

Transition metal catalyzed asymmetric hydrogenation of enamides[1] is a powerful method to prepare chiral amines, which are important building blocks in organic synthesis.[2] With the development of many effective chiral ligands,[1] a variety of prochiral enamides such as 1,[3] 2,[3a,c,d,e,g] 3,[4] and 4,[5] have been hydrogenated in excellent enantioselectivities (Figure 1). However, asymmetric hydrogenation of 5 was rarely studied. To our knowledge, the only result was reported to give moderate enantioselectivity (50% ee) for the hydrogenation of 5a (Ar = Ph) with a Rh/DIPAMP complex.[6] In this paper, we prepared a series of enamides of both (Z)-5 and (E)-5 and tested them in Rh-catalyzed asymmetric hydrogenation with several chiral ligands. Excellent ee’s (up to 99% ee) have been achieved for those (Z)-configured enamide with Rh/TangPhos catalytic system.
Figure 1.
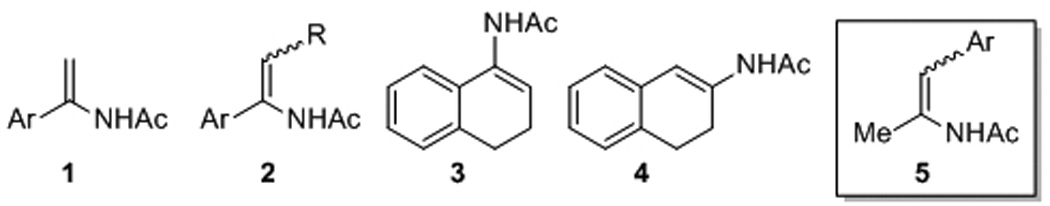
Prochiral enamide substrates for asymmetric hydrogenation.
A number of methods for the preparation of enamides have been reported, including rearrangement reactions,[6, 7] reduction of nitro alkenes[8] or ketoximes,[9] acylation of imines,[10] and direct condensation of ketone and amide.[11] Recently, a Merck group has developed an efficient Pd-catalyzed amidation reaction leading to a diverse array of enamides.[12] Under optimized condition, good selectivity for (Z)-enamide such as 6 was achieved (Scheme 1). To gain quick access to the desired substrates 5a-5i, we chose the direct condensation method due to its operational simplicity. As shown in Table 1, both isomers of 5 can be separated from the concentrated reaction mixture via flash column chromatography; in most cases, more of (Z)-enamide was obtained than the corresponding (E)-isomer. Although the moderate yields remain to be optimized, we found the present method is well suited for quick synthesis of both (Z)-5 and (E)-5 from inexpensive starting materials at laboratory scale. In addition, a diaryl enamide 5i was prepared in acceptable yield (entry 9, Table 1), which complements Pd-catalyzed amidation for this bulky substrate.[12b]
Scheme 1.
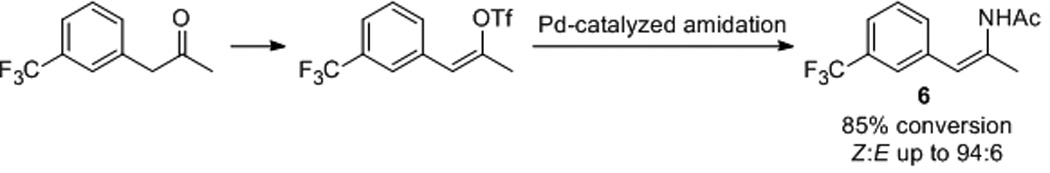
Pd-catalyzed amidation for the synthesis of (Z)-enamide 6.[12]
Table 1.
Preparation of (Z)- and (E)-5 via direct condensation of 7 with acetamide.[a]
 | ||||||
|---|---|---|---|---|---|---|
| Entry | Ketone[b] | R1 | R2 | Enamide | Yield (%)[c] | Z:E |
| 1 | 7a | Ph | Me | 5a | 24.1 | 1.8:1 |
| 2 | 7b | o-MeO-Ph | Me | 5b | 29.3 | 2:1 |
| 3 | 7c | p-MeO-Ph | Me | 5c | 29.3 | 2.8:1 |
| 4 | 7d | m-MeO-Ph | Me | 5d | 35.1 | 1.3:1 |
| 5 | 7e | m-Me-Ph | Me | 5e | 22.2 | 1.3:1 |
| 6 | 7f | p-Me-Ph | Me | 5f | 24.0 | 0.9:1 |
| 7 | 7g | o-Cl-Ph | Me | 5g | 15.6 | 1.7:1 |
| 8 | 7h | 1-Np-Ph | Me | 5h | 56.3 | 0.7:1 |
| 9 | 7i | Ph | Ph | 5i | 44.2 | 3.7:1 |
All reactions were carried out by refluxing a toluene solution (50 mL) of 7 (25 mmol), acetamide (125 mmol), and the catalyst TsOH (2.5 mmol) in a Dean-Stark apparatus for 24 h.
See supporting information for their preparation.
Combined yield of isolated (Z)- and (E)-product.
With the synthesis of a set of enamides 5, we compared Rh-catalyzed asymmetric hydrogenation of (Z)-5a and (E)-5a with four widely used chiral ligands, including TangPhos (L1),[13a] DuanPhos (L2),[13b] Et-DuPhos (L3),[13c] and Binapine (L4).[13d] Notably, under the same reaction condition each ligand showed a striking difference in enantio-differentiating ability toward the two isomers (Table 2). For (Z)-5a, excellent ee’s were obtained by the use of all ligands except L4, with TangPhos giving the best result. Change of solvent had little effect on enantioselectivity. In contrast, much lower ee’s were observed for (E)-configured 5a in EtOAc, albeit with the same sense of product chirality as from (Z)-5a. Change of solvent gave no improvement of enantioselectivity. Therefore, unlike asymmetric hydrogenation of isomeric mixture of β-substituted α-aryl enamides 2,[3a,c,d,e,g] the configuration of double bond in 5 has a dramatic influence on enantioselectivity.[14] To achieve excellent enantioselectivity with current ligands, (Z)-configured substrates need to be used.
Table 2.
Rh-catalyzed asymmetric hydrogenation of (Z)-5a and (E)-5a with different ligands.[a]
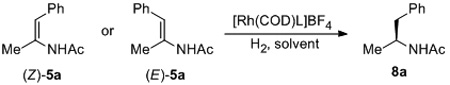 | |||||
|---|---|---|---|---|---|
| Entry | Substrate | Ligand[b] | Solvent | Ee (%)[c] | Config.[d] |
| 1 | (Z)-5a | L1 | EtOAc | 99.3 | S |
| 2 | (Z)-5a | L2 | EtOAc | 98.1 | S |
| 3 | (Z)-5a | L3 | EtOAc | 95.7 | S |
| 4 | (Z)-5a | L4 | EtOAc | 57.5 | S |
| 5 | (Z)-5a | L1 | CH2Cl2 | 98.3 | S |
| 6 | (Z)-5a | L1 | Acetone | 98.6 | S |
| 7 | (Z)-5a | L1 | MeOH | 98.6 | S |
| 8 | (Z)-5a | L1 | THF | 98.1 | S |
| 9 | (Z)-5a | L1 | Toluene | 97.5 | S |
| 10 | (E)-5a | L1 | EtOAc | 31.5 | S |
| 11 | (E)-5a | L2 | EtOAc | 23.7 | S |
| 12 | (E)-5a | L3 | EtOAc | 46.8 | S |
| 13 | (E)-5a | L4 | EtOAc | 26.3 | S |
All reactions were carried out with a substrate/catalyst ratio of 100:1 at room temperature under 30 bar hydrogen pressure for 20 h. In all cases, 100% conversion was observed.
L1 = (1S, 1S’, 2R, 2R’)-TangPhos, L2 = (SC, RP)-DuanPhos, L3 = (R, R)-Et-DuPhos, L4 = (S’)-Binapine.
Determined by chiral GC.
The absolute configuration was assigned by comparison of the observed optical rotation with reported data.
We further tested other substrates (Z)-5b-5i with Rh/TangPhos catalytic system under the optimized condition. As shown in Table 3, all substrates gave excellent ee’s. Substitution pattern on the phenyl ring generally has no appreciable effect on enantioselectivity (entries 2–4). Even hindered enamides (Z)-5h and (Z)-5i were hydrogenated with excellent results (entries 8, 9). At reduced catalyst loading (TON = 1000), (Z)-5a was still converted to 8a with almost unchanged ee (entry 10). Hence current hydrogenation route is a practical way for the preparation of various amines in this category (Figure 2).[15] For example, deacylation of the chiral product 8a leads directly to (S)-Amphetamine 9, which is a useful stimulant with strong biological and physiological effects.[16] Further modification will result in Selegiline 10 for the treatment of Alzheimer’s disease.[17] Asymmetric hydrogenation of (Z)-5c will also provide a practical access to important chiral drugs such as Formoterol 11[18] and Tamsulosin 12.[19]
Table 3.
Asymmetric hydrogenation of (Z)-5 with Rh/TangPhos catalytic system.[a]
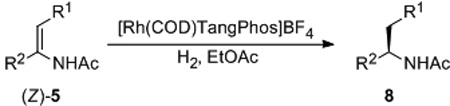 | ||||||
|---|---|---|---|---|---|---|
| Entry | Substrate | R1 | R2 | Product | Ee (%)[b] | Config.[c] |
| 1 | (Z)-5a | Ph | Me | 8a | 99.3 | S |
| 2 | (Z)-5b | o-MeO-Ph | Me | 8b | 99.0 | S |
| 3 | (Z)-5c | p-MeO-Ph | Me | 8c | 96.6 | S |
| 4 | (Z)-5d | m-MeO-Ph | Me | 8d | 99.1 | S |
| 5 | (Z)-5e | m-Me-Ph | Me | 8e | 99.1 | S |
| 6 | (Z)-5f | p-Me-Ph | Me | 8f | 98.8 | S |
| 7 | (Z)-5g | o-Cl-Ph | Me | 8g | > 99.9 | S |
| 8 | (Z)-5h | 1-Np-Ph | Me | 8h | 99.1 | S |
| 9 | (Z)-5i | Ph | Ph | 8i | 98.3 | S |
| 10d | (Z)-5a | Ph | Me | 8a | 98.7 | S |
Unless mentioned otherwise, All reactions were carried out with a substrate/catalyst ratio (S/C) of 100:1 in EtOAc at room temperature under 30 bar hydrogen pressure for 20 h. In all cases, 100% conversion was observed.
Determined by chiral GC.
The absolute configuration was assigned by comparison of the observed optical rotation with reported data.
S/C = 1000.
Figure 2.
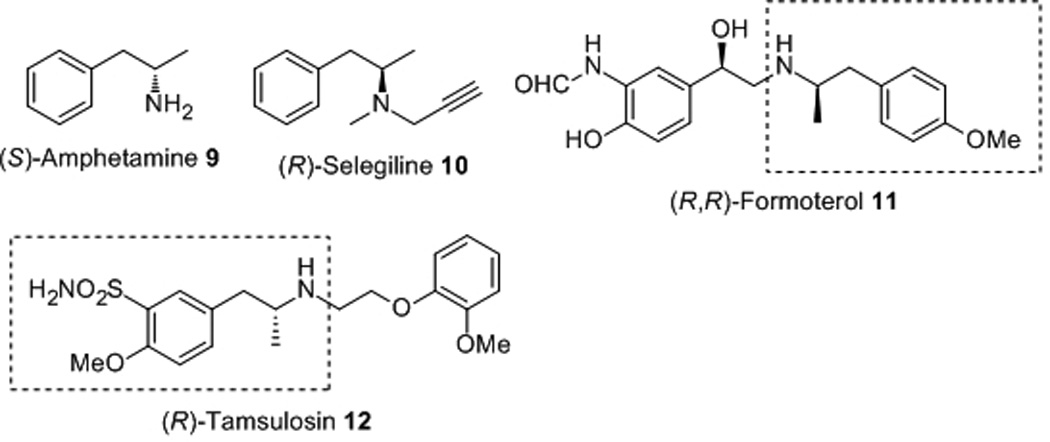
Chiral drugs bearing β-aryl isopropylamine fragment.
In this communication, we showed that β-aryl isopropylamines, an important class of chiral compounds with valuable pharmaceutical applications, can be prepared via highly efficient asymmetric hydrogenation. Excellent enantioselecitivity was obtained for (Z)-enamides, which were easily prepared via acid-catalyzed condensation of β-aryl ketone with acetamide. Alternatively, these substrates can be synthesized via Pd-catalyed amidation that exhibits better preference for the formation of (Z)-configured substrates.[12] Expansion of this methodology to other structurally relevant enamides is currently in progress and will be reported in a due course.
Experimental Section
General procedure for the substrate preparation: A toluene solution (50 mL) of 7 (25 mmol), acetamide (125 mmol), and the catalyst TsOH (2.5 mmol) was charged in a Dean-Stark apparatus and refluxed for 24 h. After cooled to room temperature, the solvent was evaporated and the concentrated mixture was passed through a flash chromatography column filled with silica gel (eluent: EtOAc/hexane). The product of (Z)- and (E)-configuration was collected as colorless oil or white powder.
General procedure for the hydrogenation: a stock solution was made by mixing [Rh(COD)2]BF4 with TangPhos at a 1:1.1 molar ratio in EtOAc at room temperature for 10 min in a nitrogen-filled glovebox. Then a specified amount of catalyst solution (0.5 mL, 0.001 mmol) was transferred by syringe into the vials charged with different substrates (0.1 mmol for each) in EtOAc (2.5 mL). All the vials were placed in a steel autoclave into which hydrogen gas (30 bar) was charged. After stirring at room temperature for 20 h, the hydrogen was released slowly and the solution was concentrated and subjected to a short column of silica gel to remove the metal complex. The purified solution was analyzed by chiral GC to determine the ee value.
Footnotes
This work was supported by National Institutes of Health (GM58832) and China Scholarship Council. Mass spectrometry was provided by the Washington University Mass Spectrometry Resource, an NIH Research Resource (Grant P41RR0954).
Contributor Information
Jian Chen, Department of Chemistry and Chemical Biology & Department of Pharmaceutical Chemistry, Rutgers, The State University of New Jersey, Piscataway, New Jersey 08854, USA; College of Chemistry and Molecular Sciences, Wuhan University, P.R. China.
Weicheng Zhang, Department of Chemistry and Chemical Biology & Department of Pharmaceutical Chemistry, Rutgers, The State University of New Jersey, Piscataway, New Jersey 08854, USA.
Huiling Geng, Department of Chemistry and Chemical Biology & Department of Pharmaceutical Chemistry, Rutgers, The State University of New Jersey, Piscataway, New Jersey 08854, USA; Northwest Agriculture and Forestry University, Yangling, Shanxi, P.R. China.
Yan Chen, Department of Bioengineering, Wuhan Bioengineering Institute, P.R. China.
Wei Li, Department of Chemistry and Chemical Biology & Department of Pharmaceutical Chemistry, Rutgers, The State University of New Jersey, Piscataway, New Jersey 08854, USA.
Guohua Hou, Department of Chemistry and Chemical Biology & Department of Pharmaceutical Chemistry, Rutgers, The State University of New Jersey, Piscataway, New Jersey 08854, USA.
Aiwen Lei, Email: aiwenlei@whu.edu.cn, College of Chemistry and Molecular Sciences, Wuhan University, P.R. China.
Xumu Zhang, Department of Chemistry and Chemical Biology & Department of Pharmaceutical Chemistry, Rutgers, The State University of New Jersey, Piscataway, New Jersey 08854, USA; College of Chemistry and Molecular Sciences, Wuhan University, P.R. China.
References
- 1.a) Jacobsen EN, Pfaltz A, Yamamoto H, editors. Comprehensive Asymmetric Catalysis I–III. Berlin: Springer; 1999. [Google Scholar]; b) Noyori R. Asymmetric Catalysis in Organic Synthesis. New York: Wiley; 1994. [Google Scholar]; c) Ojima I. Catalytic Asymmetric Synthesis. 2nd ed. New York: Wiley-VCH; 2000. [Google Scholar]; d) Tang W, Zhang X. Chem. Rev. 2003;103:3029. doi: 10.1021/cr020049i. [DOI] [PubMed] [Google Scholar]
- 2.Nugent TC. In: Process Chemistry in the Pharmaceutical Industry, Volumn 2. Gadamasetti K, Braish T, editors. Boca Raton, Fla: CRC Press LLC; 2008. p. pp137. [Google Scholar]
- 3.For representative references, see Burk MJ, Wang YM, Lee JR. J. Am. Chem. Soc. 1996;118:5142. Gridnev ID, Yasutake M, Higashi N, Imamoto T. J. Am. Chem. Soc. 2001;123:5268. doi: 10.1021/ja010161i. Tang W, Zhang X. Angew. Chem. Int. Ed. 2002;41:1612. doi: 10.1002/1521-3773(20020503)41:9<1612::aid-anie1612>3.0.co;2-h. Hu A-G, Fu Y, Xie J-H, Zhou H, Wang L-X, Zhou Q-L. Angew. Chem. Int. Ed. 2002;41:2348. doi: 10.1002/1521-3773(20020703)41:13<2348::AID-ANIE2348>3.0.CO;2-K. Bernsmann H, van den Berg M, Hoen R, Minnaard AJ, Mehler G, Reetz MT, De Vries JG, Feringa BL. J. Org. Chem. 2005;70:943. doi: 10.1021/jo048374o. Liu Y, Ding K. J. Am. Chem. Soc. 2005;127:10488. doi: 10.1021/ja052749l. Zhang W, Zhang X. Angew. Chem. Int. Ed. 2006;45:5515. doi: 10.1002/anie.200601501. Huang J-D, Hu X-P, Duan Z-C, Zeng Q-H, Yu S-B, Deng J, Wang D-Y, Zheng Z. Org. Lett. 2006;8:4367. doi: 10.1021/ol0617749. Gridnev ID, Imamoto T, Hoge G, Kouchi M, Takahashi H. J. Am. Chem. Soc. 2008;130:2560. doi: 10.1021/ja076542z.
- 4.a) Zhang Z, Zhu G, Jiang Q, Xiao D, Zhang X. J. Org. Chem. 1999;64:1774. doi: 10.1021/jo9824605. [DOI] [PubMed] [Google Scholar]; b) Tang W, Chi Y, Zhang X. Org. Lett. 2002;4:1695. doi: 10.1021/ol0258435. [DOI] [PubMed] [Google Scholar]
- 5.a) Tschaen DM, Abramson L, Cai D, Desmond R, Dolling U-H, Frey L, Karady S, Shi Y-J, Verhoeven TR. J. Org. Chem. 1995;60:4324. [Google Scholar]; b) Dupau P, Le Gendre P, Bruneau C, Dixneuf PH. Synlett. 1999:1832. [Google Scholar]; c) Jiang X-B, Lefort L, de Vries AHH, van Leeuwen PWNM, de Vries JG, Reek JNH. Angew. Chem. Int. Ed. 2006;45:1223. doi: 10.1002/anie.200503663. [DOI] [PubMed] [Google Scholar]
- 6.Bachman GL, Vineyard BD. Ger. Offen. 1977 DE 2638072. [Google Scholar]
- 7.Smith PAS, Horwitz Jerome P. J. Am. Chem. Soc. 1950;72:3718. [Google Scholar]
- 8.Laso NM, Quiclet-Sire B, Zard SZ. Tetrahedron Lett. 1996;37:1605. [Google Scholar]
- 9.a) Burk MJ, Casy G, Johnson NB. J. Org. Chem. 1998;63:6084. doi: 10.1021/jo9809332. [DOI] [PubMed] [Google Scholar]; (b) Zhang Z, Zhu G, Jiang Q, Xiao D, Zhang X. J. Org. Chem. 1999;64:1774. doi: 10.1021/jo9824605. [DOI] [PubMed] [Google Scholar]; c) Zhao H, Vandenbossche CP, Koenig SG, Singh SP, Bakale RP. Org. Lett. 2008;10:505. doi: 10.1021/ol7028788. [DOI] [PubMed] [Google Scholar]
- 10.Zhu G, Chen Z, Zhang X. J. Org. Chem. 1999;64:6907. doi: 10.1021/jo990565h. [DOI] [PubMed] [Google Scholar]
- 11.a) Elaridi J, Thaqi A, Prosser A, Jackson WR, Robinson AJ. Tetrahedron: Asymmetry. 2005;16:1309. [Google Scholar]; b) Guin J, Muck-Lichtenfeld C, Grimme S, Studer A. J. Am. Chem. Soc. 2007;129:4498. doi: 10.1021/ja0692581. [DOI] [PubMed] [Google Scholar]
- 12.a) Wallace DJ, Klauber DJ, Chen C-Y, Volante RP. Org. Lett. 2003;5:4749. doi: 10.1021/ol035959g. [DOI] [PubMed] [Google Scholar]; b) Klapars A, Campos KR, Chen C-Y, Volante RP. Org. Lett. 2005;7:1185. doi: 10.1021/ol050117y. [DOI] [PubMed] [Google Scholar]
- 13. a) ref 3c); Liu D, Zhang X. Eur. J. Org. Chem. 2005:646. Burk MJ. Acc. Chem. Res. 2000;33:363. doi: 10.1021/ar990085c. Tang W, Wang W, Chi Y, Zhang X. Angew. Chem. Int. Ed. 2003;42:3509. doi: 10.1002/anie.200351465.
- 14.Notably, distinct reactivities of (Z)- and (E)-enamides were reported in Ru/BINAPcatalyzed asymmetric hydrogenation of 1,2,3,4-tetrahydroisoquinoline derived enamides: whereas (Z)-enamides were hydrogenated smoothly with excellent ee’s, the corresponding (E)-enamides showd no reactivity under the same hydrogenation conditions see Noyori R, Ohta M, Hsiao Y, Kitamura M, Ohta T, Takaya H. J. Am. Chem. Soc. 1986;108:7117. Kitamura M, Hsiao Y, Ohta M, Tsukamoto M, Ohta T, Takaya H, Noyori R. J. Org. Chem. 1994;59:297.
- 15.For another asymmetric catalytic synthesis of this class of amines, see Sayyed IA, Sudalai A. Tetrahedron: Asymmetry. 2004;15:3111. Talluri SK, Sudalai A. Tetrahedron. 2007;63:9758.
- 16.a) Snyder SH, Tayler KM. Science. 1970;168:1487. doi: 10.1126/science.168.3938.1487. [DOI] [PubMed] [Google Scholar]; b) Hajos GT, Garattin SJ. Pharm. Pharmacol. 1973;25:418. doi: 10.1111/j.2042-7158.1973.tb10041.x. [DOI] [PubMed] [Google Scholar]
- 17.Sano M, Ernesto C, Thomas RC, Klauber MRN. Engl. J. Med. 1997;336:1216. doi: 10.1056/NEJM199704243361704. [DOI] [PubMed] [Google Scholar]
- 18.Pawels R. Formoterol, a new generation of beta-2-agonist. Toronto: Hogrefe and Huber Pub; 1990. [Google Scholar]
- 19.Roehrborn CG, Siami P, Barkin et al J. J. Urol. 2008;179:616. doi: 10.1016/j.juro.2007.09.084. [DOI] [PubMed] [Google Scholar]