

Abstract
Hermansky–Pudlak syndrome (HPS) is an autosomal recessive disorder characterized by oculocutaneous albinism, bleeding tendency, and lysosomal accumulation of ceroid-like material, with occasional development of interstitial pneumonia (IP). Nine genetically distinct subtypes of HPS are known in humans; IP develops primarily in types 1 and 4. Most reported cases of HPS with IP are type 1, and there are no published reports of type 4 in Japanese individuals. A 58-year-old man with congenital oculocutaneous albinism and progressive dyspnea for 1 month was admitted to our hospital. We administered high-dose corticosteroids on the basis of a diagnosis of acute exacerbation of interstitial pneumonia. Respiratory symptoms and the findings of high-resolution computed tomography (CT) showed improvement. He was diagnosed with HPS type 4 with interstitial pneumonia on the basis of gene analysis. He has been receiving pirfenidone for 1 year and his condition is stable. This is the first report on the use of pirfenidone for HPS with IP caused by a novel mutation in the HPS4 gene. We conclude that HPS should be suspected in patients with albinism and interstitial pneumonia. High-dose corticosteroid treatment may be useful in cases of acute exacerbation of interstitial pneumonia due to HPS-4, and pirfenidone may be useful and well tolerated in patients with HPS-4.
Keywords: Pulmonary fibrosis, Oculocutaneous albinism, Hermansky–Pudlak syndrome, Pirfenidone, Corticosteroid
1. Introduction
Hermansky–Pudlak syndrome (HPS) is a genetic multisystem disorder characterized by oculocutaneous albinism, a bleeding tendency secondary to platelet dysfunction, and lysosomal ceroid accumulation.1 Ceroid, which accumulates sporadically and slowly in the lungs, is not crucial for a diagnosis of HPS, but primarily accounts for the significant associated morbidity. Pulmonary fibrosis, usually manifesting in the third and fourth decades of life, accounts for premature death in 50% of HPS patients, which generally occurs by the fifth decade.2,3
Nine different genes cause HPS in humans.4,5 Among them, the HPS-1 gene shows the highest frequency of mutation. HPS-1 mutation represents the classical disease, which manifests with all the typical complications of HPS. HPS-4 disease is considered to resemble HPS-1, but only a few case of HPS-4 have been reported.6–9 HPS-1 and HPS-4 individuals show the greatest degree of pulmonary involvement, with an estimated 80% of HPS-1 subtypes afflicted.6,10 Here, we report a case of HPS presenting with interstitial pneumonia and exhibiting a novel HPS-4 gene mutation.
2. Case report
A 58-year-old man was admitted to our hospital because of progressive dyspnea. He had experienced shortness of breath for years, but had been well until 4 weeks earlier. He showed congenital albinism, amblyopia, and photodermatosis, but had received no previous medication for the condition. He was a massager, drank distilled spirits about 250 ml/day, and had smoked occasionally. The patient's family history revealed that his parents were cousins. There is no history of albinism in his family, but his father died of pulmonary disease at approximately 50 years.
On admission, body temperature was normal range, pulse rate was 95 beats per minute, blood pressure was 110/60 mmHg, respiratory rate was 25 ∼ 30 breaths per minute, and oxygen saturation was 82% in ambient air, increasing to 95% with oxygen supplementation (3 l) by nasal cannula. Physical examination on admission revealed blond hair, blond body hair, pale white skin, and erythema on the upper and lower extremities and neck (Fig. 1). The patient had amblyopia, strabismus, and horizontal nystagmus. Fine crackles in the bilateral lower lung fields were detected, but heart sounds were normal. He had neither clubbed finger nor edema. The results of abdominal and neurological examinations were normal.
Fig. 1.
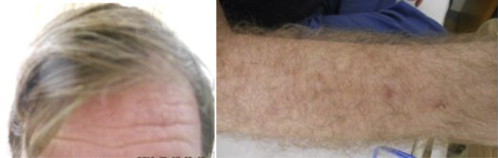
Patient's appearance. Left, note the blond hair and albinism. Right, the blond body hair and erythema at sites of exposure to sunlight.
Laboratory results on admission are shown in Table 1. Although the patient was anemic, white blood cell and platelet counts were normal, and congealing fibrinogenolysis was also normal. Blood chemistry showed elevation of lactate dehydrogenase (LDH), sialylated carbohydrate antigen (KL-6) levels. Chest radiography showed bilateral volume loss, and the lower lobes showed dominantly diffuse linear and reticular opacity with ground-glass opacity in both lung fields. Chest CT showed bilateral diffuse ground-glass opacity associated with mild traction bronchiectasis and reticulation which was consistent with acute exacerbation on chronic fibrosing interstitial pneumonia (Figs. 2 and 3). A transthoracic echocardiogram showed mild right ventricular hypokinesis, but left ventricular function was normal. There were no clinical signs of pulmonary infection.
Table 1.
Laboratory findings on admission.
| Blood analysis | Blood gas analysis (room air) | |
| WBC 6300/μL | pH 7.476 | |
| Hb 10.8 g/dL | PaO2 40.8 mmHg | |
| Ht 37.3% | PaCO2 35.6 mmHg | |
| Plt 28.6 × 104/μL | HCO3 25.9 mmol/L | |
| PT (%) 76.7% | ||
| APTT (%) 72.2% | Pulmonary function test (analyzed on day8) | |
| AT III 91% | VC | 2.28 L |
| FDP 3.0 μg/mL | %VC | 62% |
| Bleeding time 3 min | FEV1.0 | 2.06 L |
| BUN 7.7 mg/dL | FEV1.0%-G | 93.7% |
| CRE 0.89 mg/dL | (%DLCO was not examined because of respiratory insufficiency) | |
| AST 37 IU/L | ||
| ALT 36 IU/L | Platelet aggregation test | |
| LDH 371 IU/L | ADP (3.0 μM) | 76% |
| ESR 1 h 50 mm, 2 h 73 mm | Collagen (2.0 μg/mL) | 44% |
| CRP 1.82 mg/dL | ||
| β-D-glucan < 5.0 pg/mL | ||
| IgG 1383 mg/dL | ||
| IgA 740 mg/dL | ||
| IgM 87 mg/dL | ||
| BNP 78.5 pg/mL | ||
| SP-D 168 ng/mL | ||
| KL-6 1550 U/mL | ||
| ANA (−) | ||
| PR3-ANCA, MPO-ANCA (−) | ||
Fig. 2.
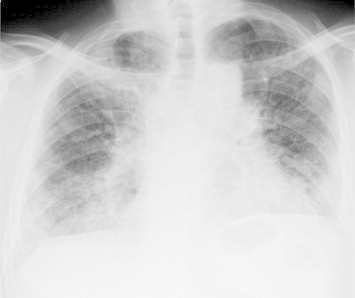
Chest radiograph showing bilateral volume loss and lower lobes with dominantly diffuse linear and reticular opacity, with ground-glass opacity in both lung fields.
Fig. 3.
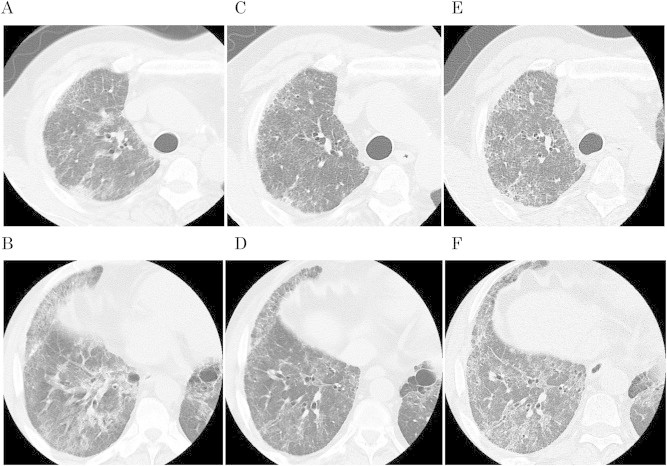
A–B, CT scan of right lung at time of admission. C–D, same slices of right lung 2 months later, showing improvement of ground-glass opacity and reticulonodular pattern as the result of treatment. E–F, same slices of right lung at 1 year after admission, showing gradual progression of disease.
We diagnosed acute exacerbation of interstitial pneumonia, and treated with high-dose corticosteroid (methylprednisolone, 1000 mg/day for three days followed by oral prednisone at a dose of 40 mg/day). His clinical symptoms and findings on high-resolution CT slowly improved; therefore, additional corticosteroid pulse therapy and pirfenidone were administered for fibrosing interstitial pneumonia. Subsequently, his breathing condition, clinical marker levels, and chest imaging results stabilized, but he needed long-term oxygen therapy.
Because the patient had interstitial pneumonia with albinism, we investigated the possibility of HPS. Therefore, bone marrow biopsy, platelet aggregation test, and genetic diagnosis were performed to diagnose HPS. A platelet aggregation test revealed a lack of secondary wave aggregation with normal first aggregation of ADP (Table 1). Bone marrow biopsy showed that macrophages contained a ceroid-like material (Fig. 4). We performed mutation analysis of HPS-1 (GenBank accession no. NM_000195.3) and HPS-4 (GenBank accession no. NM_022081.4) gene (Supplementary File). We identified a novel mutation, c.1858C > T (p.Q620X), in HPS4 homozygously in the patient and heterozygously in his mother, his sister and his daughter (Fig. 5). The diagnosis of HPS-4 was confirmed by this gene analysis.
Fig. 4.
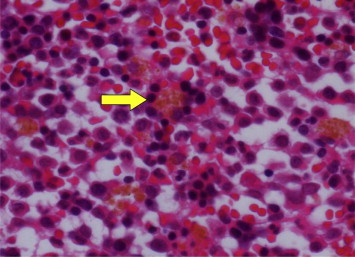
Bone marrow biopsy showing macrophages containing ceroid-like materials.
Fig. 5.
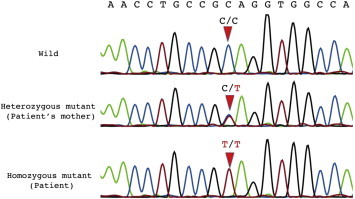
Sequence analysis of exon 13 in HPS4 gene using genomic DNA samples from the patient and his mother reveals the novel mutation, c.1858C > T (p.Q620X), homozygously in the patient and heterozygously in his mother. The mutation was also detected in his sister and his daughter (data not shown).
The corticosteroid dose was gradually tapered and then discontinued as the patient's condition became stable. Pirfenidone dosage was increased gradually without harmful adverse effects. Although chest CT performed after one year of treatment demonstrated progression of bilateral diffuse ground-glass opacity with mild traction bronchiectasis (Fig. 3), response to therapy was considered “stable” according to the ATS/ERS criteria by using pulmonary functional indices (Table 2).11
Table 2.
Clinical course over 1 year.
| Blood analysis | ||||||
|---|---|---|---|---|---|---|
| Reference range | Admission | 3 Months | 6 Months | 9 Months | 12 Months | |
| LDH (IU/L) | 112–213 | 371 | 367 | 358 | 318 | 330 |
| KL-6 (U/mL) | <500 | 1550 | 1250 | 2520 | 1790 | 1620 |
| Pulmonary function tests | ||||
|---|---|---|---|---|
| Day 8 | 4 Months | 8 Months | 12 Months | |
| VC (L) | 2.28 | 2.71 | 2.27 | 2.37 |
| %DLCO (%) | No data | 36.9 | 25.2 | 28.2 |
3. Discussion
We present a rare case of HPS-4 with interstitial pneumonia. High-dose corticosteroid treatment was effective for acute exacerbation of IP and pirfenidone has been used for 1 year.
HPS is a rare autosomal recessive disorder. It is characterized by oculocutaneous albinism, platelet dysfunction, and accumulation of ceroid-like materials and is associated with interstitial pneumonia. HPS is common only in northwest Puerto Rico. However, it is extremely rare in other regions, occurring with a prevalence of 1 in 500,000–1,000,000 persons.12 Among these cases, nearly all patients show HPS-1, and HPS-4 is extremely rare, so HPS-4 has never been reported in Japan.
The pathogenesis of HPS has been currently investigated, but many parts remain unexplained.13–17
The discovery of the HPS-4 subtype in 2002 was followed by examination of the HPS-4 mouse mutant.8 Bachli et al. have reported the only case of HPS-4 with IP.9 According to previous reports, the phenotype of HPS-4 is similar to that of HPS-1, and the mean age of onset of pulmonary symptoms in patients with mutations in the HPS1 gene is about 30–40 years.18 In our case, onset was relatively late and severity of fibrosis was moderate. Anderson et al. reported clinical features of 7 patients with documented HPS-4 mutations, ranging in age from 3 to 61 years.6 Three of the 7 patients had restrictive lung disease; one had severe pulmonary fibrosis and died of lung disease. According to Avila et al., patients with mutations in the HPS-1 gene had more severe lung disease than patients without HPS-1 gene mutations.3 The clinical course of HPS-4 may be slightly different from that of HPS-1 based on the previous reports and ours.
Regarding the treatment of interstitial pneumonia associated with HPS, corticosteroids or other immunosuppressive drugs generally have not been effective,18 but there have been no trials examining the efficacy of steroid treatment. In our case, respiratory status and findings on CT scans clearly improved in the first 2 weeks. We believe that high-dose steroid therapy contributed to the patient's improvement after the initial deterioration of IP. High-dose steroid therapy should be considered as a treatment option in these situations, because there is no alternative effective therapy during an acute exacerbation of IP.
Subsequently, we added pirfenidone and gradually reduced the corticosteroid dose. Gahl et al. reported that pirfenidone may slow the progression of pulmonary fibrosis due to HPS-1.19 On the other hand, recently, a small number of randomized controlled trials has been published that did not show slowing of decline in pulmonary function in small numbers.20 Thus, the utility of pirfenidone for HPS with IP remains controversial. Nevertheless, to our knowledge, ours is the first case in which pirfenidone was used for genetically proven HPS-4. Further study is needed whether pirfenidone may be effective in interstitial pneumonia associated with HPS. Finally, the only definitive treatment for pulmonary fibrosis related to HPS-1 is lung transplantation,21 but this was not an option in our case due to the patient's age.
When encountering a patient with oculocutaneous albinism with interstitial pneumonia, HPS should be considered. Although lung biopsy could not be performed in our patient because of respiratory insufficiency, it is a preferred diagnostic technique to reveal characteristic macrophages containing ceroid pigments and foamy swelling of pneumocytes. Further investigation of genetic analysis and enrollment of these cases is indispensable for appropriate treatment strategies.
In conclusion, our findings suggest that HPS should be suspected in patients with albinism and interstitial pneumonia. High-dose corticosteroid may be useful in cases of acute exacerbation of interstitial pneumonia due to HPS-4, and pirfenidone might be effective in treatment for progressive HPS-4 pulmonary fibrosis.
Acknowledgment
We greatly acknowledge the assistance of Takihiro Kamio in the Division of Pathology Medicine, Saiseikai Kumamoto Hospital. Gene analysis of this case was performed by Department of Dermatology, Nagoya University Graduate School of Medicine.
Appendix A. Supplementary data
References
- 1.Hermansky F., Pudlak P. Albinism associated with hemorrhagic diathesis and unusual pigmented reticular cells in the bone marrow. Blood. 1959;14:162–169. [PubMed] [Google Scholar]
- 2.Shotelersuk V., Dell'Angelica E.C., Hartnell L., Bonifacino J.S., Gahl W.A. A new variant of Hermansky–Pudlak syndrome due to mutations in a gene responsible for vesicle formation. Am J Med. 2000;108:423–427. doi: 10.1016/s0002-9343(99)00436-2. [DOI] [PubMed] [Google Scholar]
- 3.Gahl W.A., Brantly M., Troendle J., Avila N.A., Padua A., Montalvo C. Effect of pirfenidone on the pulmonary fibrosis of Hermansky–Pudlak syndrome. Mol Genet Metab. 2002;76:234–242. doi: 10.1016/s1096-7192(02)00044-6. [DOI] [PubMed] [Google Scholar]
- 4.Huizing M., Gahl W.A. Disorders of vesicles of lysosomal lineage: the Hermansky–Pudlak syndromes. Curr Mol Med. 2002;2:451–467. doi: 10.2174/1566524023362357. [DOI] [PubMed] [Google Scholar]
- 5.Anderson P.D., Huizing M., Claassen D.A., White J., Gahl W.A. Hermansky–Pudlak syndrome type4 (HPS-4): clinical and molecular characteristics. Hum Genet. 2003;113:10–17. doi: 10.1007/s00439-003-0933-5. [DOI] [PubMed] [Google Scholar]
- 6.Wei M.L. Hermansky–Pudlak syndrome: a disease of protein trafficking and organelle function. Pigment Cell Res. 2006;19:19–42. doi: 10.1111/j.1600-0749.2005.00289.x. [DOI] [PubMed] [Google Scholar]
- 7.Cullinane A.R., Curry J.A., Carmona-Rivera C., Summers C.G., Ciccone C., Cardillo N.D. A BLOC-1 mutation screen reveals that PLDN is mutated in Hermansky–Pudlak syndrome type 9. Am J Hum Genet. 2011;88:778–787. doi: 10.1016/j.ajhg.2011.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 8.Gahl W.A., Brantly M., Kaiser-Kupfer M.I., Iwata F., Hazelwood S., Shotelersuk V. Genetic defects and clinical characteristics of patients with a form of oculocutaneous albinism (Hermansky–Pudlak syndrome) N Eng J Med. 1998;338:1258–1264. doi: 10.1056/NEJM199804303381803. [DOI] [PubMed] [Google Scholar]
- 9.Suzuki T., Li W., Zhang Q., Karim A., Novak E.K., Sviderskaya E.V. Hermansky–Pudlak syndrome is caused by mutations in HPS4, the human homolog of the mouse light ear gene. Nat Genet. 2003;30:321–324. doi: 10.1038/ng835. [DOI] [PubMed] [Google Scholar]
- 10.Bachli E.B., Brack T., Eppler E., Stallmach T., Trüeb R.M., Huizing M. Hermansky–Pudlak syndrome type 4 in a patient from Sri Lanka with pulmonary fibrosis. Am J Med Genet A. 2004;127A:201–207. doi: 10.1002/ajmg.a.20683. [DOI] [PubMed] [Google Scholar]
- 11.Idiopathic pulmonary fibrosis: diagnosis and treatment: international consensus statement. American Thoracic Society (ATS), and European Respiratory Society (ERS) Am J Respir Crit Care Med. 2000;161:646–664. doi: 10.1164/ajrccm.161.2.ats3-00. [DOI] [PubMed] [Google Scholar]
- 12.Witkop C.J., Nuñez Babcock M., Rao G.H., Gaudier F., Summers C.G., Shanahan F. Albinism and Hermansky–Pudlak syndrome in Puerto Rico. Bol Asoc Med P R. 1990;82:333–339. [PubMed] [Google Scholar]
- 13.Huizing M., Helip-Wooley A., Westbroek W., Gunay-Aygun M., Gahl W.A. Disorders of lysosome-related organelle biogenesis: clinical and molecular genetics. Annu Rev Genomics Hum Genet. 2008;9:359–386. doi: 10.1146/annurev.genom.9.081307.164303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Falcón-Pérez J.M., Nazarian R., Sabatti C., Dell'Angelica E.C. Distribution and dynamics of Lamp1-containing endocytic organelles in fibroblasts deficient in BLOC-3. J Cell Sci. 2005;118:5243–5255. doi: 10.1242/jcs.02633. [DOI] [PubMed] [Google Scholar]
- 15.Young L.R., Pasula R., Gulleman P.M., Deutsch G.H., McCormack F.X. Susceptibility of Hermansky–Pudlak mice to bleomycin-induced type II cell apoptosis and fibrosis. Am J Respir Cell Mol Biol. 2007;37:67–74. doi: 10.1165/rcmb.2006-0469OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yoshioka Y., Kumasaka T., Ishidoh K., Kominami E., Mitani K., Hosokawa Y. Inflammatory response and cathepsins in silica-exposed Hermansky–Pudlak syndrome model pale ear mice. Pathol Int. 2004;54:322–331. doi: 10.1111/j.1440-1827.2004.01626.x. [DOI] [PubMed] [Google Scholar]
- 17.Mahavadi P., Korfei M., Henneke I., Liebisch G., Schmitz G., Gochuico B.R. Epithelial stress and apoptosis underlie Hermansky–Pudlak syndrome-associated interstitial pneumonia. Am J Respir Crit Care Med. 2010;182:207–219. doi: 10.1164/rccm.200909-1414OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pierson D.M., Ionescu D., Qing G., Yonan A.M., Parkinson K., Colby T.C. Pulmonary fibrosis in Hermansky–Pudlak syndrome: a case report and review. Respiration. 2006;73:382–395. doi: 10.1159/000091609. [DOI] [PubMed] [Google Scholar]
- 19.O'Brien K., Troendle J., Gochuico B.R., Markello T.C., Salas J., Cardona H. Pirfenidone for the treatment of Hermansky–Pudlak syndrome pulmonary fibrosis. Mol Genet Metab. 2011;103:128–134. doi: 10.1016/j.ymgme.2011.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lederer D.J., Kawut S.M., Sonett J.R., Vakiani E., Seward S.L., Jr., White J.G. Successful bilateral lung transplantation for pulmonary fibrosis associated with the Hermansky–Pudlak syndrome. J Heart Lung Transplant. 2005;24:1697–1699. doi: 10.1016/j.healun.2004.11.015. [DOI] [PubMed] [Google Scholar]
- 21.Avila N.A., Brantly M., Premkumar A., Huizing M., Dwyer A., Gahl W.A. Hermansky–Pudlak Syndrome: radiography and CT of the chest compared with pulmonary function tests and genetic studies. Am J Roentgenol. 2002;179:887–892. doi: 10.2214/ajr.179.4.1790887. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.