

Abstract
AChE enzymatic inhibition is a core focus of pharmacological intervention in Alzheimer's disease (AD). Yet, AChE has also been ascribed non-hydrolytic functions, which seem related to its appearance in various isoforms. Neuronal AChE presents as a tailed form (AChE-T) predominantly found on the neuronal synapse, and a facultatively expressed readthough form (AChE-R), which exerts short to medium-term protective effects. Notably, this latter form is also found in the periphery. While these non-hydrolytic functions of AChE are most controversially discussed, there is evidence for them being additional targets of AChE inhibitors. This review aims to provide clarification as to the role of these AChE splice variants and their interplay with other cholinergic parameters and their being targets of AChE inhibition: AChE-R is particularly involved in the mediation of (anti-)apoptotic events in cholinergic cells, involving adaptation of various cholinergic parameters and a time-dependent link to the expression of neuroprotective factors. The AChE-T C-terminus is central to AChE activity regulation, while isolated AChE-T C-terminal fragments mediate toxic effects via the α7 nicotinic acetylcholine receptor. There is direct evidence for roles of AChE-T and AChE-R in neurodegeneration and neuroprotection, with these roles involving AChE as a key modulator of the cholinergic system: in vivo data further encourages the use of AChE inhibitors in the treatment of neurodegenerative conditions such as AD since effects on both enzymatic activity and the enzyme's non-hydrolytic functions can be postulated. It also suggests that novel AChE inhibitors should enhance protective AChE-R, while avoiding the concomitant up-regulation of AChE-T.
Keywords: AChE, readthrough, alternative splicing, donepezil, Alzheimer's disease, non-hydrolytic, non-enzymatic, neurodegeneration, nicotinic ACh receptor, amyloid
AChE: from gene to structure and neuronal splice variants
AChE is responsible for the termination of cholinergic transmission, that is, the enzymatic breakdown of ACh (Radic et al., 1997). It is known as one of the fastest enzymes of our body (Nair et al., 1994), degrading, as a tetramer, about 25 000 molecules of ACh per second. The AChE gene is located on chromosome 7 and spans seven kilobases. It contains six exons, three of which (E2, E3 and E4) encode for the core peptide that is common to all enzyme variants and harbours the information for the enzyme's activity. The AChE pre-mRNA is susceptible to alternative splicing (Taylor and Radic, 1994), which leads to three post-transcriptional species that, however, all derive from the same gene (Marsh et al., 1984; Aziz-Aloya et al., 1993; Grisaru et al., 1999). The C-terminal domains of these different splice variants determine the post-translational processing and, thus, location (both within the organism and the single cell) as well as role of the enzyme (Massoulie et al., 1998; Massoulie, 2002; Camp et al., 2010; Hicks et al., 2011). Further AChE variants originate from different promoter use and, thus, N-terminal alterations (Meshorer and Soreq, 2006; Toiber et al., 2008;2009), but these forms are less extensively characterized.
Two splice variants are particularly relevant to neuronal tissue. Specifically, the synaptic form of AChE is the one predominantly expressed in the CNS and muscle tissue (Massoulie and Millard, 2009). It is formed by splicing of exons 4 to 6, yielding a E1-E2-E3-E4-E6 transcript. The translation of this mRNA leads to the C-terminal extension of the common core by a peptide containing a cysteine, which favours dimerization; in view of this ‘tailed’ extension, this variant is also denominated tailed AChE (AChE-T; Liang et al., 2009). This C-terminal extension leads to AChE-T post-translational modifications involving, in nervous tissue, the link between a proline-rich membrane anchor (PRiMA) and a WAT domain on the very AChE-T C-terminus (Inestrosa and Perelman, 1989; Xie et al., 2010; Chen et al., 2011). This membrane-tethered form of AChE presents the bulk of AChE activity in neuronal tissue.
The other AChE splice variant relevant to neuronal events is the readthrough form of AChE (AChE-R), which obtains its name from the continuous transcription through intron I4, which yields the E1-E2-E3-E4-I4-E5 transcript. The extension of its C-terminus over the common core is shorter and lacks cysteine (Li et al., 1991; Camp et al., 2010). Hence, AChE-R is destined to remain monomeric, with its consequential solubility and swift distribution in tissue being closely linked to its apparent, though highly debated role in stress-related conditions (Soreq and Seidman, 2001; Massoulie et al., 2008): as both in vitro and in vivo work shows, powerful inductors for AChE-R are stress, for example, forced swim stress, continuous use as well as toxic concentrations of AChE inhibitors or inflammation (Kaufer et al., 1998; Nijholt et al., 2004; Dori et al., 2007; Evron et al., 2007), with stress-inducible changes in AChE gene expression being mediated by histone deacetylase (Sailaja et al., 2012). Figure 1 offers a scheme of these different AChE splice variants and their C-terminal peptides.
Figure 1.
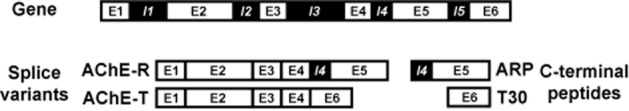
AChE splice variants and their C-terminal peptides. The figure shows the AChE gene (top; omitting the promoter region), AChE splice variant transcripts (bottom left) and the transcripts from which the translated C-terminal moieties that contain the AChE peptides are derived. E, exon; I, intron.
Among the splicing factors involved in the alteration of the AChE expression pattern are SC35 (Meshorer et al., 2005) and hnRNPA1 (Berson et al., 2012), with the latter having recently been demonstrated to assemble into self-seeding fibrils, revealing prion-like activities in an array of neurodegenerative conditions (Kim et al., 2013). This observation adds to deliberations on the striking mechanistic commonalities among neurodegenerative disorders, including, for example, alternative splicing and ubiquitination (Fotuhi et al., 2009; Ferraiuolo et al., 2011; Robberecht and Philips, 2013).
Given that cholinergic neurotransmitter levels drastically decrease in conditions like Alzheimer's disease (AD; Arendt et al., 1992; Mufson et al., 2008), AChE is usually discussed in relation to enzyme inhibition since this strategy can temporarily increase ACh levels. In fact, since the FDA approval of tacrine (1993) and donepezil (1997) as the first AChE inhibitors for the treatment of AD, the efficacy and suitability of these drugs have been considered in countless reviews (a PubMed research for ‘acetylcholinesterase inhibitors’ yields nearly 3000 hits in the ‘review’ category), and several more agents have reached the market since, among them rivastigmine and galantamine.
The discussion regarding cholinergic deficits as being centrally involved in the pathogenesis of neurodegenerative conditions such as AD (Francis et al., 1999; Mufson et al., 2008; Schliebs and Arendt, 2011) particularly focuses on the loss of cholinergic neurons and the ensuing decrease of neurotransmitter levels and the enzymatic machinery responsible for its synthesis, that is, ChAT and high-affinity choline uptake (HACU), taking also into account alterations of glutamaterigic signalling (Francis et al., 2012). In the light of the more recently discussed non-hydrolytic functions of AChE, this review specifically focusses on the role of AChE inhibitors in the context of AChE alternative splicing. Moreover, in discussing the role of AChE splice variant expression in conditions of acute and chronic neurodegeneration, the review broaches the issue of the impact of AChE inhibitors on AChE alternative splicing, deliberates on the potential limited usefulness of AChE inhibitors and hopes to instigate a discussion on concepts for future drug development.
Non-hydrolytic functions of AChE: first clues for the use of AChE inhibitors
The enzyme's complex structural polymorphism supports the theory that the different AChE molecular forms play distinct physiological roles not only in the cholinergic system: AChE has been detected in adult non-cholinergic neurons, for example, substantia nigra and cerebellar cells (Greenfield et al., 1983), as well as hematopoietic, osteogenic and even various neoplastic cells (Zakut et al., 1992; Karpel et al., 1994; Small et al., 1996; Grisaru et al., 2001; Soreq and Seidman, 2001; Deutsch et al., 2002). In addition, AChE is known for its widespread occurence during early development (Vogel-Hopker et al., 2012). Further roles may be inferred from the non-neuronal functions of ACh itself in sustaining barrier functions (Yoshida et al., 2006; Kurzen et al., 2007; Kummer et al., 2008). In addition, non-neuronal roles of ACh in conditions like inflammation (Fujii and Kawashima, 2001; Pavlov and Tracey, 2005; del Rey and Besedovsky, 2008) may give further clues as to non-enzymatic roles of AChE. This is particularly true since neuronal, stress-induced increase in interleukin-1 seems to mediate stress-related increased AChE expression (Li et al., 2000b).
In fact, various AChE inhibitors are successfully used to support the recovery in elderly, cognitively impaired stroke patients (Oldenbeuving et al., 2008; Whyte et al., 2008; Hong et al., 2012). For example, donepezil was tested in a phase II trial as an adjuvant therapy to standard medical care of stroke patients (Barrett et al., 2011) and supported cognitive improvement in a pilot study (Chang et al., 2011). In this context, the degree of recovery from stroke has been correlated to stroke-induced effects on the immune system that can lead to infections (Dirnagl, 2012). Such events involve a mechanism described as the inflammatory reflex (Tracey, 2002; Huston and Tracey, 2011). Since vagally released ACh is able to suppress the release of inflammatory cytokines, the positive effect of AChE inhibitor treatment in post-stroke patients may well extend to the effects of non-neuronal ACh.
In addition, non-hydrolytic functions of AChE are suggested by the spatio-temporally regulated expression of AChE in early embryogenesis, embryonic neurite extension and synaptogenesis (Fitzpatrick-McElligott and Stent, 1981; Layer, 1990): the mutually exclusive expression patterns of AChE and its enzymatically less specific sister molecule butyrylcholinesterase, BuChE, (and the pre-synaptogenetic role of AChE) have been described as early as 1983 (Layer, 1983). AChE is transiently expressed in the developing nervous system, in particular, during periods of neuronal proliferation, migration and axonal outgrowth (Layer and Sporns, 1987; Layer, 1990, 1991). Exogenous, purified AChE promotes and regulates axonal and neurite growth from chick nerve cells in culture. This function is not the result of the enzymatic activity per se since this function was not attenuated by treating the cell culture with various active site inhibitors, which depressed enzymatic AChE activity (Layer et al., 1993). In line with these findings, others showed that neurite outgrowth is retarded following AChE inhibitor treatment (Dupree and Bigbee, 1994) and that a secondary, peripheral anionic site (PAS) on the AChE molecule achieves this adhesive function (Small et al., 1995).
A breadth of studies carried out in various cellular systems continued to corroborate these findings throughout the 1990s (Koenigsberger et al., 1997; Grifman et al., 1998; Bigbee et al., 2000). Yet, observations for an AChE knockout mouse seemingly casted major doubt on these insights: transgenic mice not carrying any AChE allele were alive, even though they required a liquid diet in view of muscle weakness (Xie et al., 1999). This mouse model establishes central cholinergic pathways and uses BuChE to hydrolyse the enormous quantities of ACh present in the cerebral extracellular space (Li et al., 2000a; Mesulam et al., 2002; Hartmann et al., 2007). At the same time, however, this mouse model revealed severely disturbed retinal development and neuritogenesis (Bytyqi et al., 2004), thus, further suggesting a mandatory role of AChE in development. What is more, AChE shares – as reviewed previously (Soreq and Seidman, 2001) – high homology with the extracellular domain sequence of, firstly, gliotactin, a transmembrane protein transiently expressed on peripheral glia that is required for the formation of the peripheral blood-nerve barrier (Auld et al., 1995); secondly, neurotactin, which is known for its interneuronal interactions (de la Escalera et al., 1990); and, thirdly, neuroligin, a further non-catalytic transmembrane protein with its extracellular sequence being composed of a catalytically inactive esterase domain homologous to AChE. Such evidence further underlines a likely role of AChE in development. It even suggests – as put forward earlier (Layer, 1995) – that the function of AChE in neurodegeneration may equally be attributed to ‘non-classical’ functions of the enzyme. These very functions are likely affected by AChE inhibition, with this issue being explored in the following sections.
AChE and amyloid: enzymatic activity in a new light
Nibaldo Inestrosa's work contributed significantly to our understanding of AChE as being involved in amyloid fibre assembly (Inestrosa et al., 1996), which continues to be considered – and discussed as – one of the core hallmarks of the condition (Hardy et al., 1986; Hardy and Higgins, 1992; Hardy and Selkoe, 2002; Armstrong, 2011; de la Torre, 2011; Benilova et al., 2012; Reitz, 2012). In particular, amyloid beta (Aβ) aggregation is promoted by AChE forming a complex with growing fibrils (Alvarez et al., 1997); furthermore, these stable complexes change the biochemical properties of the enzyme and increase the neurotoxicity of the Aβ fibrils (Alvarez et al., 1998); thirdly, AChE promotes peptide aggregation by a mechanism not involving the hydrolysis of the amyloid precursor protein (APP; Campos et al., 1998); and finally, the neurotoxicity of AChE Aβ peptide aggregates is dependent on the type of Aβ and the AChE concentration present in the complexes (Munoz and Inestrosa, 1999). These authors further showed, most notably and in reference to the above discussion on the role of the enzyme's PAS, that propidium, an AChE PAS ligand, could (in contrast to edrophonium, an active site inhibitor) prevent amyloid fibre assembly.
As such, one should assume that specific AChE inhibitors may prevent further aggregation of Aβ fibrils. In fact, researchers encourage future work in the area of AChE inhibitor drug synthesis to move towards inhibitors that reduce the deposition of amyloid (Pohanka, 2012). Such suggestions receive further support from the fact that a co-localization of AChE and Aβ was also reported in autopsy studies of AD patients' brains: these findings suggest that AChE activity is intimately associated with the process of amyloid formation and accumulation in senile plaques in vivo (Ulrich et al., 1990; Moran et al., 1994). The source of esterase activity of senile plaques has been ascribed to glia cells by Mesulam and his co-workers (Wright et al., 1993). Yet, they also demonstrated that BuChE was as much involved in these processes as AChE itself: firstly, AChE and BuChE activities with pH preferences and inhibitor selectivities identical to those of plaque-bound cholinesterases are found in the astrocytes and oligodendrocytes of control and AD brains; secondly, these glial-type cholinesterases are selectively inhibited by indolamines and protease inhibitors; and, thirdly, in control and AD brains, AChE-positive glia are distributed throughout the cortical layers and subcortical white matter. What is more, Sultan Darvesh and colleagues strongly emphasize the expression of BuChE in brain structures involved in cognition (Darvesh et al., 2001), and claim a central role of BuChE, alongside AChE, in the occurrence, symptoms and progression of dementia (Ballard et al., 2005), also in the light of the association of BuChE with Aβ plaques (Darvesh, 2013). These findings are not easily reconciled with Inestrosa's insights that BuChE, which lacks the PAS, does not affect amyloid formation (Inestrosa et al., 1996). Indeed, they support work advocating the use of BuChE inhibitors in the treatment of AD (Giacobini, 2001; Giacobini, 2003), especially since these compounds seem to impact on the enzyme's splice variant expression and consequential attenuation of amyloidogenesis (Greig et al., 2005; Podoly et al., 2009). Effects of AChE inhibitors on gene expression are discussed in detail further below.
Nevertheless, these discrepancies may well be owed to the fact that a good part of the above work was carried out in isolated test tube conditions, which can hardly account for the individual patient's complex physiology or cerebral pathomorphology. As recently reviewed by Inestrosa himself, BuChE is able to extend the nucleation phase of Aβ polymerization, reducing the rate of fibril formation, instead stabilizing soluble Aβ assemblies (Inestrosa et al., 2008). Whether BuChE affects Aβ oligomerization remains unclear. Still, the hypothesis persists that senile plaques may be formed from the terminals of AChE containing neurons. As such, AChE PAS inhibitors may be preferable to purely competitive enzyme inhibitors. Novel AChE inhibitors are targeted against this site (Silman and Sussman, 2005), and their use is, additionally, supported by evidence suggesting that they are involved in modulating the APP metabolism (Racchi et al., 2004; Zimmermann et al., 2005a; 2005b,; Garcia-Palomero et al., 2008).
AChE-T, its C-terminal peptides and neurodegeneration: AChE inhibitors with new functions
With respect to the non-hydrolytic roles of AChE in the context of AD-related amyloid burden, further hypotheses have been put forward for the role of AChE. In view of the non-hydrolytic actions of AChE in development, its known involvement in AD pathology and its functional parallels to APP – both are secreted from neurons, have trophic action and their levels decrease in AD (Greenfield and Vaux, 2002) – molecular similarities between the two proteins were investigated. At the same time, a role for two AChE C-terminal peptides (14 and 30 amino acids (aa) in length and, thus, denominated T14 and T30, respectively) in neurodegeneration were hypothesized (Greenfield et al., 2008). AChE-T as well as T14/T30 exhibit, depending on dose and exposure time, trophic or toxic actions. These were seen in hippocampal neurons as well as organotypic slices, and could be related to opening, specifically and selectively, the L-type Ca2+ channel (Day and Greenfield, 2002; 2003,; Bon and Greenfield, 2003; Emmett and Greenfield, 2004; Greenfield et al., 2004; Zbarsky et al., 2004).
In order to further corroborate their hypothesis, Greenfield and colleagues explored the possibility of there existing a form (and role) of AChE-T, in development and degeneration, having undergone cleavage as suggested by the metabolism of the homologous APP sequence. Such an approach was encouraged by the identification of a truncated, 543 to 547 aa long form of AChE-T in development (Saxena et al., 2003). This form is supposedly the result of proteolytic cleavage (Camp et al., 2010) – a likely assumption, given that a similar mechanism has been detected for BuChE (Blong et al., 1997). Cleaving the T30 peptide – which by itself is, as referenced above, implicated in both developmental and neurodegenerative processes in vitro – off the enzyme's C-terminus would yield a truncated enzyme (AChE-Tt) devoid of a dimerization favouring cysteine in its remaining C-terminus. Hence, this AChE-Tt would remain monomeric. Such an AChE-T C-terminal shedding seemed then further likely, when considering that the form of AChE relevant in neurodegeneration and development has been identified as being monomeric (Arendt et al., 1992; Inestrosa et al., 1994), and that it has lost its characteristic of substrate inhibition (Arendt et al., 1992; Moreno et al., 1996). In this context, work on a genetically engineered form of truncated AChE-T (Bourne et al., 1999) aimed at uncovering a mechanistic link between the supposedly cleaved, residual AChE-Tt enzyme and the free floating peptides in neurodegeneration (Zimmermann et al., 2008; 2009,).
While criticism regarding this work continues to focus on the still lacking in vivo evidence of the AChE-T C-terminal peptide(s) (Massoulie et al., 2008), progress has been made regarding evidence for their potential site of action: experiments using a range of nicotinic ACh receptor (nAChR) blockers in various model systems provided indirect proof that T14 binds selectively to an allosteric site on the α7-nAChR [abbreviations for receptors follow the British Journal of Phamacology's Guide to Receptors and Channels (Alexander et al., 2011)], thus, modulating Ca2+ influx, which is involved in short-term plasticity and chronic, long-term trophic and toxic effects (Day and Greenfield, 2002; 2003,; Emmett and Greenfield, 2004; Greenfield et al., 2004). These actions were sensitive to α7-nAChR blockade in the nanomolar range. Notably, the α7-nAChR is co-expressed along with AChE in the same transient period and within the same brain regions during developmental (Taylor et al., 1994; Broide et al., 1996; Torrao et al., 2000) and degenerative processes (Dineley et al., 2001; Fodero et al., 2004). Moreover, it can bind Aβ (Wang et al., 2000; Dineley et al., 2002; Lain et al., 2005; Dineley, 2007), with this finding, however, having been challenged in cell culture work (Small et al., 2007). All the same, the receptor has been shown to be involved in neuronal development (Liu et al., 2007) as well as neurodegenerative pathogenesis (Dineley et al., 2001; Nagele et al., 2002; Duda, 2004) and oncogenesis (Schuller, 2009). As regards degenerative processes, the role of both AChE – specifically AChE-T – and the α7-nAChR have been widely discussed (Sternfeld et al., 2000; O'Neill et al., 2002; Jin et al., 2004; Rees et al., 2005; Holzgrabe et al., 2007; Schliebs and Arendt, 2011). Recent work now conclusively reveals that the two AChE C-terminal peptides T14 and, particularly, T30 are involved in binding interactions with the α7-nAChR, modulating its mRNA as well as protein expression (Bond et al., 2009).
Always considering that the above results have been obtained in different in vitro, even test tube model systems, they may, taken together, offer a possible scenario for mechanisms active under developmental/apoptotic conditions (Figure 2): AChE-T is cleaved through proteolysis, as has been suggested previously (Saxena et al., 2003), and the C-terminal peptide, T30, is freed ( ). The resulting, AChE-Tt remains enzymatically active (
). The resulting, AChE-Tt remains enzymatically active (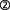 , Zimmermann et al., 2008). T30, in turn, manipulates α7-nAChR expression (
, Zimmermann et al., 2008). T30, in turn, manipulates α7-nAChR expression (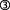 , Bond et al., 2009), and trophic-toxic actions are triggered by means of Ca2+ influx (
, Bond et al., 2009), and trophic-toxic actions are triggered by means of Ca2+ influx ( ), depending on the T30 dose (Greenfield et al., 2004). AChE-Tt activity is further enhanced by T30 (
), depending on the T30 dose (Greenfield et al., 2004). AChE-Tt activity is further enhanced by T30 (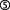 ; Zimmermann et al., 2008), on which breakdown of α7-nAChR-stimulating ACh is contingent (
; Zimmermann et al., 2008), on which breakdown of α7-nAChR-stimulating ACh is contingent (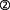 ). Such a possible scenario would be in agreement with the selective sensitivity of cholinergic neurons to a dysregulation of Ca2+ levels (Toiber and Soreq, 2005; Zundorf and Reiser, 2011). Additionally, it would fit to the mandatory role of the α7-nAChR in learning and memory (Thomsen et al., 2010), with neuroprotective effects being mediated via the downstream PI3K/pAkt pathway (Akaike et al., 2010).
). Such a possible scenario would be in agreement with the selective sensitivity of cholinergic neurons to a dysregulation of Ca2+ levels (Toiber and Soreq, 2005; Zundorf and Reiser, 2011). Additionally, it would fit to the mandatory role of the α7-nAChR in learning and memory (Thomsen et al., 2010), with neuroprotective effects being mediated via the downstream PI3K/pAkt pathway (Akaike et al., 2010).
Figure 2.
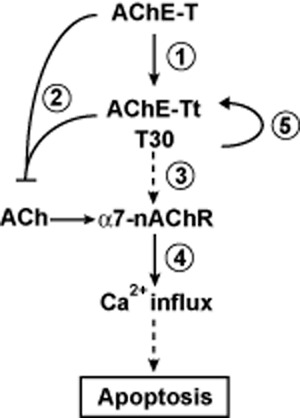
The interrelation between AChE-T and the α7-nAChR, mediated by T30. For numbers, see text;  , induction of or leading to;
, induction of or leading to;  , blocking or inhibiting;
, blocking or inhibiting; 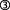 , either inducing or inhibiting, depending on concentration.
, either inducing or inhibiting, depending on concentration.
In view of the seminal role of AChE-T and α7-nAChR in degenerative processes, the in vivo identification of T30 and, to a lesser extent, T14 is eagerly awaited. Still, AChE-T itself has been described to exert toxic effects. For example, specific AChE-T overexpression is linked to programmed cell death (Greenberg et al., 2010), and mice transgenic for human AChE-T carry progressive accumulation of clustered, heat shock protein 70 immunopositive neuronal fragments as well as a high incidence of reactive astrocytes (Sternfeld et al., 2000), and show accelerated stress-related neuropathology including loss of dendritic arborization and spines (Cohen et al., 2002; Meshorer and Soreq, 2006). Likewise, N-extended forms of AChE-T lead to apoptosis (Toiber et al., 2008; 2009,), possibly as it is involved in apoptosome formation (Park et al., 2004). While these findings already suggest that AChE inhibitor treatment should avoid leading to an increase in the expression of AChE-T, I will address this question further below, in conjunction with deliberations on changes in AChE-R expression, with these being explored in the following paragraph.
AChE-R, stress-related conditions and neuroprotection: AChE and cholinergic plasticity
In comparison to AChE-T, AChE-R was shown to exert short-to medium-term beneficial effects under acute stress (Kaufer et al., 1998): mice overexpressing AChE-R display normal neuromuscular function and their brains are relatively protected from stress associated pathological hallmarks that would otherwise cause age-dependent neurodeterioration (Berson et al., 2008). Similarly, the C-terminal peptide pertaining to AChE-R, denominated C-terminal fragment of AChE-R (ARP), has been shown to promote neuronal development and plasticity (Dori and Soreq, 2006a). In addition, extensive experimental work could demonstrate a role for AChE-R in peripheral tissue as a marker of stress-related conditions. For example, modified testicular expression of AChE-R predicts male infertility (Mor et al., 2001), while sera of mothers, who just had given birth, reveal increased AChE-R levels post partum (Pick et al., 2004). Moreover, AChE-R is expressed in hematopoietic progenitors, and ARP promotes hematopoietic progenitor proliferation (Grisaru et al., 2001), while stress-induced cholinergic signalling promotes inflammation-associated thrombopoiesis (Pick et al., 2006).
However, Jean Massoulié and colleagues could not identify this form of AChE following heat shock, organophosphate AChE inhibition or stress – neither in cell culture nor in vivo (Perrier et al., 2005); in addition, immobilization stress of rodents did not lead to detectable levels of AChE-R in their hands (Perrier et al., 2006). However, their in vivo work focused on 24 and 48 h, then 7 and 14 days post-stress, while their in vitro test was ended immediately after stress. In comparison, Soreq and co-workers reported peaks of AChE activity changes 1 and 2 h post-stress in hippocampal and cortical tissue respectively (Kaufer et al., 1998). Their findings are supported by recent work exploring the role of alternative splicing of AChE under conditions of oxidative stress (Härtl et al., 2011) and other apoptotic stimuli, namely, staurosporine (Li et al., 2012a) and Aβ (Li et al., 2012b). These studies were carried out in a model of cholinergic cells (Pahlman et al., 1984; 1995,), which enabled an analysis of conditions that closely matched those in the cell type particularly lost in neurodegeneration of the Alzheimer's type. The most important findings of this cell culture-focused work were that, firstly, AChE-R levels rise significantly following exposure to mild to moderate oxidative stress (Härtl et al., 2011) or pathophysiological amounts of amyloid (Li et al., 2012b), and that, secondly, AChE-R is released into the cell media in large amounts (Härtl et al., 2011), which is in agreement with its solubility as a monomer.
In these models, controlled apoptotic events are observed, which seem connected to the expression of AChE-R and a time-dependently linked, sharp increase in Bcl-2 (Härtl et al., 2011; Li et al., 2012b). Moreover, AChE-R has been observed in the context of mitochondrial hyperactivity (Mor et al., 2008) and described as interacting with the receptor for activated C-kinase 1, RACK1 and PKC (Dori and Soreq, 2006a). Increased mitochondrial activity provides additional ACh precursor acetyl-coenzyme A. As a consequence, ACh levels rise, as also suggested by the increased choline turnover itself (Kuhar and Murrin, 1978; Jope, 1999) as well as the observed increase in ChAT activity (Li et al., 2012b). Likewise, the known interaction between RACK1 and PKC, promoted by AChE-R, may well drive choline transporter as well as ChAT activity, as they are dependent on their phosphorylation status (Dobransky et al., 2004; Gates et al., 2004; Dobransky and Rylett, 2005; Kim et al., 2006). ACh, in turn, likely exerts protective effects via muscarinic ACh receptors (mAChRs) and PKC activation (Fisher, 2007; Tiong et al., 2010).
Notably, high concentrations of Aβ destroy the balance of the cholinergic players up-regulated following the exposure to low Aβ concentrations, with AChE-R being up-regulated only transiently (Li et al., 2012b). As such, slowly increasing Aβ levels, as is the case in AD, may cause cholinergic adaptation, likely associated with AChE-R expression. Long-term accumulation of Aβ peptides, in contrast, will overwhelm the adaptive capacity of cholinergic neurons, leading to their death. This hypothesis is in agreement with the analysis of human hippocampal tissue samples that revealed a drastic decrease in functional AChE-R (Berson et al., 2008), suggesting that a high dose of Aβ, applied over an extended period of time, impairs this neuroprotective AChE-R. This biphasic phenomenon is further confirmed by work using the phosphorylation inhibitor staurosporine (Li et al., 2012a), which shows that reduced AChE activity may result only under conditions when regulatory effects involving cholinergic adaptation and AChE-R up-regulation, are exhausted, that is, once the system has tipped towards cellular necrosis.
These findings suggest that the selective increase of AChE-R levels may well be desirable in neurodegenerative conditions (Meshorer and Soreq, 2006), especially since chronic AChE-R overexpression has been shown to enhance cognitive performance in vitro (Sklan et al., 2006) and in vivo (Farchi et al., 2007). Interestingly, treatment with AChE inhibitors presented with an eightfold increase over control levels in AChE mRNA and a concomitant decrease in the mRNAs encoding for ChAT and the vesicular ACh transporter (Kaufer et al., 1999). Stress-related changes in brain microRNA expression, and microRNA-183 in particular, were shown to regulate AChE splicing and cholinergic neurotransmission (Meerson et al., 2010). Therefore, future work should address as to whether and how AChE inhibitors do impact on brain microRNA expression, and whether likely effects differ among the various drugs.
There is evidence of a shift towards AChE-R splicing following muscarinic modulation (Salmon et al., 2005), and recent studies could detect long-lasting AChE splice variant variations in AD patients treated with AChE inhibitors (Darreh-Shori et al., 2004). In particular, donepezil has been associated with decreased levels of AChE-R as compared with AChE-T, whereas rivastigmine, a pseudo-irreversible cholinesterase inhibitor, increases the AChE-R/AChE-T ratio. Even though the clinical effects of these observations still need to be established fully, several authors suggest – in the light of the beneficial effects of AChE-R outlined above – that the synthesis and design of new drugs should aim for the specific up-regulation of AChE-R, but not AChE-T (Greenberg et al., 2010; Pohanka, 2012). In addition, circadian changes in the expression of AChE-T (Erb et al., 2001) and AChE-R (Shaltiel et al., 2013) should be taken into account for therapeutic considerations.
More generally, AChE inhibitors also find application in other types of dementia. For example, rivastigmine showed modest but significant benefits in the treatment of cognitive and neuropsychiatric symptoms in Parkinson's disease dementia (PDD) and dementia with Lewy bodies (DLB; Bullock and Cameron, 2002), and also donepezil showed positive effects (Mori et al., 2012). Indeed, it is noteworthy that AChE alternative splicing has been implicated in PD and Parkinsonism (Benmoyal-Segal et al., 2012), with AChE-R confering resistance to dopaminergic cell death in a 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine mouse model (Ben Shaul et al., 2006). Likewise, the efficacy of targeting specific AChE splice variants as suitable for the therapy of chronic cholinergic malfunctioning has been highlighted for the case of myasthenia gravis: Monarsen, a 20-base antisense oligodeoxynucleotide directed against the human AChE gene is effective in improving muscle action potential in a myasthenia gravis rodent model and showed, in part, dramatic effects in patients afflicted by myasthenia gravis (Dori and Soreq, 2006b; Sussman et al., 2008), where AChE-R serum accumulation is noted (Brenner et al., 2003).
Animal models of modulated AChE activity: evidence for non-enzymatic AChE inhibitors?
Cholinergic activity increases following stress – in various cellular systems (Melo et al., 2003; Tyagi et al., 2010), in vivo (Dumont et al., 2006; Saxena et al., 2008) and in clinical studies (Correa et al., 2008) alike. At the same time, ACh itself has been shown to exert neuroprotective effects via mAChRs (Liu et al., 2011) and consequential PKC activation (Fisher, 2007; Tiong et al., 2010). In this paragraph, I would like to turn to questions as to the enzymatic impact of AChE inhibitors. In particular, I want to address whether the limited success obtained with pro-cholinergic therapeutics in conditions such as AD may be linked to their not sufficiently explored non-enzymatic effects.
In view of the drastic loss of cholinergic neurons and the consequential depression of ACh levels in conditions of neurodegeneration (Schliebs and Arendt, 2011), the therapeutic approach of choice remains the inhibition of AChE activity so as to keep neurotransmitter levels high for as long as possible (Mufson et al., 2008). Yet, both in vivo and clinical studies reveal that AChE inhibitor achieved enzyme inhibition rates are far from complete and probably remain below 50% (Giovannini et al., 1998). At the same time, advanced stages of AD present with significantly reduced levels of AChE (Perry et al., 1978; Shinotoh et al., 2000). As such, it is of note that ACh neurotransmitter levels in a genetically engineered mouse model displaying single AChE allele deletion (Xie et al., 1999) are scarcely altered when comparing with levels measured in the wild-type counterpart (Mohr et al., 2012). This data sheds further doubt on an enzyme-related effect of clinically used AChE inhibitor concentrations, especially since it was recently demonstrated that 5 to 10 mg·day−1 donepezil administered to AD patients led to only 20–25% of AChE inhibition (Bohnen et al., 2005). These findings may well explain the low efficacy of these drugs, even though individual patients report positive effects (Zimmermann, 2011). Similarly, in vivo studies detected no more than 27 to 39% reduction of enzyme activity in rats after treatment with donepezil (1 or 1.5 mg·kg−1) (Scali et al., 2002; Cerbai et al., 2007).
Notably, these models cope with reduced enzymatic function by means of cholinergic compensation. In particular, HACU and, thus, choline turnover are significantly increased (Mohr et al., 2012), suggesting an increased ACh firing rate (Jope, 1999). This finding, notably, parallels data from AD brain rapid autopsy (Bissette et al., 1996) and AChE nullizygous mice (Hartmann et al., 2007). It is then noteworthy that AChE inhibitors are more effective in enhancing ACh levels in these heterozygous mice, on a background of already low basal AChE activity (Mohr et al., 2012), which encourages the use of these drugs for the treatment of advanced cholinergic dysfunction of the AD type (Passmore et al., 2005; Roman et al., 2005). What is more, there is evidence that donepezil alleviates disturbances in energy metabolism (Zhou et al., 2001), which is significantly affected in AD (Hirono et al., 2004). In confirmation of such evidence, donepezil increased glucose levels in the AChE heterozygous mice more strongly, and also transiently pushed up choline levels (Mohr et al., 2012), hence, up-regulating the availability of both precursors of ACh, an effect that may contribute to the increase of ACh levels.
Therefore, it is not far fetched to assume that donepezil can influence cholinergic transmission independently of AChE catalytic inhibition. Yet, as an impact on AChE gene expression has to be taken into account as well as aspects of enzyme maturation and turnover, that is, protein levels as compared with actual enzyme activity (Rotundo and Fambrough, 1980; Shaked et al., 2009), enzymatic and non-enzymatic effects cannot easily be distinguished. All the same, such non-catalytic effects may well be mediated by an AChE-R increase, as suggested by the discussed in vitro work (Li et al., 2012b). In this context, it should be emphasized that a significant relative increase in AChE-R will have little impact on ACh breakdown in absolute terms, since AChE-R baseline levels are close to zero. As such, the in vivo study on AChE heterozygous mice suggests that treatment of AD-related cognitive impairment involves AChE inhibition in early to moderate stages, likely involving non-cholinergic in addition to directly enzyme activity-related effects.
Consequentially, AChE activity per se might not be the only target of the AChE inhibitor-related treatment of cholinergic degenerative processes in AD, nor an exclusive indicator of cholinergic degeneration and cell death. Yet, how can we reconcile these findings with the evidence of the role of AChE-T C-terminal fragments in triggering neurodegenerative processes? Again, genetically engineered mouse models might give clues. AChE-T is the predominant form of AChE in the CNS, and it is tethered to the neuronal plasma membrane by the small transmembrane protein PRiMA (Perrier et al., 2002; 2003,), which interacts with the WAT domain of the AChE-T C-terminus. Earlier work in a PRiMA knockout mouse had shown that the membrane anchor is necessary for targeting and stabilizing nascent AChE in neurons (Dobbertin et al., 2009). But at the same time, these mice had a phenotype similar to their wild-type counterpart in terms of weight, body temperature and ventilation (Boudinot et al., 2009), thus, further challenging the role of AChE activity in neurodegenerative processes.
Recent work on this mutant reveals a thorough adaptation of the PRiMA knockout mouse to the genetically induced excess of cholinergic neurotransmitter (Farar et al., 2012). These high levels significantly surpass the EC50 of mAChRs, but not that of most nAChRs, which may explain the cholinergic adaptation of mAChRs – both in terms of their density and functionality – as was also observed in the case of AChE knockout mice (Li et al., 2003; Volpicelli-Daley et al., 2003a,b2003b). In comparison, a corresponding adaptation of nAChRs is scarcely seen (Volpicelli-Daley et al., 2003a). As such, the question arises as to whether pharmacological intervention targeting the disposition of AChE at the neuronal surface could support cholinergic neurotransmission under circumstances where AChE activity needs to be hampered.
This question becomes even more focussed when studying a genetically engineered mouse model presenting deletion of the AChE-T exon 6, which carries the WAT domain: it similarly lacks functional AChE in the synaptic membrane (Dobbertin et al., 2009; Camp et al., 2010), but reveals a significant phenotype (Camp et al., 2010), even though the originally intended effect of preventing AChE from anchoring to the synaptic membrane by means of interaction with PRiMA corresponds to that achieved in the PRiMA knockout mouse. These findings further suggest the mandatory role of the AChE-T C-terminus in supporting the enzyme's function and the organism's physiological development. That these mice show a significantly reduced number of nAChRs (Girard et al., 2006) only further highlights the intricate relationship between AChE-T and the nAChR.
Summary, conclusion and outlook: AChE in neurodegeneration, AChE inhibitors in neuroprotection
Various hypotheses have been put forward and controversially discussed in relation to the pathogenesis of AD (Hardy et al., 1986; Hardy and Higgins, 1992; Francis et al., 1999; Hardy and Selkoe, 2002; Armstrong, 2011; Reitz, 2012; Takata and Kitamura, 2012). At the same time, recent deliberations suggest that atrophy in the cortex and hippocampus, which continue to be considered the best determinant of cognitive decline with aging (deToledo-Morrell et al., 2007; Pihlajamaki et al., 2009; Schliebs and Arendt, 2011; Li et al., 2012c), results from a combination of AD pathology, inflammation, Lewy bodies and vascular lesions (Fotuhi et al., 2009). Likewise, altered alternative splicing seems intricately involved in pathogenetic mechanisms of neurodegenerative conditions (Ferraiuolo et al., 2011; Gagliardi et al., 2012; Mills and Janitz, 2012). Against this background – and in the light of the insights collected here, there seems to be an urgent need to understand, firstly, whether and how strongly cholinergic cell death features in a broad range of neurodegenerative conditions, not least since cholinergic impairment is most convincingly related to cognitive decline (Pinto et al., 2011), and, secondly, whether and to what extent we can see a converging cholinergic picture in neurodegeneration. Conditions have to be determined, under which AChE variants trigger apoptotic processes or exert protective functions, with the pharmacologist's final aim being to further the activation of the protective variant.
Figure 3 summarises the findings discussed here and suggests both enzymatic and non-enzymatic roles for AChE inhibition: while stress-induced AChE activity ( ) depresses ACh levels (
) depresses ACh levels ( ), neurotransmitter stores are replenished by stress-mediated induction of HACU and ChAT (
), neurotransmitter stores are replenished by stress-mediated induction of HACU and ChAT ( ), as well as via AChE-R (
), as well as via AChE-R ( and
and  ) and, linked, mitochondrial hyperactivity. Neuroprotective ACh effects are mediated by both the α7-nAChR (
) and, linked, mitochondrial hyperactivity. Neuroprotective ACh effects are mediated by both the α7-nAChR ( ) and mAChRs (
) and mAChRs ( ). In the event of prolonged or enhanced stress, AChE-R-relayed synthesis of ACh via PKC and RACK1 (
). In the event of prolonged or enhanced stress, AChE-R-relayed synthesis of ACh via PKC and RACK1 ( ) is exhausted (its half-life is significantly shorter than that of AChE-T) so that protective ACh signalling (
) is exhausted (its half-life is significantly shorter than that of AChE-T) so that protective ACh signalling ( and
and  ) is not achieved any more. Apoptosis (or, in the case of high concentrations of toxic agents or prolonged exposure times, necrosis) sets in (
) is not achieved any more. Apoptosis (or, in the case of high concentrations of toxic agents or prolonged exposure times, necrosis) sets in ( ). Regardless of the ongoing debate about the actual in vivo existence of AChE-T C-terminal peptides as well as a form of AChE-Tt, the scenario set out could be complemented by data on AChE-T C-terminal fragments (see also Figure 1): a stress-related rise in AChE-T leads, following proteolysis, to increased levels of T30. The peptide induces and interacts with the α7-nAChR, which, in turn, mediates protective effects. In the event of excessively rising AChE-T levels and, hence, activity, blocking of the α7-nAChR is brought about.
). Regardless of the ongoing debate about the actual in vivo existence of AChE-T C-terminal peptides as well as a form of AChE-Tt, the scenario set out could be complemented by data on AChE-T C-terminal fragments (see also Figure 1): a stress-related rise in AChE-T leads, following proteolysis, to increased levels of T30. The peptide induces and interacts with the α7-nAChR, which, in turn, mediates protective effects. In the event of excessively rising AChE-T levels and, hence, activity, blocking of the α7-nAChR is brought about.
Figure 3.
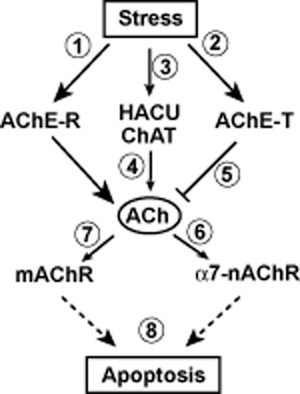
A possible scenario for how cholinergic dynamics involving AChE-R and AChE-T might stave off cell death. For numbers and the role of AChE inhibition, see text;  , induction of or leading to;
, induction of or leading to;  , blocking or inhibiting;
, blocking or inhibiting;  , either inducing or inhibiting, depending on concentration. The figure omits that a stress-related increase in AChE-T leads, following proteolysis, to increased levels of T30, which, in turn, impacts on the α7-nAChR as well.
, either inducing or inhibiting, depending on concentration. The figure omits that a stress-related increase in AChE-T leads, following proteolysis, to increased levels of T30, which, in turn, impacts on the α7-nAChR as well.
This schematic presentation is an attempt to put the individual findings collected in this review article into a coherent picture of cholinergic mechanisms. Yet, it does not want to suggest a representation of a general action network of AChE-R and AChE-T in all stress-related conditions. Nevertheless, several targets of AChE inhibitors should be considered in the light of the insights assembled here: AChE inhibition will reduce, in the early stages of the cellular demise, AChE activity, consequentially leading to ACh levels remaining high. Likewise, inhibition of AChE activity (AChE-T and AChE-Tt) reduces choline levels, which are also discussed as being implicated in α7-nAChR activation (Alkondon et al., 1997). Further effects may derive from altered AChE-R gene expression following the exposure to AChE inhibitors.
In view of the non-enzymatic mechanisms put forward for the mode of action of AChE inhibitors, it is necessary to understand whether and how they impact on the cholinergic and AChE variant scenario active in conditions such as PDD and DLB. This understanding will support the development of directly related therapeutic strategies that take into account AChE as a key modulator of the cholinergic system, both on an enzymatic and a non-hydrolytic level.
Acknowledgments
The author is grateful to J. Klein for insightful discussion; her research is supported by the Fonds der Chemischen Industrie as well as the LOEWE initiative ‘Non-neuronal cholinergic systems’.
Glossary
- aa
amino acid
- Aβ
amyloid β
- AChE-R
readthrough AChE
- AChE-T
tailed AChE
- AChE-Tt
hydrolytically cleaved AChE-T
- AD
Alzheimer's disease
- APP
amyloid precursor protein
- ARP
C-terminal fragment of AChE-R
- DLB
dementia with Lewy bodies
- HACU
high-affinity choline uptake
- mAChR
muscarinic ACh receptor
- nAChR
nicotinic ACh receptor
- PAS
peripheral anionic site
- PDD
Parkinson's disease dementia
- PRiMA
proline-rich membrane anchor
- T14 and T30
C-terminal fragments of AChE-T of 14 and 30 aa length respectively
Conflict of interest
The author has no conflict of interest to declare.
References
- Akaike A, Takada-Takatori Y, Kume T, Izumi Y. Mechanisms of neuroprotective effects of nicotine and acetylcholinesterase inhibitors: role of alpha4 and alpha7 receptors in neuroprotection. J Mol Neurosci. 2010;40:211–216. doi: 10.1007/s12031-009-9236-1. [DOI] [PubMed] [Google Scholar]
- Alexander SP, Mathie A, Peters JA. Guide to receptors and channels (GRAC), 5th edn. Br J Pharmacol. 2011;164(Suppl. 1):S1–324. doi: 10.1111/j.1476-5381.2011.01649_1.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alkondon M, Pereira EF, Cortes WS, Maelicke A, Albuquerque EX. Choline is a selective agonist of alpha7 nicotinic acetylcholine receptors in the rat brain neurons. Eur J Neurosci. 1997;9:2734–2742. doi: 10.1111/j.1460-9568.1997.tb01702.x. [DOI] [PubMed] [Google Scholar]
- Alvarez A, Opazo C, Alarcon R, Garrido J, Inestrosa NC. Acetylcholinesterase promotes the aggregation of amyloid-beta-peptide fragments by forming a complex with the growing fibrils. J Mol Biol. 1997;272:348–361. doi: 10.1006/jmbi.1997.1245. [DOI] [PubMed] [Google Scholar]
- Alvarez A, Alarcon R, Opazo C, Campos EO, Munoz FJ, Calderon FH, et al. Stable complexes involving acetylcholinesterase and amyloid-beta peptide change the biochemical properties of the enzyme and increase the neurotoxicity of Alzheimer's fibrils. J Neurosci. 1998;18:3213–3223. doi: 10.1523/JNEUROSCI.18-09-03213.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arendt T, Bruckner MK, Lange M, Bigl V. Changes in acetylcholinesterase and butyrylcholinesterase in Alzheimer's disease resemble embryonic development-a study of molecular forms. Neurochem Int. 1992;21:381–396. doi: 10.1016/0197-0186(92)90189-x. [DOI] [PubMed] [Google Scholar]
- Armstrong RA. The pathogenesis of Alzheimer's disease: a reevaluation of the ‘amyloid cascade hypothesis. Int J Alzheimers Dis. 2011;2011:630865. doi: 10.4061/2011/630865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auld VJ, Fetter RD, Broadie K, Goodman CS. Gliotactin, a novel transmembrane protein on peripheral glia, is required to form the blood-nerve barrier in Drosophila. Cell. 1995;81:757–767. doi: 10.1016/0092-8674(95)90537-5. [DOI] [PubMed] [Google Scholar]
- Aziz-Aloya R, Seidman S, Timberg R, Sternfeld M, Zakut H, Soreq H. Expression of a human acetylcholinesterase promoter-reporter construct in developing neuromuscular junctions of Xenopus embryos. Proc Natl Acad Sci U S A. 1993;90:2471–2475. doi: 10.1073/pnas.90.6.2471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballard CG, Greig NH, Guillozet-Bongaarts AL, Enz A, Darvesh S. Cholinesterases: roles in the brain during health and disease. Curr Alzheimer Res. 2005;2:307–318. doi: 10.2174/1567205054367838. [DOI] [PubMed] [Google Scholar]
- Barrett KM, Brott TG, Brown RD, Jr, Carter RE, Geske JR, Graff-Radford NR, et al. Enhancing recovery after acute ischemic stroke with donepezil as an adjuvant therapy to standard medical care: results of a phase IIA clinical trial. J Stroke Cerebrovasc Dis. 2011;20:177–182. doi: 10.1016/j.jstrokecerebrovasdis.2010.12.009. [DOI] [PubMed] [Google Scholar]
- Ben Shaul Y, Benmoyal-Segal L, Ben Ari S, Bergman H, Soreq H. Adaptive acetylcholinesterase splicing patterns attenuate 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced Parkinsonism in mice. Eur J Neurosci. 2006;23:2915–2922. doi: 10.1111/j.1460-9568.2006.04812.x. [DOI] [PubMed] [Google Scholar]
- Benilova I, Karran E, De Strooper B. The toxic Abeta oligomer and Alzheimer's disease: an emperor in need of clothes. Nat Neurosci. 2012;15:349–357. doi: 10.1038/nn.3028. [DOI] [PubMed] [Google Scholar]
- Benmoyal-Segal L, Soreq L, Ben Shaul Y, Ben Ari S, Ben Moshe T, Aviel S, et al. Adaptive alternative splicing correlates with less environmental risk of Parkinsonism. Neurodegener Dis. 2012;9:87–98. doi: 10.1159/000331328. [DOI] [PubMed] [Google Scholar]
- Berson A, Knobloch M, Hanan M, Diamant S, Sharoni M, Schuppli D, et al. Changes in readthrough acetylcholinesterase expression modulate amyloid-beta pathology. Brain. 2008;131:109–119. doi: 10.1093/brain/awm276. [DOI] [PubMed] [Google Scholar]
- Berson A, Barbash S, Shaltiel G, Goll Y, Hanin G, Greenberg DS, et al. Cholinergic-associated loss of hnRNP-A/B in Alzheimer's disease impairs cortical splicing and cognitive function in mice. EMBO Mol Med. 2012;4:730–742. doi: 10.1002/emmm.201100995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bigbee JW, Sharma KV, Chan EL, Bogler O. Evidence for the direct role of acetylcholinesterase in neurite outgrowth in primary dorsal root ganglion neurons. Brain Res. 2000;861:354–362. doi: 10.1016/s0006-8993(00)02046-1. [DOI] [PubMed] [Google Scholar]
- Bissette G, Seidler FJ, Nemeroff CB, Slotkin TA. High affinity choline transporter status in Alzheimer's disease tissue from rapid autopsy. Ann N Y Acad Sci. 1996;777:197–204. doi: 10.1111/j.1749-6632.1996.tb34419.x. [DOI] [PubMed] [Google Scholar]
- Blong RM, Bedows E, Lockridge O. Tetramerization domain of human butyrylcholinesterase is at the C-terminus. Biochem J. 1997;327(Pt 3):747–757. doi: 10.1042/bj3270747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohnen NI, Kaufer DI, Hendrickson R, Ivanco LS, Lopresti BJ, Koeppe RA, et al. Degree of inhibition of cortical acetylcholinesterase activity and cognitive effects by donepezil treatment in Alzheimer's disease. J Neurol Neurosurg Psychiatry. 2005;76:315–319. doi: 10.1136/jnnp.2004.038729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bon CL, Greenfield SA. Bioactivity of a peptide derived from acetylcholinesterase: electrophysiological characterization in guinea-pig hippocampus. Eur J Neurosci. 2003;17:1991–1995. doi: 10.1046/j.1460-9568.2003.02648.x. [DOI] [PubMed] [Google Scholar]
- Bond CE, Zimmermann M, Greenfield SA. Upregulation of alpha7 nicotinic receptors by acetylcholinesterase C-terminal peptides. PLoS One. 2009;4:e4846. doi: 10.1371/journal.pone.0004846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boudinot E, Bernard V, Camp S, Taylor P, Champagnat J, Krejci E, et al. Influence of differential expression of acetylcholinesterase in brain and muscle on respiration. Respir Physiol Neurobiol. 2009;165:40–48. doi: 10.1016/j.resp.2008.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourne Y, Taylor P, Bougis PE, Marchot P. Crystal structure of mouse acetylcholinesterase. A peripheral site-occluding loop in a tetrameric assembly. J Biol Chem. 1999;274:2963–2970. doi: 10.1074/jbc.274.5.2963. [DOI] [PubMed] [Google Scholar]
- Brenner T, Hamra-Amitay Y, Evron T, Boneva N, Seidman S, Soreq H. The role of readthrough acetylcholinesterase in the pathophysiology of myasthenia gravis. FASEB J. 2003;17:214–222. doi: 10.1096/fj.02-0609com. [DOI] [PubMed] [Google Scholar]
- Broide RS, Robertson RT, Leslie FM. Regulation of alpha7 nicotinic acetylcholine receptors in the developing rat somatosensory cortex by thalamocortical afferents. J Neurosci. 1996;16:2956–2971. doi: 10.1523/JNEUROSCI.16-09-02956.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bullock R, Cameron A. Rivastigmine for the treatment of dementia and visual hallucinations associated with Parkinson's disease: a case series. Curr Med Res Opin. 2002;18:258–264. doi: 10.1185/030079902125000813. [DOI] [PubMed] [Google Scholar]
- Bytyqi AH, Lockridge O, Duysen E, Wang Y, Wolfrum U, Layer PG. Impaired formation of the inner retina in an AChE knockout mouse results in degeneration of all photoreceptors. Eur J Neurosci. 2004;20:2953–2962. doi: 10.1111/j.1460-9568.2004.03753.x. [DOI] [PubMed] [Google Scholar]
- Camp S, Zhang L, Krejci E, Dobbertin A, Bernard V, Girard E, et al. Contributions of selective knockout studies to understanding cholinesterase disposition and function. Chem Biol Interact. 2010;187:72–77. doi: 10.1016/j.cbi.2010.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campos EO, Alvarez A, Inestrosa NC. Brain acetylcholinesterase promotes amyloid-beta-peptide aggregation but does not hydrolyze amyloid precursor protein peptides. Neurochem Res. 1998;23:135–140. doi: 10.1023/a:1022416505725. [DOI] [PubMed] [Google Scholar]
- Cerbai F, Giovannini MG, Melani C, Enz A, Pepeu G. N1phenethyl-norcymserine, a selective butyrylcholinesterase inhibitor, increases acetylcholine release in rat cerebral cortex: a comparison with donepezil and rivastigmine. Eur J Pharmacol. 2007;572:142–150. doi: 10.1016/j.ejphar.2007.06.053. [DOI] [PubMed] [Google Scholar]
- Chang WH, Park YH, Ohn SH, Park CH, Lee PK, Kim YH. Neural correlates of donepezil-induced cognitive improvement in patients with right hemisphere stroke: a pilot study. Neuropsychol Rehabil. 2011;21:502–514. doi: 10.1080/09602011.2011.582708. [DOI] [PubMed] [Google Scholar]
- Chen VP, Choi RC, Chan WK, Leung KW, Guo AJ, Chan GK, et al. The assembly of proline-rich membrane anchor (PRiMA)-linked acetylcholinesterase enzyme: glycosylation is required for enzymatic activity but not for oligomerization. J Biol Chem. 2011;286:32948–32961. doi: 10.1074/jbc.M111.261248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen O, Erb C, Ginzberg D, Pollak Y, Seidman S, Shoham S, et al. Neuronal overexpression of ‘readthrough’ acetylcholinesterase is associated with antisense-suppressible behavioral impairments. Mol Psychiatry. 2002;7:874–885. doi: 10.1038/sj.mp.4001103. [DOI] [PubMed] [Google Scholar]
- Correa MC, Maldonado P, da Rosa CS, Lunkes G, Lunkes DS, Kaizer RR, et al. Oxidative stress and erythrocyte acetylcholinesterase (AChE) in hypertensive and ischemic patients of both acute and chronic stages. Biomed Pharmacother. 2008;62:317–324. doi: 10.1016/j.biopha.2007.10.002. [DOI] [PubMed] [Google Scholar]
- Darreh-Shori T, Hellstrom-Lindahl E, Flores-Flores C, Guan ZZ, Soreq H, Nordberg A. Long-lasting acetylcholinesterase splice variations in anticholinesterase-treated Alzheimer's disease patients. J Neurochem. 2004;88:1102–1113. doi: 10.1046/j.1471-4159.2003.02230.x. [DOI] [PubMed] [Google Scholar]
- Darvesh S. Butyrylcholinesterase radioligands to image Alzheimer's disease brain. Chem Biol Interact. 2013;203:354–357. doi: 10.1016/j.cbi.2012.08.009. [DOI] [PubMed] [Google Scholar]
- Darvesh S, MacKnight C, Rockwood K. Butyrylcholinesterase and cognitive function. Int Psychogeriatr. 2001;13:461–464. doi: 10.1017/s1041610201007876. [DOI] [PubMed] [Google Scholar]
- Day T, Greenfield SA. A non-cholinergic, trophic action of acetylcholinesterase on hippocampal neurones in vitro: molecular mechanisms. Neuroscience. 2002;111:649–656. doi: 10.1016/s0306-4522(02)00031-3. [DOI] [PubMed] [Google Scholar]
- Day T, Greenfield SA. A peptide derived from acetylcholinesterase induces neuronal cell death: characterisation of possible mechanisms. Exp Brain Res. 2003;153:334–342. doi: 10.1007/s00221-003-1567-5. [DOI] [PubMed] [Google Scholar]
- Deutsch VR, Pick M, Perry C, Grisaru D, Hemo Y, Golan-Hadari D, et al. The stress-associated acetylcholinesterase variant AChE-R is expressed in human CD34(+) hematopoietic progenitors and its C-terminal peptide ARP promotes their proliferation. Exp Hematol. 2002;30:1153–1161. doi: 10.1016/s0301-472x(02)00900-1. [DOI] [PubMed] [Google Scholar]
- Dineley KT. Beta-amyloid peptide-nicotinic acetylcholine receptor interaction: the two faces of health and disease. Front Biosci. 2007;12:5030–5038. doi: 10.2741/2445. [DOI] [PubMed] [Google Scholar]
- Dineley KT, Westerman M, Bui D, Bell K, Ashe KH, Sweatt JD. Beta-amyloid activates the mitogen-activated protein kinase cascade via hippocampal alpha7 nicotinic acetylcholine receptors: in vitro and in vivo mechanisms related to Alzheimer's disease. J Neurosci. 2001;21:4125–4133. doi: 10.1523/JNEUROSCI.21-12-04125.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dineley KT, Bell KA, Bui D, Sweatt JD. beta-Amyloid peptide activates alpha 7 nicotinic acetylcholine receptors expressed in Xenopus oocytes. J Biol Chem. 2002;277:25056–25061. doi: 10.1074/jbc.M200066200. [DOI] [PubMed] [Google Scholar]
- Dirnagl U. Pathobiology of injury after stroke: the neurovascular unit and beyond. Ann N Y Acad Sci. 2012;1268:21–25. doi: 10.1111/j.1749-6632.2012.06691.x. [DOI] [PubMed] [Google Scholar]
- Dobbertin A, Hrabovska A, Dembele K, Camp S, Taylor P, Krejci E, et al. Targeting of acetylcholinesterase in neurons in vivo: a dual processing function for the proline-rich membrane anchor subunit and the attachment domain on the catalytic subunit. J Neurosci. 2009;29:4519–4530. doi: 10.1523/JNEUROSCI.3863-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobransky T, Rylett RJ. A model for dynamic regulation of choline acetyltransferase by phosphorylation. J Neurochem. 2005;95:305–313. doi: 10.1111/j.1471-4159.2005.03367.x. [DOI] [PubMed] [Google Scholar]
- Dobransky T, Doherty-Kirby A, Kim AR, Brewer D, Lajoie G, Rylett RJ. Protein kinase C isoforms differentially phosphorylate human choline acetyltransferase regulating its catalytic activity. J Biol Chem. 2004;279:52059–52068. doi: 10.1074/jbc.M407085200. [DOI] [PubMed] [Google Scholar]
- Dori A, Soreq H. ARP, the cleavable C-terminal peptide of ‘readthrough’ acetylcholinesterase, promotes neuronal development and plasticity. J Mol Neurosci. 2006a;28:247–255. doi: 10.1385/JMN:28:3:247. [DOI] [PubMed] [Google Scholar]
- Dori A, Soreq H. Neuromuscular therapeutics by RNA-targeted suppression of ACHE gene expression. Ann N Y Acad Sci. 2006b;1082:77–90. doi: 10.1196/annals.1348.004. [DOI] [PubMed] [Google Scholar]
- Dori A, Ifergane G, Saar-Levy T, Bersudsky M, Mor I, Soreq H, et al. Readthrough acetylcholinesterase in inflammation-associated neuropathies. Life Sci. 2007;80:2369–2374. doi: 10.1016/j.lfs.2007.02.011. [DOI] [PubMed] [Google Scholar]
- Duda JE. Pathology and neurotransmitter abnormalities of dementia with Lewy bodies. Dement Geriatr Cogn Disord. 2004;17(Suppl. 1):3–14. doi: 10.1159/000074677. [DOI] [PubMed] [Google Scholar]
- Dumont M, Lalonde R, Ghersi-Egea JF, Fukuchi K, Strazielle C. Regional acetylcholinesterase activity and its correlation with behavioral performances in 15-month old transgenic mice expressing the human C99 fragment of APP. J Neural Transm. 2006;113:1225–1241. doi: 10.1007/s00702-005-0373-6. [DOI] [PubMed] [Google Scholar]
- Dupree JL, Bigbee JW. Retardation of neuritic outgrowth and cytoskeletal changes accompany acetylcholinesterase inhibitor treatment in cultured rat dorsal root ganglion neurons. J Neurosci Res. 1994;39:567–575. doi: 10.1002/jnr.490390508. [DOI] [PubMed] [Google Scholar]
- Emmett SR, Greenfield SA. A peptide derived from the C-terminal region of acetylcholinesterase modulates extracellular concentrations of acetylcholinesterase in the rat substantia nigra. Neurosci Lett. 2004;358:210–214. doi: 10.1016/j.neulet.2003.12.078. [DOI] [PubMed] [Google Scholar]
- Erb C, Troost J, Kopf S, Schmitt U, Loffelholz K, Soreq H, et al. Compensatory mechanisms enhance hippocampal acetylcholine release in transgenic mice expressing human acetylcholinesterase. J Neurochem. 2001;77:638–646. doi: 10.1046/j.1471-4159.2001.00287.x. [DOI] [PubMed] [Google Scholar]
- de la Escalera S, Bockamp EO, Moya F, Piovant M, Jimenez F. Characterization and gene cloning of neurotactin, a Drosophila transmembrane protein related to cholinesterases. EMBO J. 1990;9:3593–3601. doi: 10.1002/j.1460-2075.1990.tb07570.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evron T, Greenberg D, Mor TS, Soreq H. Adaptive changes in acetylcholinesterase gene expression as mediators of recovery from chemical and biological insults. Toxicology. 2007;233:97–107. doi: 10.1016/j.tox.2006.08.018. [DOI] [PubMed] [Google Scholar]
- Farar V, Mohr F, Legrand M, d'Incamps BL, Cendelin J, Leroy J, et al. Near-complete adaptation of the PRiMA knockout to the lack of central acetylcholinesterase. J Neurochem. 2012;122:1065–1080. doi: 10.1111/j.1471-4159.2012.07856.x. [DOI] [PubMed] [Google Scholar]
- Farchi N, Shoham S, Hochner B, Soreq H. Impaired hippocampal plasticity and errors in cognitive performance in mice with maladaptive AChE splice site selection. Eur J Neurosci. 2007;25:87–98. doi: 10.1111/j.1460-9568.2006.05249.x. [DOI] [PubMed] [Google Scholar]
- Ferraiuolo L, Kirby J, Grierson AJ, Sendtner M, Shaw PJ. Molecular pathways of motor neuron injury in amyotrophic lateral sclerosis. Nat Rev Neurol. 2011;7:616–630. doi: 10.1038/nrneurol.2011.152. [DOI] [PubMed] [Google Scholar]
- Fisher A. M1 muscarinic agonists target major hallmarks of Alzheimer's disease – an update. Curr Alzheimer Res. 2007;4:577–580. doi: 10.2174/156720507783018163. [DOI] [PubMed] [Google Scholar]
- Fitzpatrick-McElligott S, Stent GS. Appearance and localization of acetylcholinesterase in embryos of the leech Helobdella triserialis. J Neurosci. 1981;1:901–907. doi: 10.1523/JNEUROSCI.01-08-00901.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fodero LR, Mok SS, Losic D, Martin LL, Aguilar MI, Barrow CJ, et al. Alpha7-nicotinic acetylcholine receptors mediate an Abeta(1-42)-induced increase in the level of acetylcholinesterase in primary cortical neurones. J Neurochem. 2004;88:1186–1193. doi: 10.1046/j.1471-4159.2003.02296.x. [DOI] [PubMed] [Google Scholar]
- Fotuhi M, Hachinski V, Whitehouse PJ. Changing perspectives regarding late-life dementia. Nat Rev Neurol. 2009;5:649–658. doi: 10.1038/nrneurol.2009.175. [DOI] [PubMed] [Google Scholar]
- Francis PT, Palmer AM, Snape M, Wilcock GK. The cholinergic hypothesis of Alzheimer's disease: a review of progress. J Neurol Neurosurg Psychiatry. 1999;66:137–147. doi: 10.1136/jnnp.66.2.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francis PT, Parsons CG, Jones RW. Rationale for combining glutamatergic and cholinergic approaches in the symptomatic treatment of Alzheimer's disease. Expert Rev Neurother. 2012;12:1351–1365. doi: 10.1586/ern.12.124. [DOI] [PubMed] [Google Scholar]
- Fujii T, Kawashima K. An independent non-neuronal cholinergic system in lymphocytes. Jpn J Pharmacol. 2001;85:11–15. doi: 10.1254/jjp.85.11. [DOI] [PubMed] [Google Scholar]
- Gagliardi S, Milani P, Sardone V, Pansarasa O, Cereda C. From transcriptome to noncoding RNAs: implications in ALS mechanism. Neurol Res Int. 2012;2012:278725. doi: 10.1155/2012/278725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Palomero E, Munoz P, Usan P, Garcia P, Delgado E, De Austria C, et al. Potent beta-amyloid modulators. Neurodegener Dis. 2008;5:153–156. doi: 10.1159/000113688. [DOI] [PubMed] [Google Scholar]
- Gates J, Jr, Ferguson SM, Blakely RD, Apparsundaram S. Regulation of choline transporter surface expression and phosphorylation by protein kinase C and protein phosphatase 1/2A. J Pharmacol Exp Ther. 2004;310:536–545. doi: 10.1124/jpet.104.066795. [DOI] [PubMed] [Google Scholar]
- Giacobini E. Do cholinesterase inhibitors have disease-modifying effects in Alzheimer's disease? CNS Drugs. 2001;15:85–91. doi: 10.2165/00023210-200115020-00001. [DOI] [PubMed] [Google Scholar]
- Giacobini E. Cholinergic function and Alzheimer's disease. Int J Geriatr Psychiatry. 2003;18:S1–S5. doi: 10.1002/gps.935. [DOI] [PubMed] [Google Scholar]
- Giovannini MG, Scali C, Bartolini L, Schmidt B, Pepeu G. Effect of subchronic treatment with metrifonate and tacrine on brain cholinergic function in aged F344 rats. Eur J Pharmacol. 1998;354:17–24. doi: 10.1016/s0014-2999(98)00429-4. [DOI] [PubMed] [Google Scholar]
- Girard E, Bernard V, Camp S, Taylor P, Krejci E, Molgo J. Remodeling of the neuromuscular junction in mice with deleted exons 5 and 6 of acetylcholinesterase. J Mol Neurosci. 2006;30:99–100. doi: 10.1385/JMN:30:1:99. [DOI] [PubMed] [Google Scholar]
- Greenberg DS, Toiber D, Berson A, Soreq H. Acetylcholinesterase variants in Alzheimer's disease: from neuroprotection to programmed cell death. Neurodegener Dis. 2010;7:60–63. doi: 10.1159/000285507. [DOI] [PubMed] [Google Scholar]
- Greenfield S, Vaux DJ. Parkinson's disease, Alzheimer's disease and motor neurone disease: identifying a common mechanism. Neuroscience. 2002;113:485–492. doi: 10.1016/s0306-4522(02)00194-x. [DOI] [PubMed] [Google Scholar]
- Greenfield SA, Grunewald RA, Foley P, Shaw SG. Origin of various enzymes released from the substantia nigra and caudate nucleus: effects of 6-hydroxydopamine lesions of the nigro-striatal pathway. J Comp Neurol. 1983;214:87–92. doi: 10.1002/cne.902140109. [DOI] [PubMed] [Google Scholar]
- Greenfield SA, Day T, Mann EO, Bermudez I. A novel peptide modulates alpha7 nicotinic receptor responses: implications for a possible trophic-toxic mechanism within the brain. J Neurochem. 2004;90:325–331. doi: 10.1111/j.1471-4159.2004.02494.x. [DOI] [PubMed] [Google Scholar]
- Greenfield SA, Zimmermann M, Bond CE. Non-hydrolytic functions of acetylcholinesterase. The significance of C-terminal peptides. FEBS J. 2008;275:604–611. doi: 10.1111/j.1742-4658.2007.06235.x. [DOI] [PubMed] [Google Scholar]
- Greig NH, Utsuki T, Ingram DK, Wang Y, Pepeu G, Scali C, et al. Selective butyrylcholinesterase inhibition elevates brain acetylcholine, augments learning and lowers Alzheimer beta-amyloid peptide in rodent. Proc Natl Acad Sci U S A. 2005;102:17213–17218. doi: 10.1073/pnas.0508575102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grifman M, Galyam N, Seidman S, Soreq H. Functional redundancy of acetylcholinesterase and neuroligin in mammalian neuritogenesis. Proc Natl Acad Sci U S A. 1998;95:13935–13940. doi: 10.1073/pnas.95.23.13935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grisaru D, Sternfeld M, Eldor A, Glick D, Soreq H. Structural roles of acetylcholinesterase variants in biology and pathology. Eur J Biochem. 1999;264:672–686. doi: 10.1046/j.1432-1327.1999.00693.x. [DOI] [PubMed] [Google Scholar]
- Grisaru D, Deutsch V, Shapira M, Pick M, Sternfeld M, Melamed-Book N, et al. ARP, a peptide derived from the stress-associated acetylcholinesterase variant, has hematopoietic growth promoting activities. Mol Med. 2001;7:93–105. [PMC free article] [PubMed] [Google Scholar]
- Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science. 2002;297:353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- Hardy JA, Higgins GA. Alzheimer's disease: the amyloid cascade hypothesis. Science. 1992;256:184–185. doi: 10.1126/science.1566067. [DOI] [PubMed] [Google Scholar]
- Hardy JA, Mann DM, Wester P, Winblad B. An integrative hypothesis concerning the pathogenesis and progression of Alzheimer's disease. Neurobiol Aging. 1986;7:489–502. doi: 10.1016/0197-4580(86)90086-2. [DOI] [PubMed] [Google Scholar]
- Härtl R, Gleinich A, Zimmermann M. Dramatic increase in readthrough acetylcholinesterase in a cellular model of oxidative stress. J Neurochem. 2011;116:1088–1096. doi: 10.1111/j.1471-4159.2010.07164.x. [DOI] [PubMed] [Google Scholar]
- Hartmann J, Kiewert C, Duysen EG, Lockridge O, Greig NH, Klein J. Excessive hippocampal acetylcholine levels in acetylcholinesterase-deficient mice are moderated by butyrylcholinesterase activity. J Neurochem. 2007;100:1421–1429. doi: 10.1111/j.1471-4159.2006.04347.x. [DOI] [PubMed] [Google Scholar]
- Hicks D, John D, Makova NZ, Henderson Z, Nalivaeva NN, Turner AJ. Membrane targeting, shedding and protein interactions of brain acetylcholinesterase. J Neurochem. 2011;116:742–746. doi: 10.1111/j.1471-4159.2010.07032.x. [DOI] [PubMed] [Google Scholar]
- Hirono N, Hashimoto M, Ishii K, Kazui H, Mori E. One-year change in cerebral glucose metabolism in patients with Alzheimer's disease. J Neuropsychiatry Clin Neurosci. 2004;16:488–492. doi: 10.1176/jnp.16.4.488. [DOI] [PubMed] [Google Scholar]
- Holzgrabe U, Kapkova P, Alptuzun V, Scheiber J, Kugelmann E. Targeting acetylcholinesterase to treat neurodegeneration. Expert Opin Ther Targets. 2007;11:161–179. doi: 10.1517/14728222.11.2.161. [DOI] [PubMed] [Google Scholar]
- Hong JM, Shin DH, Lim TS, Lee JS, Huh K. Galantamine administration in chronic post-stroke aphasia. J Neurol Neurosurg Psychiatry. 2012;83:675–680. doi: 10.1136/jnnp-2012-302268. [DOI] [PubMed] [Google Scholar]
- Huston JM, Tracey KJ. The pulse of inflammation: heart rate variability, the cholinergic anti-inflammatory pathway and implications for therapy. J Intern Med. 2011;269:45–53. doi: 10.1111/j.1365-2796.2010.02321.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inestrosa NC, Perelman A. Distribution and anchoring of molecular forms of acetylcholinesterase. Trends Pharmacol Sci. 1989;10:325–329. doi: 10.1016/0165-6147(89)90067-9. [DOI] [PubMed] [Google Scholar]
- Inestrosa NC, Moreno RD, Fuentes ME. Monomeric amphiphilic forms of acetylcholinesterase appear early during brain development and may correspond to biosynthetic precursors of the amphiphilic G4 forms. Neurosci Lett. 1994;173:155–158. doi: 10.1016/0304-3940(94)90172-4. [DOI] [PubMed] [Google Scholar]
- Inestrosa NC, Alvarez A, Perez CA, Moreno RD, Vicente M, Linker C, et al. Acetylcholinesterase accelerates assembly of amyloid-beta-peptides into Alzheimer's fibrils: possible role of the peripheral site of the enzyme. Neuron. 1996;16:881–891. doi: 10.1016/s0896-6273(00)80108-7. [DOI] [PubMed] [Google Scholar]
- Inestrosa NC, Dinamarca MC, Alvarez A. Amyloid-cholinesterase interactions. Implications for Alzheimer's disease. FEBS J. 2008;275:625–632. doi: 10.1111/j.1742-4658.2007.06238.x. [DOI] [PubMed] [Google Scholar]
- Jin QH, He HY, Shi YF, Lu H, Zhang XJ. Overexpression of acetylcholinesterase inhibited cell proliferation and promoted apoptosis in NRK cells. Acta Pharmacol Sin. 2004;25:1013–1021. [PubMed] [Google Scholar]
- Jope RS. Cholinergic muscarinic receptor signaling by the phosphoinositide signal transduction system in Alzheimer's disease. J Alzheimers Dis. 1999;1:231–247. doi: 10.3233/jad-1999-14-505. [DOI] [PubMed] [Google Scholar]
- Karpel R, Aziz-Aloya R, Sternfeld M, Ehrlich G, Ginzberg D, Tarroni P, et al. Expression of three alternative acetylcholinesterase messenger RNAs in human tumor cell lines of different tissue origins. Exp Cell Res. 1994;210:268–277. doi: 10.1006/excr.1994.1039. [DOI] [PubMed] [Google Scholar]
- Kaufer D, Friedman A, Seidman S, Soreq H. Acute stress facilitates long-lasting changes in cholinergic gene expression. Nature. 1998;393:373–377. doi: 10.1038/30741. [DOI] [PubMed] [Google Scholar]
- Kaufer D, Friedman A, Seidman S, Soreq H. Anticholinesterases induce multigenic transcriptional feedback response suppressing cholinergic neurotransmission. Chem Biol Interact. 1999;119–120:349–360. doi: 10.1016/s0009-2797(99)00046-0. [DOI] [PubMed] [Google Scholar]
- Kim AR, Rylett RJ, Shilton BH. Substrate binding and catalytic mechanism of human choline acetyltransferase. Biochemistry. 2006;45:14621–14631. doi: 10.1021/bi061536l. [DOI] [PubMed] [Google Scholar]
- Kim HJ, Kim NC, Wang YD, Scarborough EA, Moore J, Diaz Z, et al. Mutations in prion-like domains in hnRNPA2B1 and hnRNPA1 cause multisystem proteinopathy and ALS. Nature. 2013;495:467–473. doi: 10.1038/nature11922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koenigsberger C, Chiappa S, Brimijoin S. Neurite differentiation is modulated in neuroblastoma cells engineered for altered acetylcholinesterase expression. J Neurochem. 1997;69:1389–1397. doi: 10.1046/j.1471-4159.1997.69041389.x. [DOI] [PubMed] [Google Scholar]
- Kuhar MJ, Murrin LC. Sodium-dependent, high affinity choline uptake. J Neurochem. 1978;30:15–21. doi: 10.1111/j.1471-4159.1978.tb07029.x. [DOI] [PubMed] [Google Scholar]
- Kummer W, Lips KS, Pfeil U. The epithelial cholinergic system of the airways. Histochem Cell Biol. 2008;130:219–234. doi: 10.1007/s00418-008-0455-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurzen H, Wessler I, Kirkpatrick CJ, Kawashima K, Grando SA. The non-neuronal cholinergic system of human skin. Horm Metab Res. 2007;39:125–135. doi: 10.1055/s-2007-961816. [DOI] [PubMed] [Google Scholar]
- Lain E, Penke B, Delacourte A, Gundisch D, Schroder H, Witter B. Effects of Abeta1-42 fibrils and of the tetrapeptide Pr-IIGL on the phosphorylation state of the tau-protein and on the alpha7 nicotinic acetylcholine receptor in vitro. Eur J Neurosci. 2005;21:879–888. doi: 10.1111/j.1460-9568.2005.03909.x. [DOI] [PubMed] [Google Scholar]
- Layer PG. Comparative localization of acetylcholinesterase and pseudocholinesterase during morphogenesis of the chicken brain. Proc Natl Acad Sci U S A. 1983;80:6413–6417. doi: 10.1073/pnas.80.20.6413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Layer PG. Cholinesterases preceding major tracts in vertebrate neurogenesis. Bioessays. 1990;12:415–420. doi: 10.1002/bies.950120904. [DOI] [PubMed] [Google Scholar]
- Layer PG. Cholinesterases during development of the avian nervous system. Cell Mol Neurobiol. 1991;11:7–33. doi: 10.1007/BF00712798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Layer PG. Nonclassical roles of cholinesterases in the embryonic brain and possible links to Alzheimer disease. Alzheimer Dis Assoc Disord. 1995;9(Suppl. 2):29–36. doi: 10.1097/00002093-199501002-00006. [DOI] [PubMed] [Google Scholar]
- Layer PG, Sporns O. Spatiotemporal relationship of embryonic cholinesterases with cell proliferation in chicken brain and eye. Proc Natl Acad Sci U S A. 1987;84:284–288. doi: 10.1073/pnas.84.1.284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Layer PG, Weikert T, Alber R. Cholinesterases regulate neurite growth of chick nerve cells in vitro by means of a non-enzymatic mechanism. Cell Tissue Res. 1993;273:219–226. doi: 10.1007/BF00312823. [DOI] [PubMed] [Google Scholar]
- Li B, Stribley JA, Ticu A, Xie W, Schopfer LM, Hammond P, et al. Abundant tissue butyrylcholinesterase and its possible function in the acetylcholinesterase knockout mouse. J Neurochem. 2000a;75:1320–1331. doi: 10.1046/j.1471-4159.2000.751320.x. [DOI] [PubMed] [Google Scholar]
- Li B, Duysen EG, Volpicelli-Daley LA, Levey AI, Lockridge O. Regulation of muscarinic acetylcholine receptor function in acetylcholinesterase knockout mice. Pharmacol Biochem Behav. 2003;74:977–986. doi: 10.1016/s0091-3057(03)00022-4. [DOI] [PubMed] [Google Scholar]
- Li G, Gleinich A, Lau H, Zimmermann M. Staurosporine-induced apoptosis presents with unexpected cholinergic effects in a differentiated neuroblastoma cell line. Neurochem Int. 2012a;61:1011–1020. doi: 10.1016/j.neuint.2012.07.018. [DOI] [PubMed] [Google Scholar]
- Li G, Klein J, Zimmermann M. Pathophysiological amyloid concentrations induce sustained upregulation of readthrough AChE mediating anti-apoptotic effects. Neuroscience. 2012b;240:349–360. doi: 10.1016/j.neuroscience.2013.02.040. [DOI] [PubMed] [Google Scholar]
- Li J, Pan P, Huang R, Shang H. A meta-analysis of voxel-based morphometry studies of white matter volume alterations in Alzheimer's disease. Neurosci Biobehav Rev. 2012c;36:757–763. doi: 10.1016/j.neubiorev.2011.12.001. [DOI] [PubMed] [Google Scholar]
- Li Y, Camp S, Rachinsky TL, Getman D, Taylor P. Gene structure of mammalian acetylcholinesterase. Alternative exons dictate tissue-specific expression. J Biol Chem. 1991;266:23083–23090. [PubMed] [Google Scholar]
- Li Y, Liu L, Kang J, Sheng JG, Barger SW, Mrak RE, et al. Neuronal-glial interactions mediated by interleukin-1 enhance neuronal acetylcholinesterase activity and mRNA expression. J Neurosci. 2000b;20:149–155. doi: 10.1523/JNEUROSCI.20-01-00149.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang D, Blouet JP, Borrega F, Bon S, Massoulie J. Respective roles of the catalytic domains and C-terminal tail peptides in the oligomerization and secretory trafficking of human acetylcholinesterase and butyrylcholinesterase. FEBS J. 2009;276:94–108. doi: 10.1111/j.1742-4658.2008.06756.x. [DOI] [PubMed] [Google Scholar]
- Liu JJ, Li DL, Zhou J, Sun L, Zhao M, Kong SS, et al. Acetylcholine prevents angiotensin II-induced oxidative stress and apoptosis in H9c2 cells. Apoptosis. 2011;16:94–103. doi: 10.1007/s10495-010-0549-x. [DOI] [PubMed] [Google Scholar]
- Liu Z, Zhang J, Berg DK. Role of endogenous nicotinic signaling in guiding neuronal development. Biochem Pharmacol. 2007;74:1112–1119. doi: 10.1016/j.bcp.2007.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsh D, Grassi J, Vigny M, Massoulie J. An immunological study of rat acetylcholinesterase: comparison with acetylcholinesterases from other vertebrates. J Neurochem. 1984;43:204–213. doi: 10.1111/j.1471-4159.1984.tb06698.x. [DOI] [PubMed] [Google Scholar]
- Massoulie J. The origin of the molecular diversity and functional anchoring of cholinesterases. Neurosignals. 2002;11:130–143. doi: 10.1159/000065054. [DOI] [PubMed] [Google Scholar]
- Massoulie J, Millard CB. Cholinesterases and the basal lamina at vertebrate neuromuscular junctions. Curr Opin Pharmacol. 2009;9:316–325. doi: 10.1016/j.coph.2009.04.004. [DOI] [PubMed] [Google Scholar]
- Massoulie J, Anselmet A, Bon S, Krejci E, Legay C, Morel N, et al. Acetylcholinesterase: C-terminal domains, molecular forms and functional localization. J Physiol Paris. 1998;92:183–190. doi: 10.1016/s0928-4257(98)80007-7. [DOI] [PubMed] [Google Scholar]
- Massoulie J, Perrier N, Noureddine H, Liang D, Bon S. Old and new questions about cholinesterases. Chem Biol Interact. 2008;175:30–44. doi: 10.1016/j.cbi.2008.04.039. [DOI] [PubMed] [Google Scholar]
- Meerson A, Cacheaux L, Goosens KA, Sapolsky RM, Soreq H, Kaufer D. Changes in brain MicroRNAs contribute to cholinergic stress reactions. J Mol Neurosci. 2010;40:47–55. doi: 10.1007/s12031-009-9252-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melo JB, Agostinho P, Oliveira CR. Involvement of oxidative stress in the enhancement of acetylcholinesterase activity induced by amyloid beta-peptide. Neurosci Res. 2003;45:117–127. doi: 10.1016/s0168-0102(02)00201-8. [DOI] [PubMed] [Google Scholar]
- Meshorer E, Soreq H. Virtues and woes of AChE alternative splicing in stress-related neuropathologies. Trends Neurosci. 2006;29:216–224. doi: 10.1016/j.tins.2006.02.005. [DOI] [PubMed] [Google Scholar]
- Meshorer E, Bryk B, Toiber D, Cohen J, Podoly E, Dori A, et al. SC35 promotes sustainable stress-induced alternative splicing of neuronal acetylcholinesterase mRNA. Mol Psychiatry. 2005;10:985–997. doi: 10.1038/sj.mp.4001735. [DOI] [PubMed] [Google Scholar]
- Mesulam MM, Guillozet A, Shaw P, Levey A, Duysen EG, Lockridge O. Acetylcholinesterase knockouts establish central cholinergic pathways and can use butyrylcholinesterase to hydrolyze acetylcholine. Neuroscience. 2002;110:627–639. doi: 10.1016/s0306-4522(01)00613-3. [DOI] [PubMed] [Google Scholar]
- Mills JD, Janitz M. Alternative splicing of mRNA in the molecular pathology of neurodegenerative diseases. Neurobiol Aging. 2012;33:1012–1024. doi: 10.1016/j.neurobiolaging.2011.10.030. [DOI] [PubMed] [Google Scholar]
- Mohr F, Zimmermann M, Klein J. Mice heterozygous for AChE are more sensitive to AChE inhibitors but do not respond to BuChE inhibition. Neuropharmacology. 2012;67C:37–45. doi: 10.1016/j.neuropharm.2012.11.001. [DOI] [PubMed] [Google Scholar]
- Mor I, Grisaru D, Titelbaum L, Evron T, Richler C, Wahrman J, et al. Modified testicular expression of stress-associated ‘readthrough’ acetylcholinesterase predicts male infertility. FASEB J. 2001;15:2039–2041. doi: 10.1096/fj.00-0814fje. [DOI] [PubMed] [Google Scholar]
- Mor I, Sklan EH, Podoly E, Pick M, Kirschner M, Yogev L, et al. Acetylcholinesterase-R increases germ cell apoptosis but enhances sperm motility. J Cell Mol Med. 2008;12:479–495. doi: 10.1111/j.1582-4934.2008.00231.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moran MA, Mufson EJ, Gomez-Ramos P. Cholinesterases colocalize with sites of neurofibrillary degeneration in aged and Alzheimer's brains. Acta Neuropathol. 1994;87:284–292. doi: 10.1007/BF00296744. [DOI] [PubMed] [Google Scholar]
- Moreno RD, Inestrosa NC, Culwell AR, Alvarez J. Sprouting and abnormal contacts of nonmedullated axons, and deposition of extracellular material induced by the amyloid precursor protein (APP) and other protease inhibitors. Brain Res. 1996;718:13–24. doi: 10.1016/0006-8993(95)01555-8. [DOI] [PubMed] [Google Scholar]
- Mori E, Ikeda M, Kosaka K. Donepezil for dementia with Lewy bodies: a randomized, placebo-controlled trial. Ann Neurol. 2012;72:41–52. doi: 10.1002/ana.23557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mufson EJ, Counts SE, Perez SE, Ginsberg SD. Cholinergic system during the progression of Alzheimer's disease: therapeutic implications. Expert Rev Neurother. 2008;8:1703–1718. doi: 10.1586/14737175.8.11.1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munoz FJ, Inestrosa NC. Neurotoxicity of acetylcholinesterase amyloid beta-peptide aggregates is dependent on the type of Abeta peptide and the AChE concentration present in the complexes. FEBS Lett. 1999;450:205–209. doi: 10.1016/s0014-5793(99)00468-8. [DOI] [PubMed] [Google Scholar]
- Nagele RG, D'Andrea MR, Anderson WJ, Wang HY. Intracellular accumulation of beta-amyloid(1-42) in neurons is facilitated by the alpha 7 nicotinic acetylcholine receptor in Alzheimer's disease. Neuroscience. 2002;110:199–211. doi: 10.1016/s0306-4522(01)00460-2. [DOI] [PubMed] [Google Scholar]
- Nair HK, Seravalli J, Arbuckle T, Quinn DM. Molecular recognition in acetylcholinesterase catalysis: free-energy correlations for substrate turnover and inhibition by trifluoro ketone transition-state analogs. Biochemistry. 1994;33:8566–8576. doi: 10.1021/bi00194a023. [DOI] [PubMed] [Google Scholar]
- Nijholt I, Farchi N, Kye M, Sklan EH, Shoham S, Verbeure B, et al. Stress-induced alternative splicing of acetylcholinesterase results in enhanced fear memory and long-term potentiation. Mol Psychiatry. 2004;9:174–183. doi: 10.1038/sj.mp.4001446. [DOI] [PubMed] [Google Scholar]
- O'Neill MJ, Murray TK, Lakics V, Visanji NP, Duty S. The role of neuronal nicotinic acetylcholine receptors in acute and chronic neurodegeneration. Curr Drug Targets CNS Neurol Disord. 2002;1:399–411. doi: 10.2174/1568007023339166. [DOI] [PubMed] [Google Scholar]
- Oldenbeuving AW, de Kort PL, Jansen BP, Kappelle LJ, Roks G. A pilot study of rivastigmine in the treatment of delirium after stroke: a safe alternative. BMC Neurol. 2008;8:34. doi: 10.1186/1471-2377-8-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pahlman S, Ruusala AI, Abrahamsson L, Mattsson ME, Esscher T. Retinoic acid-induced differentiation of cultured human neuroblastoma cells: a comparison with phorbolester-induced differentiation. Cell Differ. 1984;14:135–144. doi: 10.1016/0045-6039(84)90038-1. [DOI] [PubMed] [Google Scholar]
- Pahlman S, Hoehner JC, Nanberg E, Hedborg F, Fagerstrom S, Gestblom C, et al. Differentiation and survival influences of growth factors in human neuroblastoma. Eur J Cancer. 1995;31A:453–458. doi: 10.1016/0959-8049(95)00033-f. [DOI] [PubMed] [Google Scholar]
- Park SE, Kim ND, Yoo YH. Acetylcholinesterase plays a pivotal role in apoptosome formation. Cancer Res. 2004;64:2652–2655. doi: 10.1158/0008-5472.can-04-0649. [DOI] [PubMed] [Google Scholar]
- Passmore AP, Bayer AJ, Steinhagen-Thiessen E. Cognitive, global, and functional benefits of donepezil in Alzheimer's disease and vascular dementia: results from large-scale clinical trials. J Neurol Sci. 2005;229–230:141–146. doi: 10.1016/j.jns.2004.11.017. [DOI] [PubMed] [Google Scholar]
- Pavlov VA, Tracey KJ. The cholinergic anti-inflammatory pathway. Brain Behav Immun. 2005;19:493–499. doi: 10.1016/j.bbi.2005.03.015. [DOI] [PubMed] [Google Scholar]
- Perrier AL, Massoulie J, Krejci E. PRiMA: the membrane anchor of acetylcholinesterase in the brain. Neuron. 2002;33:275–285. doi: 10.1016/s0896-6273(01)00584-0. [DOI] [PubMed] [Google Scholar]
- Perrier NA, Kherif S, Perrier AL, Dumas S, Mallet J, Massoulie J. Expression of PRiMA in the mouse brain: membrane anchoring and accumulation of ‘tailed’ acetylcholinesterase. Eur J Neurosci. 2003;18:1837–1847. doi: 10.1046/j.1460-9568.2003.02914.x. [DOI] [PubMed] [Google Scholar]
- Perrier NA, Salani M, Falasca C, Bon S, Augusti-Tocco G, Massoulie J. The readthrough variant of acetylcholinesterase remains very minor after heat shock, organophosphate inhibition and stress, in cell culture and in vivo. J Neurochem. 2005;94:629–638. doi: 10.1111/j.1471-4159.2005.03140.x. [DOI] [PubMed] [Google Scholar]
- Perrier NA, Salani M, Falasca C, Bon S, Augusti-Tocco G, Massoulie J. Readthrough acetylcholinesterase expression remains minor after stress or exposure to inhibitors. J Mol Neurosci. 2006;30:75–76. doi: 10.1385/JMN:30:1:75. [DOI] [PubMed] [Google Scholar]
- Perry EK, Perry RH, Blessed G, Tomlinson BE. Changes in brain cholinesterases in senile dementia of Alzheimer type. Neuropathol Appl Neurobiol. 1978;4:273–277. doi: 10.1111/j.1365-2990.1978.tb00545.x. [DOI] [PubMed] [Google Scholar]
- Pick M, Flores-Flores C, Grisaru D, Shochat S, Deutsch V, Soreq H. Blood-cell-specific acetylcholinesterase splice variations under changing stimuli. Int J Dev Neurosci. 2004;22:523–531. doi: 10.1016/j.ijdevneu.2004.07.016. [DOI] [PubMed] [Google Scholar]
- Pick M, Perry C, Lapidot T, Guimaraes-Sternberg C, Naparstek E, Deutsch V, et al. Stress-induced cholinergic signaling promotes inflammation-associated thrombopoiesis. Blood. 2006;107:3397–3406. doi: 10.1182/blood-2005-08-3240. [DOI] [PubMed] [Google Scholar]
- Pihlajamaki M, Jauhiainen AM, Soininen H. Structural and functional MRI in mild cognitive impairment. Curr Alzheimer Res. 2009;6:179–185. doi: 10.2174/156720509787602898. [DOI] [PubMed] [Google Scholar]
- Pinto T, Lanctot KL, Herrmann N. Revisiting the cholinergic hypothesis of behavioral and psychological symptoms in dementia of the Alzheimer's type. Ageing Res Rev. 2011;10:404–412. doi: 10.1016/j.arr.2011.01.003. [DOI] [PubMed] [Google Scholar]
- Podoly E, Shalev DE, Shenhar-Tsarfaty S, Bennett ER, Ben Assayag E, Wilgus H, et al. The butyrylcholinesterase K variant confers structurally derived risks for Alzheimer pathology. J Biol Chem. 2009;284:17170–17179. doi: 10.1074/jbc.M109.004952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pohanka M. Acetylcholinesterase inhibitors: a patent review (2008–present) Expert Opin Ther Pat. 2012;22:871–886. doi: 10.1517/13543776.2012.701620. [DOI] [PubMed] [Google Scholar]
- Racchi M, Mazzucchelli M, Porrello E, Lanni C, Govoni S. Acetylcholinesterase inhibitors: novel activities of old molecules. Pharmacol Res. 2004;50:441–451. doi: 10.1016/j.phrs.2003.12.027. [DOI] [PubMed] [Google Scholar]
- Radic Z, Kirchhoff PD, Quinn DM, McCammon JA, Taylor P. Electrostatic influence on the kinetics of ligand binding to acetylcholinesterase. Distinctions between active center ligands and fasciculin. J Biol Chem. 1997;272:23265–23277. doi: 10.1074/jbc.272.37.23265. [DOI] [PubMed] [Google Scholar]
- Rees TM, Berson A, Sklan EH, Younkin L, Younkin S, Brimijoin S, et al. Memory deficits correlating with acetylcholinesterase splice shift and amyloid burden in doubly transgenic mice. Curr Alzheimer Res. 2005;2:291–300. doi: 10.2174/1567205054367847. [DOI] [PubMed] [Google Scholar]
- Reitz C. Alzheimer's disease and the amyloid cascade hypothesis: a critical review. Int J Alzheimers Dis. 2012;2012:369808. doi: 10.1155/2012/369808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- del Rey A, Besedovsky HO. Sympathetic nervous system-immune interactions in autoimmune lymphoproliferative diseases. Neuroimmunomodulation. 2008;15:29–36. doi: 10.1159/000135621. [DOI] [PubMed] [Google Scholar]
- Robberecht W, Philips T. The changing scene of amyotrophic lateral sclerosis. Nat Rev Neurosci. 2013;14:248–264. doi: 10.1038/nrn3430. [DOI] [PubMed] [Google Scholar]
- Roman GC, Wilkinson DG, Doody RS, Black SE, Salloway SP, Schindler RJ. Donepezil in vascular dementia: combined analysis of two large-scale clinical trials. Dement Geriatr Cogn Disord. 2005;20:338–344. doi: 10.1159/000088494. [DOI] [PubMed] [Google Scholar]
- Rotundo RL, Fambrough DM. Synthesis, transport and fate of acetylcholinesterase in cultured chick embryos muscle cells. Cell. 1980;22:583–594. doi: 10.1016/0092-8674(80)90368-2. [DOI] [PubMed] [Google Scholar]
- Sailaja BS, Cohen-Carmon D, Zimmerman G, Soreq H, Meshorer E. Stress-induced epigenetic transcriptional memory of acetylcholinesterase by HDAC4. Proc Natl Acad Sci U S A. 2012;109:E3687–E3695. doi: 10.1073/pnas.1209990110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salmon A, Erb C, Meshorer E, Ginzberg D, Adani Y, Rabinovitz I, et al. Muscarinic modulations of neuronal anticholinesterase responses. Chem Biol Interact. 2005;157–158:105–113. doi: 10.1016/j.cbi.2005.10.015. [DOI] [PubMed] [Google Scholar]
- Saxena A, Hur RS, Luo C, Doctor BP. Natural monomeric form of fetal bovine serum acetylcholinesterase lacks the C-terminal tetramerization domain. Biochemistry. 2003;42:15292–15299. doi: 10.1021/bi030150x. [DOI] [PubMed] [Google Scholar]
- Saxena G, Singh SP, Agrawal R, Nath C. Effect of donepezil and tacrine on oxidative stress in intracerebral streptozotocin-induced model of dementia in mice. Eur J Pharmacol. 2008;581:283–289. doi: 10.1016/j.ejphar.2007.12.009. [DOI] [PubMed] [Google Scholar]
- Scali C, Casamenti F, Bellucci A, Costagli C, Schmidt B, Pepeu G. Effect of subchronic administration of metrifonate, rivastigmine and donepezil on brain acetylcholine in aged F344 rats. J Neural Transm. 2002;109:1067–1080. doi: 10.1007/s007020200090. [DOI] [PubMed] [Google Scholar]
- Schliebs R, Arendt T. The cholinergic system in aging and neuronal degeneration. Behav Brain Res. 2011;221:555–563. doi: 10.1016/j.bbr.2010.11.058. [DOI] [PubMed] [Google Scholar]
- Schuller HM. Is cancer triggered by altered signalling of nicotinic acetylcholine receptors? Nat Rev Cancer. 2009;9:195–205. doi: 10.1038/nrc2590. [DOI] [PubMed] [Google Scholar]
- Shaked I, Meerson A, Wolf Y, Avni R, Greenberg D, Gilboa-Geffen A, et al. MicroRNA-132 potentiates cholinergic anti-inflammatory signaling by targeting acetylcholinesterase. Immunity. 2009;31:965–973. doi: 10.1016/j.immuni.2009.09.019. [DOI] [PubMed] [Google Scholar]
- Shaltiel G, Hanan M, Wolf Y, Barbash S, Kovalev E, Shoham S, et al. Hippocampal microRNA-132 mediates stress-inducible cognitive deficits through its acetylcholinesterase target. Brain Struct Funct. 2013;218:59–72. doi: 10.1007/s00429-011-0376-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinotoh H, Namba H, Fukushi K, Nagatsuka S, Tanaka N, Aotsuka A, et al. Progressive loss of cortical acetylcholinesterase activity in association with cognitive decline in Alzheimer's disease: a positron emission tomography study. Ann Neurol. 2000;48:194–200. [PubMed] [Google Scholar]
- Silman I, Sussman JL. Acetylcholinesterase: ‘classical’ and ‘non-classical’ functions and pharmacology. Curr Opin Pharmacol. 2005;5:293–302. doi: 10.1016/j.coph.2005.01.014. [DOI] [PubMed] [Google Scholar]
- Sklan EH, Berson A, Birikh KR, Gutnick A, Shahar O, Shoham S, et al. Acetylcholinesterase modulates stress-induced motor responses through catalytic and noncatalytic properties. Biol Psychiatry. 2006;60:741–751. doi: 10.1016/j.biopsych.2006.03.080. [DOI] [PubMed] [Google Scholar]
- Small DH, Reed G, Whitefield B, Nurcombe V. Cholinergic regulation of neurite outgrowth from isolated chick sympathetic neurons in culture. J Neurosci. 1995;15:144–151. doi: 10.1523/JNEUROSCI.15-01-00144.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Small DH, Michaelson S, Sberna G. Non-classical actions of cholinesterases: role in cellular differentiation, tumorigenesis and Alzheimer's disease. Neurochem Int. 1996;28:453–483. doi: 10.1016/0197-0186(95)00099-2. [DOI] [PubMed] [Google Scholar]
- Small DH, Maksel D, Kerr ML, Ng J, Hou X, Chu C, et al. The beta-amyloid protein of Alzheimer's disease binds to membrane lipids but does not bind to the alpha7 nicotinic acetylcholine receptor. J Neurochem. 2007;101:1527–1538. doi: 10.1111/j.1471-4159.2006.04444.x. [DOI] [PubMed] [Google Scholar]
- Soreq H, Seidman S. Acetylcholinesterase-new roles for an old actor. Nat Rev Neurosci. 2001;2:294–302. doi: 10.1038/35067589. [DOI] [PubMed] [Google Scholar]
- Sternfeld M, Shoham S, Klein O, Flores-Flores C, Evron T, Idelson GH, et al. Excess ‘read-through’ acetylcholinesterase attenuates but the ‘synaptic’ variant intensifies neurodeterioration correlates. Proc Natl Acad Sci U S A. 2000;97:8647–8652. doi: 10.1073/pnas.140004597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sussman JD, Argov Z, McKee D, Hazum E, Brawer S, Soreq H. Antisense treatment for myasthenia gravis: experience with monarsen. Ann N Y Acad Sci. 2008;1132:283–290. doi: 10.1196/annals.1405.022. [DOI] [PubMed] [Google Scholar]
- Takata K, Kitamura Y. Molecular approaches to the treatment, prophylaxis, and diagnosis of Alzheimer's disease: tangle formation, amyloid-beta, and microglia in Alzheimer's disease. J Pharmacol Sci. 2012;118:331–337. doi: 10.1254/jphs.11r10fm. [DOI] [PubMed] [Google Scholar]
- Taylor P, Radic Z. The cholinesterases: from genes to proteins. Annu Rev Pharmacol Toxicol. 1994;34:281–320. doi: 10.1146/annurev.pa.34.040194.001433. [DOI] [PubMed] [Google Scholar]
- Taylor P, Radic Z, Kreienkamp HJ, Maeda R, Luo Z, Fuentes ME, et al. Expression and ligand specificity of acetylcholinesterase and the nicotinic receptor: a tale of two cholinergic sites. Biochem Soc Trans. 1994;22:740–745. doi: 10.1042/bst0220740. [DOI] [PubMed] [Google Scholar]
- Thomsen MS, Hansen HH, Timmerman DB, Mikkelsen JD. Cognitive improvement by activation of alpha7 nicotinic acetylcholine receptors: from animal models to human pathophysiology. Curr Pharm Des. 2010;16:323–343. doi: 10.2174/138161210790170094. [DOI] [PubMed] [Google Scholar]
- Tiong CX, Lu M, Bian JS. Protective effect of hydrogen sulphide against 6-OHDA-induced cell injury in SH-SY5Y cells involves PKC/PI3K/Akt pathway. Br J Pharmacol. 2010;161:467–480. doi: 10.1111/j.1476-5381.2010.00887.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toiber D, Soreq H. Cellular stress reactions as putative cholinergic links in Alzheimer's disease. Neurochem Res. 2005;30:909–919. doi: 10.1007/s11064-005-6963-8. [DOI] [PubMed] [Google Scholar]
- Toiber D, Berson A, Greenberg D, Melamed-Book N, Diamant S, Soreq H. N-acetylcholinesterase-induced apoptosis in Alzheimer's disease. PLoS One. 2008;3:e3108. doi: 10.1371/journal.pone.0003108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toiber D, Greenberg DS, Soreq H. Pro-apoptotic protein-protein interactions of the extended N-AChE terminus. J Neural Transm. 2009;116:1435–1442. doi: 10.1007/s00702-009-0249-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- deToledo-Morrell L, Stoub TR, Wang C. Hippocampal atrophy and disconnection in incipient and mild Alzheimer's disease. Prog Brain Res. 2007;163:741–753. doi: 10.1016/S0079-6123(07)63040-4. [DOI] [PubMed] [Google Scholar]
- Torrao AS, Carmona FM, Lindstrom J, Britto LR. Expression of cholinergic system molecules during development of the chick nervous system. Brain Res Dev Brain Res. 2000;124:81–92. doi: 10.1016/s0165-3806(00)00113-9. [DOI] [PubMed] [Google Scholar]
- de la Torre JC. Three postulates to help identify the cause of Alzheimer's disease. J Alzheimers Dis. 2011;24:657–668. doi: 10.3233/JAD-2011-101884. [DOI] [PubMed] [Google Scholar]
- Tracey KJ. The inflammatory reflex. Nature. 2002;420:853–859. doi: 10.1038/nature01321. [DOI] [PubMed] [Google Scholar]
- Tyagi E, Agrawal R, Nath C, Shukla R. Effect of melatonin on neuroinflammation and acetylcholinesterase activity induced by LPS in rat brain. Eur J Pharmacol. 2010;640:206–210. doi: 10.1016/j.ejphar.2010.04.041. [DOI] [PubMed] [Google Scholar]
- Ulrich J, Meier-Ruge W, Probst A, Meier E, Ipsen S. Senile plaques: staining for acetylcholinesterase and A4 protein: a comparative study in the hippocampus and entorhinal cortex. Acta Neuropathol. 1990;80:624–628. doi: 10.1007/BF00307630. [DOI] [PubMed] [Google Scholar]
- Vogel-Hopker A, Sperling LE, Layer PG. Co-opting functions of cholinesterases in neural, limb and stem cell development. Protein Pept Lett. 2012;19:155–164. doi: 10.2174/092986612799080266. [DOI] [PubMed] [Google Scholar]
- Volpicelli-Daley LA, Duysen EG, Lockridge O, Levey AI. Altered hippocampal muscarinic receptors in acetylcholinesterase-deficient mice. Ann Neurol. 2003a;53:788–796. doi: 10.1002/ana.10589. [DOI] [PubMed] [Google Scholar]
- Volpicelli-Daley LA, Hrabovska A, Duysen EG, Ferguson SM, Blakely RD, Lockridge O, et al. Altered striatal function and muscarinic cholinergic receptors in acetylcholinesterase knockout mice. Mol Pharmacol. 2003b;64:1309–1316. doi: 10.1124/mol.64.6.1309. [DOI] [PubMed] [Google Scholar]
- Wang HY, Lee DH, Davis CB, Shank RP. Amyloid peptide Abeta(1-42) binds selectively and with picomolar affinity to alpha7 nicotinic acetylcholine receptors. J Neurochem. 2000;75:1155–1161. doi: 10.1046/j.1471-4159.2000.0751155.x. [DOI] [PubMed] [Google Scholar]
- Whyte EM, Lenze EJ, Butters M, Skidmore E, Koenig K, Dew MA, et al. An open-label pilot study of acetylcholinesterase inhibitors to promote functional recovery in elderly cognitively impaired stroke patients. Cerebrovasc Dis. 2008;26:317–321. doi: 10.1159/000149580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright CI, Geula C, Mesulam MM. Neurological cholinesterases in the normal brain and in Alzheimer's disease: relationship to plaques, tangles, and patterns of selective vulnerability. Ann Neurol. 1993;34:373–384. doi: 10.1002/ana.410340312. [DOI] [PubMed] [Google Scholar]
- Xie HQ, Leung KW, Chen VP, Chan GK, Xu SL, Guo AJ, et al. PRiMA directs a restricted localization of tetrameric AChE at synapses. Chem Biol Interact. 2010;187:78–83. doi: 10.1016/j.cbi.2010.02.018. [DOI] [PubMed] [Google Scholar]
- Xie W, Wilder PJ, Stribley J, Chatonnet A, Rizzino A, Taylor P, et al. Knockout of one acetylcholinesterase allele in the mouse. Chem Biol Interact. 1999;119–120:289–299. doi: 10.1016/s0009-2797(99)00039-3. [DOI] [PubMed] [Google Scholar]
- Yoshida M, Inadome A, Maeda Y, Satoji Y, Masunaga K, Sugiyama Y, et al. Non-neuronal cholinergic system in human bladder urothelium. Urology. 2006;67:425–430. doi: 10.1016/j.urology.2005.08.014. [DOI] [PubMed] [Google Scholar]
- Zakut H, Lapidot-Lifson Y, Beeri R, Ballin A, Soreq H. In vivo gene amplification in non-cancerous cells: cholinesterase genes and oncogenes amplify in thrombocytopenia associated with lupus erythematosus. Mutat Res. 1992;276:275–284. doi: 10.1016/0165-1110(92)90013-y. [DOI] [PubMed] [Google Scholar]
- Zbarsky V, Thomas J, Greenfield S. Bioactivity of a peptide derived from acetylcholinesterase: involvement of an ivermectin-sensitive site on the alpha 7 nicotinic receptor. Neurobiol Dis. 2004;16:283–289. doi: 10.1016/j.nbd.2004.02.009. [DOI] [PubMed] [Google Scholar]
- Zhou J, Fu Y, Tang XC. Huperzine A and donepezil protect rat pheochromocytoma cells against oxygen-glucose deprivation. Neurosci Lett. 2001;306:53–56. doi: 10.1016/s0304-3940(01)01855-9. [DOI] [PubMed] [Google Scholar]
- Zimmermann M. Dementia in life writing: our health care system in the words of the sufferer. Neurol Sci. 2011;32:1233–1238. doi: 10.1007/s10072-010-0459-2. [DOI] [PubMed] [Google Scholar]
- Zimmermann M, Borroni B, Cattabeni F, Padovani A, Di Luca M. Cholinesterase inhibitors influence APP metabolism in Alzheimer disease patients. Neurobiol Dis. 2005a;19:237–242. doi: 10.1016/j.nbd.2005.01.002. [DOI] [PubMed] [Google Scholar]
- Zimmermann M, Gardoni F, Di Luca M. Molecular rationale for the pharmacological treatment of Alzheimer's disease. Drugs Aging. 2005b;22(Suppl. 1):27–37. doi: 10.2165/00002512-200522001-00003. [DOI] [PubMed] [Google Scholar]
- Zimmermann M, Grosgen S, Westwell MS, Greenfield SA. Selective enhancement of the activity of C-terminally truncated, but not intact, acetylcholinesterase. J Neurochem. 2008;104:221–232. doi: 10.1111/j.1471-4159.2007.05045.x. [DOI] [PubMed] [Google Scholar]
- Zimmermann M, Westwell MS, Greenfield SA. Impact of detergents on the activity of acetylcholinesterase and on the effectiveness of its inhibitors. Biol Chem. 2009;390:19–26. doi: 10.1515/BC.2009.005. [DOI] [PubMed] [Google Scholar]
- Zundorf G, Reiser G. Calcium dysregulation and homeostasis of neural calcium in the molecular mechanisms of neurodegenerative diseases provide multiple targets for neuroprotection. Antioxid Redox Signal. 2011;14:1275–1288. doi: 10.1089/ars.2010.3359. [DOI] [PMC free article] [PubMed] [Google Scholar]