

The crystal structure of the class C extended-spectrum cepahalosporinase ADC-1 has been solved to 1.2 Å resolution.
Keywords: class C β-lactamases, apoenzymes
Abstract
ADC-type class C β-lactamases comprise a large group of enzymes that are encoded by genes located on the chromosome of Acinetobacter baumannii, a causative agent of serious bacterial infections. Overexpression of these enzymes renders A. baumannii resistant to various β-lactam antibiotics and thus severely compromises the ability to treat infections caused by this deadly pathogen. Here, the high-resolution crystal structure of ADC-1, the first member of this clinically important family of antibiotic-resistant enzymes, is reported. Unlike the narrow-spectrum class C β-lactamases, ADC-1 is capable of producing resistance to the expanded-spectrum cephalosporins, rendering them inactive against A. baumannii. The extension of the substrate profile of the enzyme is likely to be the result of structural differences in the R2-loop, primarily the deletion of three residues and subsequent rearrangement of the A10a and A10b helices. These structural rearrangements result in the enlargement of the R2 pocket of ADC-1, allowing it to accommodate the bulky R2 substituents of the third-generation cephalosporins, thus enhancing the catalytic efficiency of the enzyme against these clinically important antibiotics.
1. Introduction
Over the last decade, Acinetobacter baumannii has evolved into a major nosocomial pathogen owing to its wide dissemination in clinics all over the world and its acquisition of various antibiotic-resistance determinants. At present, up to 40–60% of clinical A. baumannii isolates are multidrug-resistant, and infections caused by such bacteria result in very high mortality rates owing to the lack of adequate antibiotic treatment. A. baumannii is increasingly resistant to a variety of β-lactam antibiotics, including penicillins, extended-spectrum cephalosporins and β-lactamase inhibitors. Until recently, a group of β-lactam antibiotics, the carbapenems, were the drugs of choice for the effective treatment of Acinetobacter infections (Coelho et al., 2004 ▶). However, carbapenem-resistant A. baumannii strains are rapidly on the rise, indicating questionable efficacy of these antibiotics (Bogaerts et al., 2008 ▶; Brown & Amyes, 2006 ▶; Park et al., 2009 ▶; Poirel & Nordmann, 2006 ▶).
The production of β-lactamases by A. baumannii constitutes the major mechanism of its resistance to β-lactam antibiotics (Peleg et al., 2008 ▶), complemented by resistance arising from alterations of penicillin-binding proteins and lower expression of porins (Bou et al., 2000 ▶; Clark, 1996 ▶; Fernández-Cuenca et al., 2003 ▶). β-Lactamases of all four known classes (A, B, C and D) are widely spread in clinical Acinetobacter isolates. Among them, a group of chromosomally encoded class C β lactamases, known as ADCs (from Acinetobacter-derived cephalosporinase; Hujer et al., 2005 ▶), have been identified in A. baumannii and A. pittii. Unlike the majority of class C β-lactamases from other microorganisms, ADCs are non-inducible and are normally expressed at low levels, providing resistance to penicillins and narrow-spectrum cephalosporins (Bou & Martínez-Beltrán, 2000 ▶; Hujer et al., 2005 ▶). Acinetobacter isolates are a known reservoir of various insertion sequences (ISs), and the acquisition by ADCs of one of these IS elements, predominantly ISAba1, provides the β-lactamase genes with a strong promoter. As the result of ISAba1 insertion, ADCs are expressed at a much higher levels and are capable of conferring resistance to extended-spectrum cephalosporins such as cefotaxime and ceftazidime (Perez et al., 2007 ▶; Fig. 1 ▶). More recently, the mutant ADC derivatives ADC-33 and ADC-56, which are both capable of hydrolyzing cefepime, have been identified (Rodríguez-Martínez et al., 2010 ▶; Tian et al., 2011 ▶). ADC-56 is a single-amino-acid (R148Q) mutant derivative of ADC-30, while the ability of ADC-33 to hydrolyze cefepime has been linked to the duplication of an alanine residue at position 215. Most alarmingly, mutant ADC enzymes producing resistance to carbapenem antibiotics have recently been identified in clinical A. baumannii isolates (Lee et al., 2013 ▶).
Figure 1.

Structures of four representative cepahalosporin antibiotics, along with aztreonam (a monobactam) and imipenem (a carbapenem) for reference. The atom-numbering scheme given for cephalothin is a representation of the cephalosporins as a whole. The atom numbering for imipenem is also given.
Currently, more than 50 ADC variants have been reported. Most of these enzymes are very similar and differ by between one and 25 amino acids (Zhao & Hu, 2012 ▶). The ADC enzymes share between 35 and 45% sequence identity with other class C enzymes and retain the essential catalytic machinery comprising a conserved triad of residues: a serine, a lysine and a tyrosine. Two of these residues, the serine and the lysine, are also conserved in the two other classes of serine β-lactamases: the class A and the class D enzymes. In the class D enzymes carboxylation of this lysine is critical for activity (Golemi et al., 2001 ▶), whereas this is not the case in the class A and C enzymes. In all three classes hydrolysis of β-lactam antibiotics proceeds in two steps: (i) the formation of an acyl-enzyme intermediate at the serine side chain followed by (ii) water-activated deacylation and subsequent release of the deactivated drug. In the class A and D enzymes the mechanism is reasonably well understood; however, in the class C enzymes the specifics of the reaction mechanism are still under debate. In the acylation step, the serine residue is activated by deprotonation; early studies suggested that the general base which promotes this step was the conserved tyrosine (Dubus et al., 1996 ▶; Lobkovsky et al., 1994 ▶), but recent work suggests that the lysine abstracts the proton from the serine and the tyrosine protonates the β-lactam N5 atom (Tripathi & Nair, 2013 ▶). It is thought that activation of the deacylating water molecule is facilitated by the tyrosine (Chen et al., 2009 ▶). Despite their clinical importance and widespread occurrence in Acinetobacter isolates, ADC-type β-lactamases are poorly studied and there is currently no structural information available for these enzymes. ADC-1 was the first member of the ADC subfamily to be characterized (Bou & Martínez-Beltrán, 2000 ▶), and here we report the crystal structure of this extended-spectrum class C β-lactamase at 1.2 Å resolution.
2. Experimental procedures
2.1. Cloning of the ADC-1 gene
The gene for ADC-1 from A. baumannii (GenBank accession No. AJ009979; Bou & Martínez-Beltrán, 2000 ▶) was optimized for expression in Escherichia coli (GenScript). The predicted leader sequence of ADC-1 was replaced by the outer membrane protein (OmpA) leader sequence to facilitate the efficient transport of the enzyme to the bacterial periplasm. For determination of the minimum inhibitory concentration (MIC), the construct was cloned between the NdeI and HindIII sites of the vector pHF016 (Frase et al., 2009 ▶) and transformed into E. coli JM83. For ADC-1 expression and purification from the cytoplasm, the ADC-1 gene was amplified using the pair of primers ADC-1_Opt_Dir (5′-TATATACATATGGGCAACACGCCGAAAGATCAAG) and oHF 103 (5′-AGTGCGGCCGCAAGCTTA) to remove the OmpA leader sequence. The PCR product was cloned between the NdeI and HindIII sites of pET-24a(+) (Invitrogen) and transformed into chemically competent E. coli BL21(DE3) cells.
2.2. MIC determination
The susceptibility of E. coli JM83 harboring the gene for ADC-1 towards β-lactam antibiotics was studied using the broth microdilution technique as recommended by the CLSI guidelines (Clinical and Laboratory Standards Institute, 2009 ▶). E. coli JM83 without the vector was used as a control for the MIC determination. The cultures in Mueller–Hinton II broth (Difco) were diluted to a final inoculum of 5 × 105 CFU ml−1. The plates were incubated for 16–20 h at 37°C before the results were analyzed. The MICs were determined after repeating each experiment in triplicate.
2.3. Protein expression and purification
For expression and purification, the E. coli BL21(DE3) transformant harboring the ADC-1 gene was selected on LB agar plates supplemented with 60 µg ml−1 kanamycin. The selected clone was grown in 500 ml LB medium supplemented with 60 µg ml−1 kanamycin at 37°C until the culture reached an optical density of 0.4 at 600 nm. Expression of protein was induced by the addition of isopropyl β-d-1-thiogalactopyranoside to a final concentration of 0.4 mM and the culture was grown for a further 18 h at 22°C. The cells were harvested by centrifugation at 4000g for 15 min at 4°C and resuspended in 80 ml buffer A (20 mM Tris pH 7.0). The cells were sonicated and centrifuged at 20 000g for 30 min at 4°C. The supernatant was dialyzed against buffer A at 4°C and loaded onto a DEAE anion-exchange column (Bio-Rad) pre-equilibrated with buffer A. The enzyme was eluted with a linear NaCl gradient (0–200 mM) in buffer A. The fractions containing enzyme were eluted in 50–120 mM NaCl. The purity of the fractions was evaluated by 12% SDS–PAGE. The fractions were pooled together, dialyzed in buffer B (20 mM Tris, pH 7.0, 20 mM NaCl) and loaded onto a High S cation-exchange column (Bio-Rad) equilibrated in buffer B. ADC-1 was eluted with a linear gradient of 20–500 mM NaCl in buffer B. The purified ADC-1 was obtained from fractions containing 200–350 mM NaCl. These fractions were analyzed on 12% SDS–PAGE, pooled, concentrated and dialyzed in 20 mM Tris, pH 7.0, 10 mM NaCl. The enzyme was stored in aliquots at 4°C after the concentration of the purified ADC-1 had been evaluated spectrophotometrically using its theoretical extinction coefficient (∊280 = 46 300 M −1 cm−1; Gasteiger et al., 2003 ▶).
2.4. Enzyme kinetics
A Cary 50 spectrophotometer (Varian) was used to monitor the hydrolysis of β-lactam antibiotics in the presence of ADC-1. The reaction was carried out at 22°C in 50 mM sodium phosphate, pH 7.4, varying the concentration of the β-lactam substrate, and was supplemented with 0.04 mg ml−1 bovine serum albumin (BSA) when the final enzyme concentration was below 10 nM. The following wavelengths and extinction coefficients were used for the various substrates in the assay: ampicillin, Δ∊235 = −670 M −1 cm−1; benzylpenicillin, Δ∊232 = −1096 M −1 cm−1; oxacillin, Δ∊260 = +440 M −1 cm−1; piperacillin, Δ∊235 = −793 M −1 cm−1; cephalothin, Δ∊262 = −8610 M −1 cm−1; ceftazidime, Δ∊260 = −10 500 M −1 cm−1; cefotaxime, Δ∊265 = −6643 M −1 cm−1; ceftriaxone, Δ∊265 = −6668 M −1 cm−1. The steady-state velocity during the linear phase of the time courses of the reaction was plotted as a function of β-lactam substrate concentration to determine k cat, K m and k cat/K m. The data were fitted by nonlinear regression with the Michaelis–Menten equation using Prism 5 (GraphPad Software Inc.).
2.5. Crystallization and data collection
Initial crystallization screening was performed with PEG/Ion and PEG/Ion 2, which are commercially available from Hampton Research. Sitting-drop trials were performed in Intelli-Plates (Art Robbins Instruments) with a reservoir volume of 80 µl and drops comprising 1 µl reservoir solution mixed with 1 µl protein solution at a concentration of 24 mg ml−1. The plates were incubated at 4 and 15°C and were monitored periodically. Diffraction-quality crystals were obtained from unbuffered 0.2 M lithium nitrate, 20% PEG 3350 at 15°C.
A single ADC-1 crystal was flash-cooled in a cryoprotectant comprising crystallization buffer augmented with 30% glycerol. The crystal diffracted to 1.2 Å resolution and belonged to a primitive monoclinic space group, with unit-cell parameters a = 43.07, b = 182.15, c = 50.37 Å, β = 98.8°. The Matthews coefficient (Matthews, 1968 ▶) assuming two molecules in the asymmetric unit was 2.4 Å3 Da−1 (49% solvent content). A complete data set comprising 360 images with a rotation angle of 0.5° was collected from this crystal on beamline BL14-1 at the Stanford Synchrotron Radiation Lightsource (SSRL) using X-rays at 12 657 eV (0.98093 Å) and a Rayonix MX325 CCD detector. The data were processed with XDS (Kabsch, 2010 ▶) and scaled with SCALA from the CCP4 suite of programs (Winn et al., 2011 ▶). The program POINTLESS (Evans, 2006 ▶) indicated that the space group was P21. The final data-collection statistics are given in Table 1 ▶.
Table 1. Data-collection statistics.
Values in parentheses are for the highest resolution shell (1.25–1.20 Å).
| Maximum resolution (d min) (Å) | 1.20 |
| Observed reflections | 861438 |
| Unique reflections to d min | 227477 |
| R merge † (%) | 4.9 (64.7) |
| 〈I/σ(I)〉 | 15.1 (2.2) |
| Completeness (%) | 95.9 (91.6) |
| CC1/2 ‡ | 99.9 (65.9) |
| Multiplicity | 3.8 |
| Wilson B (Å2) | 10.7 |
| Unit-cell parameters | |
| a (Å) | 43.07 |
| b (Å) | 182.15 |
| c (Å) | 50.37 |
| β (°) | 98.8 |
R
merge = 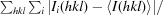
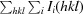 × 100, where I
i(hkl) is the observed intensity and 〈I(hkl)〉 is the mean intensity.
× 100, where I
i(hkl) is the observed intensity and 〈I(hkl)〉 is the mean intensity.
Percentage of correlation between intensities from random half-sets calculated by XDS (Kabsch, 2010 ▶).
2.6. Structure solution and refinement
The ADC-1 sequence was used in a BLAST search (Altschul et al., 1990 ▶, 1997 ▶) against the current version of the Protein Data Bank (PDB; Berman et al., 2000 ▶), resulting in 54 hits. Not surprisingly, 34 of these were with class C β-lactamases, with E-values ranging from 6 × 10−115 to 2 × 10−84. The top match was with the plasmid-encoded CMY-10 (PDB entry 1zkj, sequence identity 46%; Kim et al., 2006 ▶) and the next three were with the AmpC enzyme from Pseudomonas aeruginosa (PDB entries 3s1y, 2wzx and 4hef, sequence identity 42%; Chen et al., 2011 ▶; Blizzard et al., 2010 ▶; Lahiri et al., 2013 ▶) followed by the psychrophilic β-lactamase TAE4 from P. fluorescens (PDB entry 2qz6, sequence identity 42%; Michaux et al., 2008 ▶) and then a number of E. coli AmpC structures (sequence identity 41%). Molecular replacement (MR) was performed using CMY-10, TAE4 and AmpC from both P. aeruginosa (PDB entry 3s1y) and E. coli (PDB entry 2bls; Usher et al., 1998 ▶) as the starting models. In all cases the solvent molecules were removed and the program CHAINSAW (Stein, 2008 ▶) from the CCP4 suite (Winn et al., 2011 ▶) was used to convert the original models into a modified ADC model in which identical residues in the two sequences were retained and those which differed were truncated at the Cβ atom. For all four models, a strong MR solution gave the positions of the two ADC-1 molecules in the asymmetric unit. The four structures were refined for ten cycles using REFMAC (Murshudov et al., 2011 ▶) and at this stage the model based upon CMY-10 was chosen as the better solution (R free was 0.381) and 2F o − F c and F o − F c electron-density maps were calculated. Refinement of the structure was completed with the PHENIX suite of programs (Adams et al., 2010 ▶), and manual building of the model was performed using the molecular-graphics program Coot (Emsley & Cowtan, 2004 ▶). Water molecules were added in structurally and chemically relevant positions, and the atomic displacement parameters (ADPs) for all atoms in the structure were refined anisotropically. Final refinement statistics are given in Table 2 ▶.
Table 2. Structure-refinement statistics.
| Resolution range (Å) | 38.5–1.20 |
| R factor/R free † (%) | 15.6/18.5 |
| R all ‡ (%) | 15.7 |
| No. of atoms | |
| Total | 2950 |
| Protein | 2778 |
| Solvent | 832 |
| B factors (Å2) | |
| Protein, chain A | 15.5 |
| Protein, chain B | 24.1 |
| Solvent | 33.7 |
| R.m.s. deviations from ideality | |
| Bonds (Å) | 0.005 |
| 1–3 distances (Å) | 1.09 |
| Ramachandran plot: residues in preferred regions§ (%) | 98.2 |
R = 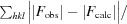
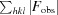 × 100. R
free was calculated with 5% of the reflections.
× 100. R
free was calculated with 5% of the reflections.
Final R factor calculated with all data using no σ cutoff.
As defined in MolProbity (Chen et al., 2010 ▶).
The atomic coordinates and structure factors for ADC-1 have been deposited in the Protein Data Bank (Berman et al., 2000 ▶) as PDB entry 4net. Superpositions were performed using the SSM procedure (Krissinel & Henrick, 2004 ▶) as implemented in Coot (Emsley & Cowtan, 2004 ▶) and the program LSQKAB in the CCP4 suite (Winn et al., 2011 ▶). Figures were generated using PyMOL (DeLano, 2002 ▶).
3. Results and discussion
3.1. The ADC-1 structure
In contrast to the class A β-lactamases, for which there is a canonical numbering scheme based upon the TEM-1 enzyme (Ambler et al., 1991 ▶), there is no specific numbering scheme for the class C enzymes. In general, however, the class C enzymes are numbered such that the catalytic triad is Ser64, Lys67 and Tyr150, based on the first reported structure of the Citrobacter freundii class C β-lactamase (PDB entry 1fr1; Oefner et al., 1990 ▶). Throughout this paper we will use the original ADC-1 numbering scheme and for important residues the C. freundii numbering will be given in italics in parentheses, unless otherwise stated.
The final refined ADC-1 model consists of two independent molecules in the asymmetric unit. Molecule A comprises 354 amino acids (residues Asn3–Lys361 in ADC-1 numbering) with five residues missing (Ser316–Arg320). Molecule B comprises 344 amino acids (residues Asn3–Lys360) with 14 residues missing (Phe203–Pro216). The protein model has a geometry close to ideal, with r.m.s.d.s of 0.005 Å and 1.09° for the bond lengths and bond angles, respectively. A Ramachandran plot calculated with the program PROCHECK (Laskowski et al., 1993 ▶) shows that 99.5% of the residues are in the most favored and additionally favored regions. The two ADC-1 molecules are related by a noncrystallographic twofold rotation axis such that a loose dimer is formed, held together by four hydrogen-bonding interactions involving residues in the loop between strand B2 and the short strand B2a, and in helix A1 (Fig. 2 ▶). The dimer interface buries only 420 Å2 of surface per monomer (approximately 3% of the total monomer surface area).
Figure 2.

Ribbon representation of ADC-1. The structure is colored according to the two major structural domains: domain 1 in green and domain 2 in magenta. The Ω-loop (yellow), the P-loop (blue), the R2-loop (red) and the P2-loop are also indicated. The parts of the structure in gray (including the R2-loop) represent the additional residues present in the class C enzymes relative to the smaller class A and class D enzymes. The three catalytic residues (Ser66, Lys69 and Tyr152) are shown in ball-and-stick representation.
The ADC-1 monomer is divided into two major structural domains (Fig. 2 ▶). Domain 1 is noncontiguous, comprising two segments (residues 3–59 and residues 309–361), and consists of a six-stranded antiparallel β-sheet (B1, B2 and B2a from the first segment and B8, B9 and B10 from the second segment), with the two terminal helices (A1 and A11) packing against the outer face of the sheet. Domain 2 is folded from a single piece of polypeptide (residues 60–308) and is a globular, predominantly α domain consisting of seven major α-helices packing against the inner face of the domain 1 β-sheet (Fig. 2 ▶).
3.2. Comparison with class A and class D β-lactamases
Compared with the two other classes of serine β-lactamases (class A and class D), the class C enzymes are substantially longer, typically by 90–110 residues, depending upon the source. Fig. 3 ▶(a) shows a topological representation of a general class C β-lactamase based on the structure of ADC-1; similar topology diagrams for the class A and class D enzymes are shown in Figs. 3 ▶(b) and 3 ▶(d). The structures of the class A and class D enzymes are represented by TEM-1 (Fig. 3 ▶ c) and OXA-23 (Fig. 3 ▶ e), respectively. The secondary-structure elements have been named such that the majority of the equivalent elements in all three classes of enzyme coincide, in particular the strands which make up the central β-sheet in domain 1 and the seven main helices in domain 2. The additional residues in the class C enzymes are highlighted. Although the overall architectures of the two structural domains are similar in the three classes of β-lactamases, the class C enzymes have an extra five-stranded antiparallel β-sheet (B4, B7a, B7b, B7c and B7d) capped by an α-helix (A10a) and a short 310-helix A10b) inserted between helices A4 and A5 in domain 2. The five additional strands extend the domain 1 β-sheet to form a twisted β-sheet running through the middle of the molecule (Fig. 2 ▶). The loop comprising helices A10a and A10b and strands B7d and B8 has been designated as the R2-loop (Jacoby, 2009 ▶; Kim et al., 2006 ▶). In the class A and D enzymes, the loop between helices H4 and H5 (equivalent to helices A4 and A5 in ADC-1) is significantly shorter, comprising only 3–4 residues.
Figure 3.

Structural relationships between the class A, class C and class D serine β-lactamases. (a) Topology diagram for ADC-1, representing the class C enzymes. Helices are colored orange and strands are colored light blue. The colored background boxes indicate the two structural domains (domain 1 in green and domain 2 in magenta), with the Ω-loop and P-loop indicated by yellow and blue boxes. Two additional loops (P2 and R2) unique to the class C enzymes are indicated. The numbering scheme for the secondary-structure elements is also given. (b) Topology diagram for the class A enzymes, using the same color scheme as in (a) to show the location of domains 1 and 2, the Ω-loop and the P-loop. (c) Ribbon representation for the class A enzyme TEM-1 (PDB entry 2blt; J. R. Knox & T. Sun, unpublished work) using the same color scheme as in Fig. 2 ▶ for the structural domains (green and magenta for domains 1 and 2) and loops (yellow and blue for the Ω-loop and the P-loop, with the latter also being indicated). (d) Topology diagram for the class D enzymes, using the same color scheme as in (a), showing the location of domains 1 and 2, the Ω-loop and the P-loop. (e) Ribbon representation for the class D enzyme OXA-23 (PDB entry 4jf4; Smith et al., 2013 ▶) using the same color scheme as in Fig. 2 ▶ and (c) for the structural domains and loops.
In addition to the elongated central β-sheet, there are several other differences between the small serine β-lactamases and the class C enzymes. A large loop with a variable structure, sometimes referred to as the P-loop (Szarecka et al., 2011 ▶), runs along the outside of domain 2 and links the long central helix (A2 in ADC-1, H2 in the class A and class D enzymes) with the three-helix cluster comprising A4, A5 and A6 (H4, H5 and H6 in the class A and class D enzymes). In the class A and D enzymes this loop is about 26–32 residues in length and is composed of two antiparallel strands and either one or two short 310-helices. The end of this loop wraps around the helical domain and projects towards the enzyme active site (Figs. 3 ▶ c and 3 ▶ e). In the class D enzymes a 310-helix at the end of this loop contains residues which have been suggested to play a key role in the selection and binding of substrates (Schneider et al., 2009 ▶; Smith et al., 2013 ▶). In contrast, the equivalent loop in ADC-1 is slightly shorter (about 24 residues), lacks the two short antiparallel β-strands and is made up of three short 310-helices (A3a, A3b and A3c; Figs. 2 ▶ and 3 ▶ a), such that two residues at the end of this loop (Leu121 and Gln122; ADC-1 numbering) are in almost identical spatial positions to two residues at the tip of the TEM-1 P-loop (Glu104 and Tyr105; Ambler numbering scheme). We have designated this loop as the P2-loop (Figs. 2 ▶ and 3 ▶ a). Moreover, a conserved YxN motif at the C-terminal end of this loop insertion, a motif that is common to the class C β-lactamases and the β-lactam-sensitive d-alanyl-d-alanine carboxypeptidases (DD-peptidases; Knox et al., 1996 ▶), provides a tyrosine residue, Tyr152 (Tyr150), that is critical in the catalytic mechanism of the class C enzymes, in which it has recently been suggested to be involved in the protonation of the β-lactam N5 atom (see Fig. 1 ▶) via the substrate carboxylate (Tripathi & Nair, 2013 ▶) and also in the activation of a deacylating water molecule (Chen et al., 2009 ▶; Dubus et al., 1996 ▶). The tyrosine is isostructural with a universally conserved serine residue in both the class A and D enzymes, such that the Oη atom of Tyr152 in ADC-1 and the Oγ atom of Ser130 in TEM-1 are only 0.7 Å apart (Fig. 4 ▶). In the class A and D enzymes, this serine residue (Ser130 in TEM-1 and Ser125 in OXA-23) is positioned in the short loop which connects helices A4 and A5, and is part of a highly conserved SxN and SxV motif in these two enzyme classes, respectively. The side chain of the serine forms an important hydrogen-bonding contact across the active-site cleft to a conserved lysine residue [Lys314 (Lys315) in ADC-1, Lys234 in TEM-1 and Lys216 in OXA-23], which is part of a highly conserved KTG motif in the three classes of serine β-lactamases.
Figure 4.

Stereoview of the superposition of ADC-1 (green) and TEM-1 (magenta). Although the polypeptide between ADC-1 helices A4 and A5 (the P2-loop comprising 40 residues unique to the class C enzymes) is substantially longer and is folded completely differently from the corresponding loop between TEM-1 helices H4 and H5 (which comprises only four residues), two residues at the end of the P2-loop (Leu121 and Gln122) occupy the same locations as the two residues at the end of the longer TEM-1 P-loop (Glu104 and Tyr105). The side chain of residue Tyr152, which is critical in the catalytic mechanism of the class C enzymes, is structurally equivalent to the conserved Ser130 side chain in the class A enzymes.
A second long loop important in the function of all serine β-lactamases (Banerjee et al., 1998 ▶), designated the Ω-loop, is adjacent to the active site and connects helices A6 and A8. Although the structure of this loop varies between the three classes of serine β-lactamases, there is considerable conservation of both sequence and structure between enzymes in the same class. In the class A enzymes this loop is about 25 residues long and comprises two short β-strands at the N-terminal and C-terminal ends of the loop (S6 and S7; Fig. 3 ▶ b), with a short 310-helix (H7) near the center of the loop. Although there is little sequence identity in helix H7 itself, the residue at the N-terminus of the helix, Glu166, is the catalytic base in the class A enzymes responsible for activation of the deacylation step (Banerjee et al., 1998 ▶). In the class D enzymes this loop is slightly shorter (20–22 residues) and although its structure resembles that in TEM-1, there is only one short β-strand (S6; Fig. 3 ▶ d) at the C-terminal end (the polypeptide at the N-terminal end does not have a β-strand configuration and diverges from the path taken by strand S6 in the class A enzymes) and the helix in the center is a single turn of regular α-helix (H7). In the class D enzymes the sequence of this helical region is very highly conserved, in particular a Trp–Leu pair. The tryptophan side chain projects down into the active site directly behind the carboxylated lysine residue and anchors the side chain of the lysine via a hydrogen-bonding interaction with one of the carboxylate O atoms. The leucine residue lies across the top of the lysine residue and is involved in a hydrophobic interaction with the conserved valine from the SxV motif in the A4/A5 loop. In the meropenem complex of OXA-23, it was observed that this leucine residue responds to the presence of the acyl-enzyme intermediate in the active site by swinging away from the valine and opening a channel for an incoming water molecule to approach the carboxylated lysine to initiate deacylation (Smith et al., 2013 ▶).
In the class C enzymes, the Ω-loop is significantly longer (approximately 44–45 residues) and much more structured, comprising in ADC-1 a short strand at the N-terminal end (B5) followed by a short 310-helix (A7a), a β-hairpin (strands B5a and B5b), a single turn of α-helix (A7b) and a final short strand (B6) antiparallel to strand B5 (Figs. 2 ▶ and 3 ▶ a). Although strands B5 and B6 are in the same location and direction as strands S6 and S7 in the class A enzymes, the rest of the class C Ω-loop takes a completely different path from that observed in the class A and D enzymes. The polypeptide makes a sharp turn just at the end of strand B5 (Fig. 2 ▶) and the loop direction is essentially reversed (Knox et al., 1996 ▶). Sequence identity in the class C Ω-loop is generally limited, although there is one highly conserved AYG motif at the C-terminus of helix A7b which projects into the active site and plays a key role in substrate recognition.
3.3. The active site of ADC-1
The active site is located in the cleft formed at the interface of domains 1 and 2. This cleft is lined by a series of highly conserved residues which play key roles in substrate recognition and binding and in enzyme catalysis. Fig. 5 ▶(a) gives an overview of the active site in ADC-1, and some representative electron density in the vicinity of the active-site Ser66 (Ser64) and the conserved Lys69 (Lys67) and Tyr152 (Tyr150) residues is shown in Fig. 5 ▶(b). Based on Fig. 5 ▶(a), the active site can be viewed as a long groove with the two sides formed by helices A4 and A5 and the P2-loop on the right and strand B8 and the N-terminus of helix A11 on the left. The two ends of the active-site cleft are formed by helix A7b from the Ω-loop at the top and helices A10a and A10b from the R2-loop at the bottom. Analysis of the structures of class C β-lactamases with bound substrates and inhibitors suggests that the active site of these enzymes can be divided into two subsites, depending upon which parts of the β-lactam substrates they interact with (Kim et al., 2006 ▶). These subsites are designated R1 and R2 because the residues which make up the two sites interact with the R1 and R2 substituents at the C7 and C3 atoms of the β-lactam substrate, respectively (see Fig. 1 ▶ for atom numbering). In ADC-1 the R1 subsite comprises Asn154 (Asn152), residues from the AYG motif on helix A7b, the tip of the P2-loop and strand B8, while the R2 subsite is composed of Tyr152 and residues from helix A10b and helix A11.
Figure 5.

The active site of ADC-1. (a) The ADC-1 molecule is shown as a molecular surface viewed down into the active-site cleft. The upper and lower ends of the cleft are formed by the Ω-loop and the R2-loop, respectively. Residues which line the active site are shown as yellow sticks. The approximate location of a cephalothin substrate (based upon the superimposed structure of AmpC; PDB entry 1kvm) is shown as a partially transparent magenta ball-and-stick model. (b) Representative final 2F o − F c electron density at 1.3σ in the vicinity of the ADC-1 active site.
The catalytic Ser66 residue and the conserved Lys69 residue are at the N-terminus of helix A2. The side chain of the catalytic serine adopts two conformations in one of the two independent ADC-1 molecules, and both conformations of the side chain form bifurcated hydrogen-bonding interactions with the Nζ atoms of Lys69 and with the Oη atom of Tyr152 at the N-terminus of helix A5 (Fig. 6 ▶ a). In the second ADC-1 molecule, in which only one Ser66 conformation is present, the serine side chain makes two traditional hydrogen bonds with Lys69 and Tyr152. The side chain of Lys69 is held in place by additional hydrogen-bonding interactions with the side chains of Tyr152 and Asn154 (Asn152) and the carbonyl O atom of Ala223 (Ala220) from the AYG motif at the C-terminus of helix A7b in the Ω-loop. The Asn154 residue is hydrogen-bonded to the side chain of the highly conserved Gln122 (Gln120), and these two residues, along with residues from strand B8 and the Ω-loop, comprise the R1 subsite of the class C β-lactamase active site (Kim et al., 2006 ▶).
Figure 6.

(a) Stereoview of the hydrogen-bonding network adjacent to the ADC-1 oxyanion hole. The water molecule which occupies the oxyanion hole in this apo form of the enzyme is indicated as W1. (b) Stereoview of a cephalothin acyl-enzyme intermediate (blue ball-and-stick representation) modeled into the ADC-1 active site (green cartoon and sticks). Potential hydrogen-bonding interactions with ADC-1 residues are indicated as dashed lines. The location of the water molecule (W1) in the ADC-1 oxyanion hole is indicated.
On the other side of the Ser66 residue is an oxyanion pocket formed by the N-terminus of helix A2 and the C-terminus of strand B8. In ADC-1 this oxyanion hole is occupied by a water molecule (W1), which is anchored in the pocket by hydrogen-bonding interactions with the main-chain amide N atoms of Ser66 and Ser317 (Ser318) from strand B8 (Fig. 6 ▶ a). W1 is part of an extensive array of water molecules that occupy the empty active-site cleft. Strand B8 forms the majority of one side of the cleft, and the C-terminus of this strand carries the highly conserved KTG motif noted above. A conserved arginine residue, Arg348 (Arg349) in helix A11, sits in a pocket just behind strand B8 in a location equivalent to a conserved arginine in the class D enzymes which forms an electrostatic interaction with the C3 carboxylate of penicillin and carbapenem substrates (Schneider et al., 2009 ▶; Smith et al., 2013 ▶). In ADC-1 and the other class C enzymes this arginine residue appears to be buried a little deeper into the pocket than the corresponding residue in the class D enzymes, owing primarily to a 1 Å shift of helix A11 away from the active site, and it seems unlikely that the Arg348 side chain could interact with the substrate C4 carboxylate of the cephalosporins. Indeed, the structures of AmpC with cephalosporins show that there is no interaction between the C4 carboxylate and this arginine (Arg349; Beadle et al., 2002 ▶; Thomas et al., 2010 ▶). However, in the structure of AmpC in complex with imipenem (PDB entry 1ll5; Beadle & Shoichet, 2002 ▶) an interaction of the imipenem carboxylate and the arginine is observed (Beadle & Shoichet, 2002 ▶). In this complex, the imipenem is an inhibitor of the enzyme and binds in a very unusual orientation with the C7 carbonyl O atom flipped out of the oxyanion hole and the molecule rotated such that the carboxylate moves towards strand B8. This rotation, caused by severe steric clashes between the α-hydroxyethyl side chain on the R1 side of the imipenem and the surrounding protein, results in the imipenem carboxylate moiety occupying a position where it is now able to form an electrostatic interaction with the arginine (Arg349). This interaction would serve to anchor the acyl-enzyme form of the imipenem in this orientation and is most likely to stabilize the inhibited form of the enzyme.
In the AmpC–cephalothin pre-acylation and acyl-enzyme complexes (PDB entries 1kvl and 1kvm, respectively; Beadle et al., 2002 ▶) and the AmpC–cefotaxime complex (PDB code 3ixh; Thomas et al., 2010 ▶), the C8 carbonyl O atom projects towards the oxyanion hole. In the AmpC–cephalothin pre-acylation state complex, the C8 carbonyl is hydrogen-bonded to both of the main-chain amide N atoms which form the hole (Beadle et al., 2002 ▶) and is in a location comparable to water W1 observed in ADC-1. In this complex the C4 carboxylate has swung towards strand B8 by approximately 3 Å compared with the AmpC imipenem complex and does not interact with Arg349 (C. freundii numbering). During acylation the six-membered dihydrothiazine ring of the cephalothin rotates approximately 100°, which moves the C8 carbonyl out of the oxyanion hole approximately 1.5 Å from its expected position (Beadle et al., 2002 ▶). The same orientation of the dihydrothiazine ring is observed in the AmpC–cefotaxime complex (Thomas et al., 2010 ▶). This rotation results in a repositioning of the C4 carboxylate towards the outer edge of the active site, where it interacts with an asparagine residue (Asn343) from the B10/A11 loop. It is at this location that ADC-1 differs from AmpC and the majority of the other class C enzymes, which have either an asparagine or a serine at this position. The equivalent residue in ADC-1 is an arginine (Arg342), the side chain of which projects across the opening of the active site (Fig. 5 ▶ a) and which may be the residue responsible for anchoring the substrate carboxylate moiety. Modeling of a cephalothin acyl-enzyme intermediate into the ADC-1 active site, based upon the superposition of the AmpC–cephalothin complex, suggests that a salt bridge could indeed form between the C4 carboxylate and Arg342 (Fig. 6 ▶ b). The modeling also suggests that the acyl-enzyme intermediate would be stabilized by similar interactions as observed in the AmpC complex, including a potential hydrophobic interaction between the dihydrothiazine ring and the side chain of Leu121 (Leu119) from the end of the P2-loop. The R1 side group would be anchored by hydrogen bonds to the side chains of Gln122 (Gln120) and Asn154 (Asn152), and the carbonyl O atom of Ser317 (Ser318) from strand B8, along with an aromatic π-stacking interaction between the thiophene ring (see Fig. 1 ▶) of the intermediate and the side chain of Tyr224 (Tyr221) from the Ω-loop (Fig. 6 ▶ b).
3.4. The class C β-lactamases
The ADC-1 enzyme was superimposed on a representative set of class C β-lactamases available in the PDB, and the results are shown in Table 3 ▶. The r.m.s.d.s for all matching Cα atoms for the ten structures used range from 1.0 to 1.4 Å, with sequence identities between 36 and 45%. The best match both structurally and by sequence is with the plasmid-encoded CMY-10 (PDB entry 1zkj). A comprehensive alignment of the sequences based upon their structures (data not shown) shows regions of very high sequence identity throughout the structure. Not surprisingly, the residues implicated in enzyme activity and located in areas adjacent to the active site show significantly greater sequence similarity than more distant areas.
Table 3. Comparison of A. baumannii ADC-1 with other class C β-lactamases.
| Enzyme | PDB code | Bacterial source | Sequence identity (%) | R.m.s.d. (Å) | Matching Cα atoms |
|---|---|---|---|---|---|
| CMY-10† | 1zkj | Enterobacter aerogenes | 44.8 | 1.0 | 337 |
| GC1† | 1gce | Enterobacter cloacae | 39.1 | 1.4 | 327 |
| AmpC | 2bls | Escherichia coli | 41.4 | 1.3 | 331 |
| GN346 | 1rgy | Citrobacter freundii | 36.1 | 1.2 | 330 |
| P99 | 1xx2 | E. cloacae | 39.2 | 1.3 | 329 |
| 908R | 1y54 | E. cloacae | 38.1 | 1.3 | 331 |
| CMY-2 | 1zc2 | Klebsiella pneumoniae | 36.3 | 1.2 | 331 |
| TAE4 | 2qz6 | Pseudomonas fluorescens | 43.3 | 1.2 | 330 |
| ACT-1 | 2zc7 | K. pneumoniae | 36.9 | 1.3 | 331 |
| 1fr1 | C. freundii | 36.3 | 1.2 | 333 |
Extended-spectrum class C β-lactamases.
Detailed analysis of the superpositions shows that the largest structural deviations among the class C enzymes occur primarily at the N- and C-termini, in the P-loop, the P2-loop and Ω-loop, the loops connecting helices A8 and A9, the loop connecting strands B7d and B8, and helix A10b (Figs. 2 ▶, 3 ▶ a and 7 ▶ a). Structural changes in two of these elements, the Ω-loop and the loop connecting strands B7d and B8, have been shown to result in extension of the substrate profile of class C enzymes towards the expanded-spectrum cephalosporins (Crichlow et al., 1999 ▶, 2001 ▶; Jacoby, 2009 ▶; Kim et al., 2006 ▶). The β-lactamase GC1 from Enterobacter cloacae is an example of an extended-spectrum class C enzyme that involves alterations in the Ω-loop. GC1 is an I16V/A88P double mutant of the E. cloacae P99 enzyme that in addition has a three-residue insertion in the Ω-loop (Crichlow et al., 1999 ▶). It has been shown that this AVR tripeptide repeat at the C-terminal end of strand B5b provides additional conformational flexibility in the Ω-loop and substantially expands the R1 subsite of the enzyme such that substrates with bulky R1 substituents (cefuroxime, ceftazidime and aztreonam) and large inhibitors can be accommodated (Crichlow et al., 2001 ▶; Nukaga et al., 2003 ▶, 2004 ▶). Analysis of the ADC-1 structure demonstrates that the four residues in the B5b/A7b loop of the enzyme form a type II β-turn constraining this part of the structure such that a large-scale conformational change as observed in GC1 could not easily occur.
Figure 7.

Comparison of ADC-1 with other class C β-lactamases. (a) Stereoview of the superposition of ADC-1 (thick blue ribbon) on CMY-10 (green ribbon) and AmpC (red ribbon). (b) Structure-based sequence alignment of the R2-loop region of ADC-1 with five representative class C β-lactamases: CMY-10, GC1, E. coli AmpC, P99 and TAE4. The R2-loop residues are indicated by the black box. (c) Stereoview of the superposition of ADC-1 (blue ribbon) with the same five class C β-lactamases as in (b): CMY-10 (green), GC1 (magenta), AmpC (red), P99 (yellow) and TAE4 (black). The R2-loop extends from the beginning of helix A10b to the beginning of strand B8.
A second important structural element in the class C enzymes which leads to extended-spectrum activity involves alterations in the loop connecting strands B7d and B8. In all of the class C structures used in the superposition this loop has essentially the same structure, with the exception of ADC-1, CMY-10 and to a lesser extent TAE4. Analysis of the sequences of the class C enzymes shows that both ADC-1 and CMY-10 have a three-residue deletion in this loop (Fig. 7 ▶ b). The shorter loop in these two enzymes gives rise to a difference in the positions of the two preceding helices (A10a and A10b), such that they move away from helix A11 by approximately 1 Å. Superposition of ADC-1 with five representative class C enzymes based only on the 30-residue part of the structure from the beginning of strand B7c to the end of strand B7d (encompassing helices A10a and A10b; Fig. 7 ▶ b) gives r.m.s.d.s ranging from 0.7 Å for CMY-10 to 1.4 Å for the majority of the enzymes. The psychrophilic enzyme TAE4 had an intermediate r.m.s.d. of 1.1 Å. Inspection of the superposition of TAE4 with ADC-1 (Fig. 7 ▶ c) showed that the movement of helix A10a was midway between the two extreme cases represented by GC1 (PDB entry 1gce; Crichlow et al., 1999 ▶) and ADC-1.
In CMY-10 the region from helix A10a to the beginning of strand B7c is designated the R2-loop (Kim et al., 2006 ▶; Jacoby, 2009 ▶), and the extended-substrate spectrum of CMY-10 is attributed primarily to a three-amino-acid deletion in the B7d/B8 loop and the resultant widening of the R2 binding pocket (Kim et al., 2006 ▶). It was suggested that the wider R2 subsite makes it more accessible to substrates with bulky R2 substituents. It is interesting to note that TAE4 has been reported to be active against ‘larger’ substrates (Michaux et al., 2008 ▶) and, although this does not suggest that the enzyme is an extended-spectrum class C β-lactamase, the intermediate position of the A10a helix could explain the increased activity of the enzyme towards larger penicillin substrates. In ADC-1, a similar three-amino-acid deletion in the R2-loop suggests that this enzyme may also possess activity towards some of the extended-spectrum cephalosporins with bulky R2 substituents. The MICs for a variety of β-lactam antibiotics were measured for ADC-1 and show that the enzyme is indeed an expanded-spectrum β-lactamase with enhanced activity against the third-generation cephalosporins ceftazidime, cefotaxime and ceftriaxone (Table 4 ▶). The enzyme-kinetic studies presented in Table 5 ▶ demonstrate that ADC-1 has a high catalytic efficiency towards penicillins (1.5 × 105 to 2 × 106 M −1 s−1), with the exception of oxacillin, which showed a significantly lower activity. The enzyme is as active towards the narrow-spectrum cephalosporin cephalothin (2 × 106 M −1 s−1) and also shows a high activity towards the third-generation cephalosporins ceftazidime, cefotaxime and ceftriaxone (4.4 × 104 to 3.6 × 105 M −1 s−1) comparable to that for the penicillins. This is in good agreement with the MIC data, which show a significant increase in resistance to these antibiotics in the presence of ADC-1. Surprisingly, the monobactam aztreonam was not a substrate of this enzyme but acted as an inhibitor with a K i of 1.7 µM. The increase in MIC values for aztreonam in the presence of ADC-1 could result from the sequestration of this antibiotic by the enzyme, as has been demonstrated for the deacylation-deficient mutant of the TEM-1 β-lactamase with ceftazidime (Antunes et al., 2011 ▶).
Table 4. MICs of β-lactam antibiotics against E. coli JM83 strain and E. coli JM83 strain producing ADC-1.
| MIC (µg ml−1) | ||
| Antimicrobial | ADC-1 | JM83 |
| Ampicillin | 2048 | 2 |
| Benzylpenicillin | 4096 | 16 |
| Oxacillin | 1024 | 256 |
| Piperacillin | 256 | 2 |
| Cephalothin | 2048 | 4 |
| Ceftazidime | 256 | 0.125 |
| Cefotaxime | 64 | 0.03 |
| Ceftriaxone | 64 | 0.03 |
| Aztreonam | 32 | 0.06 |
Table 5. Steady-state enzyme-kinetic parameters for ADC-1.
| Substrate | k cat (s−1) | K m (µM) | k cat/K m (M −1 s−1) |
|---|---|---|---|
| Ampicillin | 4.8 ± 0.18 | 32 ± 7 | (1.5 ± 0.3) × 105 |
| Benzylpenicillin | 10.26 ± 0.27 | 5.1 ± 0.3 | (2.0 ± 0.35) × 106 |
| Oxacillin | 0.046 ± 0.01 | 12.32 ± 2.9 | (3.7 ± 1.2) × 103 |
| Piperacillin | 3.1 ± 0.14 | 6.4 ± 1.4 | (4.8 ± 1.0) × 105 |
| Cephalothin | 147 ± 9 | 74 ± 8.6 | (2.0 ± 0.2) × 106 |
| Ceftazidime | 0.7 ± 0.02 | 16 ± 1.7 | (4.4 ± 0.5) × 104 |
| Cefotaxime | 0.16 ± 0.01 | 0.5 ± 0.04 | (3.2 ± 0.32) × 105 |
| Ceftriaxone | 0.24 ± 0.01 | 0.66 ± 0.13 | (3.6 ± 0.73) × 105 |
4. Conclusions
The results of the antibiotic-susceptibility testing demonstrate that ADC-1 is a bona fide extended-spectrum class C enzyme capable of conferring resistance to the third-generation cephalosporins widely used for the treatment of serious bacterial infections. Comparison of the structural information presented here for ADC-1 with the crystal structures of various class C β-lactamases indicate that the major determinant of the extended substrate profile of ADC-1 involves the deletion of three residues in the R2-loop and the subsequent enlargement of the ADC-1 active site. This allows the enzyme to accommodate the bulky R2 side chains of the expanded-spectrum cephalosporins. Mutations, deletions and insertions in the R2-loop also appear to be important in the enhancement of activity towards the third-generation cephalosporins in other class C β-lactamases (Jacoby, 2009 ▶), which demonstrates the key role that this loop plays in the activity of this family of enzymes.
Supplementary Material
PDB reference: ADC-1 β-lactamase, 4net
Acknowledgments
Portions of this research were carried out at the Stanford Synchrotron Radiation Lightsource (SSRL), a Directorate of SLAC National Accelerator Laboratory and an Office of Science User Facility operated for the US Department of Energy Office of Science by Stanford University. The SSRL Structural Molecular Biology Program is supported by the DOE Office of Biological and Environmental Research, and by the National Institutes of Health, National Institute of General Medical Sciences (including P41GM103393) and the National Center for Research Resources (P41RR001209).
References
- Adams, P. D. et al. (2010). Acta Cryst. D66, 213–221.
- Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. (1990). J. Mol. Biol. 215, 403–410. [DOI] [PubMed]
- Altschul, S. F., Madden, T. L., Schäffer, A. A., Zhang, J., Zhang, Z., Miller, W. & Lipman, D. J. (1997). Nucleic Acids Res. 25, 3389–3402. [DOI] [PMC free article] [PubMed]
- Ambler, R. P., Coulson, A. F. W., Frère, J.-M., Ghuysen, J.-M., Joris, B., Forsman, M., Levesque, R. C., Tiraby, G. & Waley, S. G. (1991). Biochem. J. 276, 269–270. [DOI] [PMC free article] [PubMed]
- Antunes, N. T., Frase, H., Toth, M., Mobashery, S. & Vakulenko, S. B. (2011). Biochemistry, 50, 6387–6395. [DOI] [PubMed]
- Banerjee, S., Pieper, U., Kapadia, G., Pannell, L. K. & Herzberg, O. (1998). Biochemistry, 37, 3286–3296. [DOI] [PubMed]
- Beadle, B. M. & Shoichet, B. K. (2002). Antimicrob. Agents Chemother. 46, 3978–3980. [DOI] [PMC free article] [PubMed]
- Beadle, B. M., Trehan, I., Focia, P. J. & Shoichet, B. K. (2002). Structure, 10, 413–424. [DOI] [PubMed]
- Berman, H. M., Westbrook, J., Feng, Z., Gilliland, G., Bhat, T. N., Weissig, H., Shindyalov, I. N. & Bourne, P. E. (2000). Nucleic Acids Res. 28, 235–242. [DOI] [PMC free article] [PubMed]
- Blizzard, T. A. et al. (2010). Bioorg. Med. Chem. Lett. 20, 918–921.
- Bogaerts, P., Cuzon, G., Naas, T., Bauraing, C., Deplano, A., Lissoir, B., Nordmann, P. & Glupczynski, Y. (2008). Antimicrob. Agents Chemother. 52, 4205–4206. [DOI] [PMC free article] [PubMed]
- Bou, G., Cerveró, G., Domínguez, M. A., Quereda, C. & Martínez-Beltrán, J. (2000). J. Clin. Microbiol. 38, 3299–3305. [DOI] [PMC free article] [PubMed]
- Bou, G. & Martínez-Beltrán, J. (2000). Antimicrob. Agents Chemother. 44, 428–432. [DOI] [PMC free article] [PubMed]
- Brown, S. & Amyes, S. (2006). J. Antimicrob. Chemother. 57, 1–3. [DOI] [PubMed]
- Chen, H. et al. (2011). Bioorg. Med. Chem. Lett. 21, 4267–4270.
- Chen, V. B., Arendall, W. B., Headd, J. J., Keedy, D. A., Immormino, R. M., Kapral, G. J., Murray, L. W., Richardson, J. S. & Richardson, D. C. (2010). Acta Cryst. D66, 12–21. [DOI] [PMC free article] [PubMed]
- Chen, Y., McReynolds, A. & Shoichet, B. K. (2009). Protein Sci. 18, 662–669. [DOI] [PMC free article] [PubMed]
- Clark, R. B. (1996). J. Antimicrob. Chemother. 45, 245–251. [DOI] [PubMed]
- Clinical and Laboratory Standards Institute (2009). Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria That Grow Aerobically; Approved Standard – Eighth Edition, M07-A8. Wayne: Clinical and Laboratory Standards Institute.
- Coelho, J., Woodford, N., Turton, J. & Livermore, D. M. (2004). J. Hosp. Infect. 58, 167–169. [DOI] [PubMed]
- Crichlow, G. V., Kuzin, A. P., Nukaga, M., Mayama, K., Sawai, T. & Knox, J. R. (1999). Biochemistry, 38, 10256–10261. [DOI] [PubMed]
- Crichlow, G. V., Nukaga, M., Doppalapudi, V. R., Buynak, J. D. & Knox, J. R. (2001). Biochemistry, 40, 6233–6239. [DOI] [PubMed]
- DeLano, W. L. (2002). PyMOL http://www.pymol.org.
- Dubus, A., Ledent, P., Lamotte-Brasseur, J. & Frère, J.-M. (1996). Proteins, 25, 473–485. [DOI] [PubMed]
- Emsley, P. & Cowtan, K. (2004). Acta Cryst. D60, 2126–2132. [DOI] [PubMed]
- Evans, P. (2006). Acta Cryst. D62, 72–82. [DOI] [PubMed]
- Fernández-Cuenca, F., Martínez-Martínez, L., Conejo, M. C., Ayala, J. A., Perea, E. J. & Pascual, A. (2003). J. Antimicrob. Chemother. 51, 565–574. [DOI] [PubMed]
- Frase, H., Shi, Q., Testero, S. A., Mobashery, S. & Vakulenko, S. B. (2009). J. Biol. Chem. 284, 29509–29513. [DOI] [PMC free article] [PubMed]
- Gasteiger, E., Gattiker, A., Hoogland, C., Ivanyi, I., Appel, R. D. & Bairoch, A. (2003). Nucleic Acids Res. 31, 3784–3788. [DOI] [PMC free article] [PubMed]
- Golemi, D., Maveyraud, L., Vakulenko, S., Samama, J.-P. & Mobashery, S. (2001). Proc. Natl Acad. Sci. USA, 98, 14280–14285. [DOI] [PMC free article] [PubMed]
- Hujer, K. M., Hamza, N. S., Hujer, A. M., Perez, F., Helfand, M. S., Bethel, C. R., Thomson, J. M., Anderson, V. E., Barlow, M., Rice, L. B., Tenover, F. C. & Bonomo, R. A. (2005). Antimicrob. Agents Chemother. 49, 2941–2948. [DOI] [PMC free article] [PubMed]
- Jacoby, G. A. (2009). Clin. Microbiol. Rev. 22, 161–182. [DOI] [PMC free article] [PubMed]
- Kabsch, W. (2010). Acta Cryst. D66, 125–132. [DOI] [PMC free article] [PubMed]
- Kim, J. Y., Jung, H. I., An, Y. J., Lee, J. H., Kim, S. J., Jeong, S. H., Lee, K. J., Suh, P.-G., Lee, H.-S., Lee, S. H. & Cha, S.-S. (2006). Mol. Microbiol. 60, 907–916. [DOI] [PubMed]
- Knox, J. R., Moews, P. C. & Frere, J.-M. (1996). Chem. Biol. 3, 937–947. [DOI] [PubMed]
- Krissinel, E. & Henrick, K. (2004). Acta Cryst. D60, 2256–2268. [DOI] [PubMed]
- Lahiri, S. D., Mangani, S., Durand-Reville, T., Benvenuti, M., De Luca, F., Sanyal, G. & Docquier, J.-D. (2013). Antimicrob. Agents Chemother. 57, 2496–2505. [DOI] [PMC free article] [PubMed]
- Laskowski, R. A., MacArthur, M. W., Moss, D. S. & Thornton, J. M. (1993). J. Appl. Cryst. 26, 283–291.
- Lobkovsky, E., Billings, E. M., Moews, P. C., Rahil, J., Pratt, R. F. & Knox, J. R. (1994). Biochemistry, 33, 6762–6772. [DOI] [PubMed]
- Lee, S.-H., Lee, J.-J., Lee, J.-J., Wu, X., Selenge, B. & Kwon, D. B. (2013). ICAAC 2013 Abstracts, Abstract C1-1585. Washington: American Society for Microbiology.
- Matthews, B. W. (1968). J. Mol. Biol. 33, 491–497. [DOI] [PubMed]
- Michaux, C., Massant, J., Kerff, F., Frère, J.-M., Docquier, J.-D., Vandenberghe, I., Samyn, B., Pierrard, A., Feller, G., Charlier, P., Van Beeumen, J. & Wouters, J. (2008). FEBS J. 275, 1687–1697. [DOI] [PubMed]
- Murshudov, G. N., Skubák, P., Lebedev, A. A., Pannu, N. S., Steiner, R. A., Nicholls, R. A., Winn, M. D., Long, F. & Vagin, A. A. (2011). Acta Cryst. D67, 355–367. [DOI] [PMC free article] [PubMed]
- Nukaga, M., Abe, T., Venkatesan, A. M., Mansour, T. S., Bonomo, R. A. & Knox, J. R. (2003). Biochemistry, 42, 13152–13159. [DOI] [PubMed]
- Nukaga, M., Kumar, S., Nukaga, K., Pratt, R. F. & Knox, J. R. (2004). J. Biol. Chem. 279, 9344–9352. [DOI] [PubMed]
- Oefner, C., D’Arcy, A., Daly, J. J., Gubernator, K., Charnas, R. L., Heinze, I., Hubschwerlen, C. & Winkler, F. K. (1990). Nature (London), 343, 284–288. [DOI] [PubMed]
- Park, Y. K., Choi, J. Y., Jung, S.-I., Park, K.-H., Lee, H., Jung, D. S., Heo, S. T., Kim, S.-W., Chang, H.-H., Cheong, H. S., Chung, D. R., Peck, K. R., Song, J. H. & Ko, K.-S. (2009). Diagn. Microbiol. Infect. Dis. 64, 389–395. [DOI] [PubMed]
- Peleg, A. Y., Seifert, H. & Paterson, D. L. (2008). Clin. Microbiol. Rev. 21, 538–582. [DOI] [PMC free article] [PubMed]
- Perez, F., Hujer, A. M., Hujer, K. M., Decker, B. K., Rather, P. N. & Bonomo, R. A. (2007). Antimicrob. Agents Chemother. 51, 3471–3484. [DOI] [PMC free article] [PubMed]
- Poirel, L. & Nordmann, P. (2006). Clin. Microbiol. Infect. 12, 826–836. [DOI] [PubMed]
- Rodríguez-Martínez, J. M., Nordmann, P., Ronco, E. & Poirel, L. (2010). Antimicrob. Agents Chemother. 54, 3484–3488. [DOI] [PMC free article] [PubMed]
- Schneider, K. D., Karpen, M. E., Bonomo, R. A., Leonard, D. A. & Powers, R. A. (2009). Biochemistry, 48, 11840–11847. [DOI] [PMC free article] [PubMed]
- Smith, C. A., Antunes, N. T., Stewart, N. K., Toth, M., Kumarasiri, M., Chang, M., Mobashery, S. & Vakulenko, S. B. (2013). Chem. Biol. 20, 1107–1115. [DOI] [PMC free article] [PubMed]
- Stein, N. (2008). J. Appl. Cryst. 41, 641–643.
- Szarecka, A., Lesnock, K. R., Ramirez-Mondragon, C. A., Nicholas, H. B. Jr & Wymore, T. (2011). Protein Eng. Des. Sel. 24, 801–809. [DOI] [PMC free article] [PubMed]
- Thomas, V. L., McReynolds, A. C. & Shoichet, B. K. (2010). J. Mol. Biol. 396, 47–59. [DOI] [PMC free article] [PubMed]
- Tian, G.-B., Adams-Haduch, J. M., Taracila, M., Bonomo, R. A., Wang, H.-N. & Doi, Y. (2011). Antimicrob. Agents Chemother. 55, 4922–4925. [DOI] [PMC free article] [PubMed]
- Tripathi, R. & Nair, N. N. (2013). J. Am. Chem. Soc. 135, 14679–14690. [DOI] [PubMed]
- Usher, K. C., Blaszczak, L. C., Weston, G. S., Shoichet, B. K. & Remington, S. J. (1998). Biochemistry, 37, 16082–16092. [DOI] [PubMed]
- Winn, M. D. et al. (2011). Acta Cryst. D67, 235–242.
- Zhao, W.-H. & Hu, Z.-Q. (2012). Crit. Rev. Microbiol. 38, 30–51. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: ADC-1 β-lactamase, 4net