

Abstract
In this study, we hypothesized that the granulomatous disorder sarcoidosis is not caused by a single pathogen, but rather results from abnormal responses of Toll-like receptors (TLRs) to conserved bacterial elements. Unsorted bronchoalveolar lavage (BAL) cells from patients with suspected pulmonary sarcoidosis and healthy non-smoking control subjects were stimulated with representative ligands of TLR-2 (in both TLR-2/1 and TLR-2/6 heterodimers) and TLR-4. Responses were determined by assessing resulting production of tumour necrosis factor (TNF)-α and interleukin (IL)-6. BAL cells from patients in whom sarcoidosis was confirmed displayed increased cytokine responses to the TLR-2/1 ligand 19-kDa lipoprotein of Mycobacterium tuberculosis (LpqH) and decreased responses to the TLR-2/6 agonist fibroblast stimulating ligand-1 (FSL)-1. Subsequently, we evaluated the impact of TLR-2 gene deletion in a recently described murine model of T helper type 1 (Th1)-associated lung disease induced by heat-killed Propionibacterium acnes. As quantified by blinded scoring of lung pathology, P. acnes-induced granulomatous pulmonary inflammation was markedly attenuated in TLR-2–/– mice compared to wild-type C57BL/6 animals. The findings support a potential role for disordered TLR-2 responses in the pathogenesis of pulmonary sarcoidosis.
Keywords: bronchoalveolar lavage (BAL), sarcoidosis, Toll-like receptors
Introduction
Sarcoidosis is a multi-system granulomatous disorder of unknown aetiology in which pulmonary disease is the most common manifestation. Active disease is characterized by lymphocytic alveolitis with a predominance of activated T helper type 1 (Th1)-like CD4+ lymphocytes [1]. The presence of granulomas in involved tissues has suggested that sarcoidosis results from chronic infection. Although multiple studies have implicated both the acid-fast aerobe Mycobacterium tuberculosis and the Gram-positive anaerobe Propionibacterium acnes as causative pathogens [2–8], no single organism has been identified consistently across varied populations of affected individuals [9–11]. These findings suggest an alternative possibility, that sarcoidosis might result from abnormal innate immune responses to conserved motifs common to multiple organisms.
Toll-like receptors (TLRs) are a family of immune recognition molecules that initiate production of proinflammatory cytokines in response to molecular patterns common to many pathogens [12]. Some of these cytokines, such as tumour necrosis factor (TNF)-α and interleukin (IL)-6, are observed in bronchoalveolar lavage (BAL) fluid of patients with active sarcoidosis and are implicated in the formation of sarcoid granulomas [1, 13]. We therefore compared TLR-induced cytokine responses of BAL cells of sarcoidosis patients to those of healthy non-smoking volunteers. Because these human studies suggested a role for TLR-2 responses in the pathogenesis of sarcoidosis, we then evaluated the impact of TLR-2 gene deletion in a murine model of Th1-associated granulomatous lung disease [14].
Materials and methods
Human studies
Subjects
Patients were recruited primarily at the time of diagnostic bronchoscopies performed at University Hospitals Case Medical Center and MetroHealth Medical Center (Cleveland, OH, USA). One patient with previously confirmed sarcoidosis underwent bronchoscopy with BAL for research purposes only. Eligibility of results for study inclusion was based on biopsy confirmation of sarcoidosis (on the basis of the presence of non-caseating granulomas with negative stains for acid-fast bacilli and fungi), non-smoking status, absence of systemic immunosuppressive therapy within the preceding 6 months and lack of significant co-morbidities. Of 44 patients referred to the study, thirty-two (aged 28–68 years) were confirmed ultimately to have sarcoidosis and otherwise to be study-eligible. Forty-one healthy non-smoking volunteers aged 18–50 years were recruited as controls. All human subjects’ protocols were approved by the Institutional Review Boards of the participating institutions.
Bronchoscopy procedures and characterization of BAL cell samples
For patients undergoing diagnostic bronchoscopy, dedicated research BAL was performed using up to four 30-ml aliquots of sterile saline. Bronchoscopies with BAL performed for research purposes only followed previously described protocols [15]. BAL cell differentials and CD4+/CD8+ T cell ratios were determined as described previously [15].
TLR ligands
For initial studies, TLR-2 was stimulated with native 19-kDa lipoprotein of M. tuberculosis (LpqH) extracted from lysates of M. tuberculosis strain H37Ra [16], whereas subsequent studies utilized a synthetic lipopeptide based on the structure of LpqH, Pam-3-Cys-SSNKSTTGSGETTTA (EMC Microcollections, Tuebingen, Germany) in concentrations of 500 and 2500 ng/ml. Pam-3-Cys-SKKKK (concentrations of 1–1000 ng/ml, tlrl-pms; InvivoGen, San Diego, CA, USA) and fibroblast stimulating ligand-1 (FSL-1) (Pam-2-Cys-GDPKHPKSF, 0·1 and 1·0 μg/ml, #L7000; EMC Microcollections) were used as ligands of TLR-2/1 and TLR-2/6 heterodimers. Lipopolysaccharide (LPS) (0·1, 1·0 and 10 ng/ml; Sigma-Aldrich, St Louis, MO, USA) served as a model TLR-4 ligand. Incubation of BAL cells with TLR-2 and-4 agonists for 2 h was followed by harvesting of culture supernatants, rinsing the plated cells to remove residual TLR ligand, and replenishing cultures with fresh medium. Because differences between TNF-α and IL-6 responses of the two subject groups were observed with regard to the magnitudes rather than durations of responses, statistical comparison was based on peak cytokine levels, observed 24 h after TLR stimulation for both cytokines.
Determination of cytokine responses to TLR stimulation
BAL cells were aliquoted into 24-well plates (400 000 cells/ml) and incubated with medium or specific TLR ligands. Supernatants were harvested and stored at −70°C for eventual enzyme-linked immunosorbent assay (ELISA)-based quantification of TNF-α (Quantikine® DTA00C; R&D Systems, Minneapolis, MN, USA) and IL-6 (Quantikine® D6050; R&D Systems).
Assessment of TLR expression via flow cytometry
Samples were incubated with anti-CD3-fluorescein isothiocyanate (FITC) (#11-0038-71; eBioscience, San Diego, CA, USA) and anti-TLR-2-antigen-presenting cells (APC) (#17-9922-71; eBioscience) in combination with either anti-TLR-1-phycoerythrin (PE) (#12-9911-71; eBioscience) or anti-TLR-6-PE (IMG-203D; Imgenex, San Diego, CA, USA). Data were acquired on an LSR-II Flow Cytometer (Becton-Dickinson, Franklin Lakes, NJ, USA). This was calibrated on a daily basis using Cytometer Tracking and Setup Beads (Becton-Dickinson) to assure reproducibility of data acquired on different days. Analysis was performed using FlowJo software (Becton-Dickinson).
Murine model of Propionibacterium acnes-induced granulomatous lung disease
Mice
Wild-type C57BL/6 mice were obtained from Jackson Laboratories (Bar Harbor, ME, USA) and TLR-2 gene-disrupted mice (on C57BL/6 background) provided generously by Drs O. Takeuchi and S. Akira of Osaka University [17]. Mice were aged 12–16 weeks at the time of study entry. Animal protocols were approved by the Institutional Animal Care and Use Committee of Case Western Reserve University.
Sensitization and challenge protocol
Wild-type mice received an intraperitoneal (i.p.) injection of 0·5 mg of P. acnes or sham sensitization with sterile saline. Intratracheal (i.t.) challenge was performed 14 days later by direct injection of 0·5 mg of P. acnes into surgically exposed tracheas of anaesthetized animals. BAL procedures and harvesting of lungs for histological examination were performed as described previously [18]. BAL cytokines were analysed using Milliplex reagents (Millipore, Billerica, MA, USA) on a Bioplex 200 reader (Biorad, Hercules, CA, USA). Histological review was performed in a blinded fashion by a single pulmonary pathologist (J.F.T.). Lung cell homogenates for specific studies were prepared using dissection from the mainstem bronchi followed by mincing with scalpels and disruption by repeated passage through an 18G needle, as described previously [19].
Statistical methods
Non-parametric cytokine responses of human BAL cells were assessed by Wilcoxon's rank sum test using sas version 9.1 software (SAS Institute, Cary, NC, USA). Expression of TLR receptors, murine cytokine results and murine pathology scoring was evaluated using t-tests, and correlations between BAL lymphocytosis and TNF-α responses utilized Spearman's rank sum testing (all performed using GraphPad Prism; GraphPad Software, San Diego, CA, USA).
Results
Clinical and immunological features of study subjects
Clinical and BAL findings of confirmed study-eligible sarcoidosis patients and healthy controls are summarized in Table 1]. Both groups included diverse subject populations; however, the sarcoidosis patients were older and more likely to be African American than were controls. As expected, patients also displayed greater BAL cell lymphocytosis and higher BAL CD4 : CD8 T cell ratios than did healthy volunteers. At the time of study entry, more than 80% of the sarcoidosis patients displayed stage 2 radiographic findings, and none had classic features of Lofgren's syndrome (i.e. acute illness characterized by fever, arthralgias, erythema nodosum and bihilar adenopathy). Of 18 confirmed sarcoidosis patients who had follow-up within our system for at least 18 months (median 38 months, range 19–69), none demonstrated complete resolution of radiographic findings. Three had some radiographic improvement without a change in radiographic stage. Three had worsening of findings leading to initiation of treatment. For 12 patients, no radiographic changes were observed, although this group remained minimally symptomatic and were generally not treated.
Table 1.
Clinical and immunological findings of study-eligible sarcoidosis patients and control subjects.
| Variable | Sarcoidosis | Controls |
|---|---|---|
| Number of subjects | 32 | 41 |
| Age, mean (range) | 45·1 (28–68) | 28·8 (18–48) |
| Sex | ||
| Female | 22 (68·8%) | 19 (46·3%) |
| Male | 10 (31·2%) | 22 (53·7%) |
| Race | ||
| Caucasian | 4 (12·5%) | 29 (70·7%) |
| African American | 26 (81·3%) | 8 (19·5%) |
| Hispanic | 2 (6·2%) | 2 (4·9%) |
| Asian | 0 (0·0%) | 2 (4·9%) |
| Radiographic stage | ||
| 1 | 5 (15·6%) | n.a. |
| 2 | 26 (81·3%) | |
| 3 | 1 (3·1%) | |
| Constitutional symptoms/extrapulmonary disease | 13 (40·6%) | n.a. |
| Hypercalcaemia | 3 (9·4%) | n.a. |
| Liver enzyme abnormalities | 9 (28·1%) | n.a. |
| Anaemia | 14 (43·8%) | n.a. |
| BAL cell differential (mean) | ||
| Lymphocytes | 7·74% | 4·63% |
| Neutrophils | 3·44% | 2·13% |
| Eosinophils | 0·83% | 0·49% |
| Macrophages | 87·58% | 91·84% |
| BAL CD4 : CD8 ratio | 2·28:1 | 1·15:1 |
BAL: bronchoalveolar lavage; n.a.: not applicable.
Sarcoidosis patients display increased cytokine responses to the TLR-2 agonist, the 19-kDa lipoprotein of M. tuberculosis and its corresponding synthetic lipopeptide, but not to the TLR-4 agonist LPS
TLR responses of alveolar macrophages (AM), the predominant cells in BAL, have been characterized most extensively with regard to stimulation of TLR-2 and-4 [12]. Our initial studies focused therefore on responses to a model TLR-2 agonist, the 19-kDa lipoprotein of M. tuberculosis (LpqH), and bacterial LPS, an agonist of TLR-4.
TLR-2 stimulation using 500 ng/ml of the native 19-kDa lipoprotein of M. tuberculosis or a synthetic lipopeptide based on its structure (Pam-3-Cys-SSNKSTTGSGETTTA) elicited significantly greater production of both TNF-α and IL-6 by BAL cells of sarcoidosis patients than did those of controls (P < 0·006 and P = 0·002, respectively, by Wilcoxon's rank sum test, Fig. 1a,b). Because the BAL cell responses of healthy controls to the 19-kDa lipoprotein of M. tuberculosis were unexpectedly low, we also performed a limited number of additional studies using a higher concentration of the ligand. In these studies, mean TNF-α responses of control subjects to 500 ng/ml of the 19-kDa lipoprotein was 559·1 ± 300·3 pg/ml; this increased to 2021·9 ± 141·3 pg/ml following incubation with 2500 ng/ml of the ligand. In contrast, BAL cells of sarcoidosis patients produced TNF-α levels of 2226·8 ± 3101·2 pg/ml in response to 500 ng/ml of LpgH, and 10 310·3 ± 8952·5 pg/ml in response to 2500 ng/ml of the ligand. These findings suggest that the two subject groups display distinct dose–response relationships for this agonist.
Figure 1.

Compared to those of healthy non-smokers, bronchoalveolar lavage (BAL) cells from sarcoidosis patients display greater cytokine responses to the Toll-like receptor (TLR)-2 ligand 19-kDa lipoprotein of Mycobacterium tuberculosis, but not to the TLR-4 ligand lipopolysaccharide (LPS). In response to stimulation the 19-kDa M. tuberculosis lipoprotein LpqH and the synthetic lipopeptide based on its structure (Pam-3-Cys-SSNKSTTGSGETTTA), peak levels of tumour necrosis factor (TNF)-α (a) and interleukin (IL)-6 (b) stimulation were both significantly greater for patients than control subjects. In contrast, no significant differences were observed between tumour necrosis factor (TNF)-α and IL-6 responses of BAL cells from sarcoidosis patients and from control subjects 24 h after stimulation with several concentrations of the TLR-4 ligand LPS, as detailed in the text (c,d, respectively). For all figures, horizontal lines indicate median values and shaded boxes indicate 25–75th percentile ranges.
In contrast, studies of TLR-4 stimulation with LPS showed no differences in cytokine production by BAL cells of sarcoidosis patients and those of control subjects. As illustrated in Fig. 1c,d, stimulation with LPS in concentrations 10, 1 or 0·1 ng/ml resulted in no differences between the two groups with regard to production of TNF-α (P = 0·226, 0·535 and 0·352, respectively, Fig. 1c) or IL-6 (P = 0·083, 0·278 and 0·391, respectively, Fig. 1d).
BAL cells of patients with sarcoidosis display differing responses to TLR-2 ligands with specificities for the TLR-2/1 and TLR-2/6 heterodimer
Both the 19-kDa M. tuberculosis lipoprotein LpqH and its associated Pam-3-Cys-SSNKSTTGSGETTTA lipopeptide act upon the TLR-2/1 heterodimer [16]. To characterize further the mechanisms of abnormal TLR-2 responsiveness in patients with sarcoidosis, we evaluated responses of BAL cells from sarcoidosis patients and control subjects to a more standard TLR-2/1 ligand, Pam-3-Cys-SKKKK (Pam-3-Cys), and to FSL-1, a TLR-2/6-specific synthetic peptide derived from Mycoplasma salivarum lipoprotein LP44 (MALP) [20]. No differences in TNF-α production were observed following stimulation of BAL cells of the two subject groups with a wide range of doses of Pam-3-Cys (Fig. 2a). However, although BAL cells of the two subject groups displayed similar TNF-α responses to stimulation with 0·1 μg/ml of FSL-1 (P = 0·143), sarcoidosis patients produced significantly less TNF-α than control subjects in response to 1·0 μg/ml FSL-1 (P = 0·005, Fig. 2b). BAL cells of sarcoidosis patients therefore are not hyperresponsive to all TLR-2/1 agonists; however, the reciprocal decrease in TNF-α production following FSL-1 stimulation of TLR-2/6 suggests that a more generalized dysregulation of TLR-2 responses might be present in sarcoidosis.
Figure 2.

Bronchoalveolar lavage (BAL) cells of sarcoidosis patients and healthy control subjects have differing responsiveness to stimulation of Toll-like receptor (TLR)-2 heterodimers, but do not display differential expression of TLR-2/1 and TLR-2/6. As detailed in the text, peak tumour necrosis factor (TNF)-α responses of BAL cells from patients and control subjects did not differ following stimulation with Pam-3-Cys (a). In contrast, BAL cells from sarcoidosis patients produced significantly less TNF-α than those of healthy controls in response to 1 μg/ml of the TLR-2/6 agonist fibroblast stimulating ligand-1 (FSL-1), but not in response to a lower concentration (0·1 μg/ml) of this ligand (b). Nevertheless, no differences were observed between expression of TLR-2, TLR-1 or TLR-6 on alveolar macrophages (AM) of control subjects and patients, as detailed in the text (c). For all figures, horizontal lines again indicate median results, whereas shaded boxes illustrate the 25–75th percentile range for each assessment.
Expression of TLR-2 heterodimers on BAL cells from patients with sarcoidosis does not differ from that of healthy control subjects
To determine whether disordered BAL cell cytokine responses to TLR-2 stimulation reflect skewing of TLR-2 heterodimer expression in sarcoidosis, we compared expression of TLR-2,-1 and-6 expression on BAL cells from sarcoidosis patients and control subjects using flow cytometry, as expressed as mean fluorescence intensity. No significant differences were observed, however (P = 0·340, P-0·714 and P = 0·354, respectively, by t-test, Fig. 2c).
BAL cell composition and responses to LpqH
Because BAL cells of sarcoidosis often display a higher percentage of lymphocytes than those observed in healthy individuals, we evaluated the relationship between lymphocytosis and BAL cell TNF-α responses to the 19 kDa M. tuberculosis lipoprotein (LpqH). No correlation was observed between percentage of lymphocytes in BAL and LpqH-induced TNF-α production by subjects with sarcoidosis (correlation coefficient −0·048 by Spearman's rank sum test, P = 0·840, Fig. 3a). Further, differences in TNF-α production by BAL cells from sarcoidosis patients and those of control subjects were observed both when lymphocytosis was observed (>8% BAL lymphocytes) and when it was not (<8% lymphocytes, Fig. 3b).
Figure 3.
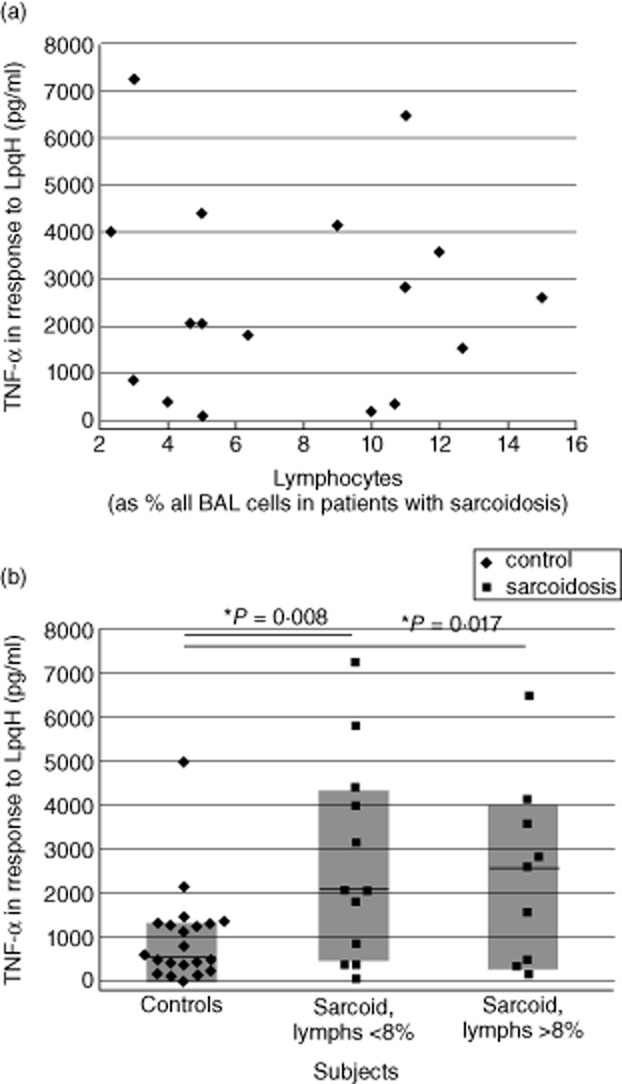
Increased LpgH-induced production of tumour necrosis factor (TNF)-α by bronchoalveolar lavage (BAL) cells in sarcoidosis is independent of BAL cell lymphocytosis. As illustrated, BAL cell production of TNF-α shows no correlation with the percentage of lymphocytes present within BAL, (a). Further, division of these results into those observed for patients in whom lymphocytes accounted for greater or fewer than 8% of total BAL cells indicates that both groups display TNF-α responses that are distinct from those of control subjects (b).
TLR-2 gene-disruption alters BAL cytokine responses of sensitized mice to P. acnes challenge
To evaluate the potential importance of TLR-2-dependent immune responses in the development of pulmonary sarcoidosis, we utilized a recently described murine model of Th1-associated granulomatous lung disease induced by i.p. sensitization followed 14 days later by i.t. challenge with heat-killed P. acnes [14]. BAL fluid from sensitized wild-type and TLR2–/– mice was then obtained 24 h after i.t. instillation of P. acnes. As displayed in Fig. 4a, TNF-α responses following i.t. P. acnes were reduced markedly in TLR-2–/– mice compared to wild-type C57BL/6 animals (P = 0·022 by t-test). To confirm that impaired TNF-α production was not a generalized phenomenon in these mice, we also incubated lung homogenates of non-sensitized TLR-2–/– mice overnight with medium alone, heat-killed P. acnes and LPS (Fig. 4b). As illustrated, although lung homogenates of TLR-2–/– mice did not display significant TNF-α production in response to P. acnes, they did produce TNF-α appropriately in response to the TLR-4 ligand LPS (P = 0·027 by t-test).
Figure 4.
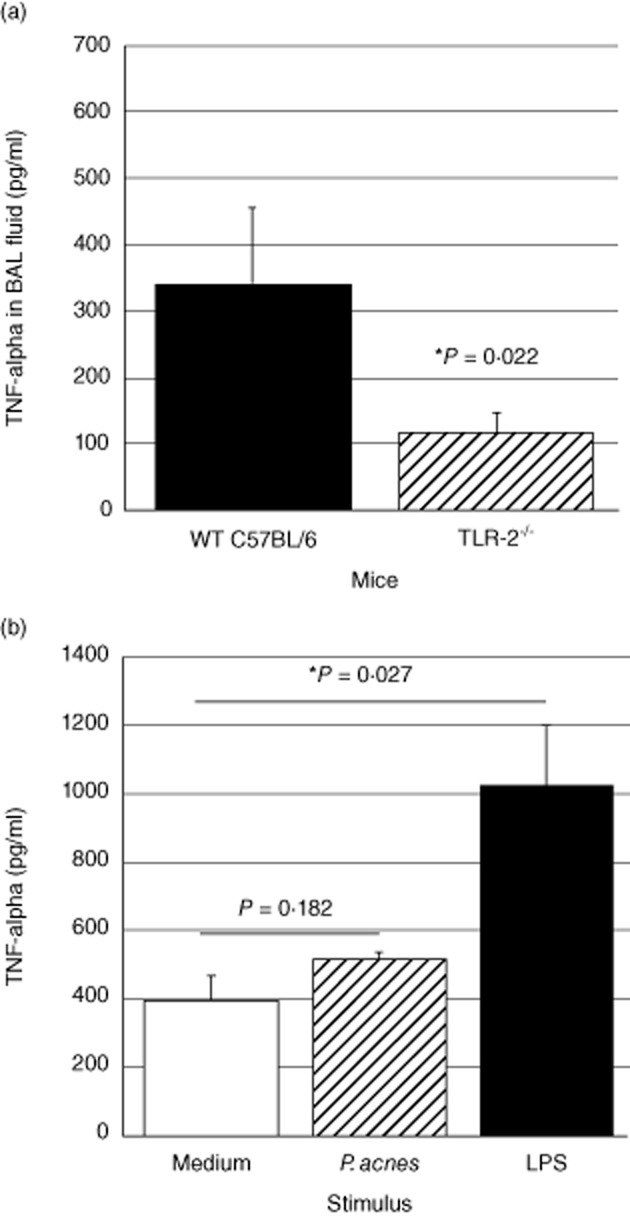
Pulmonary tumour necrosis factor (TNF)-α responses to heat-killed Propionibacterium acnes are reduced in Toll-like receptor (TLR)-2–/– animals. Compared to similarly treated wild-type C57BL/6 animals, TLR-2–/– mice in which intratracheal (i.t.) P. acnes challenge was performed 14 days after initial intraperitoneal (i.p.) sensitization displayed significantly lower concentrations of TNF-α in bronchoalveolar lavage (BAL) fluid at 24 h. The data represent mean ± standard deviation (s.d.) of studies of six wild-type and seven TLR-2–/– animals (a). The specificity of this finding was clarified by in-vitro studies of lung homogenates from non-sensitized TLR-2–/– mice. As detailed in the text, stimulation with P. acnes did not induce significant TNF-α responses at 24 h, but stimulation with the TLR-4 ligand lipopolysaccharide (LPS) resulted in significant TNF-α production by these cells. Results represent mean ± s.d. of findings following incubation of three pools of homogenized lungs, each obtained from three TLR-2–/– mice (b).
TLR-2 gene disruption results in marked reduction of granulomatous pulmonary inflammation
Histological assessments were performed in three groups of animals: (1) wild-type C57BL/6 (sham i.p. sensitization/P. acnes i.t. challenge), wild-type C57BL/6 (P. acnes i.p. sensitization, P. acnes i.t. challenge) and TLR-2–/– C57BL/6 (P. acnes-i.p. sensitization/P. acnes i.t. challenge). All histology specimens were reviewed and scored in a blinded fashion by a single pathologist (J.F.T.). Upon unblinding, lungs of non-sensitized mice revealed minimal or no inflammation following i.t. P. acnes challenge (not shown). In contrast, wild-type mice treated with i.p. sensitization followed by i.t. challenge with P. acnes displayed substantial granulomatous inflammation (Fig. 5a–d), whereas lung pathology was much more limited in i.p.-sensitized, i.t.-challenged TLR-2–/– mice (Fig. 5e–h).
Figure 5.
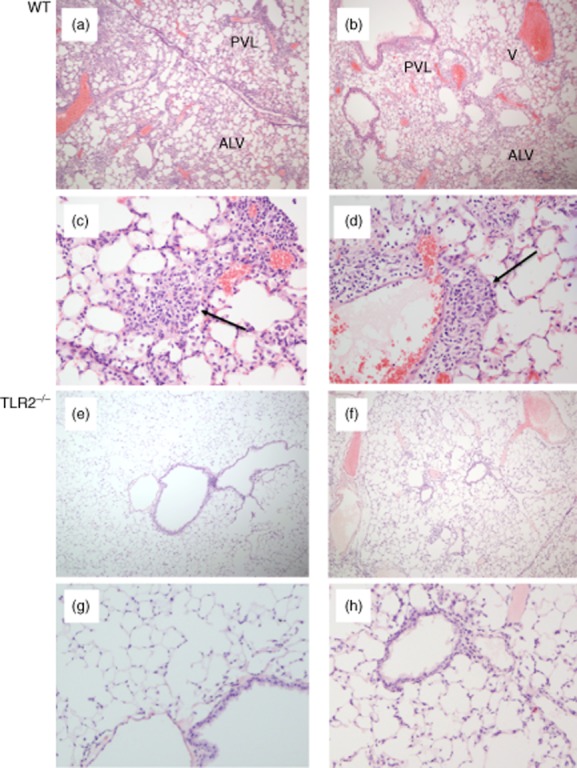
Propionibacterium acnes-induced granulomatous pulmonary inflammation is reduced markedly in Toll-like receptor (TLR)-2–/– mice compared to that observed in wild-type C57BL/6 animals. Both groups of animals received intraperitoneal (i.p.) sensitization followed by intratracheal (i.t.) challenge with P. acnes. The images show representative histology of two wild-type animals at low (a,b) and high magnification (c,d). As illustrated, wild-type animals displayed a marked influx of lymphocytes and macrophages that were primarily adjacent to bronchovascular-lymphatic bundles (PVL) and large veins (V). Varying degrees of intra-alveolar and interstitial (ALV) inflammation were also observed. Magnified views show loosely formed granulomas typical of those observed in mice (indicated by arrows). In contrast, P. acnes induced relatively little inflammation in TLR-2–/– mice, as illustrated in (e) and (f) and the corresponding higher-power images (g) and (h). In these animals, peribronchovascular and lymphatic inflammation was minimal and intra-alveolar/interstitial regions were largely normal, as illustrated.
The pathology of each histological lung section was scored in a blinded fashion on a scale of 0–3 with regard to degree of inflammation observed overall and in specific anatomical regions of the lung. As illustrated in Fig. 6a, lungs of P. acnes-sensitized and challenged TLR-2–/– mice displayed significantly less inflammation than did sensitized, challenged wild-type mice in peribronchovascular regions (P = 0·007 by t-test), alveolar regions (P = 0·007) and the interstitium (P < 0·001). Overall inflammation scores for the TLR-2–/– mice were similarly significantly lower than those of sensitized wild-type C57BL/6 mice (P < 0·001). The percentage of peribronchovascular–lymphatic bundles and veins involved in the inflammatory response to P. acnes was significantly lower in TLR-2–/– mice than in sensitized wild-type animals (P = 0·027, Fig. 6b). As illustrated in Fig. 6c, the overall proportion of the lung tissue involved in the inflammatory response of TLR-2–/– mice was remarkably lower than that of wild-type mice (6·65 ± 8·39% versus 40·17 ± 16·98%, P < 0·001) and, in fact, did not differ significantly from that observed in non-sensitized wild-type mice (0·33 ± 0·52%, P = 0·117). Sarcoid-like granulomatous lung disease in C57BL/6 mice was thus attenuated markedly in TLR-2-deficient animals, supporting a role for TLR-2 stimulation in the development of this process.
Figure 6.
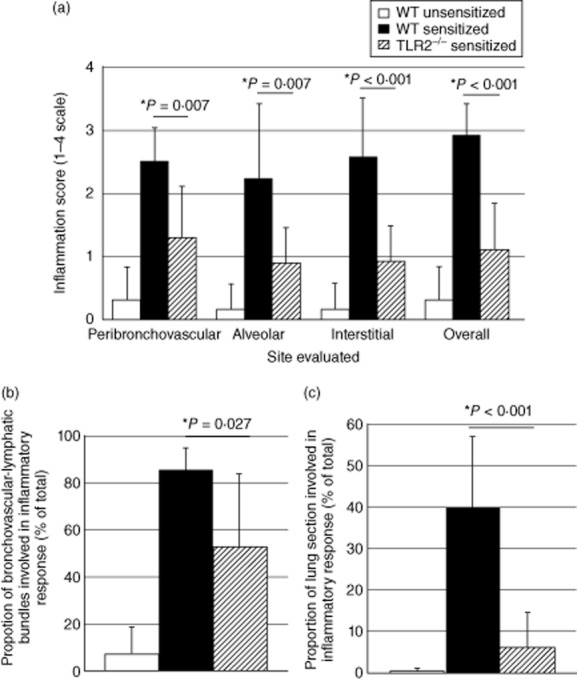
Detailed histological grading confirms marked reduction of pulmonary inflammation Toll-like receptor (TLR)-2 gene-disrupted animals following intratracheal (i.t.) challenge with Propionibacterium acnes. Pathology of non-sensitized and sensitized wild-type mice (n = 6 in each case) and of sensitized TLR-2–/– animals (n = 10) was evaluated. As detailed in the text, lungs of P. acnes-sensitized and challenged TLR-2–/– mice displayed significantly less inflammation than did sensitized, challenged wild-type mice in peribronchovascular, alveolar and interstitial regions. Overall inflammation scores were also significantly lower in TLR-2–/– mice (a). The percentage of peribronchovascular-lymphatic bundles and veins involved in the inflammatory response to P. acnes was significantly lower in TLR-2–/– mice than in sensitized wild-type animals (b). Similarly, the overall proportion of the lung tissue involved in the inflammatory response of TLR-2–/– mice was remarkably lower than that of wild-type mice and, in fact, did not differ significantly from that observed in non-sensitized wild-type mice (c). In all figures, findings in non-sensitized wild-type animals are represented by unshaded bars, sensitized wild-type animals by shaded bars and sensitized TLR-2–/– mice by striped bars.
Discussion
In this study, we evaluated the hypothesis that sarcoidosis could result from abnormal responsiveness to TLR stimulation. Compared to healthy control subjects, BAL cells of patients with sarcoidosis displayed increased production of proinflammatory cytokines (TNF-α and IL-6) in response to stimulation with the TLR-2/1 ligand the 19-kDa M. tuberculosis lipoprotein (LpqH). Patients also displayed decreased responsiveness to a standard ligand of the TLR-2/6 heterodimer, FSL1. These responses were not associated with differences in AM expression of TLR-2,-1 or-6, nor did they correlate with BAL lymphocytosis. Subsequent studies using a murine model of Th1-associated granulomatous lung disease (induced by i.p. sensitization followed by i.t. challenge with heat-killed P. acnes) indicated that resulting pulmonary inflammation was markedly attenuated in TLR-2–/– mice compared to wild-type animals. This combination of findings supports the possibility that abnormal host responsiveness to certain TLR-2 ligands might predispose to the development of pulmonary sarcoidosis.
Previous investigations of TLRs in sarcoidosis have focused primarily on assessment of gene polymorphisms [21–25]. Association between sarcoidosis and TLR-2 polymorphisms in particular was not observed in Japanese subjects [23], but was noted in a subset of Dutch patients, in whom they correlated with chronicity of disease [24]. Our patient population, being predominantly African American, was ethnically distinct from those of these previous studies. Clinical presentation and outcomes of our population were also distinct. Notably, Lofgren's syndrome, which is associated with the haplotype human leucocyte antigen (HLA)-DRB1*0301 [26], was observed in greater than one-third of Dutch patients [24], but in none of the patients in our study. Although HLA typing was not performed on our subjects, DRB1*0301 has been reported as being fourfold less frequent in African Americans than in American Caucasians [27], which may be consistent with the lack of fulminant disease observed in the current study. Conversely, persistent disease, as determined by lack of resolution of radiographic findings, was a much more common outcome in our population. In addition, our observation that BAL cells from sarcoidosis patients display contrasting responses to ligands of TLR-2/1 and TLR-2/6 heterodimers emphasizes that polymorphisms of TLR-1 and/or TLR-6 may also impact responses to TLR-2 stimulation, as has been evaluated more recently by Veltkamp and colleagues [25].
Altered TLR-2 responses observed in sarcoidosis could also be mediated at a non-genomic level. However, the current studies did not identify differences in expression of total TLR-2 or of specific TLR-2 heterodimers between sarcoidosis patients and controls to provide another explanation of these findings. Similarly, no relationship was observed between intensity of inflammation (as manifested by BAL lymphocytosis) and responses to TLR-2 stimulation. In contrast, a previous study observed increased AM TLR-2 expression in a sarcoidosis patient population that displayed greater BAL cell lymphocytosis than that observed currently [28]. As TLR-2 expression is known to be up-regulated during inflammation [29, 30], our finding of increased TLR-2 responsiveness in this group of sarcoidosis patients may support more strongly the possibility that these responses represent an underlying cause, rather than a result, of pulmonary inflammation in sarcoidosis.
The observed differences in BAL cell TLR responses of sarcoidosis patients and control subjects were largely dose-dependent, and therefore subject to consideration of how the observed responses correlate with those induced by physiological exposures. For example, several previous studies that reported differences in LPS-induced cytokine production by sarcoidosis patients compared to control subjects utilized much higher LPS concentrations than those of our studies [31–32]. The nanogram doses we selected were based specifically on previous assessments of TLR-4 activation in macrophages [34–35]. Further, LPS concentrations within the epithelial lining fluid of patients with active sarcoidosis, while higher than those of controls, do not approach the microgram doses evaluated previously [37]. It is possible that additional differences in TLR-induced responses of sarcoidosis patients and control subjects could be observed for other ligands if more detailed dose–responses were assessed; however, performance of these studies was necessarily limited by BAL cell numbers obtainable from human subjects.
Recent studies indicate that the peptide structures of lipoprotein ligands of TLR-2 impact independently their capacity to induce TLR-2 activation [38], and therefore imply that differences in the peptide structures of Pam-3-Cys-based TLR-2 ligands might alter the magnitudes of resulting TLR-2 responses. These findings offer a possible explanation for our unexpected observation that BAL cells of sarcoidosis patients display increased cytokine responses to the LpqH-based lipopeptide Pam-3-Cys-SSNKSTTGSGETTTA, but not to the more standard TLR-2/1 ligand, Pam-3-Cys SKKKK. Reports by Drake and colleagues have demonstrated interferon (IFN)-γ responses to multiple mycobacterial proteins in specimens obtained from patients with sarcoidosis. This cytokine production was TLR-2-dependent, however, in the case of the two proteins [early secreted antigenic target-6 (ESAT-6) and mycobacterial catalase–peroxidase (mKatG)] for which the issue was assessed [6]. We cannot exclude the possibility that the differences in BAL cell responses of sarcoidosis patients to the 19-kDa M. tuberculosis lipoprotein and Pam-3-Cys SKKKK could reflect antigen-specific responses induced by prior mycobacterial exposure. However, this explanation appears less likely, given the reciprocal decrease in responsiveness to TLR-2/6 activation displayed by these patients, and the demonstration of the TLR-2-dependence of granulomatous lung disease in an animal model based on a completely distinct TLR-2-inducing pathogen, P. acnes.
The murine model of Th1-associated granulomatous lung inflammation was optimized in previous studies by McCaskill et al. [14], who determined that P. acnes induced robust granulomatous pulmonary inflammation in C57BL/6 mice but not in BALB/C animals. In parallel studies, Staphylococcus epidermidis induced markedly less pulmonary inflammation and only minimal granuloma formation, despite recent reports that S. epidermidis proteins also serve as TLR-2 ligands [39]. In this context, the previous murine studies support the concept that, even in hosts predisposed genetically to development of sarcoid-like pulmonary inflammation, not all TLR-2 agonists have equal capacity to induce this pathology. The current murine findings augment our human studies by indicating that development of Th1-mediated granulomatous lung disease can be largely TLR-2-dependent, despite its induction by a whole organism capable of activating multiple TLRs [40, 41].
A potential role of TLR-2 in the pathogenesis of sarcoidosis was reported previously by Chen et al. in studies implicating serum amyloid A (SAA) as a sarcoidosis-specific autoantigen. Intravenous SAA intensified the granulomatous response to (mKatG)-coated beads in a rat model of disease, and treatment with anti-TLR-2 prior to exposure to mKatG and SAA markedly reduced this response [28]. Our findings suggest that hyperresponsiveness to TLR-2/1 stimulation may itself represent an immunophenotype that confers susceptibility to the development of sarcoidosis. In this scenario, abnormally robust cytokine responses to TLR-2/1 stimulation may predispose to the development of lung pathology following exposure to organisms with particularly potent TLR-2/1 activity. This same immunophenotype could also promote the subsequent amplification of granulomatous inflammation in response to autologous TLR-2 ligands, including SAA.
In summary, our evaluation of functional responses of BAL cells from sarcoidosis patients to TLR stimulation demonstrated dysregulation of local TLR-2 heterodimer responses in pulmonary sarcoidosis. Parallel murine studies suggest that TLR-2 plays a central role in the development of Th1-mediated granulomatous lung disease in hosts predisposed to development of this pathology. The pathogenesis of sarcoidosis might therefore be clarified further by investigations of the mechanisms that underlie the abnormally robust cytokine responses induced by some TLR-2/1 ligands in individuals with sarcoidosis, as well as the capacities of certain bacterial lipoproteins and autoantigens to provide unusually potent stimulation of TLR-2.
Acknowledgments
The authors thank Dr Steven L. Tilley of the University of North Carolina for guiding our efforts to recreate the murine model of sarcoidosis, and Dr Rosana Eisenberg, currently of Vanderbilt University, for evaluating lung pathology in our preliminary studies using this model. They also thank Dr Michael Drage and Mr Scott Reba of CWRU for providing native 19-kD lipoprotein of M. tuberculosis and for technical assistance with studies of murine lung homogenates, respectively. In addition, they thank Drs W. Henry Boom and Mitchell Drumm of CWRU for helpful discussions. Funding was provided by an American Thoracic Society-Foundation for Sarcoidosis Research Partnership Grant and a US Department of Veterans’ Affairs Merit Review Award (to R.F.S.), and by NIH T32 training grant ‘Pulmonary Host Defense Mechanisms’ T32-HL07889 (to M.I.G.), NIH R01s HL075422 (to J.A.K.) and AI034343 and AI069085 (to C.V.H.). Research bronchoscopies were performed in the Clinical Research Units of the Clinical and Translational Science Collaborative at CWRU with the support of NIH UL1 RR024989.
Disclosure
The authors have no conflicts of interest to report relating to this manuscript.
References
- 1.Zissel G, Prasse A, Muller-Quernheim J. Immunologic response of sarcoidosis. Semin Respir Crit Care Med. 2010;321:390–403. doi: 10.1055/s-0030-1262208. [DOI] [PubMed] [Google Scholar]
- 2.Saboor SA, Johnson NM, McFadden J. Detection of mycobacterial DNA in sarcoidosis and tuberculosis with polymerase chain reaction. Lancet. 1992;339:1012–1015. doi: 10.1016/0140-6736(92)90535-b. [DOI] [PubMed] [Google Scholar]
- 3.Almenoff PL, Johnson A, Lesser M, Mattman LH. Growth of acid fast L forms from the blood of patients with sarcoidosis. Thorax. 1996;51:530–533. doi: 10.1136/thx.51.5.530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Drake WP, Pei Z, Pride DT, Collins RD, Cover TL, Blaser MJ. Molecular analysis of sarcoidosis tissues for mycobacterium species DNA. Emerg Infect Dis. 2002;11:1334–1341. doi: 10.3201/eid0811.020318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Song Z, Marzilli L, Greenlee BM, et al. Mycobacterial catalase–peroxidase is a tissue antigen and target of the adaptive immune response in systemic sarcoidosis. J Exp Med. 2005;201:755–767. doi: 10.1084/jem.20040429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Oswald-Richter KA, Culver DA, Hawkins C, et al. Cellular responses to mycobacterial antigens are present in bronchoalveolar lavage fluid used in the diagnosis of sarcoidosis. Infect Immun. 2009;77:3740–3748. doi: 10.1128/IAI.00142-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ebe Y, Ikushima S, Yamaguchi T, et al. Proliferative response of peripheral blood mononuclear cells and levels of antibody to recombinant protein from Propionibacterium acnes DNA expression library in Japanese patients with sarcoidosis. Sarcoidosis Vasc Diffuse Lung Dis. 2000;17:256–265. [PubMed] [Google Scholar]
- 8.Yamada T, Eishi Y, Ikeda S, et al. In situ localization of Propionibacterium acnes DNA in lymph nodes from sarcoidosis patients by signal amplification with catalysed reporter deposition. J Pathol. 2002;198:541–547. doi: 10.1002/path.1243. [DOI] [PubMed] [Google Scholar]
- 9.Bocart D, Lecossier D, De Lassence A, Valelyre D, Battesti J, Hance AJ. A search for mycobacterial DNA in granulomatous tissues from patients with sarcoidosis using the polymerase chain reaction. Am Rev Respir Dis. 1992;145:1142–1148. doi: 10.1164/ajrccm/145.5.1142. [DOI] [PubMed] [Google Scholar]
- 10.Brown ST, Brett I, Almenoff PL, Lesser M, Terrin M, Teirstein AS. Recovery of cell wall-deficient organisms from blood does not distinguish between patients with sarcoidosis and control subjects. Chest. 2003;123:413–417. doi: 10.1378/chest.123.2.413. [DOI] [PubMed] [Google Scholar]
- 11.Ishige I, Eishi Y, Takemura T, et al. Propionibacterium acnes is the most common bacterium commensal in peripheral lung tissue and mediastinal lymph nodes from subjects without sarcoidosis. Sarcoidosis Vasc Diffuse Lung Dis. 2005;22:33–42. [PubMed] [Google Scholar]
- 12.Basu S, Fenton M. Toll-like receptors: function and roles in lung disease. Am J Physiol Lung Cell Mol Physiol. 2004;286:L887–892. doi: 10.1152/ajplung.00323.2003. [DOI] [PubMed] [Google Scholar]
- 13.Ishioka S, Saito T, Hiyama K, et al. Increased expression of tumor necrosis factor-alpha, interleukin-6, platelet-derived growth factor B and granulocyte-macrophage colony stimulating factor mRNA in cells of bronchoalveolar lavage fluides from patients with sarcoidosis. Sarcoidosis Vasc Diffuse Lung Dis. 1996;13:139–145. [PubMed] [Google Scholar]
- 14.McCaskill JG, Chason KD, Hua X, et al. Pulmonary immune responses to Propionibacterium acnes in C57BL/6 and BALB/c mice. Am J Respir Cell Mol Biol. 2006;35:347–356. doi: 10.1165/rcmb.2005-0285OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Silver RF, Li Q, Zukowski L, et al. Recruitment of antigen-specific Th1-like responses to the human lung following segmental antigen challenge with purified protein derivative of M. tuberculosis. Am J Respir Cell Mol Biol. 2003;29:117–123. doi: 10.1165/rcmb.4840. [DOI] [PubMed] [Google Scholar]
- 16.Drage MG, Pecora ND, Hise AG, et al. TLR2 and its co-receptors determine responses of macrophages and dendritic cells to liproproteins of Mycobacterium tuberculosis. Cell Immunol. 2009;258:29–37. doi: 10.1016/j.cellimm.2009.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Takeuchi O, Hoshino K, Kawai T, et al. Differential roles of TLR2 and TLR4 in recognition of Gram-negative and Gram-positive bacterial cell wall components. Immunity. 1999;11:443–451. doi: 10.1016/s1074-7613(00)80119-3. [DOI] [PubMed] [Google Scholar]
- 18.Nethery DE, Moore BB, Minowada G, Carroll J, Faress JA, Kern JA. Expression of mutant human epidermal receptor 3 attenuates lung fibrosis and improves survival in mice. J Appl Physiol. 2005;99:298–307. doi: 10.1152/japplphysiol.01360.2004. [DOI] [PubMed] [Google Scholar]
- 19.Kutchey J, Fulton SA, Reba SM, Harding CV, Boom WH. Interferon-αβ mediates partial control of early pulmonary Mycobacterium bovis bacillus Calmette–Guérin infection. Immunology. 2006;118:39–49. doi: 10.1111/j.1365-2567.2006.02337.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Okusawa T, Fujita M, Nakamura J, et al. Relationship between structures and biological activities of mycoplasmal diacylated lipopeptides and their recognition by Toll-like receptors 2 and 6. Infect Immun. 2004;72:1657–1665. doi: 10.1128/IAI.72.3.1657-1665.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pabst S, Baumgarten G, Stremmel A, et al. Toll-like receptor (TLR) 4 polymorphisms are associated with a chronic course of sarcoidosis. Clin Exp Immunol. 2006;143:420–426. doi: 10.1111/j.1365-2249.2006.03008.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Veltkamp M, Grutters JC, van Moorsel CH, Ruven HJ, van den Bosch JM. Toll-like receptor (TLR) 4 polymorphism Asp299Gly is not associated with disease course in Dutch sarcoidosis patients. Clin Exp Immunol. 2006;145:215–218. doi: 10.1111/j.1365-2249.2006.03127.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sato M, Kawagoe T, Meguro A, et al. Toll-like receptor 2 (TLR2) gene polymorphisms are not associated with sarcoidosis in the Japanese population. Mol Vis. 2011;17:731–736. [PMC free article] [PubMed] [Google Scholar]
- 24.Veltkamp M, Wijnen PA, van Moorsel CH, et al. Linkage between Toll-like receptor (TLR) 2 promotor and intron polymorphisms: functional effects and relevance to sarcoidosis. Clin Exp Immunol. 2007;149:453–462. doi: 10.1111/j.1365-2249.2007.03428.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Veltkamp M, van Moorsel CHM, Rijkers GT, Ruven HJT, Grutters JC. Genetic variation in the Toll-like receptor gene cluster (TLR10–TLR1–TLR6) influences disease course in sarcoidosis. Tissue Antigens. 2011;79:25–32. doi: 10.1111/j.1399-0039.2011.01808.x. [DOI] [PubMed] [Google Scholar]
- 26.Wennerstrom A, Pietinalho A, Vaukonen H, et al. HLA-DRB1 allele frequencies and C4 copy number variation in Finnish sarcoidosis patients and association with disease prognosis. Hum Immunol. 2012;73:93–100. doi: 10.1016/j.humimm.2011.10.016. [DOI] [PubMed] [Google Scholar]
- 27.Howson JMM, Roy MS, Stevens H, Todd JA. HLA class II gene associations in African American Type 1 diabetes reveal a protective HLA-DRB1*03 haplotype. Diabet Med. 2013;6:710–716. doi: 10.1111/dme.12148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen ES, Song Z, Willett MH, et al. Serum amyloid A regulates granulomatous inflammation in sarcoidosis through Toll-like receptor-2. Am J Respir Crit Care Med. 2010;181:360–373. doi: 10.1164/rccm.200905-0696OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Szebeni B, Veres G, Dezsofi A, et al. Increased expression of Toll-like receptor (TLR)-2 and TLR-4 in the colonic mucosa of children with inflammatory bowel disease. Clin Exp Immunol. 2008;151:34–41. doi: 10.1111/j.1365-2249.2007.03531.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hopkins PA, Pridmore AC, Ellmerich S, et al. Increased surface Toll-like receptor 2 expression in superantigen shock. Crit Care Med. 2008;36:1267–1276. doi: 10.1097/CCM.0b013e31816a0a78. [DOI] [PubMed] [Google Scholar]
- 31.Homolka J, Muller-Quernheim J. Increased interleukin 6 production by bronchoalveolar lavage cells in patients with active sarcoidosis. Lung. 1993;171:173–183. doi: 10.1007/BF00183946. [DOI] [PubMed] [Google Scholar]
- 32.Pueringer RJ, Schwartz DA, Dayton CS, Gilbert SR, Hunninghake GW. The relationship between alveolar macrophage TNF, IL-1, and PGE2 release, alveolitis, and disease severity in sarcoidosis. Chest. 1993;103:832–838. doi: 10.1378/chest.103.3.832. [DOI] [PubMed] [Google Scholar]
- 33.Shigehara K, Shijubo N, Ohmichi M, et al. IL-12 and IL-18 are increased and stimulate IFN-gamma production in sarcoid lungs. J Immunol. 2001;166:642–649. doi: 10.4049/jimmunol.166.1.642. [DOI] [PubMed] [Google Scholar]
- 34.Turner JD, Langley RS, Johnston KL, Egerton G, Wanji S, Taylor MJ. Wolbachia endosymbiotic bacteria of Brugia malayi mediate macrophage tolerance to TLR-and CD40-specific stimuli in a MyD88/TLR2-dependent manner. J Immunol. 2006;177:1240–1249. doi: 10.4049/jimmunol.177.2.1240. [DOI] [PubMed] [Google Scholar]
- 35.Kawamoto T, Ii M, Kitazaki T, Iizawa Y, Kimura H. TAK-242 selectively suppresses Toll-like receptor 4-signaling mediated by the intracellular domain. Eur J Pharmacol. 2008;584:40–48. doi: 10.1016/j.ejphar.2008.01.026. [DOI] [PubMed] [Google Scholar]
- 36.Laplante P, Amireault P, Subang R, Dieudé M, Levine JS, Rauch J. Interaction of β2-glycoprotein I with lipopolysaccharide leads to Toll-like receptor 4 (TLR4)-dependent activation of macrophages. J Biol Chem. 2011;286:42494–42503. doi: 10.1074/jbc.M111.230383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kelly DM, Greene CM, Meachery G, et al. Endotoxin up-regulates interleukin-18: potential role for Gram-negative colonization in sarcoidosis. Am J Respir Crit Care Med. 2005;172:1299–1307. doi: 10.1164/rccm.200411-1594OC. [DOI] [PubMed] [Google Scholar]
- 38.Buwitt-Beckmann U, Heine H, Weismuller K-H, Jung G, Brock R, Ulmer AJ. Lipopeptide structure determines TLR2-dependent cell activation level. FEBS J. 2005;272:6354–6364. doi: 10.1111/j.1742-4658.2005.05029.x. [DOI] [PubMed] [Google Scholar]
- 39.Strunk T, Power Coombs MR, Currie AJ, et al. TLR2 mediates recognition of live Staphylococcus epidermidis and clearance of bacteremia. PLoS ONE. 2010;9:e10111. doi: 10.1371/journal.pone.0010111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kim J, Ochoa MT, Krutzik SR, et al. Activation of Toll-like receptor 2 in acne triggers inflammatory cytokine responses. J Immunol. 2002;169:1535–1541. doi: 10.4049/jimmunol.169.3.1535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kalis C, Gumenscheimer M, Freudenberg N, et al. Requirement for TLR9 in the immunomodulatory activity of Propionibacterium acnes. J Immunol. 2005;174:4295–4300. doi: 10.4049/jimmunol.174.7.4295. [DOI] [PubMed] [Google Scholar]