

Abstract
Bacterial cell division occurs in conjunction with the formation of a cytokinetic Z-ring structure comprised of FtsZ subunits. Agents that can disrupt Z-ring formation have the potential, through this unique mechanism, to be effective against several of the newly emerging multi-drug resistant strains of infectious bacteria. 1- and 12-Aryl substituted benzo[c]phenanthridines have been identified as antibacterial agents that could exert their activity by disruption of Z-ring formation. Substituted 4- and 5-amino-1-phenylnaphthalenes represent substructures within the pharmacophore of these benzo[c]phenanthridines. Several 4- and 5-substituted 1-phenylnaphthalenes were synthesized and evaluated for antibacterial activity against Staphylococcus aureus and Enterococcus faecalis. The impact of select compounds on the polymerization dynamics of S. aureus FtsZ was also assessed.
Keywords: Antibacterial, 1-Phenylnaphthalenes, FtsZ-targeting, Enterococcus faecalis, Staphylococcus aureus
1. Introduction
Nosocomial infections associated with multi-drug resistant pathogens such as methicillin-resistant Staphylococcus aureus (MRSA) and vancomycin-resistant enterococci (VRE) represent a major health concern. Bacterial resistance to conventional antibiotics has created a critical need for the identification of novel therapeutic antibacterial targets [1,2]. FtsZ is an essential and highly conserved bacterial protein involved in cytokinesis [3–8]. Cell division in bacteria occurs at the site of formation of a cytokinetic Z-ring polymeric structure comprised of FtsZ subunits [3]. Several recent reviews have discussed the potential of antibacterial agents that target FtsZ, as well as recent advances in the discovery of small molecules that perturb processes in which FtsZ is involved [9–13]. FtsZ-targeting antibacterial agents can exert their disruptive effects on the Z-ring by either enhancing or inhibiting FtsZ self-polymerization [14–21].
Sanguinarine (a) (Fig. 1) is a plant alkaloid that has been identified as a small molecule that alters FtsZ Z-ring formation and has modest antibacterial activity [15]. Studies in our laboratory have also shown that chelerythrine (b) has similar effects on S. aureus and Bacillus subtilis. In addition, the presence of hydrophobic functionality at either the 12- or 1-position of benzo[c]phenanthridines, as in the case of (c) and (d), significantly enhanced antibacterial activity relative to either (a) or (b) [22,23]. These compounds (a–d) have a constitutive cationic charge that can adversely affect their desired pharmacokinetic properties. 4- and 5-Substituted 1-phenylnaphthalenes represent a truncated form within the core structure of these alkaloids. Similar results were observed within a series of substituted dibenzo [a,g] quinolizinium derivatives [24]. In the present study, we examined the relative structure–activity relationships associated with the potential antibacterial activity of several 4- and 5-substituted 1-phenylnaphthalene derivatives (Fig. 1). Unlike sanguinarine and chelerythrine, the 4- and 5-substituted 1-phenylnaphthalene derivatives selected for synthesis and evaluation did not possess a constitutive cationic charge. Instead, they contained functionality at the site in proximity to that of the iminium cation of compounds (a–d)), which would to some extent be protonated at physiological pH. Using aminomethyl, alkylamidinomethyl, or alkyl guanidinomethyl derivatives, the functionality of different base strengths was assessed. Increasing the alkyl linkage to two carbons from the naphthyl-ring further increases basic properties and the degree to which these derivatives will be protonated under physiological conditions.
Fig. 1.

The structure of sanguinarine (a), chelerythrine (b), 2,3,7,8-tetramethoxy-5-methyl-12-phenylbenzo[c]phenathridin-5-ium chloride (c), and 2,3,7,8-tetramethoxy-5-methyl-1-phenylbenzo[c]phenathridin-5-ium chloride (d).
2. Chemistry
The methods used for preparation of the 4-substituted-1-phenylnaphthalenes listed in Table 1 are outlined in Scheme 1. Treatment of 4-hydroxy-1-naphthaldehyde with triflic anhydride followed by Suzuki coupling with 4-(t-butyl)phenylboronic acid provided 4-(4-(t-butyl)phenyl)-1-naphthaldehyde. This intermediate was reduced with NaBH4 to its hydroxymethyl derivative, which was converted to its azidomethyl derivative allowing for reduction with PPh3 to give 1. Alternatively, this benzyl alcohol intermediate was treated under Mitsunobu conditions to provide the bis-N-Boc protected guanidinomethyl intermediate, which was treated with trifluoroacetic acid to yield 3. Condensation of 4-(4-(t-butyl)phenyl)-1-naphthaldehyde with nitromethane provided the 2-nitroethylene intermediate, which was reduced with LAH to give 2. Treatment of 2 with 1,3-di-Boc-2-(trifluoromethylsulfonyl)guanidine followed by treatment with TFA provided the 2-guanidinoethyl derivative, 4.
Table 1.
4- and 5-substituted 1-phenylnaphthalenes.
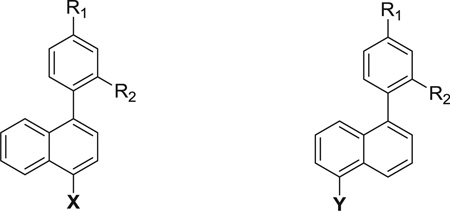 | ||||
|---|---|---|---|---|
| X | Y | R1 | R2 | |
| 1 | CH2NH2 | H | H | t-butyl |
| 2 | CH2CH2NH2 | H | H | t-butyl |
| 3 | CH2NC(NH2)2 | H | H | t-butyl |
| 4 | CH2CH2NC(NH2)2 | H | H | t-butyl |
| 5 | H | CH2NH2 | H | t-butyl |
| 6 | H | CH2CH2NH2 | H | t-butyl |
| 7 | H | CH2CNH(NH2) | H | t-butyl |
| 8 | H | CH2NC(NH2)2 | H | t-butyl |
| 9 | H | CH2CH2NC(NH2)2 | H | t-butyl |
| 10 | H | CH2NC(NH2)2 | H | cyclopropyl |
| 11 | H | CH2NC(NH2)2 | H | Cl |
| 12 | H | CH2NC(NH2)2 | H | Br |
| 13 | H | CH2CNH(NH2) | H | CF3 |
| 14 | H | CH2NC(NH2)2 | H | CF3 |
| 15 | H | CH2CH2NC(NH2)2 | H | CF3 |
| 16 | H | CH2CH2NH2 | OCH3 | CF3 |
| 17 | H | CH2CH2NH2 | OH | CF3 |
| 18 | H | CH2NC(NH2)2 | OCH3 | CF3 |
| 19 | H | CH2CH2NC(NH2)2 | OCH3 | CF3 |
Scheme 1.

Preparation of 1-(4-t-butylphenyl)-4-naphthalene derivatives. Reagents and conditions: (a) Tf2O, Et3N, DCM, −78 °C; (b) 4-t-butylphenylboronic acid, Pd(OAc)2, XPhos, K2CO3, ACN/H2O, 90 °C; (c) NaBH4, EtOH, rt; (d) DPPA, DBU, THF, 0 °C to rt; (e) PPh3 polymer bound, THF/H2O, rt; (f) CH3NO2, NH4OAc, reflux; (g) LAH, THF, rt; (h) 1,3-di-Boc-2-(trifluoromethylsulfonyl)guanidine, Et3N, CH2Cl2, rt; (i) TFA, CH2Cl2, 0 °C to rt; (j) 1,3-bis(t-butoxycarbonyl)guanidine, PPh3, DIAD, toluene, 0 °C to rt.
The 5-substituted-1-phenylnaphthalenes listed in Table 1 were synthesized as outlined in Schemes 2–4. 1-Naphthaldehyde was converted to 5-bromo-1-naphthaldehyde as previously described [25]. 5-Bromo-1-naphthaldehydewas subjected to a Suzuki coupling reaction to form the appropriate 5-phenyl-1-naphthaldehyde, which was then reduced to its hydroxymethyl derivative with NaBH4. These 5-phenyl-1-hydroxymethylnaphthalenes were then treated under Mitsunobu conditions with 1,3-bis(t-butoxycarbonyl)guanidine followed by treatment with TFA to provide the guanidinomethyl derivatives, 8 and 14. Alternatively, 5-bromo-1-hydroxymethylnaphthalene was converted first to its bis-(N-Boc)guanidinomethyl derivative and subsequently subjected to a Suzuki coupling followed by treatment with TFA to provide the 1-guanidinomethyl-5-phenyl substituted naphthalene, 18. In the case of 5-(4-t-butylphenyl)-1-hydroxymethylnaphthalene, formation of its mesylate followed by treatment with sodium azide and reduction with triphenylphosphine provided the 1-aminomethyl derivative, 5.
Scheme 2.

Methods used in the preparation of 5-substituted 1-(4-t-butylphenyl)naphthalenes, 5, 7, and 8, and 1-(4-trifluoromethylphenyl)naphthalene derivatives, 13, 14, and 18. Reagents and conditions: (a) R1R2-phenylboronic acid, Pd(OAc)2, XPhos, K2CO3, ACN/H2O, 90 °C; (b) NaBH4, EtOH, rt; (c) 1,3-bis(t-butoxycarbonyl)guanidine, PPh3, DIAD, toluene, 0 °C to rt; (d) TFA/CH2Cl2, 0 °C to rt; (e) PBr3, CH2Cl2, 0 °C to rt; (f) KCN, DMF, rt; (g) (i) 4 N HCl/dioxane, Et2O, MeOH, 0 °C; (ii) NH4Cl, EtOH, 85 °C [26]; (h) NaN3, PPh3, DMF/CCl4, 90 °C; (i) polymer-bound PPh3, THF/H2O, rt.
Scheme 4.

Methods used in the preparation of 5-substituted 1-phenylnaphthalenes, 10–12. Reagents and conditions: (a) borane ester, Pd(OAc)2, KOAc, DMF, 80 °C; (b) 4-bromo-X-benzene [X = Cl or I], Pd(PPh3)4, K2CO, dioxane/H2O, 100 °C; (c) cyclopropylboronic acid, Pd(OAc)2, Pcy3, K3PO4, toluene/H2O, 100 °C; (d) NaBH4, EtOH, rt; (e) 1,3-bis(t-butoxycarbonyl)guanidine, PPh3, DIAD, toluene, 0 °C to rt; (f) TFA/CH2Cl2, 0 °C to rt.
The 5-[(4-trifluoromethyl)phenyl]- and 5-[(4-t-butyl)phenyl]-1-amidinomethylnaphthalenes, 7 and 13, were synthesized from their respective bromomethylnaphthalenes, which were synthesized by treatment of their 1-hydroxymethylnaphthalene derivatives with PBr3.
The 1-(2-aminoethyl)-5-phenylnaphthalenes, 6 and 16, were prepared as outlined in Scheme 3. In one instance, the 1-naphthaldehyde intermediates could be condensed with nitromethane and then reduced with LAH. Alternatively, the 1-hydroxymethyl intermediates could be converted to their mesylates, reacted with NaCN and then reduced with LAH. Treatment of the requisite 2-aminoethyl derivative with 1,3-di-Boc-2-(trifluoromethylsulfonyl)guanidine followed by TFA provided the 2-guanidinoethyl derivatives, 9, 15 and 19.
Scheme 3.

Methods used in the preparation of 5-substituted 1-(4-t-butylphenyl)naphthalenes, 6 and 9, and 1-(4-trifluoromethylphenyl)naphthalene derivatives, 15–17 and 19. Reagents and conditions: (a) R1R2-phenylboronic acid, Pd(OAc)2, XPhos, K2CO3, ACN/H2O, 90 °C; (b) NaBH4, EtOH, rt; (c) MsCl, Et3N, CH2Cl2, 0 °C to rt; (d) KCN, DMF, rt; (e) LAH, ether; (f) 1,3-di-Boc-2-(trifluoromethylsulfonyl)-guanidine, Et3N, CH2Cl2, rt; (g) TFA/CH2Cl2; (h) BBr3, CH2Cl2, 0 °C to rt; (i) 4-trifluoromethylphenylboronic acid, Pd(OAc)2, XPhos, K2CO3, ACN/H2O, 90 °C; (j) nitromethane, NH4OAc, 100 °C.
Preparation of 10 was accomplished by coupling 5-(4-bromophenyl)-1-naphthaldehyde with cyclopropylboronic acid, yielding an aldehyde that was reduced to its hydroxymethyl derivative, which was converted to the bis-(N-Boc)guanidinomethyl derivative and then treated with TFA. The preparation of 11 and 12 was accomplished by conversion of 5-bromo-1-naphthaldehyde to its borane ester, which was Suzuki-coupled with either 1-bromo-4-chlorobenzene or 1-bromo-4-iodobenzene. The resulting 5-(4-chlorophenyl)-1-naphthaldehyde and 5-(4-bromophenyl)-1-naphthaldehyde were reduced to their hydroxymethyl derivatives, treated under Mitsunobu conditions and subsequently treated with TFA.
3. Results and discussion
3.1. In vitro antibacterial activity
The antibiotic activities of several 4- and 5-substituted 1-phenylnaphthalenes are provided in Table 2.
Table 2.
Antistaphylococcal and antienterococcal activities of 4- and 5-substituted 1-phenylnaphthalenes.
| Compound | aMIC (µg/mL) | |||
|---|---|---|---|---|
|
S. aureus 8325-4 (MSSA) |
S. aureus ATCC 33591 (MRSA) |
E. faecalis ATCC 19433 (VSE) |
E. faecalis ATCC 51575 (VRE) |
|
| 1 | 4.0 | 8.0 | 8.0 | 8.0 |
| 2 | 4.0 | 4.0 | 4.0 | 4.0 |
| 3 | 2.0 | 2.0 | 2.0 | 2.0 |
| 4 | 2.0 | 4.0 | 2.0 | 4.0 |
| 5 | 4.0 | 4.0 | 8.0 | 8.0 |
| 6 | 2.0 | 4.0 | 2.0 | 4.0 |
| 7 | 4.0 | 4.0 | 4.0 | 8.0 |
| 8 | 0.5 | 2.0 | 2.0 | 4.0 |
| 9 | 1.0 | 2.0 | 2.0 | 2.0 |
| 10 | 1.0 | 8.0 | 8.0 | 8.0 |
| 11 | 2.0 | 8.0 | 8.0 | 8.0 |
| 12 | 2.0 | 8.0 | 8.0 | 8.0 |
| 13 | 4.0 | 8.0 | 4.0 | 8.0 |
| 14 | 2.0 | 8.0 | 8.0 | 16 |
| 15 | 1.0 | 4.0 | 4.0 | 4.0 |
| 16 | 8.0 | 16 | 8.0 | 8 |
| 17 | 8.0 | 16 | 16 | 16 |
| 18 | 2.0 | 8.0 | 4.0 | 4.0 |
| 19 | 2.0 | 8.0 | 4.0 | 4.0 |
| Sanguinarine | 2.0 | 2.0 | 8.0 | 16 |
| Chelerythrine | 4.0 | 4.0 | 32 | 32 |
| Oxacillin | 0.06 | >64 | 8.0 | >64 |
| Vancomycin | 1.0 | 2.0 | 1.0 | >64 |
| Erythromycin | 0.13 | >64 | 1.0 | >64 |
| Tetracycline | 0.06 | 64 | 0.5 | >64 |
| Clindamycin | 0.03 | >64 | 2.0 | >64 |
MIC is defined as the lowest compound concentration at which bacterial growth is ≥90% inhibited.
Minimum inhibitory concentration (MIC) assays were conducted in accordance with Clinical Laboratory Standards Institute (CLSI) guidelines for broth microdilution [27].
The data in Table 2 include the relative activities of (4-t-butyl)phenylnaphthalenes with identical substituents at either the 4- or 5-position (1–9). Among the pairs of similarly substituted derivatives are the aminomethyl derivatives, 1 and 5; 2-aminoethyl derivatives, 2 and 6; guanidinomethyl derivatives, 3 and 8 and; 2-guanidinoethyl derivatives, 4 and 9. The only notable difference in activity is that the 5-substituted guanidinoalkyl derivatives, 8 and 9, tend to be slightly more active than their comparable 4-substituted isomers, 3 and 4, in S. aureus. The data suggest that, at either the 4- or 5-position, the more basic guanidine derivatives (3, 8 and 4, 9) have a tendency to be more potent than their corresponding aminomethyl (1, 5) and 2-aminoethyl (2, 6) derivatives. While less active than the 5-guanidinomethyl derivative, 8, the 5-amidinomethyl derivative, 7, exhibited antibacterial activity comparable to 5 and 6.
Compound 8 was most active against MSSA of the 1-(4-t-butylphenyl)naphthalenes (1–9), with an MIC of 0.5 µg/mL. Compounds 1–9 are significantly less active against MRSA, VSE and VRE. These data prompted further assessment of various 1-phenyl-5-guanidinomethylnaphthalene derivatives wherein the phenyl moiety had in place of the p-(t-butyl) substituent a cyclopropyl (10), chloro (11), bromo (12), or trifluoromethyl group (14–19). 5-Amidinomethyl-1-(4-trifluoromethyl)phenylnaphthalene (13) is 8-fold less active against MSSA and 4-fold less active against MRSA than 8. 5-(2-Guanidinoethyl)-1-(4-trifluoromethyl)phenylnaphthalene (15) had comparable activity to 9 against MSSA, but is less active against MRSA, VSE and VRE. The addition of an o-methoxyl group to the 1-phenyl substituent did slightly improve the activity of 18 relative to 14 against Enterococcus faecalis, but slightly decreased the potency of 19 relative to 15 against S. aureus. As anticipated from the previous structure–activity studies, the 5-(2-aminoethyl) derivatives, 16 and 17, are less active than similarly substituted 5-(guanidinoalkyl)-1-(4-trifluoromethyl)phenylnaphthalenes (15 and 19).
All of these compounds were more active than chelerythrine against both E. faecalis strains and only in the case of the vancomycin-sensitive E. faecalis did one compound, 17, exhibit less active antibacterial activity than sanguinarine. Several of these compounds (8–10, and 15) were more active against methicillin-sensitive S. aureus than either sanguinarine or chelerythrine. When evaluated for their antibacterial activity against methicillin-resistant S. aureus, the more potent 1-phenylnaphthalene derivatives had comparable or slightly reduced potency relative to sanguinarine.
The various 4- and 5-susbtituted 1-phenylnaphthalenes did not exhibit pronounce cross-resistance between MSSA and MRSA, as well as between VSE and VRE, strains of bacteria. This is particularly apparent among the various 1-(t-butylphenyl)naphthalene derivatives (1–9) wherein each instance there is less than a one well difference in potency between pairs of sensitive and resistant bacterial strains. The potency of these compounds against resistant bacteria is clearly evident in a comparison of their activity to that observed with the five known antibiotics used as control agents in this study. Compounds 3, 8, and 9 had comparable MICs to van-comycin against MRSA. Compounds 1–9 all had lower MIC values (2.0–8.0 µg/mL) against VRE than the clinical antibiotics evaluated in this study.
3.2. FtsZ polymerization assays
The selection of various 4- and 5-susbtituted 1-phenylnaphthalenes evaluated in this study was based upon the reported activity of sanguinarine to alter FtsZ Z-ring formation, as well as the observation that chelerythrine has a similar effect in S. aureus and B. subtilis [15,22]. It has been speculated that agents that alter or perturb the formation of FtsZ Z-ring could be effective against bacteria that have shown multi-drug resistance, such as MRSA and VRE [6–9]. Recent studies have shown that agents that stimulate FtsZ polymerization can cause an inhibition of FtsZ Z-ring formation [14,17,18]. To determine whether this mechanism of antibacterial activity may be associated with the antibacterial activity observed for these 4- and 5-substituted 1-phenylnaphthalenes, we examined the effects of select compounds on the dynamics of FtsZ self-polymerization. To this end, we used a turbidity assay to monitor the self-polymerization of purified S. aureus FtsZ (SaFtsZ). In this assay, FtsZ polymerization is detected in solution by a time-dependent increase in solution turbidity, as reflected by a corresponding increase in solution absorbance at 340 nm (A340).
As illustrative examples, Fig. 2 shows the time-dependent A340 profiles of SaFtsZ in the absence and presence of 6, 8, or the non-FtsZ-targeting control antibiotic vancomycin at a concentration of 40 µg/mL. As expected, vancomycin exerts a negligible impact on SaFtsZ polymerization since its antibacterial target is the bacterial cell wall. In striking contrast to vancomycin, compounds 6 and 8 markedly increase both the kinetics and extent of SaFtsZ polymerization. Stimulation of FtsZ polymerization has been shown to be the antibacterial mechanism of action for agents such as the substituted benzamide class of compounds (e.g., PC190723) [14,17,18]. Our polymerization results suggest that the antibacterial activity of the phenylnaphthalenes is associated with the stimulatory impact of the compounds on the dynamics of FtsZ polymerization.
Fig. 2.

Impact of select phenylnaphthalenes on the polymerization of S. aureus FtsZ (SaFtsZ), as determined by monitoring time-dependent changes in absorbance at 340 nm (A340). Polymerization profiles of SaFtsZ (10 µM) in the presence of DMSO vehicle (violet) or 40 µg/mL of either 6 (black), 8 (red), or the non-FtsZ-targeting comparator antibiotic vancomycin (green) are depicted.
4. Conclusions
The antistaphylococcal activities of several 4- and 5-substituted 1-phenylnaphthalenes, as measured by their MIC values, are significant. Particularly noteworthy are the antibacterial activities observed against methicillin-resistant S. aureus, which in several instances ranged between 2.0 and 4.0 µg/mL. These values are comparable to that observed with vancomycin and are well below the MICs observed with several clinical antibiotics evaluated in this study. Similar activity against MRSA was observed for sanguinarine or chelerythrine. As these alkaloids have a constitutive cationic charge, the physico-chemical properties of several of these 1-phenylnaphthalenes, would be expected to favor their in vivo absorption and distribution. In addition, the relative MIC values for several of these 1-phenylnaphthalenes against vancomycin-resistant E. faecalis (VRE) are lower than the clinical antibiotics or the two alkaloids evaluated in this study. Our studies with FtsZ polymerization suggest that the antibacterial activity of the phenylnaphthalenes is associated with the stimulatory impact of the compounds on the dynamics of FtsZ polymerization. These data suggest that compounds that interfere with FtsZ polymerization may prove to be useful in the development of antibiotics against multi-drug resistant bacteria.
5. Experimental
5.1. Chemistry: general methods
All reactions, unless otherwise stated, were done under nitrogen atmosphere. Reaction monitoring and follow-up were done using aluminum backed Silica G TLC plates (UV254 – Sorbent Technologies), visualizing with ultraviolet light. Flash column chromatography was done on a Combi Flash Rf Teledyne ISCO using hexane, ethyl acetate, dichloromethane, and methanol. The 1H (400 MHz or 300 MHz) and 13C (100 MHz) NMR spectra were done in CDCl3, Methanol-d4, and DMSO-d6 and recorded on a Bruker Avance III (400 MHz) Multinuclear NMR Spectrometer or a Varian Unity INOVA (300 MHz) NMR Spectrometer. Data is expressed in parts per million relative to the residual nondeuterated solvent signals, spin multiplicities are given as s (singlet), d (doublet), dd (doublet of doublets), t (triplet), dt (doublet of triplets), q (quartet), m (multiplet), bs (broad singlet), and bt (broad triplet), and coupling constants (J) are reported in Hertz. Melting points were determined using Mel-temp II apparatus and are uncorrected. HRMS experiments were conducted by Washington University Resource for Biomedical and Bioorganic Mass Spectrometry Department of Chemistry. All compounds tested for biological activity were analyzed for purity using a Shimadzu LC-20AT Prominence chromatograph equipped with an SPD-20A UV–VIS detector monitoring absorbances at both 254 and 280 nm. Compounds were analyzed using a PrincetonSPHER-100 5 µM C18 reverse-phase column (150 mm × 4.6 mm), using gradients of 0.1% TFA in water with increasing percentages of 0.1% TFA in acetonitrile. Under these solvent conditions, compounds eluted within 12 min using a flow rate of 1.0 mL/min and a gradient from 10 to 100% of acetonitrile containing 0.1% TFA.
5.2. General procedure for the synthesis of compound (1)
5.2.1. 4-Formylnaphthalen-1-yl trifluoromethanesulfonate
4-Hydroxy-1-naphthaldehyde (250 mg, 1.45 mmol) and Et3N (0.4 mL, 2.9 mmol) in anhydrous DCM (20 mL) were cooled to −78 °C. Tf2O (0.3 mL, 1.74 mmol) was slowly added to the mixture and was stirred for 30 min at −78 °C. Reaction was then quickly diluted with additional DCM and washed with saturated NaHCO3. Organic layer was dried over sodium sulfate and concentrated. Chromatography using ISCO max gradient 30% EtOAc/hexane yielded product as a brown oil (192 mg, 44% yield); 1H NMR (400 MHz) (CDCl3) δ 10.28 (s, 1H), 9.18 (d, J = 8.0 Hz, 1H), 8.06–8.04 (m, 1H), 7.90 (d, J = 8.0 Hz, 1H), 7.71–7.61 (m, 2H), 7.51 (d, J = 8.0 Hz, 1H); 13C NMR (100 MHz) (CDCl3) δ 191.8, 149.5, 135.6, 132.2, 131.2, 130.4, 128.7, 126.5, 125.2, 121.2, 120.2–117.1 (m), 116.8.
5.2.2. 4-(4-(t-Butyl)phenyl)-1-naphthaldehyde
4-Formylnaphthalen-1-yl trifluoromethanesulfonate (190 mg, 0.625 mmol), 4-t-butylphenylboronic acid (134 mg, 0.75 mmol), Pd(OAc)2 (14 mg, 0.0625 mmol), XPhos (60 mg, 0.125 mmol), and K2CO3 (259 mg, 1.875 mmol) in dioxane (6 mL) and H2O (3 mL) were combined in a flask and degassed. Reaction was refluxed at 100 °C for 1 h. Reaction was then cooled to RT, diluted with EtOAc, and washed with saturated NaHCO3. Organic layer was collected, dried over sodium sulfate, and concentrated. Chromatography using ISCO max gradient 30% EtOAc/hexane yielded product as a sticky golden oil (158 mg, 88% yield); 1H NMR (400 MHz) (CDCl3) δ 10.45 (s, 1H), 9.41 (d, J = 8.0 Hz, 1H), 8.08–8.05 (m, 2H), 7.75–7.72 (m, 1H), 7.63–7.55 (m, 4H), 7.47 (d, J = 8.0 Hz, 2H), 1.46 (s, 9H); 13C NMR (100 MHz) (CDCl3) δ 193.3, 151.2, 147.6, 136.9, 136.3, 132.1, 131.2, 130.5, 129.6, 128.8, 127.0, 126.9, 126.1, 125.4, 125.1, 34.8, 31.4; HRMS (ESI): calcd for C21H21O (M + H)+ 289.1587, found 289.1582.
5.2.3. (4-(4-(t-Butyl)phenyl)naphthalen-1-yl)methanol
NaBH4 (48 mg, 1.26 mmol) was carefully added to a solution of 4-(4-(t-butyl)phenyl)-1-naphthaldehyde (120 mg, 0.42 mmol) in EtOH (5 mL). Reaction was stirred at RT for 1 h then quenched with acetone (5 drops), filtered, and concentrated. Chromatography using ISCO max gradient 40% EtOAc/hexane yielded product as a glassy white solid (95 mg, 78% yield); mp 114–115 °C; 1H NMR (400 MHz) (CDCl3) δ 8.23 (d, J = 8.0 Hz, 1H), 8.05 (d, J = 8.5 Hz, 1H), 7.62–7.38 (m, 8H), 5.22 (s, 2H), 1.46 (s, 9H); 13C NMR (100 MHz) (CDCl3) δ 150.2, 140.9, 137.7, 135.5, 132.2, 131.6, 129.8, 127.1, 126.5, 126.2, 125.9, 125.2, 125.1, 123.9, 63.8, 34.7, 31.5; HRMS (ESI): calcd for C21H22NaO (M + Na)+ 313.1563, found 313.1560.
5.2.4. 1-(Azidomethyl)-4-(4-(t-butyl)phenyl)naphthalene
(4-(4-(t-Butyl)phenyl)naphthalen-1-yl)methanol (95 mg, 0.328 mmol) in anhydrous THF (3 mL) was cooled to 0 °C. DPPA (0.14 mL, 0.656 mmol) was then added drop-wise followed by DBU (0.1 mL, 0.656 mmol). Reaction was kept stirring over ice and was warmed to RT overnight. Reaction mixture was then poured into 0.5 N HCl and extracted with EtOAc. Organic layer was collected, dried over sodium sulfate, and concentrated. Chromatography using ISCO max gradient 20% EtOAc/hexane yielded product as a white solid (78 mg, 76% yield); mp 80–81 °C; 1H NMR (400 MHz) (CDCl3) δ 8.13 (d, J = 8.0 Hz, 1H), 8.06 (d, J = 8.0 Hz, 1H), 7.65–7.61 (m, 1H), 7.58–7.50 (m, 4H), 7.49–7.44 (m, 3H), 4.85 (s, 2H), 1.46 (s, 9H); 13C NMR (100 MHz) (CDCl3) δ 150.4, 141.7, 137.4, 132.3, 131.7, 130.2, 129.7, 127.3, 127.0, 126.6, 126.2, 126.1, 125.3, 123.7, 53.3, 34.7, 31.5.
5.2.5. (4-(4-(t-Butyl)phenyl)naphthalen-1-yl)methanamine (1)
1-(Azidomethyl)-4-(4-(t-butyl)phenyl)naphthalene (70 mg, 0.222 mmol) was combined in a flask with polymer supported (3 mmol/g loading) PPh3 (111 mg, 0.333 mmol), THF (5 mL), and H2O (0.5 mL). Reaction was stirred overnight at RT. Resin was then filtered off and filtrate concentrated. Chromatography using ISCO max gradient 10% MeOH/DCM yielded product as a golden oil (43 mg, 67% yield); 1H NMR (400 MHz) (CD3OD) δ 8.12 (d, J = 8.0 Hz, 1H), 7.89 (d, J = 8.0 Hz, 1H), 7.56–7.49 (m, 4H), 7.42–7.38 (m, 1H), 7.34–7.32 (m, 3H), 4.30 (s, 2H), 1.39 (s, 9H); 13C NMR (100 MHz) (CD3OD) δ 151.4, 141.2, 139.2, 138.1, 133.4, 132.8, 130.8, 128.0, 127.6, 127.2, 126.7, 126.3, 125.5, 124.4, 43.9, 35.5, 31.9; HRMS (ESI): calcd for C21H24N (M + H)+ 290.1903, found 290.1903.
5.3. General procedure for the synthesis of compound (2)
5.3.1. 1-(4-(t-Butyl)phenyl)-4-(2-nitrovinyl)naphthalene
4-(4-(t-Butyl)phenyl)-1-naphthaldehyde (5.2.2) (168 mg, 0.583 mmol) and NH4OAc (52 mg, 0.67 mmol) in nitromethane (4 mL) were refluxed at 102 °C for 2 h. Solvents were then evaporated. Chromatography using ISCO max gradient 10% EtOAc/hexane yielded product as a yellow solid (160 mg, 83% yield); mp 151 °C; 1H NMR (400 MHz) (CDCl3) δ 8.84 (d, J = 12.0 Hz, 1H), 8.13 (d, J = 8.0 Hz, 1H), 7.97 (d, J = 8.0 Hz, 1H), 7.74 (d, J = 4.0 Hz, 1H), 7.61 (d, J = 4.0 Hz, 1H), 7.60–7.56 (m, 1H), 7.48–7.44 (m, 3H), 7.40 (d, J = 8.0 Hz, 1H), 7.37–7.34 (m, 2H), 1.34 (s, 9H); 13C NMR (100 MHz) (CDCl3) δ 151.1, 145.1, 138.3, 136.8, 136.3, 132.2, 132.1, 129.6, 127.5, 126.7, 126.6, 126.2, 126.0, 124.4, 123.2, 34.7, 31.4; HRMS (ESI): calcd for C22H21NO2 (M)+ 331.1572, found 331.1569.
5.3.2. 2-(4-(4-(t-Butyl)phenyl)naphthalen-1-yl)ethanamine (2)
To a cooled solution of 1-(4-(t-butyl)phenyl)-4-(2-nitrovinyl)naphthalene (160 mg, 0.483 mmol) in anhydrous THF (5 mL) was carefully added LAH (55 mg, 1.45 mmol) at 0 °C. Reaction was then stirred at RT overnight and quenched with H2O over an ice bath. Reaction mixture was then diluted with EtOAc and washed with 1 N NaOH solution. Organic layer was collected, dried over sodium sulfate, and concentrated. Chromatography using ISCO max gradient 10% MeOH/DCM/0.1% NH4OH yielded product as a tan solid (75 mg, 51% yield); mp 101–102 °C; 1H NMR (400 MHz) (CD3OD) δ 8.18 (d, J = 12.0 Hz, 1H), 7.90–7.88 (m, 1H), 7.55–7.50 (m, 3H), 7.43–7.39 (m, 2H), 7.37–7.34 (m, 2H), 7.31 (d, J = 8.0 Hz, 1H), 3.30 (t, J = 16.0 Hz, 2H), 3.07–3.03 (m, 2H), 1.41 (s, 9H); 13C NMR (100 MHz) (CD3OD) δ 151.3, 140.6, 139.4, 136.1, 133.6, 130.8, 127.9, 127.5, 127.4, 126.9, 126.6, 126.2, 125.0, 43.4, 37.1, 35.5, 31.9; HRMS (ESI): calcd for C22H26N (M + H)+ 304.2060, found 304.2052.
5.4. General procedure for the synthesis of compound (3)
5.4.1. 1,3-Di-Boc-2-((4-(4-(t-butyl)phenyl)naphthalen-1-yl)methyl)guanidine
(4-(4-(t-Butyl)phenyl)naphthalen-1-yl)methanol (5.2.3) (40 mg, 0.14 mmol), PPh3 (60 mg, 0.23 mmol), and 1,3-bis(t-butoxycarbonyl)guanidine (70 mg, 0.27 mmol) in anhydrous toluene (3 mL) at 0 °C was added diisopropylazodicarboxylate (0.04 mL, 0.20 mmol) drop-wise over 15 min. Reaction was stirred at RT overnight then concentrated to an oil residue. Chromatography using ISCO max gradient 30% EtOAc/hexane yielded product as a white fluffy solid (70 mg, 97% yield); mp 88–90 °C; 1H NMR (400 MHz) (CDCl3) δ 9.46 (bs, 1H), 9.39 (bs, 1H), 7.97 (d, J = 8.4 Hz, 1H), 7.91 (d, J = 8.0 Hz, 1H), 7.46–7.41 (m, 3H), 7.37–7.28 (m, 3H), 7.28 (d, J = 7.4 Hz, 1H), 7.12 (d, J = 7.4 Hz, 1H), 5.67 (s, 2H), 1.37 (s, 9H), 1.33 (s, 9H), 1.10 (s, 9H); 13C NMR (100 MHz) (CDCl3) δ 163.8, 161.0, 155.2, 150.1, 139.2, 137.8, 133.7, 131.8, 131.0, 129.8, 127.0, 126.3, 125.7, 125.5, 125.1, 123.1, 121.6, 84.1, 79.0, 45.3, 34.6, 31.5, 28.3, 27.6; HRMS (ESI): calcd for C32H42N3O4 (M + H)+ 532.3175, found 532.3185.
5.4.2. 2-((4-(4-(t-Butyl)phenyl)naphthalen-1-yl)methyl)guanidine (3)
To a cooled solution of 1,3-di-Boc-2-((4-(4-(t-butyl)phenyl)naphthalen-1-yl)methyl)guanidine (70 mg, 0.13 mmol) in anhydrous DCM (0.5 mL) was slowly added TFA (0.5 mL). Reaction was stirred at RT overnight then solvents were evaporated. Chromatography using ISCO max gradient 10% MeOH/DCM/0.1% NH4OH yielded product as a white solid (35 mg, 80% yield); mp 85–87 °C; 1H NMR (400 MHz) (CD3OD) δ 8.07 (d, J = 8.4 Hz, 1H), 7.95 (d, J = 8.5 Hz, 1H), 7.65–7.61 (m, 1H), 7.57–7.48 (m, 4H), 7.43–7.37 (m, 3H), 4.93 (s, 2H), 1.42 (s, 9H); 13C NMR (100 MHz) (CD3OD) δ 158.9, 151.7, 142.6, 138.9, 133.6, 132.8, 131.9, 130.8, 128.1, 127.7, 127.4, 127.2, 126.4, 126.3, 124.3, 44.5, 35.5, 31.9; HRMS (ESI): calcd for C22H26N3 (M + H)+ 332.2121, found 332.2137.
5.5. General procedure for the synthesis of compound (4)
5.5.1. 1,3-Di-Boc-2-(2-(4-(4-(t-butyl)phenyl)naphthalen-1-yl)ethyl)guanidine
2-(4-(4-(t-Butyl)phenyl)naphthalen-1-yl)ethanamine (2) (25 mg, 0.08 mmol), 1,3-di-Boc-2-(trifluoromethylsulfonyl)-guanidine (40 mg, 0.1 mmol), and Et3N (0.03 mL, 0.22 mol) in anhydrous DCM (4 mL) were stirred at RT overnight then concentrated to an oil residue. Chromatography using ISCO max gradient 10% EtOAc/hexane yielded product as a white solid (38 mg, 84% yield); 1H NMR (300 MHz) (CDCl3) δ 8.50 (m, 1H), 8.35 (d, J = 9.0 Hz, 1H), 8.18 (d, J = 6.0 Hz, 1H), 7.56–7.34 (m, 7H), 3.82 (m, 2H), 3.42 (t, J = 6.0 Hz, 2H), 1.55 (s, 9H), 1.49 (s, 9H), 1.42 (s, 9H).
5.5.2. 2-(2-(4-(4-(t-Butyl)phenyl)naphthalen-1-yl)ethyl)guanidine (4)
To a cooled solution of 1,3-di-Boc-2-(2-(4-(4-(t-butyl)phenyl)naphthalen-1-yl)ethyl)guanidine (36 mg, 0.07 mmol) in anhydrous DCM (0.8 mL) was slowly added TFA (0.8 mL). Reaction was stirred at RT overnight then solvents were evaporated. Chromatography using ISCO max gradient 10% MeOH/DCM/0.1% NH4OH yielded product as a white solid (21 mg, 91% yield); mp 115–116 °C; 1H NMR (300 MHz) (CD3OD) δ 8.16 (d, J = 6.0 Hz, 1H), 7.92 (d, J = 6.0 Hz, 1H), 7.57 (m, 3H), 7.47 (m, 2H), 7.38 (m, 3H), 3.66 (t, J = 6.0 Hz, 2H), 3.44 (t, J = 6.0 Hz, 2H), 1.43 (s, 9H); 13C NMR (100 MHz) (CD3OD) δ 158.7, 151.5, 141.2, 139.2, 134.6, 133.6, 133.5, 130.8, 128.1, 127.7, 127.6, 127.2, 126.8, 126.3, 124.6, 43.1, 35.5, 33.0, 31.9; HRMS (ESI): calcd for C23H28N3 (M + H)+ 346.2278, found 346.2284.
5.6. General procedure for the synthesis of compound (5)
5.6.1. 5-(4-(t-Butyl)phenyl)-1-naphthaldehyde
5-Bromo-1-naphthaldehyde (500 mg, 2.1 mmol), 4-t-butyl-phenylboronic acid (470 mg, 2.6 mmol), and K2CO3 (880 mg, 6.4 mmol) in dioxane (15 mL) and H2O (3 mL) were degassed and Pd(PPh3)4 (73 mg, 0.06 mmol) was quickly added. Reaction was then refluxed at 100 °C for 5 h then cooled to RT. Solution was dried over magnesium sulfate, filtered, and concentrated. Chromatography using ISCO max gradient 50% EtOAc/hexane yielded product as a glassy white residue (490 mg, 88% yield); 1H NMR (300 MHz) (CDCl3) δ 10.45 (s, 1H), 9.29 (d, J = 6.0 Hz, 1H), 8.24 (d, J = 9.0 Hz, 1H), 8.00 (d, J = 9.0 Hz, 1H), 7.76–7.70 (m, 1H), 7.60–7.51 (m, 4H), 7.42–7.39 (m, 2H), 1.42 (s, 9H); HRMS (ESI): calcd for C21H20NaO (M + Na)+ 311.1412, found 311.1406.
5.6.2. (5-(4-(t-Butyl)phenyl)naphthalen-1-yl)methanol
A mixture of 5-(4-(t-butyl)phenyl)-1-naphthaldehyde (750 mg, 2.6 mmol) and NaBH4 (70 mg, 1.85 mmol) in 95% EtOH/MeOH (20 mL) was stirred at RT for 1 h. Reaction was then filtered and filtrate was concentrated. Residue was taken back up in EtOAc and washed with saturated NaHCO3 solution followed by brine. Organic layer was collected, dried over magnesium sulfate, and concentrated. Chromatography using ISCO max gradient 30% EtOAc/hexane yielded product as a white solid (740 mg, 99% yield); mp 81–83 °C; 1H NMR (400 MHz) (CDCl3) δ 8.18 (d, J = 8.5 Hz, 1H), 7.96 (d, J = 8.6 Hz, 1H), 7.64–7.60 (m, 1H), 7.57–7.52 (m, 3H), 7.49 (dd, J = 7.0 Hz, J = 1.2 Hz, 1H), 7.46–7.40 (m, 3H), 5.22 (s, 2H), 1.45 (s, 9H); 13C NMR (100 MHz) (CDCl3) δ 150.2, 141.0, 137.9, 136.4, 132.2, 131.6, 129.8, 128.6, 127.0, 126.9, 125.9, 125.4, 125.2, 123.0, 64.0, 34.6, 31.5; HRMS (ESI): calcd for C21H22NaO (M + Na)+ 313.1568, found 313.1565.
5.6.3. 1-(Azidomethyl)-5-(4-(t-butyl)phenyl)naphthalene
(5-(4-(t-Butyl)phenyl)naphthalen-1-yl)methanol (66 mg, 0.23 mmol), NaN3 (18 mg, 0.27 mmol), and PPh3 (125 mg, 0.48 mmol) in DMF (4 mL) and CCl4 (1 mL) were heated to 90 °C overnight. Solvents were then evaporated without further purification yielded product as a clear oil (50 mg, 69% yield); 1H NMR (400 MHz) (CDCl3) δ 7.95 (d, J = 8.5 Hz, 1H), 7.79 (d, J = 8.6 Hz, 1H), 7.54–7.50 (m, 1H), 7.44–7.38 (m, 4H), 7.35–7.28 (m, 3H), 4.72 (s, 2H), 1.33 (s, 9H); 13C NMR (100 MHz) (CDCl3) δ 150.3, 141.2, 137.7, 132.3, 131.8, 131.1, 129.8, 127.8, 127.3, 127.2, 126.2, 125.2, 125.1, 122.8, 53.4, 34.7, 31.5.
5.6.4. (5-(4-(t-Butyl)phenyl)naphthalen-1-yl)methanamine (5)
1-(Azidomethyl)-5-(4-(t-butyl)phenyl)naphthalene (50 mg, crude material) was combined in a flask with polymer supported (3 mmol/g loading) PPh3 (300 mg), THF (5 mL), and H2O (0.5 mL). Reaction was stirred overnight at RT. Resin was then filtered off and filtrate concentrated. Chromatography using ISCO max gradient 10% MeOH/DCM yielded product as an oily residue (34 mg, 74% yield); 1H NMR (400 MHz) (CDCl3) δ 8.03 (d, J = 8.0 Hz, 1H), 7.80 (d, J = 8.0 Hz, 1H), 7.51 (m, 1H), 7.44–7.29 (m, 7H), 4.31 (s, 2H), 1.58 (bs, 2H), 1.34 (s, 9H); 13C NMR (100 MHz) (CD3OD) δ 151.5, 142.5, 139.5, 138.6, 133.5, 132.9, 130.8, 127.9, 127.0, 126.8, 126.5, 126.2, 126.1, 123.5, 44.0, 35.5, 31.9; HRMS (ESI): calcd for C21H23N (M + H)+ 290.1903, found 290.1891.
5.7. General procedure for the synthesis of compound (6)
5.7.1. 2-(5-(4-(t-Butyl)phenyl)naphthalen-1-yl)acetonitrile
To a solution of (5-(4-(t-butyl)phenyl)naphthalen-1-yl)methanol (5.6.2) (1.0 g, 3.4 mmol) and Et3N (0.8 mL, 5.8 mmol) in DCM (25 mL) at 0 °C was slowly added MsCl (0.33 mL, 4.3 mmol). The reaction was stirred at 0 °C for 30 min then quenched with saturated NaHCO3 and extracted with EtOAc. Organic layer was collected, dried over magnesium sulfate, and concentrated to an oil. To this oil residue was added KCN (310 mg, 4.8 mmol) in DMSO (10 mL). The reaction was stirred at RT overnight then diluted with H2O and extracted with EtOAc. Extract was then washed with additional H2O followed by brine. Organic layer was collected, dried over magnesium sulfate, and concentrated. Chromatography using ISCO max gradient 50% EtOAc/hexane yielded product as a pale oil (510 mg, 50% yield); 1H NMR (400 MHz) (CDCl3) δ 8.01 (d, J = 8.6 Hz, 1H), 7.91 (d, J = 8.5 Hz, 1H), 7.70–7.63 (m, 2H), 7.57–7.53 (m, 3H), 7.46–7.42 (m, 3H), 4.21 (s, 2H), 1.45 (s, 9H); 13C NMR (100 MHz) (CDCl3) δ 150.5, 141.5, 137.5, 132.2, 131.2, 129.8, 127.5, 127.4, 126.6, 126.4, 125.9, 125.4, 125.2, 121.7, 117.7, 34.7, 31.4, 22.1; HRMS (ESI): calcd for C22H21N (M)+ 299.1674, found 299.1672.
5.7.2. 2-(5-(4-(t-Butyl)phenyl)naphthalen-1-yl)ethanamine (6)
LAH (1.0M/THF, 0.9 mL) was added drop-wise to a cooled 0 °C solution of 2-(5-(4-(t-butyl)phenyl)naphthalen-1-yl)acetonitrile (85 mg, 0.28 mmol) in anhydrous THF (5 mL). The resultant mixture was then refluxed for 7 h. Reaction was cooled to 0 °C and carefully treated with aqueous NaOH solution and extracted with EtOAc. Organic layer was collected, dried over magnesium sulfate, and concentrated. Chromatography using ISCO max gradient 20% 1.5% NH4OH/MeOH/DCM solution yielded product as a pale oil (15 mg, 17% yield); 1H NMR (300 MHz) (CDCl3) δ 8.99 (d, J = 6.0 Hz, 1H), 7.77 (d, J = 6.0 Hz, 1H), 7.50–7.41 (m, 3H), 7.34 (m, 3H), 7.29 (m, 2H), 3.21 (t, J = 6.0 Hz, 2H), 3.08 (t, J = 6.0 Hz, 2H), 1.92 (bs, 2H), 1.34 (s, 9H); 13C NMR (100 MHz) (CD3OD) δ 151.3, 140.6, 139.4, 136.1, 133.6, 130.8, 127.9, 127.5, 127.4, 126.9, 126.2, 125.0, 43.5, 37.1, 35.5, 31.9; HRMS (ESI): calcd for C22H25N (M + H)+ 304.2060, found 304.2046.
5.8. General procedure for the synthesis of compound (7)
5.8.1. 1-(Bromomethyl)-5-(4-(t-butyl)phenyl)naphthalene
(5-(4-(t-Butyl)phenyl)naphthalen-1-yl)methanol (5.6.2) (600 mg, 2.0 mmol) in anhydrous DCM (15 mL) was cooled to 0 °C and PBr3 (0.4 mL) was added drop-wise. The reaction was stirred at 0 °C for 1 h then quenched with saturated NaHCO3 and extracted with DCM. Organic layer was collected, dried over magnesium sulfate, and concentrated to yield product as a pale oil (670 mg, 86% yield); 1H NMR (300 MHz) (CDCl3) δ 8.21 (d, J = 9.0 Hz, 1H), 8.00 (d, J = 9.0 Hz, 1H), 7.69 (m, 2H), 7.60–7.50 (m, 3H), 7.46–7.36 (m, 3H), 5.06 (s, 2H), 1.45 (s, 9H).
5.8.2. 2-(5-(4-(t-Butyl)phenyl)naphthalen-1-yl)acetonitrile
1-(Bromomethyl)-5-(4-(t-butyl)phenyl)naphthalene (200 mg, 0.57 mmol) and KCN (55 mg, 0.85 mmol) in DMSO (6 mL) were stirred at RT overnight. Reaction was then diluted with H2O and extracted with ether. Organic layer was collected, dried over magnesium sulfate, and concentrated. Chromatography using ISCO max gradient 20% EtOAc/hexane yielded product as a pale oil (120 mg, 74% yield); 1H NMR (400 MHz) (CDCl3) δ 8.01 (d, J = 8.6 Hz, 1H), 7.91 (d, J = 8.5 Hz, 1H), 7.70–7.63 (m, 2H), 7.57– 7.53 (m, 3H), 7.46–7.42 (m, 3H), 4.21 (s, 2H), 1.45 (s, 9H); 13C NMR (100 MHz) (CDCl3) δ 150.5, 141.5, 137.5, 132.2, 131.2, 129.8, 127.5, 127.4, 126.6, 126.4, 125.9, 125.4, 125.2, 121.7, 117.7, 34.7, 31.4, 22.1; HRMS (ESI): calcd for C22H21N (M)+ 299.1674, found 299.1672.
5.8.3. 2-(5-(4-(t-Butyl)phenyl)naphthalen-1-yl)acetimidamide (7)
A solution of 2-(5-(4-(t-butyl)phenyl)naphthalen-1-yl)acetonitrile (120 mg, 0.4 mmol) in anhydrous Et2O (2 mL) was cooled to 0 °C and treated with 4 N HCl/dioxane solution (2 mL). Reaction was stirred at 0 °C for 4 h. The mixture was then placed in the refrigerator overnight. The precipitated solids were filtered out then treated with NH4OH/EtOH (6 mL) and heated to reflux for 6 h. Solvents were then evaporated. Chromatography using ISCO max gradient 20% 3% NH4OH/MeOH/DCM solution yielded product as pale solid (37 mg, 29% yield); mp 266–269 °C; 1H NMR (300 MHz) (CD3OD) δ 7.96 (m, 2H), 7.68 (m, 1H), 7.59–7.57 (m, 3H), 7.52–7.45 (m, 3H), 7.40 (m, 1H), 4.41 (s, 2H), 1.44 (s, 9H); 13C NMR (100 MHz) (CD3OD) δ 171.8, 151.8, 142.9, 139.0, 133.7, 133.5, 130.8, 129.6, 129.5, 128.4, 127.6, 127.4, 126.7, 126.3, 123.2, 54.8, 35.5, 31.8; HRMS (ESI): calcd for C22H25N2 (M + H)+ 317.2012, found 317.2019.
5.9. General procedure for the synthesis of compound (8)
5.9.1. 1,3-Di-Boc-2-((5-(4-(t-butyl)phenyl)naphthalen-1-yl)methyl)guanidine
(5-(4-(t-Butyl)phenyl)naphthalen-1-yl)methanol (5.6.2) (106 mg, 0.364 mmol), PPh3 (100 mg, 0.38 mmol), and 1,3-bis(t-butoxycarbonyl)guanidine (132 mg, 0.52 mmol) in anhydrous toluene (3 mL) at 0 °C was added diisopropylazodicarboxylate (0.1 mL, 0.52 mmol) drop-wise over 15 min. Reaction was stirred at RT overnight then 2 drops H2O were added, and the solution was concentrated. Chromatography using ISCO max gradient 30% EtOAc/hexane yielded product as a yellow solid (104 mg, 54% yield); 1H NMR (400 MHz) (CDCl3) δ 9.55 (bs, 1H), 9.50 (bs, 1H), 8.06 (d, J = 8.0 Hz, 1H), 7.87 (d, J = 8.0 Hz, 1H), 7.59–7.51 (m, 3H), 7.47–7.43 (m, 2H), 7.39–7.37 (m, 2H), 7.18 (d, J = 4.0 Hz, 1H), 5.77 (s, 2H), 1.46 (s, 9H), 1.43 (s, 9H), 1.20 (s, 9H).
5.9.2. 2-((5-(4-(t-Butyl)phenyl)naphthalen-1-yl)methyl)guanidine (8)
1,3-Di-Boc-2-((5-(4-(t-butyl)phenyl)naphthalen-1-yl)methyl)guanidine (20 mg, 0.07 mmol) was stirred at RT in a mixture of anhydrous DCM (0.5 mL) and TFA (0.5 mL) overnight. Solvents were then evaporated. Chromatography using ISCO max gradient 20% 3% NH4OH/MeOH/DCM solution yielded product as a white solid (17 mg, 74% yield); mp 160–163 °C; 1H NMR (400 MHz) (CD3OD) δ 8.00–7.60 (m, 5H), 7.56–7.36 (m, 5H), 4.89 (s, 2H), 1.41 (s, 9H); 13C NMR (100 MHz) (CD3OD) δ 158.8, 151.7, 142.7, 139.1, 133.6, 132.9, 132.8, 130.8, 128.3, 128.2, 127.3, 126.7, 126.4, 126.3, 126.3, 44.7, 35.5, 31.8; HRMS (ESI): calcd for C22H25N3 (M + H)+ 332.2121, found 332.2104.
5.10. General procedure for the synthesis of compound (9)
5.10.1. 1,3-Di-Boc-2-(2-(5-(4-(t-butyl)phenyl)naphthalen-1-yl)ethyl)guanidine
2-(5-(4-(t-Butyl)phenyl)naphthalen-1-yl)ethanamine (5.7.2) (35 mg, 0.12 mmol), 1,3-di-Boc-2-(trifluoromethylsulfonyl)-guanidine (55 mg, 0.14 mmol), and Et3N (0.04 mL, 0.29 mmol) in anhydrous DCM (6 mL) were stirred at RT overnight. Solvents were then evaporated. Chromatography using ISCO max gradient 20% EtOAc/hexane yielded product as a white solid (51 mg, 81% yield); 1H NMR (300 MHz) (CDCl3) δ 8.50 (t, J = 3.0 Hz, 1H), 8.30 (d, J = 9.0 Hz, 1H), 7.87 (dd, J = 6.0 Hz, J = 3.0 Hz, 1H), 7.62–7.50 (m, 3H), 7.46–7.33 (m, 4H), 3.80 (m, 2H), 3.43 (t, J = 6.0 Hz, 2H), 1.56 (s, 9H), 1.43 (s, 9H), 1.41 (s, 9H).
5.10.2. 2-(2-(5-(4-(t-Butyl)phenyl)naphthalen-1-yl)ethyl)guanidine (9)
1,3-Di-Boc-2-(2-(5-(4-(t-butyl)phenyl)naphthalen-1-yl)ethyl)guanidine (40 mg, 0.09 mmol) was stirred at RT in a mixture of anhydrous DCM (0.8 mL) and TFA (0.8 mL) overnight. Solvents were then evaporated. Chromatography using ISCO max gradient 15% 3% NH4OH/MeOH/DCM solution yielded product as a white solid (29 mg, 96% yield); mp 185 °C; 1H NMR (300 MHz) (CD3OD) δ 8.13 (d, J = 9.0 Hz, 1H), 7.81 (d, J = 9.0 Hz, 1H), 7.65–7.55 (m, 3H), 7.45–7.36 (m, 5H), 3.66 (t, J = 6.0 Hz, 2H), 3.46 (t, J = 6.0 Hz, 2H), 1.43 (s, 9H); 13C NMR (100 MHz) (CD3OD) δ 158.7, 151.6, 142.6, 139.4, 135.5, 133.7, 133.6, 130.8, 128.1, 127.9, 126.8, 126.7, 126.5, 126.3, 123.7, 43.2, 35.5, 33.3, 31.9; HRMS (ESI): calcd for C23H28N3 (M + H)+ 346.2278, found 346.2281.
5.11. General procedure for the synthesis of compound (10)
5.11.1. 5-(4,4,5,5-Tetramethyl-1,3,2-dioxaborolan-2-yl)-1-naphthaldehyde
To a stirred mixture of 5-bromo-naphthaldehyde (500 mg, 2.21 mmol), diborane (810 mg, 3.19 mmol), and KOAc (800 mg, 6.3 mmol) in anhydrous DMF (10 mL) was added Pd(OAc)2 (20 mg, 0.09 mmol). Reaction mixture was degassed then heated at 80 °C for 6 h. Reaction was cooled to RT and diluted with EtOAc and washed with brine. Organic layer was collected, dried over magnesium sulfate, and concentrated. Chromatography using ISCO max gradient 15% EtOAc/hexane yielded product as a clear oil (410 mg, 68% yield); 1H NMR (400 MHz) (CDCl3) δ 10.40 (s, 1H), 9.42 (d, J = 8.0 Hz, 1H), 9.12 (d, J = 8.0 Hz, 1H), 8.21 (d, J = 8.0 Hz, 1H), 8.01 (d, J = 8.0 Hz, 1H), 7.73–7.69 (m, 2H), 1.46 (s, 12H).
5.11.2. 5-(4-Bromophenyl)-1-naphthaldehyde
A flask containing 5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1-naphthaldehyde (209 mg, 0.7 mmol), 4-bromoiodobenzene (230 mg, 0.8 mmol), and K2CO3 (200 mg, 1.4 mmol) in dioxane (8 mL) and H2O (1.5 mL) was purged with N2. Pd(PPh3)4 (40 mg, 0.03 mmol) was then quickly added and resultant mixture was heated at 100 °C for 5 h. Reaction was cooled to RT, dried over magnesium sulfate, and concentrated. Chromatography using ISCO max gradient 50% EtOAc/hexane yielded product as a light yellow solid (142 mg, 65% yield); 1H NMR (300 MHz) (CDCl3) δ 10.50 (s, 1H), 8.15 (d, J = 9.0 Hz, 1H), 8.05 (d, J = 9.0 Hz, 1H), 7.76 (m, 2H), 7.69–7.60 (m, 3H), 7.55 (d, J = 9.0 Hz, 1H), 7.39–7.36 (m, 2H).
5.11.3. (5-(4-Bromophenyl)naphthalen-1-yl)methanol
5-(4-Bromophenyl)-1-naphthaldehyde (140 mg, 0.45 mmol) and NaBH4 (12 mg, 0.32 mmol) in 95% EtOH/MeOH (8 mL) were stirred at RT for 2 h. Reaction mixture was then filtered and filtrate concentrated. Chromatography using ISCO max gradient 30% EtOAc/hexane yielded product as a clear oil (140 mg, quantitative); 1H NMR (300 MHz) (CDCl3) δ 8.11 (d, J = 9.0 Hz, 1H), 7.74 (d, J = 9.0 Hz, 1H), 7.56–7.47 (m, 4H), 7.36–7.26 (m, 4H), 5.14 (s, 2H).
5.11.4. (5-(4-Cyclopropylphenyl)naphthalen-1-yl)methanol
(5-(4-Bromophenyl)naphthalen-1-yl)methanol (130 mg, 0.415 mmol), cyclopropylboronic acid (71 mg, 0.83 mmol), Pd(OAc)2 (5 mg, 0.021 mmol), tricyclohexylphosphine (12 mg, 0.0415 mmol), and K3PO4 (308 mg, 1.45 mmol) were combined in a flask with toluene (3 mL) and H2O (1 mL) and degassed. Reaction mixture was then refluxed at 100 °C for 3 h. Solution was cooled to RT then diluted with EtOAc and washed with saturated NaHCO3. Organic layer was dried over sodium sulfate and concentrated. Chromatography using ISCO max gradient 35% EtOAc/hexane yielded product as a brown oil (86 mg, 75% yield); 1H NMR (400 MHz) (CDCl3) δ 8.17–8.15 (m, 1H), 7.96–7.94 (m, 1H), 7.61 (dd, J = 8.0 Hz, J = 8.0 Hz, 1H), 7.56–7.53 (m, 2H), 7.49–7.47 (m, 1H), 7.43–7.40 (m, 2H), 7.25–7.23 (m, 2H), 5.19 (s, 2H), 2.08–2.01 (m, 1H), 1.11–1.06 (m, 2H), 0.87–0.83 (m, 2H); 13C NMR (100 MHz) (CDCl3) δ 143.2, 138.0, 136.5, 132.2, 131.6, 130.1, 130.1, 138.3, 126.8, 125.8, 125.6, 125.4, 125.3, 123.0, 63.9, 15.3, 9.4; HRMS (ESI): calcd for C20H18NaO (M + Na)+ 297.1250, found 297.1251.
5.11.5. 2-((5-(4-Cyclopropylphenyl)naphthalen-1-yl)methyl)guanidine (10)
To a solution of (5-(4-cyclopropylphenyl)naphthalen-1-yl)methanol (80 mg, 0.292 mmol), PPh3 (115 mg, 0.584 mmol), and 1,3-bis(t-butoxycarbonyl)guanidine (151 mg, 0.438 mmol) in anhydrous toluene (3 mL) at 0 °C was added diisopropylazodicarboxylate (0.09 mL, 0.438 mmol) drop-wise over 15 min. Reaction was stirred for 3 h at RT then 2 drops H2O were added, and the solution was concentrated. Solid was then dissolved in DCM and passed through silica column and resulting crude product was then re-dissolved in anhydrous DCM (1.5 mL) and cooled to 0 °C. TFA (1.5 mL) was then added. Reaction was taken off ice bath and stirred at RT for 2 h then solvents were evaporated. Solid was then taken back up in DCM and precipitate was filtered off yielded product as a gummy tan solid (18 mg, 20% yield over 2 steps); 1H NMR (400 MHz) (CD3OD) δ 8.02–8.00 (m, 1H), 7.90–7.88 (m, 1H), 7.67–7.63 (m, 1H), 7.54–7.50 (m, 2H), 7.47–7.42 (m, 1H), 7.33–7.31 (m, 2H), 7.24–7.22 (m, 2H), 4.93 (s, 2H), 2.06–1.99 (m, 1H), 1.06–1.02 (m, 2H), 0.79–0.76 (m, 2H); 13C NMR (100 MHz) (CD3OD) δ 158.9, 144.9, 142.7, 139.1, 133.6, 132.8, 131.0, 129.4, 128.2, 128.1, 127.3, 126.7, 126.6, 126.4, 123.3, 44.7, 16.0, 9.7; HRMS (ESI): calcd for C21H22N (M + H)+ 316.1808, found 316.1805.
5.12. General procedure for the synthesis of compound (11)
5.12.1. 5-(4-Chlorophenyl)-1-naphthaldehyde
A flask containing 5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1-naphthaldehyde (200 mg, 0.7 mmol), 4-chlorobromobenzene (160 mg, 0.8 mmol), and K2CO3 (200 mg, 1.4 mmol) in dioxane (6 mL) and H2O (1.5 mL) was purged with N2. Pd(PPh3)4 (40 mg, 0.03 mmol) was then quickly added and resultant mixture was heated at 100 °C for 5 h. Reaction was cooled to RT, dried over magnesium sulfate, and concentrated. Chromatography using ISCO max gradient 15% EtOAc/hexane yielded product as a pale solid (160 mg, 84% yield); 1H NMR (300 MHz) (CDCl3) δ 10.50 (s, 1H), 8.15 (d, J = 9.0 Hz, 1H), 8.06 (d, J = 9.0 Hz, 1H), 7.77 (m, 1H), 7.63 (m, 1H), 7.56–7.50 (m, 4H), 7.44–7.14 (m, 2H).
5.12.2. (5-(4-Chlorophenyl)naphthalen-1-yl)methanol
5-(4-Chlorophenyl)-1-naphthaldehyde (160 mg, 0.6 mmol) and NaBH4 (16 mg, 0.42 mmol) in 95% EtOH/MeOH (10 mL) were stirred at RT for 1 h. Reaction mixture was then filtered and filtrate concentrated. Residue was then taken back up in DCM and washed with saturated NaHCO3 solution followed by brine. Organic layer was collected, dried over magnesium sulfate, and concentrated. Chromatography using ISCO max gradient 30% EtOAc/hexane yielded product as a clear oil (160 mg, quantitative); 1H NMR (300 MHz) (CDCl3) δ 8.21 (d, J = 9.0 Hz, 1H), 7.84 (d, J = 9.0 Hz, 1H), 7.66–7.58 (m, 2H), 7.52–7.48 (m, 6H), 5.23 (s, 2H); 13C NMR (100 MHz) (CDCl3) δ 139.7, 139.4, 136.6, 133.4, 131.9, 131.6, 131.4, 128.5, 127.0, 126.3, 125.8, 125.7, 125.4, 123.6, 63.8; HRMS (ESI): calcd for C17H14ClO (M + H)+ 291.0547, found 291.0541.
5.12.3. 1,3-Di-Boc-2-((5-(4-chlorophenyl)naphthalen-1-yl)methyl)guanidine
To a solution of (5-(4-chlorophenyl)naphthalen-1-yl)methanol (50 mg, 0.186 mmol), PPh3 (73 mg, 0.278 mmol), and 1,3-bis(t-butoxycarbonyl)guanidine (96 mg, 0.37 mmol) in anhydrous toluene (3 mL) at 0 °C was added diisopropylazodicarboxylate (0.06 mL, 0.31 mmol) drop-wise over 15 min. Reaction was stirred at RT overnight, and the solution was then concentrated to an oil residue. Chromatography using ISCO max gradient 30% EtOAc/hexane yielded product as a pale solid (46 mg, 48% yield); 1H NMR (300 MHz) (CDCl3) δ 8.06 (d, J= 9.0 Hz, 1H), 7.75 (d, J=9.0 Hz, 1H), 7.61–7.37 (m, 7H), 7.21 (d, J = 6.0 Hz, 1H), 5.80 (s, 2H), 1.47 (s, 9H), 1.21 (s, 9H).
5.12.4. 2-((5-(4-Chlorophenyl)naphthalen-1-yl)methyl)guanidine (11)
1,3-Di-Boc-2-((5-(4-chlorophenyl)naphthalen-1-yl)methyl)guanidine (46 mg, 0.09 mmol) in a mixture of anhydrous DCM (0.8 mL) and TFA (0.8 mL) was stirred at RT overnight. Solvents were then evaporated. Chromatography using ISCO max gradient 20% 3% NH4OH/MeOH/DCM solution yielded product as a white solid (27 mg, 78% yield); mp 106–109 °C; 1H NMR (300 MHz) (DMSO-d6) δ 8.08 (m, 2H), 7.75–7.59 (m, 4H), 7.53–7.46 (m, 4H), 4.89 (d, J = 3.0 Hz, 2H); 13C NMR (100 MHz) (CD3OD) δ 158.9, 141.3, 140.7, 134.7, 133.3, 132.8, 132.6, 129.6, 128.4, 127.6, 127.3, 126.8, 123.9, 44.6; HRMS (ESI): calcd C18H17ClN3 (M + H)+ 310.1106, found 310.1110.
5.13. General procedure for the synthesis of compound (12)
5.13.1. 1,3-Di-Boc-2-((5-(4-bromophenyl)naphthalen-1-yl)methyl)guanidine
To a solution of (5-(4-bromophenyl)naphthalen-1-yl)methanol (5.11.3) (40 mg, 0.128 mmol), PPh3 (60 mg, 0.23 mmol), and 1,3-bis(t-butoxycarbonyl)guanidine (66 mg, 0.26 mmol) in anhydrous toluene (3 mL) at 0 °C was added diisopropylazodicarboxylate (0.04 mL, 0.20 mmol) drop-wise over 15 min. Reaction was stirred at RT overnight, and the solution was then concentrated to an oil residue. Chromatography using ISCO max gradient 30% EtOAc/hexane yielded product as a pale solid (46 mg, 47% yield); 1H NMR (300 MHz) (CDCl3) δ 9.60 (bs, 1H), 9.50 (bs, 1H), 8.06 (d, J = 9.0 Hz, 1H), 7.75 (d, J = 9.0 Hz, 1H), 7.66–7.37 (m, 7H), 7.21 (d, J = 6.0 Hz, 1H), 5.78 (s, 2H), 1.47 (s, 9H), 1.22 (s, 9H).
5.13.2. 2-((5-(4-Bromophenyl)naphthalen-1-yl)methyl)guanidine (12)
1,3-Di-Boc-2-((5-(4-bromophenyl)naphthalen-1-yl)methyl)guanidine (34 mg, 0.06 mmol) in a mixture of anhydrous DCM (0.8 mL) and TFA (0.8 mL) was stirred at RT overnight. Solvents were then evaporated. Chromatography using ISCO max gradient 20% 3% NH4OH/MeOH/DCM solution yielded product as a white solid (18 mg, 75% yield); mp 124–127 °C; 1H NMR (300 MHz) (DMSO-d6) δ 8.02 (m, 2H), 7.75–7.67 (m, 2H), 7.52 (m, 2H), 7.12 (m, 4H), 4.90 (s, 2H); 13C NMR (100 MHz) (CD3OD) δ 158.9, 141.3, 141.2, 133.2, 133.1, 132.9, 132.8, 132.6, 131.0, 128.3, 127.6, 127.3, 126.8, 124.0, 122.7, 44.6; HRMS (ESI): calcd for C18H17BrN3 (M + H)+ 354.0600, found 354.0599.
5.14. General procedure for the synthesis of compound (13)
5.14.1. 5-(4-(Trifluoromethyl)phenyl)-1-naphthaldehyde
5-Bromo-1-naphthaldehyde (500 mg, 2.1 mmol), 4-trifluoromethylphenylboronic acid (620 mg, 3.2 mmol), and K2CO3 (880 mg, 6.4 mmol) in dioxane (15 mL) and H2O (3 mL) were degassed and Pd(PPh3)4 (73 mg, 0.06 mmol) was quickly added. Reaction was then refluxed at 100 °C for 5 h then cooled to RT. Solution was dried over magnesium sulfate, filtered, and concentrated. Chromatography using ISCO max gradient 30% EtOAc/hexane yielded product as a pale solid (560 mg, 88% yield); mp 78–80 °C; 1H NMR (400 MHz) (CDCl3) δ 10.45 (s, 1H), 9.35 (d, J = 8.0 Hz, 1H), 8.08–8.01 (m, 2H), 7.78–7.73 (m, 3H), 7.62–7.58 (m, 3H), 7.52 (d, J = 8.0 Hz, 1H); 13C NMR (100 MHz) (CDCl3) δ 193.4, 144.1, 139.3, 136.6, 132.7, 131.7, 131.0, 130.5, 128.4, 128.0, 125.4, 125.3, 125.2, 125.1, 124.9, 120.3.
5.14.2. (5-(4-(Trifluoromethyl)phenyl)naphthalen-1-yl)methanol
A mixture of 5-(4-(trifluoromethyl)phenyl)-1-naphthaldehyde (350 mg, 1.16 mmol) and NaBH4 (31 mg, 0.82 mmol) in 95% EtOH/MeOH (15 mL) was stirred at RT for 1 h. Reaction was then filtered and filtrate was concentrated. Residue was taken back up in EtOAc and washed with saturated NaHCO3 solution followed by brine. Organic layer was collected, dried over magnesium sulfate, and concentrated. Chromatography using ISCO max gradient 30% EtOAc/hexane yielded product as a white solid (350 mg, quantitative); mp 98 °C; 1H NMR (400 MHz) (CDCl3) δ 8.23 (d, J = 8.5 Hz, 1H), 7.79 (t, J = 15.0, 3H), 7.66–7.58 (m, 4H), 7.48–7.43 (m, 2H), 5.22 (s, 2H); 13C NMR (100 MHz) (CDCl3) δ 144.7, 139.5, 136.7, 131.7–131.6 (m), 130.4, 129.8, 129.4, 127.0, 126.2, 125.9, 125.8, 125.5, 125.3–125.2 (m), 124.0, 123.0, 63.9.
5.14.3. 2-(5-(4-(Trifluoromethyl)phenyl)naphthalen-1-yl)acetonitrile
(5-(4-(Trifluoromethyl)naphthalen-1-yl)methanol (300 mg, 0.99 mmol) in anhydrous DCM (10 mL) was cooled to 0 °C and PBr3 (0.14 mL) was added drop-wise. The reaction was stirred at 0 °C for 1 h then quenched with saturated NaHCO3 and extracted with DCM. Organic layer was collected, dried over magnesium sulfate, and concentrated to yield crude product as a pale gum which was taken forward without further purification. This crude 1-(bromomethyl)-5-(4-(trifluoromethyl)phenyl)naphthalene (300 mg, 0.82 mmol) and KCN (100 mg, 1.5 mmol) in DMSO were stirred at RT overnight. Reaction was then diluted with H2O and extracted with ether. Organic layer was collected, dried over magnesium sulfate, and concentrated. Chromatography using ISCO max gradient 20% EtOAc/hexane yielded product as a white solid (260 mg, 85% yield over two steps); mp 44–45 °C; 1H NMR (400 MHz) (CDCl3) δ 7.87 (d, J = 8.6 Hz, 1H), 7.73 (d, J = 8.6 Hz, 1H), 7.69–7.68 (m, 2H), 7.62–7.58 (m, 1H), 7.56 (d, J = 7.2 Hz, 1H), 7.52–7.51 (m, 2H), 7.43–7.34 (m, 2H), 4.12 (s, 2H); 13C NMR (100 MHz) (CDCl3) δ 144.2, 140.0, 131.8, 131.2, 130.4, 130.0, 129.7, 127.5, 126.8, 126.7, 126.5, 126.3, 125.9, 125.3, 122.7, 117.5, 22.1; HRMS (ESI): calcd for C19H12F3NNa (M + Na)+ 334.0820, found 334.0823.
5.14.4. 2-(5-(4-(Trifluoromethyl)phenyl)naphthalen-1-yl)acetimidamide (13)
A solution of 2-(5-(4-(trifluoromethyl)phenyl)naphthalen-1-yl)acetonitrile (100 mg, 0.32 mmol) in anhydrous Et2O (2 mL) was cooled to 0 °C and treated with 4 N HCl/dioxane solution (2 mL). Reaction was stirred at 0 °C for 4 h. The mixture was then placed in the refrigerator overnight. The precipitated solids were filtered out then treated with NH4OH/EtOH (5 mL) and heated to reflux for 6 h. Solvents were then evaporated. Chromatography using ISCO max gradient 20% 3% NH4OH/MeOH/DCM solution yielded product as a pale gummy solid (37 mg, 29% yield); mp 1H NMR (300 MHz) (CDCl3) δ 8.06 (d, J = 6.0 Hz, 1H), 7.86 (d, J = 9.0 Hz, 2H), 7.76–7.67 (m, 3H), 7.62–7.50 (m, 4H), 4.44 (s, 2H); 13C NMR (100 MHz) (CD3OD) δ 171.7, 141.2, 133.5, 133.2, 131.8, 130.2, 130.0, 129.8, 129.4, 128.6, 127.8, 127.6, 127.4, 127.3, 126.4–126.3 (m), 124.2, 37.2; HRMS (ESI): calcd for C19H16F3N2 (M + H)+ 329.1260, found 329.1244.
5.15. General procedure for the synthesis of compound (14)
5.15.1. 1,3-Di-Boc-2-((5-(4-(Trifluoromethyl)phenyl)naphthalen-1-yl)methyl)guanidine
To a solution of (5-(4-(trifluoromethyl)phenyl)naphthalen-1-yl)methanol (5.14.2) (40 mg, 0.13 mmol), PPh3 (55 mg, 0.21 mmol), and 1,3-bis(t-butoxycarbonyl)guanidine (70 mg, 0.27 mmol) in anhydrous toluene (3 mL) at 0 °C was added diisopropylazodicarboxylate (0.04 mL, 0.20 mmol) drop-wise over 15 min. Reaction was stirred at RT overnight, and the solution was then concentrated to an oil residue. Chromatography using ISCO max gradient 30% EtOAc/hexane yielded product as a white glassy residue (56 mg, 80% yield); 1H NMR (400 MHz) (CDCl3) δ 9.44 (bs, 2H), 8.00 (d, J = 8.0 Hz, 1H), 7.67 (d, J = 8.0 Hz, 2H), 7.61 (d, J = 8.0 Hz, 1H), 7.53–7.48 (m, 3H), 7.35–7.28 (m, 2H), 7.12 (d, J = 4.0 Hz, 1H), 5.68 (s, 2H), 1.37 (s, 9H), 1.12 (s, 9H); 13C NMR (100 MHz) (CDCl3) δ 163.8, 160.9, 155.1, 144.8, 139.4, 134.4, 131.4, 131.0, 130.5, 129.7, 126.8, 125.7, 125.6, 125.3, 125.2–125.1 (m), 124.6, 123.1, 122.1, 84.1, 79.0, 45.4, 28.2, 27.7; HRMS (ESI): calcd for C29H33F3N3O4 (M + H)+ 544.2423, found 544.2431.
5.15.2. 2-((5-(4-(Trifluoromethyl)phenyl)naphthalen-1-yl)methyl)guanidine (14)
1,3-Di-Boc-2-((5-(4-(Trifluoromethyl)phenyl)naphthalen-1-yl)methyl)guanidine (56 mg, 0.1 mmol) in a mixture of anhydrous DCM (0.8 mL) and TFA (0.8 mL) was stirred at RT overnight. Solvents were then evaporated. Chromatography using ISCO max gradient 20% 3% NH4OH/MeOH/DCM solution yielded product as a white solid (20 mg, 51% yield); mp 198–200 °C; 1H NMR (400 MHz) (CD3OD) δ 8.10 (d, J = 8.6 Hz, 1H), 7.85–7.79 (m, 3H), 7.73–7.69 (m, 1H), 7.65 (d, J = 8.0 Hz, 2H), 7.58–7.47 (m, 3H), 4.96 (s, 2H); 13C NMR (100 MHz) (CD3OD) δ 158.9, 146.2, 141.0, 133.2, 133.1, 132.8, 131.8, 130.9, 128.5, 127.4, 127.3, 127.0, 126.9, 126.4–126.3 (m), 124.5, 124.4, 44.6; HRMS (ESI): calcd for C19H17F3N3 (M + H)+ 344.1369, found 344.1352.
5.16. General procedure for the synthesis of compound (15)
5.16.1. 1-(2-Nitrovinyl)-5-(4-(trifluoromethyl)phenyl)naphthalene
5-(4-(Trifluoromethyl)phenyl)-1-naphthaldehyde (200 mg, 0.67 mmol) and NH4OAc (60 mg, 0.78 mmol) in nitromethane (3 mL) were heated to reflux for 6 h. Solvents were then evaporated. Chromatography using ISCO max gradient 10% EtOAc/hexane yielded product as a yellow solid (210 mg, 91% yield); 1H NMR (400 MHz) (CDCl3) δ 8.82 (d, J = 13.4 Hz, 1H), 8.12 (d, J = 8.6 Hz, 1H), 7.88 (d, J = 8.6 Hz, 1H), 7.70 (d, J = 7.9 Hz, 3H), 7.66–7.58 (m, 2H), 7.52 (d, J = 8.0 Hz, 2H), 7.42–7.40 (m, 2H); 13C NMR (100 MHz) (CDCl3) δ 143.9, 139.9, 138.9, 136.3, 131.9, 131.8, 130.4, 130.1, 127.9, 127.6, 127.1, 126.5, 125.8, 125.4, 125.3, 123.3.
5.16.2. 2-(5-(4-(Trifluoromethyl)phenyl)naphthalen-1-yl)ethanamine
LAH (1.0M/THF, 2 mL) was added drop-wise to a cooled 0 °C solution of 1-(2-nitrovinyl)-5-(4-(trifluoromethyl)phenyl)naphthalene (210 mg, 0.62 mmol) in anhydrous THF (10 mL). The resultant mixture was then refluxed for 7 h. Reaction was cooled to 0 °C and carefully treated with aqueous NaOH solution and diluted with EtOAc. Solids were filtered off and filtrate washed with 6 N HCl to extract out amine. 6 N HCl solution was then basified with 4 N NaOH solution back to pH 9 and extracted with CHCl3. Organic layer was then washed with brine, collected, dried over magnesium sulfate, and concentrated yielded product as a pale solid requiring no further purification (50 mg, 26% yield); 1H NMR (300 MHz) (CDCl3) δ 8.16 (d, J = 9.0 Hz, 1H), 7.80–7.71 (m, 3H), 7.63–7.57 (m, 3H), 7.44–7.37 (m, 3H), 3.36 (m, 2H), 3.23 (m, 2H).
5.16.3. 1,3-Di-Boc-2-(2-(5-(4-(trifluoromethyl)phenyl)naphthalen-1-yl)ethyl)guanidine
2-(5-(4-(Trifluoromethyl)phenyl)naphthalen-1-yl)ethanamine (25 mg, 0.08 mmol), 1,3-di-Boc-2-(trifluoromethylsulfonyl)-guanidine (40 mg, 0.1 mmol), and Et3N (0.03 mL, 0.22 mmol) in anhydrous DCM (6 mL) were stirred at RT overnight. Solvents were then evaporated. Chromatography using ISCO max gradient 30% EtOAc/hexane yielded product as a white solid (20 mg, 44% yield); mp 134–137 °C; 1H NMR (300 MHz) (CDCl3) δ 8.50–8.40 (m, 1H), 7.78–7.60 (m, 6H), 7.44–7.35 (m, 3H), 3.81 (m, 2H), 3.44 (t, J = 6.0 Hz, 2H), 1.56 (s, 9H), 1.50 (s, 9H); 13C NMR (100 MHz) (CDCl3) δ 163.7, 156.2, 153.2, 144.9, 139.4, 135.2, 132.4, 131.8, 130.5, 131.8, 127.0, 126.9, 125.9, 125.7, 125.5, 125.2–125.1 (m), 124.9, 124.5, 83.1, 79.1, 41.8, 33.0, 28.4, 28.1; HRMS (ESI): calcd for C30H35F3N3O4 (M + H)+ 558.2580, found 558.2590.
5.16.4. 2-(2-(5-(4-(Trifluoromethyl)phenyl)naphthalen-1-yl)ethyl)guanidine (15)
1,3-Di-Boc-2-(2-(5-(4-(trifluoromethyl)phenyl)naphthalen-1-yl)ethyl)guanidine (20 mg, 0.036 mmol) was stirred at RT in a mixture of anhydrous DCM (0.5 mL) and TFA (0.5 mL) overnight. Solvents were then evaporated. Chromatography using ISCO max gradient 20% 3% NH4OH/MeOH/DCM solution yielded product as a white solid (11 mg, 85% yield); mp 240 °C; 1H NMR (300 MHz) (CD3OD) δ 8.21 (d, J = 6.0 Hz, 1H), 7.84 (d, J = 6.0 Hz, 2H), 7.73–7.64 (m, 4H), 7.51–7.43 (m, 3H), 3.66 (t, J = 6.0 Hz, 2H), 3.46 (t, J = 6.0 Hz, 2H); 13C NMR (100 MHz) (CD3OD) δ 158.7, 146.4, 140.9, 135.9, 133.5, 133.1, 131.7, 128.4, 128.1, 127.1, 126.8, 126.3, 126.2, 126.1, 124.8, 124.5, 43.1, 33.2; HRMS (ESI): calcd for C20H19F3N3 (M + H)+ 358.1526, found 358.1535.
5.17. General procedure for the synthesis of compound (16)
5.17.1. (5-Bromonaphthalen-1-yl)methanol
To a solution of 5-bromo-1-naphthaldehyde (500 mg, 2.13 mmol) in EtOH (20 mL) was slowly added NaBH4 (243 mg, 6.39 mmol) and reaction was stirred at RT for 30 min. Acetone (2 mL) was then added and solution was filtered through filter paper. Filtrate was concentrated then re-dissolved in DCM and washed with H2O. Organic layer was dried over sodium sulfate and concentrated to yield pure product as a white solid (473 mg, 94% yield); mp 100–101 °C; 1H NMR (400 MHz) (CDCl3) δ 8.19–8.17 (m, 1H), 8.04 (d, J = 8.0 Hz, 1H), 7.75–7.73 (m, 1H), 7.51–7.46 (m, 2H), 7.33–7.29 (m, 1H), 5.08 (d, J = 8.0 Hz, 2H); 13C NMR (100 MHz) (CDCl3) δ 136.7, 132.6, 132.4, 130.1, 127.8, 126.8, 126.6, 126.2, 123.6, 63.6.
5.17.2. (5-(2-Methoxy-4-(trifluoromethyl)phenyl)naphthalen-1-yl)methanol
(5-Bromonaphthalen-1-yl)methanol (150 mg, 0.63 mmol), 2-methoxy-4-trifluromethylphenylboronic acid (167 mg, 0.76 mmol), Pd(OAc)2 (14 mg, 0.063 mmol), XPhos (60 mg, 0.13 mmol), and K2CO3 (262 mg, 1.9 mmol) were combined in a flask with dioxane (5 mL) and H2O (1.6 mL) and degassed. Reaction mixture was then refluxed at 100 °C for 2 h. Solution was cooled to RT then diluted with EtOAc and washed with saturated NaHCO3. Organic layer was dried over sodium sulfate and concentrated. Chromatography using ISCO max gradient 35% EtOAc/hexane yielded product as a clear oil (180 mg, 86% yield); 1H NMR (400 MHz) (CDCl3) δ 8.24–8.22 (m, 1H), 7.64 (dd, J = 8.0 Hz, J = 8.0 Hz, 1H), 7.55 (d, J = 8.0 Hz, 1H), 7.51 (d, J = 8.0 Hz, 1H), 7.45–7.37 (m, 4H), 7.29 (s, 1H), 5.22 (s, 2H), 3.76 (s, 3H); 13C NMR (100 MHz) (CDCl3) δ 157.5, 136.5, 136.3, 133.5, 132.2, 132.2, 131.5, 131.3, 131.2, 127.3, 126.6, 125.8, 125.5–125.4 (m), 123.9, 122.8, 117.5–117.4 (m), 107.7, 66.0, 55.8; HRMS (ESI): calcd for C19H15F3NaO2 (M + Na)+ 355.0916, found 355.0919.
5.17.3. 2-(5-(2-Methoxy-4-(trifluoromethyl)phenyl)naphthalen-1-yl)acetonitrile
To a solution of (5-(2-methoxy-4-(trifluoromethyl)phenyl)naphthalen-1-yl)methanol (180 mg, 0.54 mmol) and Et3N (0.15 mL, 1.08 mmol) in DCM (5 mL) was added MsCl (0.06 mL, 0.81 mmol), and the reaction mixture was stirred at RT overnight. Reaction was then diluted with DCM and washed with saturated NaHCO3. Organic layer was dried over sodium sulfate and concentrated without further purification. This crude (5-(2-methoxy-4-(trifluoromethyl)phenyl)naphthalen-1-yl)methyl methanesulfonate (150 mg, 0.37 mmol) was combined with KCN (48 mg, 0.73 mmol) in anhydrous DMF (3 mL) and stirred at RT overnight. Reaction mixture was then diluted with EtOAc and washed with 10% LiCl solution. Organic layer was dried over sodium sulfate and concentrated. Chromatography using ISCO max gradient 30% EtOAc/hexane yielded product as clear oil (125 mg, 68% yield over two steps); 1H NMR (400 MHz) (CDCl3) δ 7.97 (d, J = 12.0 Hz, 1H), 7.72–7.68 (m, 1H), 7.63 (d, J = 8.0 Hz, 1H), 7.56 (d, J = 8.0 Hz, 1H), 7.47 (d, J = 4.0 Hz, 1H), 7.44–7.40 (m, 3H), 7.30 (s, 1H), 4.21 (s, 2H), 3.77 (s, 3H); 13C NMR (100 MHz) (CDCl3) δ 157.4, 136.8, 133.0, 132.2, 132.2, 130.9, 127.8, 127.2, 126.6, 126.5, 126.1, 125.5, 122.6, 117.7, 117.5, 117.4, 107.8, 107.7, 55.8, 22.0; HRMS (ESI): calcd for C20H14F3NO (M)+ 341.1022, found 341.1019.
5.17.4. 2-(5-(2-Methoxy-4-(trifluoromethyl)phenyl)naphthalen-1-yl)ethanamine (16)
A flask was charged with LAH (15.6 mg, 0.41 mmol) in anhydrous ether (3 mL). To the suspension was added 2-(5-(2-methoxy-4-(trifluoromethyl)phenyl)naphthalen-1-yl)acetonitrile (70 mg, 0.21 mmol) in anhydrous ether (2 mL) drop-wise. Reaction was stirred at RT for 30 min then placed on an ice bath. H2O (10 drops) was carefully added in to quench remaining LAH then 1 M NaOH was added to increase pH > 9. Solution was then diluted with additional ether and organic layer was extracted from aqueous layer. Organic layer was dried over sodium sulfate and concentrated. Chromatography using ISCO max gradient 10% MeOH/DCM yielded product as clear oil (31 mg, 44% yield); 1H NMR (400 MHz) (CDCl3) δ 8.15 (d, J = 8.0 Hz, 1H), 7.62–7.58 (m, 1H), 7.43–7.32 (m, 6H), 7.28 (s, 1H), 3.77 (s, 3H), 3.33–3.29 (m, 2H), 3.19 (t, J = 12.0 Hz, 2H); 13C NMR (100 MHz) (CDCl3) δ 157.5, 136.3, 135.9, 133.7, 132.3, 132.2, 132.0, 127.0, 126.8, 125.5, 125.2, 125.0, 123.9, 117.4, 117.3, 107.7, 55.8, 42.9, 37.5; HRMS (ESI): calcd for C20H19F3NO (M + H)+ 346.1413, found 346.1415.
5.18. General procedure for the synthesis of 2-(5-(2-aminoethyl)naphthalen-1-yl)-5-(trifluoromethyl)phenol (17)
To a cooled solution of 2-(5-(2-methoxy-4-(trifluoromethyl)phenyl)naphthalen-1-yl)ethanamine (28 mg, 0.081 mmol) in anhydrous DCM (4 mL) was added BBr3 (1.0 M in DCM, 0.41 mL) drop-wise. The solution was then stirred at RT for 3 h and reaction was placed back over ice. H2O was slowly dripped into the flask to quench the remaining BBr3 and mixture was then diluted with additional DCM and washed with NaHCO3. Organic layer was dried over sodium sulfate and concentrated. Chromatography using ISCO max gradient 10% MeOH/DCM to yield product as a clear oil (13 mg, 48% yield); 1H NMR (400 MHz) (CD3OD) δ 8.05 (d, J = 8.0 Hz, 1H), 7.48 (dd, J = 8.0 Hz, J = 8.0 Hz, 1H), 7.35 (d, J = 8.0 Hz, 1H), 7.29–7.26 (m, 2H), 7.24–7.20 (m, 2H), 7.12–7.10 (m, 2H), 3.20 (t, J = 12.0 Hz, 2H), 2.96 (t, J = 12.0 Hz, 2H); 13C NMR (100 MHz) (CD3OD) δ 157.0, 138.1, 136.5, 133.8, 133.6, 133.5, 133.4, 132.3, 132.0, 128.3, 127.9, 127.0, 126.6, 126.4, 124.8, 116.8, 113.4–113.3 (m), 43.2, 36.5; HRMS (ESI): calcd for C19H17F3NO (M + H)+ 332.1257, found 332.1259.
5.19. General procedure for the synthesis of compound (18)
5.19.1. 1,3-Di-Boc-2-((5-bromonaphthalen-1-yl)methyl)guanidine
(5-Bromonaphthalen-1-yl)methanol (5.17.1) (275 mg, 1.16 mmol), PPh3 (456 mg, 1.74 mmol), and 1,3-bis(t-butoxycarbonyl)guanidine (601 mg, 2.32 mmol) in anhydrous toluene (5 mL) at 0 °C was added diisopropylazodicarboxylate (0.34 mL, 1.74 mmol) drop-wise over 15 min. Reaction was stirred for 3 h at RT then 2 drops H2O were added, and the solution was concentrated. Chromatography using ISCO max gradient 20% EtOAc/hexane yielded product as a white solid (493 mg, 90% yield); mp 164–165 °C; 1H NMR (400 MHz) (CDCl3) δ 9.47 (bs, 2H), 8.11 (d, J = 8.0 Hz, 1H), 7.91 (d, J = 8.0 Hz, 1H), 7.73 (d, J = 8.0 Hz, 1H), 7.46 (t, J = 16.0 Hz, 1H), 7.28 (t, J = 16.0 Hz, 1H), 7.16 (d, J = 8.0 Hz, 1H), 5.62 (s, 2H), 1.36 (s, 9H), 1.07 (s, 9H); 13C NMR (100 MHz) (CDCl3) δ 160.8, 155.0, 135.0, 132.0, 129.9, 126.7, 126.3, 126.2, 123.7, 122.9, 122.7, 84.2, 79.0, 45.1, 28.2, 27.6; HRMS (ESI): calcd for C22H29BrN3O4 (M + H)+ 478.1336, found 478.1344.
5.19.2. 1,3,-Di-Boc-2-((5-(2-methoxy-4-(trifluoromethyl)phenyl)naphthalen-1-yl)methyl)guanidine
1,3-Di-Boc-2-((5-bromonaphthalen-1-yl)methyl)guanidine (100 mg, 0.21 mmol), 2-methoxy-4-trifluromethylphenylboronic acid (55 mg, 0.252 mmol), Pd(OAc)2 (5 mg, 0.021 mmol), XPhos (20 mg, 0.042 mmol), and K2CO3 (87 mg, 0.63 mmol) were combined in a flask with dioxane (3 mL) and H2O (1 mL) and degassed. Reaction mixture was then refluxed at 100 °C for 2 h. Solution was cooled to RT then diluted with EtOAc and washed with saturated NaHCO3. Organic layer was dried over sodium sulfate and concentrated. Chromatography using ISCO max gradient 15% EtOAc/hexane yielded product as a clear oil (55 mg, 46% yield); 1H NMR (400 MHz) (CDCl3) δ 9.55 (bs, 1H), 9.48 (bs, 1H), 8.08 (d, J = 8.4 Hz, 1H), 7.61–7.57 (m, 1H), 7.42–7.33 (m, 5H), 7.27 (s, 1H), 7.19 (d, J = 4.0 Hz, 1H), 5.86–5.70 (m, 2H), 3.75 (s, 3H), 1.47 (s, 9H), 1.21 (s, 9H); 13C NMR (100 MHz) (CDCl3) δ 163.8, 160.9, 157.4, 155.1, 136.2, 134.7, 132.2, 131.9, 127.0, 125.3, 125.0, 123.0, 121.9, 84.0, 79.0, 55.8, 45.4, 28.3, 27.6; HRMS (ESI): calcd for C30H35F3N3O5 (M + H)+ 574.2523, found 574.2541.
5.19.3. 2-((5-(2-methoxy-4-(trifluoromethyl)phenyl)naphthalen-1-yl)methyl)guanidine (18)
To a cooled solution of 1,3-di-Boc-2-((5-(2-methoxy-4-(trifluoromethyl)phenyl)naphthalen-1-yl)methyl)guanidine (55 mg, 0.1 mmol) in anhydrous DCM (1.5 mL) was added TFA (1.5 mL). Reaction was taken off ice bath and stirred at RT for 2 h then solvents were evaporated. Chromatography using ISCO max gradient 10% MeOH/DCM yielded product as a tan gummy solid (31 mg, 86% yield); 1H NMR (400 MHz) (CD3OD) δ 7.96 (d, J = 8.0 Hz, 1H), 7.59–7.55 (m, 1H), 7.43–7.36 (m, 2H), 7.34–7.28 (m, 5H), 4.84–4.82 (m, 2H), 3.62 (s, 3H); 13C NMR (100 MHz) (CD3OD) δ 159.1, 158.8, 138.1, 135.0, 133.7, 133.2, 132.9, 132.4, 128.6, 128.0, 127.3, 127.0, 126.8, 126.5, 124.1, 118.5, 118.4, 108.9, 56.3, 44.8; HRMS (ESI): calcd for C20H19F3N3O (M + H)+ 374.1475, found 374.1470.
5.20. General procedure for the synthesis of compound (19)
5.20.1. 1,3-Di-Boc-2-(2-(5-(2-methoxy-4-(trifluoromethyl)phenyl)naphthalen-1-yl)ethyl)guanidine
2-(5-(2-Methoxy-4-(trifluoromethyl)phenyl)naphthalen-1-yl)ethanamine (25 mg, 0.073 mmol), 1,3-di-Boc-2-(trifluoromethylsulfonyl)-guanidine (34 mg, 0.087 mmol), and Et3N (0.01 mL, 0.087 mmol) in anhydrous DCM (2.5 mL) were stirred at RT overnight. Reaction mixture was then diluted with DCM and washed with NaHCO3. Organic layer was dried over sodium sulfate and concentrated. Chromatography using ISCO max gradient 30% EtOAc/hexane yielded product as a clear oil (37 mg, 88% yield); 1H NMR (400 MHz) (CDCl3) δ 11.50 (bs, 1H), 8.50 (bt, J = 12.0 Hz, 1H), 8.38 (d, J = 8.0 Hz, 1H), 7.62 (dd, J = 8.0 Hz, J = 8.0 Hz, 1H), 7.43–7.36 (m, 5H), 7.35–7.31 (m, 1H), 7.27 (s, 1H), 3.85–3.79 (m, 2H), 3.76 (s, 3H), 3.50–3.37 (m, 2H), 1.57 (s, 9H), 1.51 (s, 9H); 13C NMR (100 MHz) (CDCl3) δ 163.7, 157.5, 156.1, 153.2, 136.2, 135.0, 133.8, 132.2, 132.1, 127.1, 126.8, 125.5, 125.4, 125.3, 124.3, 117.3, 83.0, 79.1, 55.8, 41.8, 33.0, 28.4, 28.1; HRMS (ESI): calcd for C31H37F3N3O5 (M + H)+ 588.2680, found 588.2698.
5.20.2. 2-(2-(5-(2-Methoxy-4-(trifluoromethyl)phenyl)naphthalen-1-yl)ethyl)guanidine (19)
To a cooled solution of 1,3-di-Boc-2-(2-(5-(2-methoxy-4-(trifluoromethyl)phenyl)naphthalen-1-yl)ethyl)guanidine (35 mg, 0.06 mmol) in anhydrous DCM (1 mL) was added TFA (1 mL). Reaction was taken off ice bath and stirred at RT for 2 h then solvents were evaporated. Chromatography using ISCO max gradient 10% MeOH/DCM yielded product as a clear oil (19 mg, 83% yield); 1H NMR (400 MHz) (CD3OD) δ 8.16 (d, J = 8.6 Hz, 1H), 7.66–7.62 (m, 1H), 7.44–7.34 (m, 7H), 3.74 (s, 3H), 3.65 (t, J = 16.0 Hz, 2H), 3.46–3.42 (m, 2H); 13C NMR (100 MHz) (CD3OD) δ 159.1, 158.7, 138.0, 135.5, 135.3, 133.7, 133.3, 133.2, 128.3, 128.1, 127.0, 126.8, 126.7, 126.6, 124.5, 118.4, 118.3, 108.8, 56.2, 43.2, 33.1; HRMS (ESI): calcd for C21H21F3N3O (M + H)+ 388.1631, found 388.1630.
5.21. Minimum inhibitory concentration (MIC) assays
MIC assays were conducted in accordance with Clinical Laboratory Standards Institute (CLSI) guidelines for broth microdilution [26]. The following bacterial strains were included in these assays: S. aureus 8325-4 (MSSA); S. aureus ATCC 33591 (MRSA); E. faecalis ATCC 19433 (VSE) and; E. faecalis ATCC 51575 (VRE). Log-phase bacteria were added to 96-well microtiter plates (at 105 CFU/mL) containing 2-fold serial dilutions of compound or comparator drug (at concentrations ranging from 64 to 0.031 µg/mL) in cation-adjusted Mueller-Hinton (CAMH) broth (for the S. aureus assays) or brain-heart infusion (BHI) broth (for the E. faecalis assays). In the MRSA assays, the CAMH broth was supplemented with 2% NaCl. The final volume in each well was 0.1 mL and the microtiter plates were incubated aerobically for 24 h at 37 °C. Bacterial growth was then monitored by measuring OD600 using a VersaMax® plate reader (Molecular Devices, Inc.), with the MIC being defined as the lowest compound concentration at which growth was ≥90% inhibited.
5.22. Expression and purification of S. aureus FtsZ (SaFtsZ)
The FtsZ gene from S. aureus was amplified by polymerase chain reaction (PCR) from the S. aureus genome obtained from ATCC (ATCC 33591-D). The amplified gene product was then ligated into the pET-22b(+) cloning vector (Novagen-EMD Chemicals, Inc.), with the sequence of the final recombinant plasmid (pETSAFtsZ) being verified by sequence analysis and used to transform E. coli BL21 (DE3) cells.
A single colony of pETSAFtsZ-transformed E. coli cells was used to inoculate Luria Bertani media containing 100 µg/mL of ampicillin (LB-amp) and grown overnight at 37 °C. 20 mL of the culture was diluted into 4 L of LB-amp and grown until an optical density at 600 nm (OD600) of 0.4, at which point FtsZ production was induced by addition of isopropyl β-D-1-thiogalactopyranoside (IPTG) to a final concentration of 1 mM. Following addition of IPTG, the cultures were incubated for an additional 3 h at 37 °C. Cells were then harvested by centrifugation at 4 °C and 4000×g in a swinging bucket Sorvall RC-3BP+ centrifuge with rotor H-6000 for 20 min. The bacterial cell pellet was washed with ice cold 50 mM Tris·HCl (pH 8.0), 0.5 M NaCl and re-pelleted by centrifugation as described above. The washed bacterial pellet was stored at −20 °C.
The dialyzed material was loaded onto a MonoQ 10/100 anion exchange column at 1 mL/min and washed with 5 column volumes of TKEGE buffer at 1 mL/min. The column was eluted at 4 °C with a 0–60% (v/v) linear gradient (120 mL total volume) of the following two buffers: (i) TKEGE (containing 50 mM KCl) and (ii) TKEGE containing 1 MKCl instead of 50 mM KCl. The rate of elution was maintained at 1 mL/min. Fractions were assayed for FtsZ by SDS-PAGE analysis on a 10–15% Tris·HCl polyacrylamide gel. FtsZ elutes at 250–350 mM KCl. The FtsZ containing fractions were pooled and dialyzed against 2 L of TKEGE buffer. The dialyzed fractions were concentrated, if necessary, to 5 mL and loaded onto a Superdex-200 size exclusion column, with TKEGE buffer as the running buffer at 0.25 mL/min. SaFtsZ-containing fractions were detected by SDS-PAGE as above. Peak fractions containing the pure SaFtsZ were pooled and concentrated using Amicon® Ultra centrifugal filters (Millipore Corp.). Quantitation was performed spectrophotometrically at 595 nm using a colorimetric protein assay kit (Bio-Rad) and bovine serum albumin as the standard. The final FtsZ concentration was ~8 mg/mL and the protein was ~90% pure as determined by SDS-PAGE analysis.
5.23. FtsZ polymerization assays
Polymerization of SaFtsZ was monitored using a microtiter plate-based turbidity assay. Experiments were conducted at 25 °C in solution containing 50 mM Tris· HCl (pH 7.4), 50 mM KCl, 2 mM magnesium acetate and 1 mM GTP. GTP was combined with vehicle, compound, or control drug and the reactions were initiated by addition of the protein at a final concentration of 10 µM. The reactions (100 µL total volume) were assembled in half-volume, flat-bottom 96-well microtiter plates and their absorbances at 340 nm were continuously monitored using a VersaMax® plate reader over a time period of 300 min.
Acknowledgments
This study was supported by research agreements between TAXIS Pharmaceuticals, Inc. and both Rutgers, The State University of New Jersey (E.J.L.) and the University of Medicine and Dentistry of New Jersey (D.S.P). We would like to thank Drs. Steve Tuske and Eddy Arnold (Center for Advanced Biotechnology and Medicine, Rutgers University) for their assistance with the expression and purification of S. aureus FtsZ protein. We also appreciate the editorial review by Greg Mario. S. aureus 8325-4 was the generous gift of Dr. Glenn W. Kaatz (John D. Dingell VA Medical Center, Detroit, MI). The Brucker Avance III 400 MHz NMR spectrometer used in this study was purchased with funds from NCRR Grant No. 1S10RR23698-1A1. Mass spectrometry was provided by the Washington University Mass Spectrometry Resource with support from the NIH National Center for Research Resources Grant No. P41RR0954.
References
- 1.Leavis HL, Willems RJL, Top J, Spalburg E, Mascini EM, Fluit AC, Hoepelman A, de Neeling AJ, Bonten MJM. Emerg. Infect. Dis. 2003;9:1108–1115. doi: 10.3201/eid0909.020383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Klevens RM, Morrison MA, Nadle J, Petit S, Gershman K, Ray S, Harrison LH, Lynfield R, Dumyati G, Townes JM, Craig AS, Zell ER, Fosheim GE, McDougal LK, Carey RB, Fridkin SK. JAMA. 2007;298:1763–1771. doi: 10.1001/jama.298.15.1763. [DOI] [PubMed] [Google Scholar]
- 3.Addinall SG, Holland B. J. Mol. Biol. 2002;318:219–236. doi: 10.1016/S0022-2836(02)00024-4. [DOI] [PubMed] [Google Scholar]
- 4.Margolin W. Nat. Rev. Mol. Cell. Biol. 2005;6:862–871. doi: 10.1038/nrm1745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Addinall SG, Bi E, Lutkenhaus J. J. Bacteriol. 1996;178:3877–3884. doi: 10.1128/jb.178.13.3877-3884.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pinho MG, Errington J. Mol. Microbiol. 2003;50:871–881. doi: 10.1046/j.1365-2958.2003.03719.x. [DOI] [PubMed] [Google Scholar]
- 7.Lutkenhaus J, Addinall SG. Annu. Rev. Biochem. 1997;66:93–116. doi: 10.1146/annurev.biochem.66.1.93. [DOI] [PubMed] [Google Scholar]
- 8.Lowe J, van den Ent F, Amos LA. Annu. Rev. Biophys. Biomol. Struct. 2004;33:177–198. doi: 10.1146/annurev.biophys.33.110502.132647. [DOI] [PubMed] [Google Scholar]
- 9.Lock RL, Harry EJ. Nat. Rev. 2008;7:324–338. doi: 10.1038/nrd2510. [DOI] [PubMed] [Google Scholar]
- 10.Kapoor S, Panda D. Expert Opin. Ther. Targets. 2009;13:1037–1051. doi: 10.1517/14728220903173257. [DOI] [PubMed] [Google Scholar]
- 11.Foss MH, Eun Y-J, Weibel DB. Biochemistry. 2011;50:7719–7734. doi: 10.1021/bi200940d. [DOI] [PubMed] [Google Scholar]
- 12.Schaffner-Barbero C, Martin-Fontecha M, Chacon P, Andreu JM. ACS Chem. Biol. 2012;7:269–277. doi: 10.1021/cb2003626. [DOI] [PubMed] [Google Scholar]
- 13.Margalit DN, Romberg L, Mets RB, Hebert AM, Mitchision TJ, Kirschner MW, RayChaudhuri D. Proc. Natl. Acad. Sci. U.S.A. 2004;101:11821–11826. doi: 10.1073/pnas.0404439101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Andreu JM, Schaffner-Barbero C, Huecas S, Alonso D, Lopez-Rodriguez ML, Ruiz-Avila LB, Núñez-Ramírez R, Llorca O, Martín-Galiano AJ. J. Biol. Chem. 2010;285:14239–14246. doi: 10.1074/jbc.M109.094722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Beuria TK, Santra MK, Panda D. Biochemistry. 2005;44:16584–16593. doi: 10.1021/bi050767+. [DOI] [PubMed] [Google Scholar]
- 16.Domadia PN, Bhunia A, Sivaraman J, Swarup S, Dasgupta D. Biochemistry. 2008;47:3225–3234. doi: 10.1021/bi7018546. [DOI] [PubMed] [Google Scholar]
- 17.Haydon DJ, Bennett JM, Brown D, Collins I, Galbraith G, Lancett P, Macdonald R, Stokes NR, Chauhan PK, Sutariya JK, Nayal N, Srivastava A, Beanland J, Hall R, Henstock V, Noula C, Rockley C, Czaplewski L. J. Med. Chem. 2010;53:3927–3936. doi: 10.1021/jm9016366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Haydon DJ, Stokes NR, Ure R, Galbraith G, Bennett JM, Brown DR, Baker PJ, Barynin VV, Rice DW, Sedelnikova SE, Heal JR, Sheridan JM, Aiwale ST, Chauhan PK, Srivastava A, Taneja A, Collins I, Errington J, Czaplewski LG. Science. 2008;321:1673–1675. doi: 10.1126/science.1159961. [DOI] [PubMed] [Google Scholar]
- 19.Kumar K, Awasthi D, Lee SY, Zanardi I, Ruzsicska B, Knudson S, Tonge PJ, Slayden RA, Ojima I. J. Med. Chem. 2011;54:374–381. doi: 10.1021/jm1012006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Läppchen T, Hartog AF, Pinas VA, Koomen GJ, den Blaauwen T. Biochemistry. 2005;44:7879–7884. doi: 10.1021/bi047297o. [DOI] [PubMed] [Google Scholar]
- 21.Urgaonkar S, La Pierre SH, Meir I, Lund H, RayChaudhuri D, Shaw JT. Org. Lett. 2005;7:5609–5612. doi: 10.1021/ol052269z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Parhi A, Kelley C, Kaul M, Pilch DS, LaVoie EJ. Bioorg. Med. Chem. Lett. 2012;22:7080–7083. doi: 10.1016/j.bmcl.2012.09.097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.(a) LaVoie EJ, Parhi A, Pilch DS, Kaul M. 20120022061 U.S. Patent. ; (b) LaVoie EJ, Parhi A, Pilch DS, Kaul M. 20120059026 U.S. Patent.
- 24.Parhi A, Lu S, Kelley C, Kaul M, Pilch DS, LaVoie EJ. Bioorg. Med. Chem. Lett. 2012;22:6962–6966. doi: 10.1016/j.bmcl.2012.08.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ruggli P, Preuss R. Helv. Chim. Acta. 1941;24:1345–1359. [Google Scholar]
- 26.Kotian PL, Krishnan R, Rowland S, El-Kattan Y, Saini SK, Upshaw R, Bantia S, Arnold S, Babu YS, Chand P. Bioorg. Med. Chem. 2009;17:3934–3958. doi: 10.1016/j.bmc.2009.04.013. [DOI] [PubMed] [Google Scholar]
- 27.CLSI Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria that Grow Aerobically; Approved Standard-Eighth Edition. Wayne, PA: Clinical and Laboratory Standards Institute; 2009. [Google Scholar]