

Abstract
Background and Purpose
Hypoesthesia is a clinical feature of neuropathic pain. The feature is partly explained by the evidence of epigenetic repression of Nav1.8 sodium channel in the dorsal root ganglion (DRG).
Experimental Approach
We investigated the possibility of trichostatin A (TSA), valproic acid (VPA) and suberoylanilide hydroxamic acid (SAHA) to reverse the unique C-fibre sensitivity observed following partial ligation of sciatic nerve in mice.
Key Results
Nerve injury-induced down-regulation of DRG Nav1.8 sodium channel and C-fibre-related hypoesthesia were reversed by TSA, VPA and SAHA treatments, which inhibit histone deacetylase (HDAC), and increase histone acetylation at the regulatory sequence of Nav1.8.
Conclusions and Implications
Taken together, these studies provide the evidence that hypoesthesia and underlying down-regulation of Nav1.8, negative symptoms observed in nerve injury-induced neuropathic pain models are regulated by an epigenetic chromatin remodelling through HDAC-related machineries.
Keywords: epigenetics, HDAC inhibitor, Nav1.8, hypoesthesia, neuropathic pain
Introduction
Neural plasticity in sensory neurons following nerve injury contributes to pain hypersensitivity (Tracey and Mantyh, 2007; Woolf, 2011). Transcriptional changes in ion channels, algesic substances, chemokines and their cognate receptors at sensory nerve termini have been reported to play key roles in this plasticity (Woolf and American Physiological Society, 2004; Liu and Wood, 2011). Recent studies have focused that gene expression is epigenetically regulated through chromatin remodelling as well as altered transcriptional regulation (Borrelli et al., 2008). In line with this position, the roles of epigenetic effectors such as neuron-restrictive silencer factor (NRSF; Uchida et al., 2010a), methyl-CpG-binding protein 2/MeCP2 (Wang et al., 2011), histone deacetylase (HDAC; Zhang et al., 2011) and DNA-methyl transferases (DNMTs; Tochiki et al., 2012) have been reported as key molecules to be related to neuropathic pain. Although these epigenetic modulators are supposed to regulate the expression of several genes, it remains to be solved which target genes are important to cause abnormal pain behaviours in neuropathic pain condition.
We have firstly reported that epigenetic silencing is closely related to the mechanisms underlying unique abnormal hypoesthesia and loss of peripheral morphine analgesia observed in nerve injury-induced neuropathic pain model. Epigenetic silencing of Nav1.8 and μ-opioid receptor genes is the representative examples in the dorsal root ganglion (DRG; Uchida et al., 2010a). Another example is the finding that the epigenetic silencing is closely related to the binding activity of NRSF on the neuron-restrictive silencing elements (NRSE) of Nav1.8, Kav4.3 and μ-opioid receptor genes through chromatin immunoprecipitation (ChIP). Binding of NRSF to NRSE within the promoter regions of genes recruit the complex of other epigenetic effector such as HDAC and Mecp2 (Vaissière et al., 2008) thus, providing the molecular basis for anti-hyperalgesia roles of HDAC inhibitors (Bai et al., 2010).
Here, we demonstrate the evidence for the applicability of chemical inhibitors of histone deacetylase (HDACi) such as trichostatin A (TSA), valproic acid (VPA) and suberoylanilide hydroxamic acid (SAHA) in the restoration of electrophysiological sensitivity of C-fibre and recovery of Nav1.8 sodium channel transcription through inhibition of HDAC.
Methods
Animals and surgery
Male C57BL/6J mice weighing 20–25 g were used. They were kept in a room with a temperature of 21 ± 2°C with ad libitum access to a standard laboratory diet and tap water. All procedures were approved by the Nagasaki University Animal Care Committee (Nagasaki, Japan) and complied with the recommendations of the International Association for the Study of Pain (Zimmermann, 1983). All studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (Kilkenny et al., 2010; McGrath et al., 2010). Partial ligation of the sciatic nerve was performed as previously described (Uchida et al., 2010a).
Nociception tests
Thermal and mechanical paw withdrawal tests were used to evaluate hyperalgesia and allodynia responses while electrical stimulation-induced paw withdrawal (EPW) was used to characterize the contribution of the individual sensory fibres. In thermal paw withdrawal test, latency of paw withdrawal upon a thermal stimulus was performed as previously reported by Hargreaves et al. (1988). Unanesthetized animals were placed in Plexiglas cages on top of a glass sheet, and an adaptation period of 1 h was allowed. The thermal stimulator (IITC Inc., Woodland Hills, CA, USA) was positioned under the glass sheet and the focus of the projection bulb was aimed exactly at the middle of the plantar surface of the animal. A mirror attached to the stimulator permitted visualization of the plantar surface. A cut-off time of 20 s was set to prevent tissue damage. The mechanical paw pressure test was performed as described previously (Matsumoto et al., 2006). Briefly, mice were placed in a Plexiglas chamber on a 6 × 6 mm wire mesh grid floor and allowed to acclimatize for a period of 1 h. A mechanical stimulus was then delivered to the middle of the plantar surface of the right hind-paw using a transducer indicator (model 1601; IITC Inc.). The pressure needed to induce a flexor response was defined as the pain threshold. A cut-off pressure of 20 g was also set to avoid tissue damage. During mechanical and thermal tests, the thresholds were determined from three repeated tests at 10 min intervals.
EPW test used to characterize the contribution of the individual sensory fibres. EPW test was performed as reported previously (Matsumoto et al., 2008; Ueda, 2008). Electrodes were fastened to the plantar surfaces and insteps of mice. Transcutaneous nerve stimuli with each of the three sine-wave pulses (5, 250 and 2000 Hz) were applied using a Neurometer CPT/C system (Neurotron Inc., Baltimore, MD, USA). The minimum intensity (μA) at which each mouse withdrew its paw was defined as the current stimulus threshold. Investigators blind to drug treatments performed.
Drug treatments
VPA sodium salt (Sigma-Aldrich, St. Louis, MO, USA) was dissolved in physiological saline while TSA (Wako Pure Chemical Industries, Ltd., Osaka, Japan) was dissolved in 10% ethanol in physiological saline. SAHA (Focus Biomolecules, Plymouth Meeting, PA, USA) was dissolved in 5% dimethyl sulfoxide (DMSO) in physiological saline. Physiological saline was used for control experiments. Intraplantar injections (i.pl.) were administered using a Hamilton microsyringe connected to hypodermic needle. A-803467 (Biomol, Hamburg, Germany), a selective blocker for Nav1.8, was dissolved in DMSO and diluted 28-fold for i.pl injection in physiological saline. The EPW test was performed 30 min after A-803467 injection (30 nmol, i.pl.) at day 3 post-injury. VPA (200 mg·kg−1, i.p.) and TSA (1 mg·kg−1, i.p.; except for dose-dependent investigation (Figure 1A) where graded doses between 0.1 and 1.0 mg·kg−1 were administered) were administered 60 min before injury (day 0) and once daily after injury for 7 days. SAHA (5 mg·kg−1, i.p.) was administered once daily from day 3 to day 9 after nerve injury.
Figure 1.
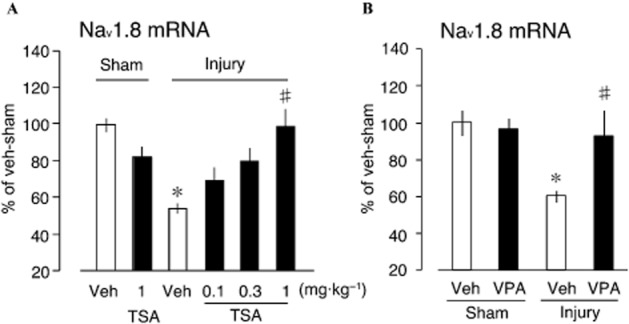
Prevention of decreased Nav1.8 gene expression by TSA and VPA. (A) Graded doses of VPA (0.1–1.0 mg·kg−1) administered 30 min before injury and once daily for 3 days post-injury dose dependently restored Nav1.8 sodium channel expression following its down-regulation by nerve injury. (B) 200 mg·kg−1 dose of VPA administered 30 min before injury and once daily for 3 days post-injury restored Nav1.8 sodium channel expression following its down-regulation by nerve injury. The mRNA expression levels were assessed using RT and real-time PCR and normalized to that of GAPDH mRNA (samples were post-injury day 3, plots represent mean ± SEM of at least 4–8 independent samples; *P < 0.05 vs. vehicle-sham, #P < 0.05 vs. injury-vehicle).
Reverse transcription (RT) and quantitative real-time PCR
Total RNA was extracted from L4-6 DRGs using TRIzol (Invitrogen, Carlsbad, CA, USA) and 500 ng of RNA was used for cDNA synthesis. Quantitative real-time PCR was performed with qPCR MasterMix Plus for SYBR Green I (Eurogentec, Seraing, Belgium) using an ABI Prism 7000 Sequence Detection system (Applied Biosystems, Tokyo, Japan) and LightCycler 480 system II (Roche Diagnostics K.K., Basel, Schweiz). The primers used for the amplification of Nav1.8 NRSF, Transient receptor potential cation channel, subfamily A, member 1 (TRPA1), transient receptor potential cation channel subfamily M member 8 (TRPM8), calcitonin gene related peptide (CGRP) and GAPDH (internal control) have been previously published (Uchida et al., 2010a). Single-peak melting curve and linear amplification validation was performed for all samples.
ChIP assay
ChIP assays were performed as previously described (Uchida et al., 2010b). For each assay, the L4-6 DRGs from two mice were pooled, homogenized in ice-cold cell lysis buffer [10 mM Tris-HCl, pH 8.0, 10 mM NaCl, 0.2% Nonidet P-40, 1 μM p-Amidinophenyl Methansulfonyl Fluoride (p-APMSF)] and cross-linked in PBS containing 1% formaldehyde at 37°C for 5 min. The cross-linking reaction was terminated with glycine (0.125 M) and, after repeated washing with PBS, the samples were resuspended in SDS lysis buffer (50 mM Tris-HCl, pH 8.1, 10 mM EDTA, 1% SDS, 1 μM p-APMSF). Sample chromatin was sheared by sonication into 200–500 bp fragments. Ten percent of each lysate was used as the input control for normalization. The sheared chromatin was diluted 10-fold in ChIP dilution buffer (16.7 mM Tris-HCl, pH 8.1, 1.2 mM EDTA, 167 mM NaCl, 1.1% Triton X-100, 0.01% SDS, 1 μM p-APMSF) and then pre-cleared with protein A-agarose beads (Millipore, Billerica, MA, USA) for 45 min at 4°C with rotation. The supernatant was incubated overnight at 4°C with anti-acetyl-H3 (5 μg; Millipore, Catalogue Number #06–599), anti-acetyl-H4 antibodies (5 μL; Millipore, Catalogue Number #06–866) or normal rabbit IgG (5 μg; Santa Cruz Biotechnology, Santa Cruz, CA, USA). Complexes were precipitated for 2 h using protein A-agarose beads. Following washing and elution steps, DNA was purified by using the Chelex-100 and dissolved in 180 μL of distilled water. The amplicons were also validated as described for quantitative real-time PCR.
Statistical analysis
The differences between multiple groups were analysed using a one-way anova with Tukey–Kramer multiple-comparison post hoc. The criterion of significance was set at P < 0.05. All results are expressed as means ± SEM.
Results
Prevention of decreased Nav1.8 gene expression by TSA and VPA
Chronic transcriptional repression of several genes including Nav1.8 gene in DRG and its epigenetic control following partial sciatic nerve ligation (nerve injury), have been previously reported (Uchida et al., 2010a). Nav1.8 mRNA expression levels were quantified using RT and real-time PCR. TSA treatment is associated with slight but statistically insignificant change by itself (Figure 1A). The injury-induced decrease in Nav1.8 expression was reversed by TSA in a dose-dependent manner. At 1.0 mg·kg−1 TSA treatment, Nav1.8 was expressed at pre-injury levels. To determine the class of HDACs potentially invoked following nerve injury, TSA (class I and IIa HDAC inhibitor) was replaced with VPA (predominantly class IIa HDAC inhibitor; Bai et al., 2010). VPA treatment also restored the reduced Nav1.8 expression (Figure 1B), indicating the potential involvement of classes I and IIa HDAC in nerve injury-mediated down-regulation of Nav1.8 expression.
Blockade of nerve injury-induced C-fibre hypoesthesia by TSA and VPA
DRG contains soma originating from different sensory fibres and capable of detecting wide spectrum of painful stimuli. To determine which of these fibres becomes dysfunctional in TSA-and VPA-reversible manner, we utilized EPW test using Neurometer CPT/C system. The Neurometer is clinically used device for measuring the threshold of responses to sensory stimuli. Many studies have indicated the usefulness of this device in the quantification of nerve dysfunction in patients. Furthermore, we have established the method for measurement of three subsets of nerve fibres (innocuous Aβ, noxious Aδ-fibres and C-fibres) using the Neurometer in pharmacological experiments in laboratory animals (Kiso et al., 2001; Koga et al., 2005; Matsumoto et al., 2008; Ueda, 2008). Indeed, mice exposed to electrical stimuli at 2000 and 250 Hz, which otherwise stimulate innocuous Aβ and noxious Aδ-fibres, respectively, become hypersensitive following nerve injury, while those given with 5 Hz (noxious C-fibres) become hyposensitive (Uchida et al., 2010a). This type of hypoesthesia/hypoalgesia was reversed by the pretreatments with anti-sense oligodeoxynucleotide (AS-ODN) for NRSF, while the injury-induced thermal hyperalgesia or mechanical allodynia was not. In the present study, the treatment with TSA has no effect on the electrical response to 2000 Hz Aβ-and 250 Hz Aδ-stimulation, whereas it significantly reversed the hyposensitivity to the 5 Hz C-fibre stimulation following the injury (Figure 2A–C). As in the case with AS-ODN for NRSF, the TSA treatment did not reverse the thermal hyperalgesia or mechanical allodynia (Figure 2D and E), indicating that both A-fibre dysfunctions are not associated with repression of Nav1.8.
Figure 2.
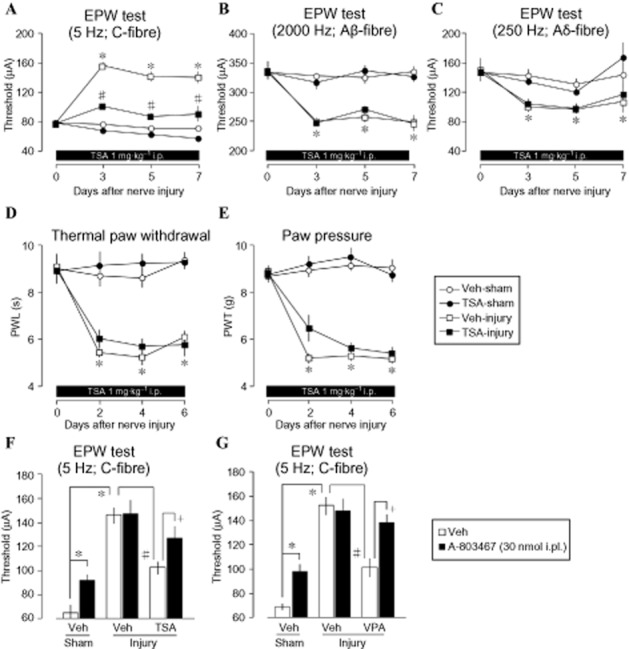
Blockade of nerve injury-induced C-fibre hypoesthesia but not A-fibre hyperalgesia. (A) 1.0 mg·kg−1 (b.w.) administered once 30 min prior to and once daily after nerve injury for 7 days reversed nerve injury-induced C-fibre (5 Hz) hyposensitivity. (B and C) Similar treatment has no observable restorative effect on the nerve injury-induced A-fibre hypersensitivity to electrical stimulation at 2000 Hz (Aβ) and 250 Hz (Aδ). (D and E) Thermal hyperalgesia (D) and mechanical allodynia (E) accompanying nerve injury did not improve following chronic TSA treatment. (F and G) Nav1.8 sodium channel blocker (A-803467) treatment caused C-fibre hypoesthesia in sham-operated mice. In nerve-injured mice, recovery of nerve injury-induced loss of peripheral A-803467 hypoesthesia by TSA and VPA (EPW experiments, thermal paw withdrawal and paw pressure tests were conducted 30 min after treatment. Lines represent mean ± SEM of at least 3–8 independent experiments: *P < 0.05 vs. vehicle-sham, #P < 0.05 vs. injury-vehicle, +P < 0.05 vs. injury-TSA). PWL, Paw withdrawal latency; PWT, Paw withdrawal threshold.
The i.pl. treatment with A-803467, a Nav1.8 blocker, significantly increased the C-fibre threshold, but showed no further increase in the C-fibre threshold, which was elevated by nerve injury (Figure 2F). When the injury-induced hyposensitivity was reversed by TSA, the A-803467-induced increase in the C-fibre threshold appeared again. Similar results were also obtained with VPA treatment (Figure 2G). Together, these data provide the strong evidence for epigenetic repression of Nav1.8 gene as key factor in C-fibre hypoesthesia and that HDAC class I or IIa inhibitors are potential drug candidates for the treatment of C-fibre-related hypoesthesia following nerve injury.
Prevention of histone hypoacetylation by TSA treatment
Given the complexity of biological system, we sought to determine whether the chromatin modification occurred directly on the Nav1.8 gene or through indirect mechanisms. We precipitated cross-linked DRG samples of injured or sham-operated groups treated with TSA or vehicle using AcH3 (acetylated H3) or AcH4 (acetylated H4) antibodies followed by the amplification of the NRSE II (+30005/+30025) region of Nav1.8 (Otto et al., 2007), which is reported as the conserved reverse-oriented sequence within intron-10 capable of assembling histone-modifying protein complex (Mori et al., 2002; Uchida et al., 2010a). Our data showed that nerve injury invoked extensive deacetylation Lys14 of histone-3 (H3)-bound Nav1.8-NRSE II as indicated by significantly decreased Nav1.8-NRSE in vehicle-treated nerve-injury group (vs. sham-operated vehicle treatment, P < 0.05; Figure 3A). Recovery of Nav1.8-NRSE copy to pre-injury state following TSA treatment in nerve-injury group provide clear evidence for the direct deacetylation of N-terminal lysine residues (Lys14) of H3 following nerve injury as basis for Nav1.8 gene repression. Similar pattern of recovered acetylation was observed in acetyl-H4 antibody (recognizes acetyl-Lysine 5, 8, 12 and 16) precipitated samples following TSA treatment (Figure 3B). In both cases of anti-AcH3 and anti-AcH4 precipitation conditions, significant reduction in the copy number of Nav1.8-NRSE in vehicle treatment of injury groups (vs. vehicle sham) provide evidence of increased HDAC activity following nerve injury, explaining the rationale for improvement of some neuropathic pain symptoms following HDAC inhibitor treatment (Agrawal et al., 2009; Chiechio et al., 2009; Zhang et al., 2011).
Figure 3.
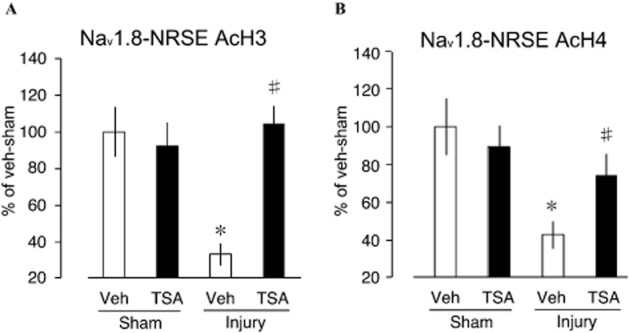
Prevention of histone 3 and 4 hypoacetylation by TSA. (A) Following nerve injury, N-terminal acetylated (Lys) histone 3-bound Nav1.8-NRSE II promoter was significantly reduced in TSA treatment reversible manner. (B) TSA treatment similarly increased the population of acetylated histone 4-bound Nav1.8-NRSE II promoter (plots represent mean ± SEM of 6–8 independent samples; *P < 0.05 vs. vehicle-sham, #P < 0.05 vs. injury-vehicle, +P < 0.05 vs. injury-TSA).
Reversal of decreased C-fibre sensitivity by SAHA
Our data provide compelling evidence in support of epigenetic role in C-fibre hypoesthesia following nerve injury. Therefore, to establish the clinical relevance of HDAC inhibitors in restoration of C-fibre functions following nerve injury, we performed additional experiments using SAHA treatment. SAHA (tradename: Zolinza) has been approved for the treatment of cancer via HDAC inhibition mechanisms (Marks et al., 2001). Starting at day 3 following nerve injury, examination of C-fibre sensitivity at day 1 of SAHA treatment (day 3 of injury) until day 9 of injury showed that SAHA treatment significantly restored C-fibre sensitivity at days 7 and 9 of injury at the ipsilateral side (Figure 4A), but has no detectable effect on the contralateral side (Figure 4B). This data therefore demonstrated that that the actions of VPA and TSA are via HDAC inhibition and that HDAC inhibitors may hold clinical benefits for the treatment of C-fibre hypoesthesia following nerve injury.
Figure 4.
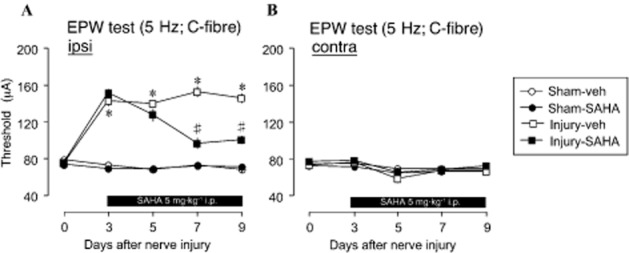
Blockade of C-fibre hypoesthesia by SAHA. (A) 5 mg·kg−1 i.p. Dose of SAHA starting at post-injury day 3 significantly reversed ipsilateral C-fibre hyposensitivity at day 7 of injury (day 3 of treatment) through day 9 of injury (treatment day 6). (B) The contralateral side showed no observable difference in C-fibre response to electrical stimulation following injury or SAHA treatment (plots represent mean ± SEM of 6–8 independent experiments; *P < 0.05 vs. vehicle-sham, #P < 0.05 vs. injury-vehicle, +P < 0.05 vs. injury-SAHA).
Restoration of decreased Nav1.8, TRPA1 and TRPM8 but not NRSF and CGRP gene expression by SAHA
In the ipsilateral side, injury-induced repression of Nav1.8 gene following nerve injury was reversed following 5 mg·kg−1 treatment of SAHA at day 3 to 9 of injury (5Figure A). This data explained the recovery of C-fibre sensitivity. As a control, the contralateral side showed no difference in the expression levels of Nav1.8 both in the nerve-injured or sham-operated groups. Down-regulation of Nav1.8 mRNA is therefore consequent to nerve injury without mirrored effects and that SAHA treatment has Nav1.8 mRNA up-regulation response only in injured tissues (Figure 5B). We further examined the mechanisms involved in the assembly of histone modifying complex following HDAC. At the ipsilateral side, SAHA-independent up-regulation of NRSF mRNA following nerve injury was observed (Figure 5C), confirming our earlier report that NRSF is key to epigenetic down-regulation of key genes mediating nociceptive response following nerve injury (Uchida et al., 2010a). The finding that up-regulated NRSF binds to Nav1.8-NRSE recruits and assembles co-repressor complexes including HDAC I and II (Ooi and Wood, 2007) accounts for the restorative actions of VPA, TSA and SAHA on the expression of Nav1.8.
Figure 5.
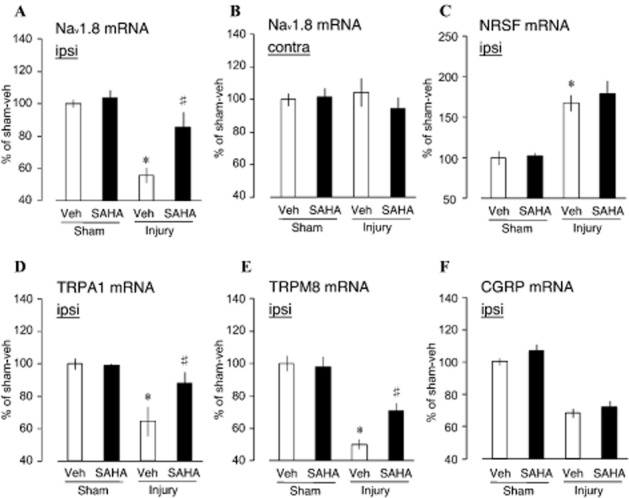
Restoration of Nav1.8, TRPA1 and TRPM8 gene expression but not NRSF and CGRP by SAHA. (A) 5 mg·kg−1 i.p. daily single dose of SAHA starting at post-injury day 3 to day 9 significantly restored ipsilateral Nav1.8 sodium channel expression in DRG. (B) At the contralateral side, SAHA treatment had no effect on the expression of Nav1.8 sodium channel. (C) NRSF, (D) TRPA1, (E) TRPM8 and (F) CGRP mRNA expressions in the DRG after nerve injury. The mRNA expression levels were assessed using RT and real-time PCR and normalized to that of GAPDH mRNA (plots represent mean ± SEM of 5–12 independent samples; *P < 0.05 vs. vehicle-sham, #P < 0.05 vs. injury-vehicle, +P < 0.05 vs. injury-SAHA).
Apart from Nav1.8, we further sought to establish whether SAHA has any effect on the expression levels of other key molecules involved in C-fibre nociception. We quantified TRPA1 and TRPM8 expression levels using RT and real-time PCR. Indeed, nerve injury-induced repression of TRPA1 (Caspani et al., 2009) was also reversed by SAHA (Figure 5D). Similarly, the down-regulated transcription of TRPM8 gene was also significantly reversed SAHA treatment (Figure 5E). Another key gene whose expression is down-regulated in the C-fibre following nerve injury is CGRP (Uchida et al., 2010a). The injury-induced down-regulation of CGRP expression was not recovered by SAHA treatment (Figure 5F), being consistent with the previous finding that NRSF AS-ODN treatments did not affect this down-regulation (Uchida et al., 2010a).
Discussion
Neuropathic pain condition following nerve injury presents two clinically observable sensations: the positive sensations, such as allodynia and hyperalgesia, and negative sensation observed as hypoesthesia (Ueda, 2006; Devigili et al., 2008). The plastic changes in the injured nerves and non-neuron cells that account for these clinical observations have been under intensive investigation (Inoue et al., 2004; Ma et al., 2009). The current understanding indicates that loss of C-fibre sensitivity evokes the negative symptoms (Fields et al., 1998; Uchida et al., 2010a) and the most obvious question is what transcriptional response is the response for the loss of C-fibre sensitivity. We have provided evidence for the transcriptional down-regulation of TRPA1, TRPM8 and Nav1.8 (Uchida et al., 2010a). Next, investigations were conducted into the mechanisms underlying the transcriptional repression of these genes. Interestingly, we found long-lasting up-regulation of NRSF following nerve injury negatively correlates with TRPA1, TRPM8, Nav1.8 sodium channel genes expression and AS-ODN-mediated knockdown of NRSF protected against this abnormal down-regulation and rescued the injury-induced behavioural phenotypes (Uchida et al., 2010a). Arguably, increased firing and ectopic discharge activities in sensory fibres following nerve injury may have driven sustained ERK1/2 phosphorylation (Xie et al., 2005) in the injured nerves. Downstream ERK1/2 activation, AP-1 (c-fos/c-jun) within the promoter region of NRSF is activated through ERK1/2-mediated c-jun phosphorylation resulting in long-lasting NRSF expression (Koenigsberger et al., 2000).
Accumulating intranuclear NRSF binds to Nav1.8-NRSE (Otto et al., 2007) and possible homologous sequence on TRPA1 and TRPM8, where other histone deacetylation complex are recruited and assembled (Bruce et al., 2004). Deacetylation of histone around these gene regulatory regions is required for gene silencing through chromatin modification and the mechanism involves the hydrolysis of acetyl-L-Lysine side chains of acetylated histones (Lombardi et al., 2011). ChIP data presented here strongly suggest extensive histone deacetylation of histone 3 and histone 4 bound to Nav1.8 sodium channel gene around the NRSE (+30005/+30025) region resulting in a more condensed heterochromatin structure (Xiao et al., 2003), which is essentially inaccessible to RNA polymerases for transcription resulting in transcriptional repression as observed in Nav1.8 sodium channel, TRPA1 and TRPM8. Consistent with the foregoing, following nerve injury, TSA administration altered histone acetylation by inhibiting histone deacetylase (Vanhaecke et al., 2004), providing a more RNA polymerase-accessible chromatin structure with resulting increased transcription. Of interest are the findings that injury-induced insensitivity of C-fibre was reversed by HDAC inhibitors, the epigenetic control might not fully explain the altered transcriptional response following nerve injury, as SAHA did not recover the reduced CGRP mRNA expression. This specificity may be important for future design of therapeutic epigenetic medicines with minimized side effects for chronic pain.
HDAC inhibitors also exhibited robust potential to provide much needed progress in the treatment of the behavioural phenotypes.
Acknowledgments
We thank Hitoshi Uchida for technical help. This work was supported by the Uehara Memorial Foundation and AstraZeneca Foundation.
Glossary
- ChIP
Chromatin immunoprecipitation
- CGRP
Calcitonin, gene-related peptide
- DRG
Dorsal root ganglion
- HDAC
Histone deacetylase
- p-APMSF
p-Amidinophenyl Methansulfonyl Fluoride
- PWL
Paw withdrawal latency
- PWT
Paw withdrawal threshold
- SAHA
Suberoylanilide hydroxamic acid
- TRPA1
Transient receptor potential cation channel, subfamily A, member 1
- TRPM8
Transient receptor potential cation channel, subfamily M, member 8
- TSA
Trichostatin
- VPA
Valproic acid
Conflict of interest
None.
References
- Agrawal RP, Goswami J, Jain S, Kochar DK. Management of diabetic neuropathy by sodium valproate and glyceryl trinitrate spray: a prospective double-blind randomized placebo-controlled study. Diabetes Res Clin Pract. 2009;83:371–378. doi: 10.1016/j.diabres.2008.12.018. [DOI] [PubMed] [Google Scholar]
- Bai G, Wei D, Zou S, Ren K, Dubner R. Inhibition of class II histone deacetylases in the spinal cord attenuates inflammatory hyperalgesia. Mol Pain. 2010;6:51. doi: 10.1186/1744-8069-6-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borrelli E, Nestler EJ, Allis CD, Sassone-Corsi P. Decoding the epigenetic language of neuronal plasticity. Neuron. 2008;60:961–974. doi: 10.1016/j.neuron.2008.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruce AW, Donaldson IJ, Wood IC, Yerbury SA, Sadowski MI, Chapman M, et al. Genome-wide analysis of repressor element 1 silencing transcription factor/neuron-restrictive silencing factor (REST/NRSF) target genes. Proc Natl Acad Sci U S A. 2004;101:10458–10463. doi: 10.1073/pnas.0401827101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caspani O, Zurborg S, Labuz D, Heppenstall PA. The contribution of TRPM8 and TRPA1 channels to cold allodynia and neuropathic pain. PLoS ONE. 2009;4:e7383. doi: 10.1371/journal.pone.0007383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiechio S, Zammataro M, Morales ME, Busceti CL, Drago F, Gereau RW, et al. Epigenetic modulation of mGlu2 receptors by histone deacetylase inhibitors in the treatment of inflammatory pain. Mol Pharmacol. 2009;75:1014–1020. doi: 10.1124/mol.108.054346. [DOI] [PubMed] [Google Scholar]
- Devigili G, Tugnoli V, Penza P, Camozzi F, Lombardi R, Melli G, et al. The diagnostic criteria for small fibre neuropathy: from symptoms to neuropathology. Brain. 2008;131:1912–1925. doi: 10.1093/brain/awn093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fields HL, Rowbotham M, Baron R. Postherpetic neuralgia: irritable nociceptors and deafferentation. Neurobiol Dis. 1998;5:209–227. doi: 10.1006/nbdi.1998.0204. [DOI] [PubMed] [Google Scholar]
- Hargreaves K, Dubner R, Brown F, Flores C, Joris J. A new and sensitive method for measuring thermal nociception in cutaneous hyperalgesia. Pain. 1988;32:77–88. doi: 10.1016/0304-3959(88)90026-7. [DOI] [PubMed] [Google Scholar]
- Inoue M, Rashid MH, Fujita R, Contos JJ, Chun J, Ueda H. Initiation of neuropathic pain requires lysophosphatidic acid receptor signaling. Nat Med. 2004;10:712–718. doi: 10.1038/nm1060. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG. Animal research: Reporting in vivo experiments: The ARRIVE guidelines. Br J Pharmacol. 2010;160:1577–1579. doi: 10.1111/j.1476-5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiso T, Nagakura Y, Toya T, Matsumoto N, Tamura S, Ito H, et al. Neurometer measurement of current stimulus threshold in rats. J Pharmacol Exp Ther. 2001;297:352–356. [PubMed] [Google Scholar]
- Koenigsberger C, Chicca JJ, Amoureux MC, Edelman GM, Jones FS. Differential regulation by multiple promoters of the gene encoding the neuron-restrictive silencer factor. Proc Natl Acad Sci U S A. 2000;97:2291–2296. doi: 10.1073/pnas.050578797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koga K, Furue H, Rashid MH, Takaki A, Katafuchi T, Yoshimura M. Selective activation of primary afferent fibers evaluated by sine-wave electrical stimulation. Mol Pain. 2005;1:13. doi: 10.1186/1744-8069-1-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu M, Wood JN. The roles of sodium channels in nociception: implications for mechanisms of neuropathic pain. Pain Med. 2011;12(Suppl. 3):S93–S99. doi: 10.1111/j.1526-4637.2011.01158.x. [DOI] [PubMed] [Google Scholar]
- Lombardi PM, Cole KE, Dowling DP, Christianson DW. Structure, mechanism, and inhibition of histone deacetylases and related metalloenzymes. Curr Opin Struct Biol. 2011;21:735–743. doi: 10.1016/j.sbi.2011.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma L, Uchida H, Nagai J, Inoue M, Chun J, Aoki J, et al. Lysophosphatidic acid-3 receptor-mediated feed-forward production of lysophosphatidic acid: an initiator of nerve injury-induced neuropathic pain. Mol Pain. 2009;5:64. doi: 10.1186/1744-8069-5-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath J, Drummond G, McLachlan E, Kilkenny C, Wainwright C. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marks P, Rifkind RA, Richon VM, Breslow R, Miller T, Kelly WK. Histone deacetylases and cancer: causes and therapies. Nat Rev Cancer. 2001;1:194–202. doi: 10.1038/35106079. [DOI] [PubMed] [Google Scholar]
- Matsumoto M, Inoue M, Hald A, Xie W, Ueda H. Inhibition of paclitaxel-induced A-fiber hypersensitization by gabapentin. J Pharmacol Exp Ther. 2006;318:735–740. doi: 10.1124/jpet.106.103614. [DOI] [PubMed] [Google Scholar]
- Matsumoto M, Xie W, Ma L, Ueda H. Pharmacological switch in Abeta-fiber stimulation-induced spinal transmission in mice with partial sciatic nerve injury. Mol Pain. 2008;4:25. doi: 10.1186/1744-8069-4-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori N, Mizuno T, Murai K, Nakano I, Yamashita H. Effect of age on the gene expression of neural-restrictive silencing factor NRSF/REST. Neurobiol Aging. 2002;23:255–262. doi: 10.1016/s0197-4580(01)00286-x. [DOI] [PubMed] [Google Scholar]
- Ooi L, Wood IC. Chromatin crosstalk in development and disease: lessons from REST. Nat Rev Genet. 2007;8:544–554. doi: 10.1038/nrg2100. [DOI] [PubMed] [Google Scholar]
- Otto SJ, McCorkle SR, Hover J, Conaco C, Han JJ, Impey S, et al. A new binding motif for the transcriptional repressor REST uncovers large gene networks devoted to neuronal functions. J Neurosci. 2007;27:6729–6739. doi: 10.1523/JNEUROSCI.0091-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tochiki K, Cunningham J, Hunt S, Géranton S, Tochiki KK, Hunt SP, et al. The expression of spinal methyl-CpG-binding protein 2, DNA methyltransferases and histone deacetylases is modulated in persistent pain states. Mol Pain. 2012;8:14. doi: 10.1186/1744-8069-8-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tracey I, Mantyh PW. The cerebral signature for pain perception and its modulation. Neuron. 2007;55:377–391. doi: 10.1016/j.neuron.2007.07.012. [DOI] [PubMed] [Google Scholar]
- Uchida H, Ma L, Ueda H. Epigenetic gene silencing underlies C-fiber dysfunctions in neuropathic pain. J Neurosci. 2010a;30:4806–4814. doi: 10.1523/JNEUROSCI.5541-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchida H, Sasaki K, Ma L, Ueda H. Neuron-restrictive silencer factor causes epigenetic silencing of Kv4.3 gene after peripheral nerve injury. Neuroscience. 2010b;166:1–4. doi: 10.1016/j.neuroscience.2009.12.021. [DOI] [PubMed] [Google Scholar]
- Ueda H. Molecular mechanisms of neuropathic pain-phenotypic switch and initiation mechanisms. Pharmacol Ther. 2006;109:57–77. doi: 10.1016/j.pharmthera.2005.06.003. [DOI] [PubMed] [Google Scholar]
- Ueda H. Peripheral mechanisms of neuropathic pain – involvement of lysophosphatidic acid receptor-mediated demyelination. Mol Pain. 2008;4:11. doi: 10.1186/1744-8069-4-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaissière T, Sawan C, Herceg Z. Epigenetic interplay between histone modifications and DNA methylation in gene silencing. Mutat Res. 2008;659:40–48. doi: 10.1016/j.mrrev.2008.02.004. [DOI] [PubMed] [Google Scholar]
- Vanhaecke T, Papeleu P, Elaut G, Rogiers V. Trichostatin A-like hydroxamate histone deacetylase inhibitors as therapeutic agents: toxicological point of view. Curr Med Chem. 2004;11:1629–1643. doi: 10.2174/0929867043365099. [DOI] [PubMed] [Google Scholar]
- Wang Y, Liu C, Guo QL, Yan JQ, Zhu XY, Huang CS, et al. Intrathecal 5-azacytidine inhibits global DNA methylation and methyl-CpG-binding protein 2 expression and alleviates neuropathic pain in rats following chronic constriction injury. Brain Res. 2011;1418:64–69. doi: 10.1016/j.brainres.2011.08.040. [DOI] [PubMed] [Google Scholar]
- Woolf CJ. Central sensitization: implications for the diagnosis and treatment of pain. Pain. 2011;152:S2–15. doi: 10.1016/j.pain.2010.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolf CJ American Physiological Society. Pain: moving from symptom control toward mechanism-specific pharmacologic management. Ann Intern Med. 2004;140:441–451. doi: 10.7326/0003-4819-140-8-200404200-00010. [DOI] [PubMed] [Google Scholar]
- Xiao B, Jing C, Wilson JR, Walker PA, Vasisht N, Kelly G, et al. Structure and catalytic mechanism of the human histone methyltransferase SET7/9. Nature. 2003;421:652–656. doi: 10.1038/nature01378. [DOI] [PubMed] [Google Scholar]
- Xie W, Strong JA, Meij JT, Zhang JM, Yu L. Neuropathic pain: early spontaneous afferent activity is the trigger. Pain. 2005;116:243–256. doi: 10.1016/j.pain.2005.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Cai YQ, Zou F, Bie B, Pan ZZ. Epigenetic suppression of GAD65 expression mediates persistent pain. Nat Med. 2011;17:1448–1455. doi: 10.1038/nm.2442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmermann M. Ethical guidelines for investigations of experimental pain in conscious animals. Pain. 1983;16:109–110. doi: 10.1016/0304-3959(83)90201-4. [DOI] [PubMed] [Google Scholar]