

Abstract
Anti-microbial peptides might influence the pathogenesis and course of inflammatory bowel disease (IBD). We sought to clarify the role of the anti-microbial glycoprotein lipocalin 2 (LCN2) in the colon by determining its localization and regulation in IBD. Following a microarray gene expression study of colonic biopsies from a large IBD population (n = 133), LCN2 was localized using immunohistochemistry and in-situ hybridization. Moreover, we examined the regulation of LCN2 in HT-29 cells with a panel of pattern recognition receptors (PRRs) and sought evidence by immunohistochemistry that the most relevant PRR, the Toll-like receptor (TLR)-3, was indeed expressed in colonic epithelium in IBD. LCN2 was among the 10 most up-regulated genes in both active ulcerative colitis (UCa) and active Crohn's disease (CDa) versus healthy controls. LCN2 protein was found in both epithelial cells and infiltrating neutrophils, while mRNA synthesis was located solely to epithelial cells, indicating that de-novo synthesis and thus regulation of LCN2 as measured in the gene expression analysis takes place in the mucosal epithelial cells. LCN2 is a putative biomarker in faeces for intestinal inflammation, different from calprotectin due to its epithelial site of synthesis. LCN2 release from the colonic epithelial cell line HT-29 was enhanced by both interleukin (IL)-1β and the TLR-3 ligand poly(I:C), and TLR-3 was shown to be expressed constitutively in colonic epithelial cells and markedly increased during inflammation.
Keywords: Crohn's disease, inflammatory bowel disease, lipocalin 2, TLR-3, ulcerative colitis
Introduction
The pathogenesis of inflammatory bowel disease (IBD) involves both genetic susceptibility and environmental factors. Genetic studies suggest an important role for the innate immune system in IBD pathogenesis [1].
The normal intestine shows a balance between factors activating host immunity, such as gut bacteria, dietary antigens, endogenous inflammatory stimuli and host defence. The host responds to maintain mucosal integrity and down-regulate the inflammatory response to avoid excessive inflammation [2]. Perturbed homeostasis between commensal bacteria and mucosal immunity is a critical determinant in the development of inflammation in IBD [3]. Although the precise dysfunction remains unclear, emerging evidence has revealed that host-derived anti-microbial peptides play a key role in determining the composition of gut commensal bacteria, and there is accumulating evidence of a dysregulated expression of anti-microbial peptides (e.g. defensins) in intestinal epithelial cells in IBD [4, 5].
Lipocalin 2 (LCN2), known formerly as neutrophil gelatinase-associated lipocalin (NGAL), is an anti-microbial glycoprotein. It has high affinity for secreted bacterial siderophores, which scavenge for iron. By binding siderophores, LCN2 reduces available iron for bacterial growth and plays an important role in bacterial colonization [6, 7]. LCN2 was found originally in neutrophil granules, but respiratory and intestinal epithelial cells, endothelial cells and renal tubular cells also express LCN2 during inflammation and injury [8–11]. LCN2 has properties of an acute phase protein and in several inflammatory and infectious diseases is released rapidly into the systemic circulation [7, 9, 12]. Some studies have suggested a role for LCN2 as a disease activity marker in IBD [10, 13, 14].
From our large study on colonic mucosal gene expression in IBD, we show that LCN2 is among the most over-expressed genes in active ulcerative colitis (UC) and Crohn's disease (CD). This led us to examine the localization and regulation of LCN2 in colonic biopsies. Because Toll-like receptors (TLRs) are key mediators of intestinal innate host defence, we examined the effect of various TLR-and nucleotide-binding oligomerization domain (NOD) ligands on LCN2 release from colonic epithelial cell lines.
Materials and methods
Patient material
Patients undergoing colonoscopy for known or suspected IBD at the Gastrointestinal Endoscopy Unit, St Olav's University Hospital, Trondheim, Norway were included into the study. Healthy controls were recruited among people undergoing colonoscopy due to gastrointestinal symptoms and who had no signs of gastrointestinal disease.
Four adjacent endoscopic pinch biopsies were taken from non-inflamed mucosa of IBD patients or healthy controls at the hepatic flexure and at the site of maximally inflamed mucosa, if found. Three biopsies were snap-frozen and kept on liquid nitrogen for molecular analyses, and one was formalin-fixed. Inflammation was confirmed on haematoxylin and eosin-stained slides by an expert pathologist before including the sample in the analysis.
Blood was drawn and serum prepared by 30-min coagulation at ambient temperature before centrifugation at 2000 g, 4°C for 10 min and stored at −80°C.
Ethical considerations
All subjects gave informed written consent. The study was approved by the Regional Medical Research Ethics Committee (ref. no. 5.2007.910) and registered in the Clinical Trials Protocol Registration System (identifier NCT00516776).
Gene expression analysis
Gene expression analysis has been described previously [15]. A full data set from microarray analysis is available at ArrayExpress E-MTAB-184.
Histological examination, immunostaining and in-situ hybridization
Formalin-fixed, paraffin-embedded biopsies were cut into 4-μm-thick sections for routine histology, immunohistochemical (IHC) examination and in-situ hybridization. LCN2 and TLR-3 immunohistochemistry was performed on 25 randomly selected biopsies, including healthy controls, inactive UC (UCi), active UC (UCa), inactive CD (CDi) and active CD (CDa) (five from each group). Primary antibody, rabbit polyclonal anti-human LCN2 (antibody 41105) was diluted 1:500. Primary antibody, mouse monoclonal anti-human TLR-3 (antibody 13915) was diluted 1:50. Primary antibodies were from Abcam (Cambridge, UK). Secondary antibody was from Dako Real Envision (rabbit/mouse) and detection was performed using diaminobenzidine (DAB)+ chromogen (Dako, Glostrup, Denmark). Two independent examiners assessed the epithelium staining for both LCN2 and TLR-3 as no to little staining, or moderate to strong staining. Fisher's test was used to detect group differences. Three different types of negative control were made for the TLR-3 staining: first, by merely excluding primary antibody; secondly, by replacing primary antibody with non-human-immunized antibody of the same isotype as the primary antibody for TLR-3 used (mouse, monoclonal IgG1; X0931; Dako); and thirdly, blocking primary antibody with recombinant human TLR-3 peptide (Abnova P0506, Heidelberg, Germany).
In-situ hybridization for LCN2 mRNA was performed on the same colonic biopsies as the immunohistochemical staining, using a custom RNAscope (Advanced Cell Diagnostics, Hayward, CA, USA) kit, according to the manufacturer's protocol.
Culture, stimulation and small interfering RNA transfection of intestinal epithelial cells
The human intestinal cell lines HT-29, HCT 116 (colorectal adenocarcinoma) and SW620 (lymph node metastasis of a colorectal adenocarcinoma) were used [cat. no. HTB-38, CCL247 and CCL227, respectively; American Type Culture Collection (ATCC), Manassas, VA, USA]. The medium for HT-29 and SW620 was RPMI-1640 with 10% fetal calf serum, glutamine 2 mM and gentamicin 0·05%, and for HCT 116 we used McCoy's medium (ATCC) with 10% fetal calf serum, 1% L-glutamine and 1% penicillin–streptomycin. Cells were cultured at 37°C, 5% CO2. Trypsin/ethylenediamine tetraacetic acid (EDTA) was used to detach the cells from the culture flasks. Cells were counted using the Countess Automated Cell Counter (Life Technologies, Grand Island, NY, USA). Stimulation was performed in triplicate with 20 000 or 30 000 cells per well on 96-well plates overnight. The medium was then replaced and ligand added. The ligands were the lipopeptide Pam3CysSK4 (P3C) (TLR-2/1) 300 ng/ml, lipomannan (LM) (TLR-2/6) 30 ng/ml, synthetic double-stranded RNA mimic polyinosinic:polycytidylic acid (poly(I:C)) (TLR-3) 0·5, 5 or 50 μg/ml, lipopolysaccharide (LPS) (TLR-4) 100 ng/ml, flagellin (TLR-5) 100 ng/ml, the anti-viral compound R848 (TLR-7/8) 100 ng/ml [all from InvivoGen, Toulouse, France, except poly(I:C) from Amersham Bioscience, Piscataway, NJ, USA], unmethylated cytosine-phosphate-guanosine (CpG) dinucleotides (TLR-9) 10 μM (TibMolBiol, Berlin, Germany), the peptidoglycan component muramyl dipeptide (MDP) (NOD2) 1 μg/ml (InvivoGen, Toulouse, France) and the recombinant human cytokines interleukin (IL)-10, 100 ng/ml and IL-1β, 100 ng/ml (both from PeproTech, Rocky Hill, NJ, USA), CXCL8 50 ng/ml (Invitrogen, Paisley, UK), tumour necrosis factor (TNF)-α 100 ng/ml and interferon (IFN)-γ 1, 10 and 100 ng/ml (both from PeproTech). The cells were stimulated for 20 h before supernatant was harvested and stored at −20°C. LCN2 was analysed in supernatant using a commercially available enzyme-linked immunosorbent assay (ELISA) kit, DY 1757 (R&D Systems, Abingdon, UK). The cell viability was assessed by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)-assay, as described previously [15].
To validate the poly(I:C)-induced TLR-3 response, HT-29 cells were transfected for 24 h using lipofectamin, RNAiMAX (Ambion, Invitrogen Dynal, Oslo, Norway) and TLR3 small interfering RNA (siRNA) (sense-GAACUGGAUAUCUUUGCATT, anti-sense-UGGCAAAGAUAUCCAGUUCTT) (5 nM) or a control siRNA RNA (sense-UUCUCCGAACGUGUCACGUdTdT, anti-sense-ACGUGACACGUUCGGAGAAdTdT (5 nM) (Qiagen, Solentuna, Sweden). The cells were then stimulated with poly(I:C) 5 μg/ml for 20 h as described. The supernatant was collected and stored at −20°C for analysis of LCN2 using ELISA. The remaining cells were either lysed and used for TLR3 quantitative reverse transcription–polymerase chain reaction (qRT-PCR) or analysed by MTT-assay.
RNA isolation and TLR3 qRT–PCR
RNA isolation and TLR-3 qRT–PCR was performed as described previously [15] using TaqMan gene expression assays for TLR3 (Hs01551078_m1) and for the housekeeping gene GAPDH (Hs99999905_m1).
Serum analyses
LCN2 was measured in serum using a LCN2 ELISA kit based on the polyclonal rabbit antibody (antibody 954) developed by T. Flo [16]. Samples were diluted 1:100.
A commercially available assay was used for high sensitivity measurement of C-reactive protein (CRP) in serum (Tina-quant, Roche, Indianapolis, IN, USA).
In the LCN2 analysis in serum and plasma from healthy volunteers, serum was prepared by 30-min coagulation at ambient temperature before centrifugation at 2000 g, 4°C for 10 min. Plasma was obtained by immediate centrifugation of citrate blood at 2200 g, 4°C for 10 min. LCN2 was analysed using a commercially available ELISA kit, DY 1757 (R&D Systems).
Statistical analysis
Microarray data analysis was performed as described previously [17]. Other data were assessed for normality using the Shapiro–Wilk test. The data from serum and supernatant were not distributed normally and were thus tested by Kruskal–Wallis test; if significances were detected, the Mann–Whitney U-test was used to detect group differences. Correlation between serum protein levels was performed with the non-parametric Spearman's rank correlation. Calculations were performed by PASW Statistics version 20 and GraphPad Prism version 5.0. Differences of P < 0·05 (two-sided) were considered significant.
Results
Clinical material
The 133 colonic biopsies were taken from 112 subjects and have been described previously [15]. Serum samples were from 223 subjects partly overlapping the biopsy material. There were no differences in patient characteristics between the groups (Table 1). However, aminosalisylic acid/sulphasalazine (5-ASA/S-ASA) was used more often in the UC group and CD patients used more systemic steroids, as expected from clinical practice.
Table 1.
Characteristics of subjects enrolled in serum lipocalin 2 (LCN2) analysis
| Controls | UC | CD | P | |
|---|---|---|---|---|
| Number of subjects (total 223) | 23 | 119 | 81 | |
| Age (range) | 43 (19–71) | 42 (19–76) | 42 (19–71) | n.s. |
| Female sex (%) | 13 (55·6%) | 56 (47·1%) | 35 (43·2) | n.s. |
| Duration of disease(range) | – | 12 (0–40) | 10 (3–28) | n.s. |
| 5-ASA/S-ASA (%) | 0 | 94 (79·0%) | 27 (33·3%) | 0·000α |
| Systemic corticosteroids (%) | 0 | 15 (12·6%) | 24 (29·6%) | 0·007β |
| hsCRP (range) | 1·9 (0·3–12·8) | 2·3 (0·3–96·4) | 4·3 (0·3–67·6) | 0·000γ |
Age, duration of disease and high sensitivity C-reactive protein (hsCRP) are given as median. Gender and medication are given as numbers.
Significantly higher use of 5-ASA/salazopyrine in UC versus CD subjects;
significantly higher use of systemic steroids in CD versus UC subjects;
significantly higher hsCRP in CD versus both controls and UC subjects. CD: Crohn's disease; UC: ulcerative colitis; n.s.: not significant; 5-ASA/S-ASA: aminosalisylic acid/sulphasalazine.
LCN2 mRNA and protein expression in colonic biopsies
The microarray analysis showed a remarkable over-expression of LCN2 in biopsies from UCa and CDa compared to controls, or to biopsies from UCi or CDi (Fig. 1). LCN2 was among the 10 most over-expressed genes comparing UCa or CDa to healthy controls [17]. The log2 difference between UCa and controls was 4·10 (fold change 17·15), between UCa and UCi was 3·56 (fold change 11·79), between CDa and controls was 3·85 (fold change 14·42) and between CDa and CDi was 3·17 (fold change 9·00). These were highly significant. There was no significant difference in LCN2 expression UCi or CDi versus controls, but a tendency to higher LCN2 abundances could be seen.
Figure 1.
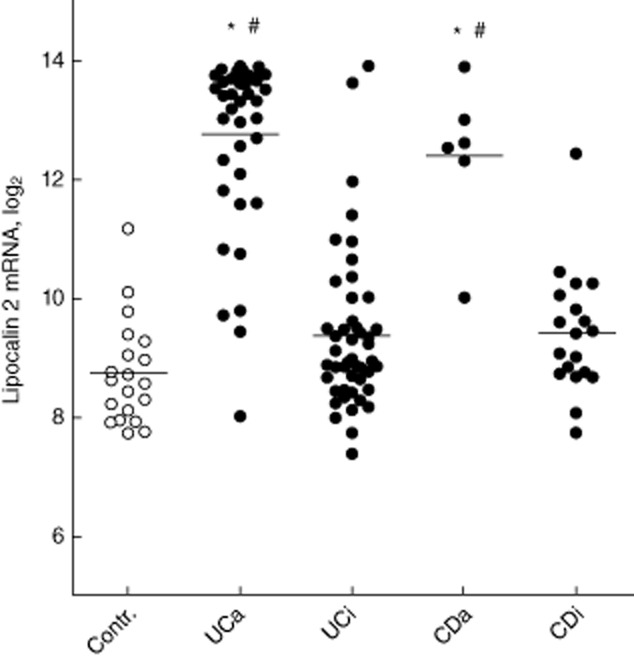
The results of microarray-derived gene expression levels of lipocalin 2 (LCN2) mRNA in colonic biopsies are shown as log2. Mean and individual values plotted. UC: ulcerative colitis; CD: Crohn's disease; a, active; i, inactive. *P < 0·001 versus control, #P < 0·001 versus inactive disease.
The mRNA expression levels of IL-1β, CXCL8, TNF-α and IFN-γ were also over-expressed in both UCa and CDa, correlating strongly with LCN2. For IL-1β, rho = 0·748, CXCL8, rho = 0·729, TNF-α, rho = 0·542 and IFN-γ, rho = 0·618 (all correlations P < 0·001).
LCN2 immunohistochemistry of colonic biopsies showed strong staining of the epithelium in UCa and CDa (Fig. 2a), both enterocytes and goblet cells. Interestingly, goblet cell mucus stained positive in active disease. Moreover, LCN2-positive polymorphonuclear leucocytes were seen. Colonic epithelial cells in UCi or CDi were negative or weakly positive for LCN2. LCN2 epithelial staining was assessed as significantly increased in both UCa and CDa versus controls, and also in UCa versus UCi and CDa versus CDi (all P < 0·05, two-sided). LCN2 immunostaining in epithelium was not significantly different in UCi versus control or CDi versus control.
Figure 2.
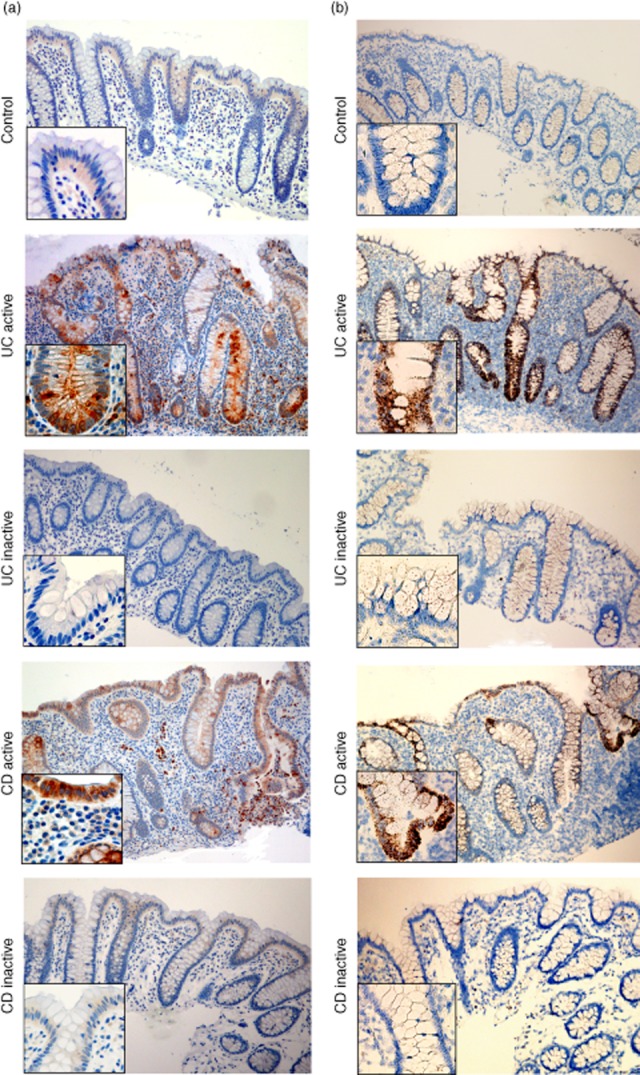
(a) Immunohistochemical staining of lipocalin 2 (LCN2) in colonic biopsies from controls, active and inactive ulcerative colitis (UC), active and inactive Crohn's disease (CD). LCN2 is located in the infiltrating neutrophils and the epithelial cells, both with the appearance of enterocytes and goblet cells. We found significantly increased LCN2 expression in epithelium of active ulcerative colitis (UC) and active Crohn's disease (CD) versus controls. (b) In-situ hybridization of LCN2 in the same biopsies as immunohistochemical staining. The LCN2 mRNA is located only in the epithelial cells, indicating that these cells are responsible for de-novo synthesis. Also, we found significant expression of LCN2 mRNA in the epithelium of active disease versus healthy control mucosa. Original magnification ×10 and ×40.
In-situ hybridization revealed LCN2 induction in colonic epithelial cells in the same biopsies positive for LCN2 by immunohistochemistry (Fig. 2b). No infiltrating immune cells or other cells of the lamina propria or submucosa were positive for LCN2.
Stimulation of colonic epithelial cell lines and LCN2 release
The dynamics of LCN2 regulation was studied further in the cell lines HT-29, HCT 116 and SW620. The LCN2 response to pathogen associated molecular patterns (PAMPs) is particularly interesting, thus we used a ligand panel covering TLR-1–9 and included NOD2 due to its role in IBD. The HT-29 and SW620 cell lines were tested for the full panel, HCT116 with TLR-3 ligand. We found that the HT-29 cells release LCN2 constitutively, and the release was enhanced markedly by the TLR-3 ligand poly(I:C) (Fig. 3). SW620 did not release LCN2 at all. The HCT 116 cell line also released LCN2 dose-dependently after poly(I:C) stimulation (Supporting information, Fig. S1).
Figure 3.
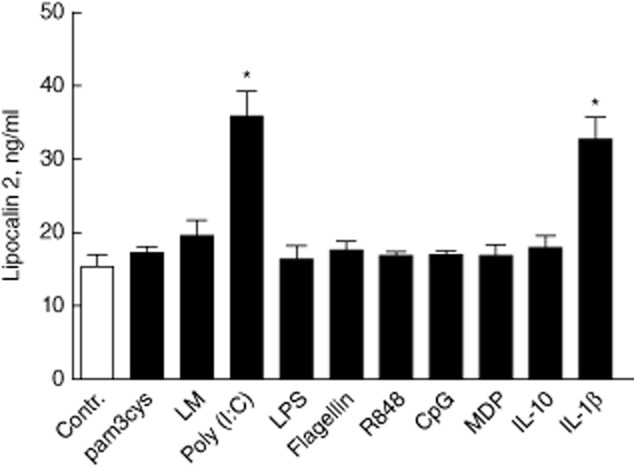
Lipocalin 2 (LCN2) release from HT-29 cells stimulated with pattern recognition receptor (PRR) ligands; from left to right, Toll-like receptors (TLR)-1–9, nucleotide-binding oligomerization domain (NOD)2 and the cytokines interleukin (IL)-10 and IL-1β for 20 h. We found a considerable constitutive release of LCN2. * and #P < 0·05 versus medium. Mean ± standard deviation is shown.
The correlation between LCN2 and mRNA of the proinflammatory cytokines IL-1β, CXCL-8, TNF-α and IFN-γ in the microarray data was studied further by examining whether or not these cytokines release LCN2. IL-1β potently induced LCN2 release from HT-29 cells, and this effect was additive to poly(I:C) (Fig. 4). Neither CXCL8, TNF-α or IFN-γ induced LCN2 release. HT-29 cells did not produce IL-1β, either constitutively or upon stimulation with the same ligands as used when assessing LCN2 release.
Figure 4.
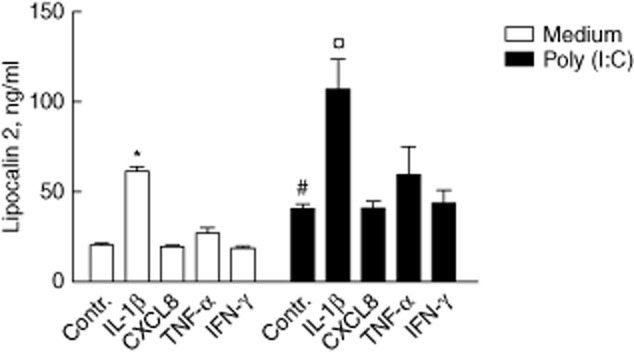
Lipocalin 2 (LCN2) release from HT-29 cells after 20 h stimulation with proinflammatory cytokines with and without poly(I:C). Poly(I:C) and interleukin (IL)-1β both induce LCN2 release, but in an additive pattern. * and #P < 0·05 versus medium control. ¤P < 0·05 versus poly(I:C) control. Mean ± standard deviation.
Silencing of TLR-3 in poly(I:C)-stimulated HT-29 cells
Besides TLR-3, dsRNA can also signal via at least three other sensors: melanoma differentiation-associated gene 5 (MDA-5), retinoic acid inducible gene-I (RIG1) and protein kinase R (PKR). The TLR-3-mediated response to poly(I:C) in HT-29 cells was thus explored further using small interfering RNA (siRNA) for TLR3. Poly(I:C) increased LCN2 release twofold compared to the constitutive release from HT-29 cells, and TLR-3 siRNA almost abolished this response (Fig. 5).
Figure 5.

The poly(I:C) effect on lipocalin 2 (LCN2) release was silenced in HT-29 cells by transfection with Toll-like receptor (TLR)-3 siRNA. The cells were transfected for 24 h using TLR-3 siRNA or control/non-signalling siRNA and then stimulated with poly(I:C) or left unstimulated (medium) for 20 h. * and #P < 0·05. Mean ± s.d. of triplicates.
In the same experiment, poly(I:C) increased TLR3 9·53-fold. Further, siRNA transfection of poly(I:C)-stimulated cells attenuated TLR3 to 33·1% of cells transfected with non-silencing siRNA. HT-29 cell viability was unaltered by poly(I:C) or the transfecting reagents as assessed by MTT assay (Supporting information, Fig. S1).
Localization of TLR-3 in the colonic mucosa
Having observed the TLR-3-induced LCN2 response, colonic biopsies were examined by immunohistochemistry for TLR-3, and TLR-3 positivity was seen in the epithelium (Fig. 6). TLR-3 positivity increased significantly in UCa and CDa versus controls (P < 0·05, two-sided), but there was no significant difference in TLR-3 staining of the epithelium between UCi or CDi versus controls. The TLR-3 staining was located in the nuclei/perinucleic areas. To ensure the staining specificity, we added three different types of negative control in a repeated IHC experiment for TLR-3 as described in Materials and methods. The TLR-3 staining pattern was reproduced, but could not be visualized in any of the three controls, as expected (Supporting information, Fig. S1). This nuclear/perinuclear pattern of staining for TLR-3 has been found previously by others in fibrosarcoma cells [18].
Figure 6.
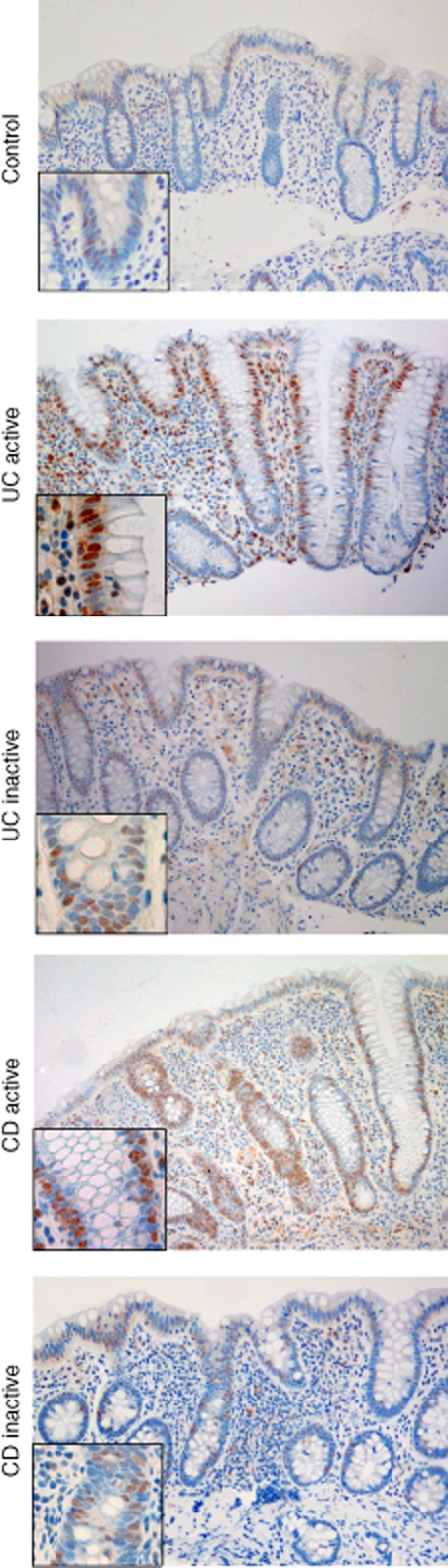
Immunohistochemical staining of Toll-like receptor (TLR)-3 in colonic biopsies from controls, active and inactive ulcerative colitis (UC), active and inactive Crohn's disease (CD). TLR-3 staining is found in the epithelium and also in various cell types in lamina propria. The TLR-3 expression of the epithelium was enhanced in both active UC and active CD versus healthy controls. Original magnification ×10 and ×40.
Serum levels of LCN2
As mucosal LCN2 was increased markedly in active IBD, it is potentially a marker of disease activity.
Serum levels of LCN2 were increased significantly in CD patients compared to controls (Fig. 7a), but not in UC patients. Because the results in both the UC and CD groups are from patients with both active and inactive disease these were recalculated, restricting the data to patients with endoscopic results from the time of blood sampling, and a significant increase in serum LCN2 in UCa versus UCi (Fig. 7b) was found. In a further analysis of serum LCN2 in all subjects included we found a correlation to CRP, rho = 0·315 and P < 0·001 (two-sided).
Figure 7.
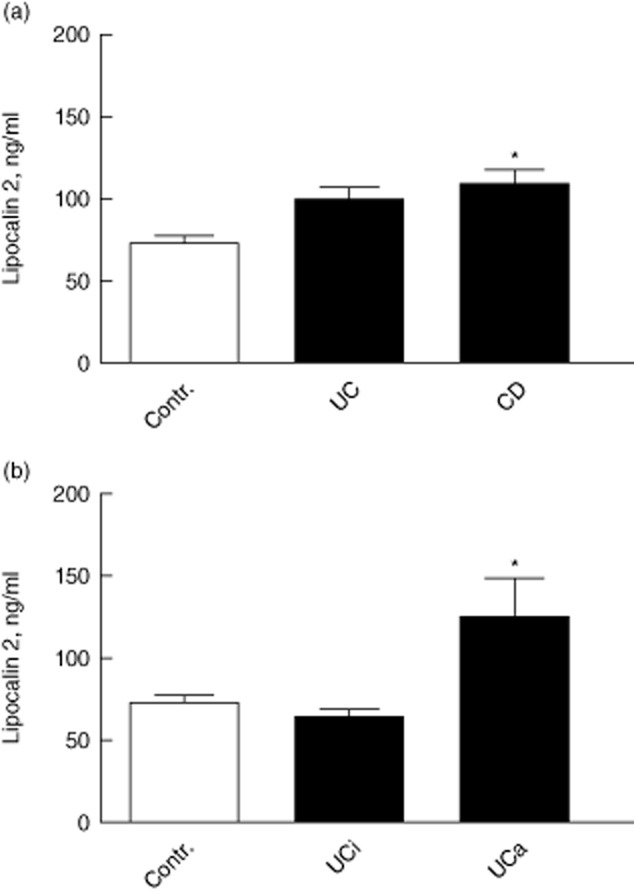
(a) Serum lipocalin 2 (LCN2) levels in ulcerative colitis (UC) and Crohn's disease (CD) patients (both active and inactive disease) versus controls. *P < 0·05, two-sided versus controls. (b) Serum LCN2 level in endoscopic verified active UC, n = 22 and inactive UC, n = 16 versus healthy controls, n = 23. *P < 0·05 two-sided, versus inactive UC. For difference between active UC and controls, P = 0·0550. In both figures mean ± standard deviation is shown.
This shows that LCN2 in serum might reflect IBD disease activity, but the strength of correlation was somewhat weak. We hypothesized that the weak correlation between serum LCN2 and CRP was caused by LCN2 leaking ex vivo from neutrophils, which store large amounts of this peptide. Thus, we examined whether technical procedures during blood sampling and preparation of serum could influence LCN2 levels. In a study in healthy volunteers (n = 3), we found that LCN2 levels in plasma were markedly lower, 50·9% of that in serum.
Discussion
The present work is based on a microarray study of a large and well-controlled material collection of colonic biopsies from an IBD population obtained in a routine clinical setting. The remarkably potent and robust LCN2 response in active IBD seen in our own material makes it likely that this anti-microbial peptide has an important role in these diseases. Over-expression of LCN2 in IBD has been shown previously to variable degrees and in materials of variable size [19],[20].
Our immunohistochemical and in-situ hybridization studies located LCN2 protein to epithelial cells and neutrophils, while ongoing synthesis was found only in the epithelial cells. Neutrophils are ‘prepacked’ with LCN2, and further synthesis does not take place as the mature cells are released into peripheral blood. These findings are in accordance with previous studies [10, 22]. From our results we conclude that the profound up-regulation of LCN2 in microarray analysis is due to LCN2 induction in the epithelium. Having localized inducible de-novo synthesis of LCN2 in colonic IBD to epithelial cells only, we wanted to explore if LCN2 could be induced by PRRs and found that TLR-3 stimulation by the dsRNA-mimicking substance poly(I:C) induced LCN2 release in vitro. Supplementary studies with siRNA inhibition of TLR-3 confirmed that the poly(I:C) response was indeed mediated via TLR-3, indicating strongly that TLR-3 has a central role in the LCN2 response in IBD.
Moreover, as the gene expression levels of several of the central proinflammatory cytokines were correlated with that of LCN2 in the microarray study, we tested these for effect on LCN2 release. IL-1β induced LCN2 release potently in both cell lines, as also found by Cowland et al. [9] in keratinocytes and a pneumocyte-derived cell line. We found the effect of IL-1β on LCN2 to be additive to poly(I:C). While the gene expression of the proinflammatory cytokines CXCL8, TNF-α and IFN-γ also correlated well with LCN2 in our initial microarray study, no effect could be shown on LCN2 release in this experimental system. These results suggest a dual mechanism of LCN2 release in vivo. The relative importance of TLR-3 signalling versus IL-1β in induction of LCN2 is unknown; however, as the cell system tested does not produce IL-1β itself, TLR-3 signalling represents a more direct effect on LCN2 induction in epithelial cells.
To substantiate further a role for TLR-3 in IBD, we examined its expression on protein level by immunohistochemistry of colonic biopsies. We found TLR-3 to be expressed constitutively in the colonic epithelium. Changes in expression level are generally difficult to assess on histological sections, and must be interpreted with caution. However, after examining sections from 25 biopsies, the results appear to be fairly solid. We found enhanced expression of TLR-3 in mucosal biopsies with active inflammation compared to healthy controls and inactive IBD. This constitutive expression of TLR-3 in colonic epithelium has been shown previously [23, 24]. In contrast to our findings, Cario and Podolsky [23] found unchanged expression of TLR-3 in active UC versus inactive UC and reduced TLR-3 in active CD versus inactive CD. They compared the fluorescence intensity in samples from a material of both inflamed and non-inflamed biopsies from ileum and colon in UC and CD patients. This heterogeneity might have biased their results. The material used in the present work is very well controlled, and our assessment of TLR-3 expression in colonic epithelium might be more robust. Thus, to the best of our knowledge, this is the first study that shows increased TLR-3 expression in epithelial cells in active IBD.
Currently, there is scarce knowledge and some controversy around the role of TLR-3 in gut inflammation in general. There is some proof of up-regulation of TLR-3 in intestinal epithelial cells upon rotavirus infection in vitro [25]. Previous studies indicate both protective and detrimental effects of TLR-3 signalling on gut inflammation [26],[27]. The presence of TLR-3 and its action in the colonic epithelium is of potentially great interest in understanding the disease mechanisms of IBD. TLR-3 senses dsRNA, which is found during replication of most viruses, in addition to the dsRNA viruses themselves. There is also evidence that TLR-3 senses endogenous mRNA from damaged tissue and might maintain inflammation independently of viral infection [27, 29]. Our studies show enhanced expression of TLR-3 and a strong regulation of LCN2 in active IBD, suggesting that TLR-3, via binding of viral RNA or endogenous mRNA, has a role in the regulation of LCN2. The obvious role for LCN2 in this setting is as an anti-microbial peptide [7]. This opens the interesting possibility of LCN2, with its anti-bacterial effect, to be a part of the mechanisms behind the altered microbiome that is associated with IBD.
A very interesting aspect of LCN2 in IBD is as a marker of inflammation. Previous studies have suggested serum LCN2 as a clinically useful marker of bowel inflammation [13]. Our results also showed that LCN2 levels in serum correlate with CRP. However, the clearly lower levels of LCN2 that we found in plasma, compared to serum, suggest strongly that LCN2 release from neutrophil leucocytes contributes significantly to LCN2 levels in blood, and that the use of LCN2 in blood as a clinical marker of inflammation will depend critically upon laboratory procedures.
Our results indicate that LCN2 in the epithelium reflects the degree of local inflammation, as it correlates well with proinflammatory cytokines in the mucosa. Moreover, we saw strong LCN2 staining in both the cytoplasm of enterocytes and in the mucus of goblet cells. This suggests that secretion of LCN2 from goblet cells is an important mechanism for LCN2 release into gut lumen. Additionally, LCN2 from neutrophil leucocytes might leak into the gut lumen. This is interesting in a clinical setting, and LCN2 in faeces has been found to be correlated with disease activity during gut inflammation in both humans and mice [14, 30]. Currently, faecal calprotectin is considered the best non-invasive marker of bowel inflammation. Calprotectin is found in neutrophils, and to some extent in monocytes, and thus reflects mucosal infiltration and shedding. As LCN2 is released from both neutrophils and activated epithelial cells it has the potential to be a more sensitive marker of disease activity than calprotectin, especially in chronic inflammation where neutrophils are scarce.
Our study has revealed that there is markedly enhanced LCN2 expression in active IBD, mediated most probably via TLR-3 signalling. TLR-3 expression is enhanced in epithelium in active IBD, suggesting the interesting view of viral or endogenous mRNA as factors contributing to altered immunological homeostasis in IBD. LCN2 is an extremely interesting protein in clinical practice, with potential as a marker of active IBD, measured in plasma or faeces.
Acknowledgments
We thank Kari Slørdahl, Bjørn Munkvold, Liv Ryan and Unni Nonstad at Department of Cancer Research and Molecular Medicine (IKM), Norwegian University of Science and Technology (NTNU), who provided excellent technical assistance. The microarray and bioinformatics work were provided by Genomics Core Facility at NTNU supported by the Functional Genomics Programme (FUGE) of the Norwegian Research Council (RCN). This study was supported by grants from the Liaison Committee between the Central Norway Regional Health Authority and NTNU (A.E.Ø.), the Norwegian Cancer Society (N.J.N. and M.B.) and NTNU (A.v.B.G.). This work also received funding from a research grant from the Liaison Committee between St Olav's University Hospital and the Faculty of Medicine, NTNU.
Disclosure
The authors have nothing to declare.
Supporting information
Additional Supporting Information may be found in the online version of this article at the publisher's website:
Immunohistochemical staining of Toll-like receptor (TLR)-3 in colonic biopsy from actively inflamed mucosa in ulcerative colitis and three different types of negative controls in the same experiment are shown. We used serial sections from same biopsy. The first negative control was made by omitting primary antibody (no ab). In the second negative control, primary antibody was replaced by isotype immunoglobulin (Ig)G1 directed against non-human epitope (IgG1). The third control was made by blocking primary antibody with recombinant human TLR-3 peptide (rhTLR-3+TLR-3ab); ×10 and ×40 magnification for each slide are shown.
{kind=link}
References
- 1.Abraham C, Cho JH. Inflammatory bowel disease. N Engl J Med. 2009;361:2066–2078. doi: 10.1056/NEJMra0804647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rakoff-Nahoum S, Paglino J, Eslami-Varzaneh F, Edberg S, Medzhitov R. Recognition of commensal microflora by Toll-like receptors is required for intestinal homeostasis. Cell. 2004;118:229–241. doi: 10.1016/j.cell.2004.07.002. [DOI] [PubMed] [Google Scholar]
- 3.Maloy KJ, Powrie F. Intestinal homeostasis and its breakdown in inflammatory bowel disease. Nature. 2011;474:298–306. doi: 10.1038/nature10208. [DOI] [PubMed] [Google Scholar]
- 4.Muniz LR, Knosp C, Yeretssian G. Intestinal antimicrobial peptides during homeostasis, infection, and disease. Front Immunol. 2012;3:1–10. doi: 10.3389/fimmu.2012.00310. 310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wehkamp J, Schmid M, Fellermann K, Stange EF. Defensin deficiency, intestinal microbes, and the clinical phenotypes of Crohn's disease. J Leukoc Biol. 2005;77:460–465. doi: 10.1189/jlb.0904543. [DOI] [PubMed] [Google Scholar]
- 6.Goetz DH, Holmes MA, Borregaard N, Bluhm ME, Raymond KN, Strong RK. The neutrophil lipocalin NGAL is a bacteriostatic agent that interferes with siderophore-mediated iron acquisition. Mol Cell. 2002;10:1033–1043. doi: 10.1016/s1097-2765(02)00708-6. [DOI] [PubMed] [Google Scholar]
- 7.Flo TH, Smith KD, Sato S, et al. Lipocalin 2 mediates an innate immune response to bacterial infection by sequestrating iron. Nature. 2004;432:917–921. doi: 10.1038/nature03104. [DOI] [PubMed] [Google Scholar]
- 8.Kjeldsen L, Johnsen AH, Sengelov H, Borregaard N. Isolation and primary structure of NGAL, a novel protein associated with human neutrophil gelatinase. J Biol Chem. 1993;268:10425–10432. [PubMed] [Google Scholar]
- 9.Cowland JB, Sorensen OE, Sehested M, Borregaard N. Neutrophil gelatinase-associated lipocalin is up-regulated in human epithelial cells by IL-1 beta, but not by TNF-alpha. J Immunol. 2003;171:6630–6639. doi: 10.4049/jimmunol.171.12.6630. [DOI] [PubMed] [Google Scholar]
- 10.Nielsen BS, Borregaard N, Bundgaard JR, Timshel S, Sehested M, Kjeldsen L. Induction of NGAL synthesis in epithelial cells of human colorectal neoplasia and inflammatory bowel diseases. Gut. 1996;38:414–420. doi: 10.1136/gut.38.3.414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bu DX, Hemdahl AL, Gabrielsen A, et al. Induction of neutrophil gelatinase-associated lipocalin in vascular injury via activation of nuclear factor-kappaB. Am J Pathol. 2006;169:2245–2253. doi: 10.2353/ajpath.2006.050706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Eagan TM, Damas JK, Ueland T, et al. Neutrophil gelatinase-associated lipocalin: a biomarker in COPD. Chest. 2010;138:888–895. doi: 10.1378/chest.09-2718. [DOI] [PubMed] [Google Scholar]
- 13.Oikonomou KA, Kapsoritakis AN, Theodoridou C, et al. Neutrophil gelatinase-associated lipocalin (NGAL) in inflammatory bowel disease: association with pathophysiology of inflammation, established markers, and disease activity. J Gastroenterol. 2012;47:519–530. doi: 10.1007/s00535-011-0516-5. [DOI] [PubMed] [Google Scholar]
- 14.Nielsen OH, Gionchetti P, Ainsworth M, et al. Rectal dialysate and fecal concentrations of neutrophil gelatinase-associated lipocalin, interleukin-8, and tumor necrosis factor-alpha in ulcerative colitis. Am J Gastroenterol. 1999;94:2923–2928. doi: 10.1111/j.1572-0241.1999.01439.x. [DOI] [PubMed] [Google Scholar]
- 15.Ostvik AE, Vb Granlund A, Bugge M, et al. Enhanced expression of CXCL10 in inflammatory bowel disease: potential role of mucosal Toll-like receptor 3 stimulation. Inflamm Bowel Dis. 2013;19:265–274. doi: 10.1002/ibd.23034. [DOI] [PubMed] [Google Scholar]
- 16.Landro L, Damas JK, Flo TH, et al. Decreased serum lipocalin-2 levels in human immunodeficiency virus-infected patients: increase during highly active anti-retroviral therapy. Clin Exp Immunol. 2008;152:57–63. doi: 10.1111/j.1365-2249.2008.03592.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Granlund AB, Beisvag V, Torp SH, et al. Activation of REG family proteins in colitis. Scand J Gastroenterol. 2011;46:1316–1323. doi: 10.3109/00365521.2011.605463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sen GC, Sarkar SN. Transcriptional signaling by double-stranded RNA: role of TLR3. Cytokine Growth Factor Rev. 2005;16:1–14. doi: 10.1016/j.cytogfr.2005.01.006. [DOI] [PubMed] [Google Scholar]
- 19.Dooley TP, Curto EV, Reddy SP, et al. Regulation of gene expression in inflammatory bowel disease and correlation with IBD drugs: screening by DNA microarrays. Inflamm Bowel Dis. 2004;10:1–14. doi: 10.1097/00054725-200401000-00001. [DOI] [PubMed] [Google Scholar]
- 20.Lawrance IC, Fiocchi C, Chakravarti S. Ulcerative colitis and Crohn's disease: distinctive gene expression profiles and novel susceptibility candidate genes. Hum Mol Genet. 2001;10:445–456. doi: 10.1093/hmg/10.5.445. [DOI] [PubMed] [Google Scholar]
- 21.Csillag C, Nielsen OH, Vainer B, et al. Expression of the genes dual oxidase 2, lipocalin 2 and regenerating islet-derived 1 alpha in Crohn's disease. Scand J Gastroenterol. 2007;42:454–463. doi: 10.1080/00365520600976266. [DOI] [PubMed] [Google Scholar]
- 22.Borregaard N, Sehested M, Nielsen BS, Sengelov H, Kjeldsen L. Biosynthesis of granule proteins in normal human bone marrow cells. Gelatinase is a marker of terminal neutrophil differentiation. Blood. 1995;85:812–817. [PubMed] [Google Scholar]
- 23.Cario E, Podolsky DK. Differential alteration in intestinal epithelial cell expression of Toll-like receptor 3 (TLR3) and TLR4 in inflammatory bowel disease. Infect Immun. 2000;68:7010–7017. doi: 10.1128/iai.68.12.7010-7017.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Furrie E, Macfarlane S, Thomson G, Macfarlane GT. Toll-like receptors-2,-3 and-4 expression patterns on human colon and their regulation by mucosal-associated bacteria. Immunology. 2005;115:565–574. doi: 10.1111/j.1365-2567.2005.02200.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xu J, Yang Y, Wang C, Jiang B. Rotavirus and coxsackievirus infection activated different profiles of toll-like receptors and chemokines in intestinal epithelial cells. Inflamm Res. 2009;58:585–592. doi: 10.1007/s00011-009-0022-x. [DOI] [PubMed] [Google Scholar]
- 26.Vijay-Kumar M, Wu H, Aitken J, et al. Activation of Toll-like receptor 3 protects against DSS-induced acute colitis. Inflamm Bowel Dis. 2007;13:856–864. doi: 10.1002/ibd.20142. [DOI] [PubMed] [Google Scholar]
- 27.Cavassani KA, Ishii M, Wen H, et al. TLR3 is an endogenous sensor of tissue necrosis during acute inflammatory events. J Exp Med. 2008;205:2609–2621. doi: 10.1084/jem.20081370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhou R, Wei H, Sun R, Tian Z. Recognition of double-stranded RNA by TLR3 induces severe small intestinal injury in mice. J Immunol. 2007;178:4548–4556. doi: 10.4049/jimmunol.178.7.4548. [DOI] [PubMed] [Google Scholar]
- 29.Kariko K, Ni H, Capodici J, Lamphier M, Weissman D. mRNA is an endogenous ligand for Toll-like receptor 3. J Biol Chem. 2004;279:12542–12550. doi: 10.1074/jbc.M310175200. [DOI] [PubMed] [Google Scholar]
- 30.Chassaing B, Srinivasan G, Delgado MA, Young AN, Gewirtz AT, Vijay-Kumar M. Fecal lipocalin 2, a sensitive and broadly dynamic non-invasive biomarker for intestinal inflammation. PLoS ONE. 2012;7:e44328. doi: 10.1371/journal.pone.0044328. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Immunohistochemical staining of Toll-like receptor (TLR)-3 in colonic biopsy from actively inflamed mucosa in ulcerative colitis and three different types of negative controls in the same experiment are shown. We used serial sections from same biopsy. The first negative control was made by omitting primary antibody (no ab). In the second negative control, primary antibody was replaced by isotype immunoglobulin (Ig)G1 directed against non-human epitope (IgG1). The third control was made by blocking primary antibody with recombinant human TLR-3 peptide (rhTLR-3+TLR-3ab); ×10 and ×40 magnification for each slide are shown.