DOI] [PubMed] [Google Scholar]
- 10.Riesco-Eizaguirre G, Gutierrez-Martinez P, Garcia-Cabezas MA, Nistal M, Santisteban P.2006The oncogene BRAF V600E is associated with a high risk of recurrence and less differentiated papillary thyroid carcinoma due to the impairment of Na+/I− targeting to the membrane. Endocr Relat Cancer 13:257–269 [DOI] [PubMed] [Google Scholar]
- 11.Knauf JA, Ma X, Smith EP, Zhang L, Mitsutake N, Liao XH, Refetoff S, Nikiforov YE, Fagin JA.2005Targeted expression of BRAFV600E in thyroid cells of transgenic mice results in papillary thyroid cancers that undergo dedifferentiation. Cancer Res 65:4238–4245 [DOI] [PubMed] [Google Scholar]
- 12.Thomas GA, Williams ED.1991Evidence for and possible mechanisms of non-genotoxic carcinogenesis in the rodent thyroid. Mutat Res 248:357–370 [DOI] [PubMed] [Google Scholar]
- 13.Lu C, Zhao L, Ying H, Willingham MC, Cheng SY.2010Growth activation alone is not sufficient to cause metastatic thyroid cancer in a mouse model of follicular thyroid carcinoma. Endocrinology 151:1929–1939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Franco AT, Malaguarnera R, Refetoff S, Liao XH, Lundsmith E, Kimura S, Pritchard C, Marais R, Davies TF, Weinstein LS, Chen M, Rosen N, Ghossein R, Knauf JA, Fagin JA.2011Thyrotropin receptor signaling dependence of Braf-induced thyroid tumor initiation in mice. Proc Natl Acad Sci USA 108:1615–1620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sobrinho-Simoes M, Preto A, Rocha AS, Castro P, Maximo V, Fonseca E, Soares P.2005Molecular pathology of well-differentiated thyroid carcinomas. Virchows Arch 447:787–793 [DOI] [PubMed] [Google Scholar]
- 16.Nakashima M, Suzuki K, Meirmanov S, Naruke Y, Matsuu-Matsuyama M, Shichijo K, Saenko V, Kondo H, Hayashi T, Ito M, Yamashita S, Sekine I.2008Foci formation of P53-binding protein 1 in thyroid tumors: activation of genomic instability during thyroid carcinogenesis. Int J Cancer 122:1082–1088 [DOI] [PubMed] [Google Scholar]
- 17.Ward IM, Minn K, Jorda KG, Chen J.2003Accumulation of checkpoint protein 53BP1 at DNA breaks involves its binding to phosphorylated histone H2AX. J Biol Chem 278:19579–19582 [DOI] [PubMed] [Google Scholar]
- 18.Schultz LB, Chehab NH, Malikzay A, Halazonetis TD.2000p53 binding protein 1 (53BP1) is an early participant in the cellular response to DNA double-strand breaks. J Cell Biol 151:1381–1390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rappold I, Iwabuchi K, Date T, Chen J.2001Tumor suppressor p53 binding protein 1 (53BP1) is involved in DNA damage-signaling pathways. J Cell Biol 153:613–620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Anderson L, Henderson C, Adachi Y.2001Phosphorylation and rapid relocalization of 53BP1 to nuclear foci upon DNA damage. Mol Cell Biol 21:1719–1729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Naruke Y, Nakashima M, Suzuki K, Matsuu-Matsuyama M, Shichijo K, Kondo H, Sekine I.2008Alteration of p53-binding protein 1 expression during skin carcinogenesis: association with genomic instability. Cancer Sci 99:946–951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mitsutake N, Knauf JA, Mitsutake S, Mesa C, Jr, Zhang L, Fagin JA.2005Conditional BRAFV600E expression induces DNA synthesis, apoptosis, dedifferentiation, and chromosomal instability in thyroid PCCL3 cells. Cancer Res 65:2465–2473 [DOI] [PubMed] [Google Scholar]
- 23.Marians RC, Ng L, Blair HC, Unger P, Graves PN, Davies TF.2002Defining thyrotropin-dependent and -independent steps of thyroid hormone synthesis by using thyrotropin receptor-null mice. Proc Natl Acad Sci USA 99:15776–15781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Muller PY, Janovjak H, Miserez AR, Dobbie Z.2002Processing of gene expression data generated by quantitative real-time RT-PCR. Biotechniques 32:1372–1374, 1376, 1378–1379 [PubMed] [Google Scholar]
- 25.Postiglione MP, Parlato R, Rodriguez-Mallon A, Rosica A, Mithbaokar P, Maresca M, Marians RC, Davies TF, Zannini MS, De Felice M, Di Lauro R.2002Role of the thyroid-stimulating hormone receptor signaling in development and differentiation of the thyroid gland. Proc Natl Acad Sci USA 99:15462–15467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Knauf JA, Sartor MA, Medvedovic M, Lundsmith E, Ryder M, Salzano M, Nikiforov YE, Giordano TJ, Ghossein RA, Fagin JA.2011Progression of BRAF-induced thyroid cancer is associated with epithelial-mesenchymal transition requiring concomitant MAP kinase and TGFβ signaling. Oncogene 30:3153–3162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Haymart MR, Glinberg SL, Liu J, Sippel RS, Jaume JC, Chen H.2009Higher serum TSH in thyroid cancer patients occurs independent of age and correlates with extrathyroidal extension. Clin Endocrinol (Oxf) 71:434–439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Boelaert K.2009The association between serum TSH concentration and thyroid cancer. Endocr Relat Cancer 16:1065–1072 [DOI] [PubMed] [Google Scholar]
- 29.Fiore E, Rago T, Provenzale MA, Scutari M, Ugolini C, Basolo F, Di Coscio G, Berti P, Grasso L, Elisei R, Pinchera A, Vitti P.2009Lower levels of TSH are associated with a lower risk of papillary thyroid cancer in patients with thyroid nodular disease: thyroid autonomy may play a protective role. Endocr Relat Cancer 16:1251–1260 [DOI] [PubMed] [Google Scholar]
- 30.Fiore E, Vitti P.2012Serum TSH and risk of papillary thyroid cancer in nodular thyroid disease. J Clin Endocrinol Metab 97:1134–1145 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.