

Abstract
Background and Purpose
Selective MAO type B (MAO-B) inhibitors are effective in potentiation of the clinical effect of L-DOPA in Parkinson's disease, but dopamine (DA) is deaminated mainly by MAO type A (MAO-A) in rat brain. We sought to clarify the roles of MAO-A and MAO-B in deamination of DA formed from exogenous L-DOPA in rat striatum depleted of dopaminergic, or both dopaminergic and serotonergic innervations. We also studied the effect of organic cation transporter-3 (OCT-3) inhibition by decinium-22 on extracellular DA levels following L-DOPA.
Experimental Approach
Striatal dopaminergic and/or serotonergic neuronal innervations were lesioned by 6-hydroxydopamine or 5,7-dihydroxytryptamine respectively. Microdialysate DA levels after systemic L-DOPA were measured after inhibition of MAO-A or MAO-B by clorgyline or rasagiline respectively. MAO subtype localization in the striatum was determined by immunofluorescence.
Key Results
Rasagiline increased DA extracellular levels following L-DOPA to a greater extent in double-than in single-lesioned rats (2.8-and 1.8-fold increase, respectively, relative to saline treatment); however, clorgyline elevated DA levels in both models over 10-fold. MAO-A was strongly expressed in medium spiny neurons (MSNs) in intact and lesioned striata, while MAO-B was localized in glia and to a small extent in MSNs. Inhibition of OCT-3 increased DA levels in the double-more than the single-lesion animals.
Conclusions and Implications
In striatum devoid of dopaminergic and serotonergic inputs, most deamination of L-DOPA-derived DA is mediated by MAO-A in MSN and a smaller amount by MAO-B in both MSN and glia. OCT-3 plays a significant role in uptake of DA from extracellular space. Inhibitors of OCT-3 are potential future targets for anti-Parkinsonian treatments.
Keywords: Parkinson's disease, L-DOPA, rasagiline, clorgyline, 6-hydroxydopamine, glial cells, microdialysis, OCT-3, medium spiny neurons
Introduction
L-DOPA treatment of Parkinson's disease (Pd) is commonly referred to as dopamine (DA) replacement therapy; however, the precise way in which L-DOPA replaces DA lost from the Parkinsonian brain is far from clear. Because L-DOPA is decarboxylated to DA by aromatic L-amino acid decarboxylase (AAAD; EC 4.1.1.28), DA can be produced from L-DOPA not only in dopaminergic neurons, but in all cells that possess this enzyme, such as certain populations of glial cells, blood vessels, medium spiny neurons (MSN), small aspiny interneurons and serotonergic neurons (Tashiro et al., 1989; Mura et al., 1995; Lopez-Real et al., 2003; Vialou et al., 2007). Following dopaminergic deafferentation with 6-hydroxydopamine (6-OHDA) serotonergic axon density increases in striatum (Maeda et al., 2003), and DA has been detected in serotonergic axonal varicosities following L-DOPA administration in 6-OHDA-treated rats (Yamada et al., 2007). DA production from L-DOPA in 6-OHDA-treated rats is markedly reduced following serotonergic deafferentation with 5,7-dihydroxytryptamine (5,7-DHT; Tanaka et al., 1999; Navailles et al., 2010). These observations are strongly indicative of a role of serotonergic neurons in production of DA from L-DOPA in Parkinsonian striatum.
The level of DA in extracellular fluid, which is proportional to the synaptic level, is controlled by neuronal and extraneuronal uptake and metabolism by the intracellular enzymes MAO and catechol O-methyltransferase. Following dopaminergic deafferentation of the striatum in rats, levels of DA in striatal extracellular fluid after systemic L-DOPA administration (as detected by microdialysis) increase to values higher than those seen in intact brain (Miller and Abercrombie, 1999), reflecting the efficient removal of DA from the synapse by the DA transporter (DAT) in normal brain. In Parkinsonian brain, in which DAT is absent, DA formed from L-DOPA can be taken up by serotonergic neurons, since these neurons express the serotonergic transporter (SERT), which is also capable of transporting DA across the axolemma (Carta et al., 2007). Although serotonergic perikarya of the raphe nucleus express MAO-B, serotonergic axon terminals are thought to express MAO-A, because serotonin behaves as a MAO-A substrate in vivo (Green and Youdim, 1975). DA can also be taken up and deaminated in astrocytes, which express MAO type B (MAO-B; Ekblom et al., 1993), the plasma membrane noradrenaline transporter (Inazu et al., 2003), and the organic cation transporter-3 (OCT-3; Cui et al., 2009). The role of the low-affinity transporter OCT-3 in clearance of DA from the extracellular space has been little studied; however, this transporter could become more important in the clearance of L-DOPA-derived DA in Parkinsonian striatum.
In the intact rat brain, both subtypes A and B of MAO are expressed in similar proportions (Youdim and Finberg, 1983), but MAO-A is the dominant form responsible for both DA and 5-HT metabolism, as seen by a much larger effect of MAO-A than MAO-B inhibition in suppressing formation of striatal 3,4-dihydroxyphenylacetic acid (DOPAC), homovanillic acid (HVA) and 5-hydroxyindoleacetic acid (5-HIAA), and increasing DA microdialysate levels (Green and Youdim, 1975; Waldmeier et al., 1976; Wachtel and Abercrombie, 1994; Finberg et al., 1995). In early-stage Pd patients, administration of the selective MAO-B inhibitors selegiline and rasagiline in monotherapy produces a beneficial dopaminergic effect, and in advanced stage patients, MAO-B inhibition reduces fluctuations in response to L-DOPA (Parkinson Study Group, 2005). Inhibitors of MAO-A cannot be administered to PD patients treated with L-DOPA, because of the possibility of serious cardiovascular reactions.
The object of this study was to clarify the effect of MAO-A and MAO-B inhibition on L-DOPA-derived extracellular DA levels in striatum devoid of dopaminergic innervation, and both dopaminergic and serotonergic innervations. Because the extent of DA metabolism by MAO-A or-B depends on the localization of the MAO subtypes to neuronal and glial elements, we have further clarified the location of MAO subtypes in striatum by immunofluorescence, as well as their enzyme activities in striatal tissue following lesioning. In addition, we have probed the role of low-affinity uptake by OCT-3 in clearance of DA from the extracellular space in both lesion models.
Methods
Experimental animals
All procedures with animals were authorized by the Technion Animal Care and Use Committee, whose ethical standards are based on those detailed in the National Institutes of Health (Bethesda, MD, USA) Guide for the Care and Use of Laboratory Animals, and whose general procedures for animal welfare comply with Israeli law on animal experimentation. The study procedures and results are reported in compliance with the ARRIVE guidelines (Kilkenny et al., 2010; McGrath et al., 2010). All experiments were carried out on male Sprague-Dawley rats, which were obtained from Harlan Laboratories, Jerusalem, at 7 weeks of age, and were maintained in our animal facility for an additional 2 weeks before use. Animals were fed rat pellets and water ad libitum, and were caged in plastic individually ventilated cages in a non-sterile room of our animal department, under 12 h light/dark regimen at an environmental temperature of 21 ± 1°C.
Materials
5-[2-14C]-hydroxytryptamine binoxalate and β-[ethyl-1-14C]-phenylethylamine hydrochloride (14C-PEA) were purchased from Perkin Elmer (Waltham, MA, USA), alkaline phosphatase conjugated anti-rabbit antibody and p-nitrophenyl phosphate from Chemicon (Millipore, Billerica, MA, USA). Rasagiline and carbidopa were obtained from Teva Pharmaceuticals (Jerusalem, Israel). Isoflurane was purchased from Nicholas Piramal (Mumbai, India). Mouse anti-dopamine and cAMP-regulated phosphoprotein, Mr 32 kDa (DARPP-32) (sc-271111), rabbit anti-MAO-A (H-70) and goat anti-MAO-B (D-16) antibodies were obtained from Santa Cruz (Santa Cruz, CA, USA). Rabbit anti-DARPP-32 (19A3) antibody was purchased from Cell Signaling Technology (Danvers, MA, USA). Rabbit anti-OCT3 (OCT31-A) antibody was obtained from Alpha Diagnostics (San Antonio, TX, USA). Monoclonal anti-tyrosine hydroxylase and monoclonal anti-tryptophan hydrxylase antibodies were obtained from Sigma-Aldrich (Rehovot, Israel). Alexa fluor conjugated antibodies were purchased from Invitrogen (Carlsbad, NY, USA). All other compounds and antibodies were obtained from Sigma-Aldrich. Microdialysis guide cannulas and probes were purchased from CMA Microdialysis (Kista, Sweden).
Lesioning procedure
Rats were administered desipramine 10 mg·kg−1 s.c., anaesthetized with ketamine/xylazine (70/7 mg·kg−1 i.p.) and placed in a stereotaxic frame (David Kopf Instruments, Tujunga, CA, USA). A thermostatically controlled heating pad maintained the body temperature constant at 37°C and blood gases were monitored. For single (dopaminergic only) lesion, 6-OHDA (8 μg in saline with 0.1% ascorbic acid) was stereotactically injected into the left medial forebrain bundle (coordinates 4.4 mm posterior, 1.5 mm left, 8 mm ventral with respect to bregma). For additional serotonergic lesioning (double lesion), rats were injected with 5,7-DHT (150 μg free base in saline with 0.1% ascorbic acid) into the left lateral ventricle (coordinates: 0.8 mm posterior, 1.4 mm left, 3.4 mm ventral with respect to bregma) during the same operation. Sham-lesioned rats were subjected to the same procedures using 0.9% saline (with 0.1% ascorbic acid) for injections. The extent of dopaminergic lesion was indicated by injection of apomorphine (0.1 mg·kg−1 s.c.), 3 weeks after lesioning. Only rats with intensive contralateral rotation (more than 100 complete contralateral turns in the 60 min following injection, and more than 20 contralateral turns between 15 and 20 min following the injection) induced by apomorphine were used for the present experiments. These criteria have been found in our laboratory to select rats with more than 99% dopaminergic lesion, which was additionally verified by: (i) determination of striatal tissue DA levels at end of microdialysis experiments and (ii) immunohistological examination of tyrosine-hydroxylase-positive neurons in substantia nigra in an additional group of 11 double-lesioned and 7 sham-operated rats. The extent of serotonergic lesion of striatal tissue was measured: (i) by determination of striatal tissue levels of 5-HIAA at the end of microdialysis experiments and (ii) immunohistological examination of tryptophan hydroxylase-positive neurons in dorsal raphe nucleus in the same additional group of double-lesioned rats.
Determination of MAO activity
The effect of 6-OHDA and 5,7-DHT lesions on striatal tissue MAO activity was determined in a separate group of rats not administered MAO inhibitors, but which fulfilled the criteria for extensive 6-OHDA lesioning as described later for apomorphine-induced turning, and were killed by decapitation under isoflurane anaesthesia 1 week after apomorphine administration. MAO activity in rat striatum was determined in vitro using a radiometric method (Otsuka and Kobayashi, 1964), with modifications (O'Carroll et al., 1983). Briefly, 4 weeks after the induction of the lesions, and 1 week after apomorphine screening test, rats were decapitated under ketalar/xylazine anaesthesia, striata were dissected and homogenized in sucrose (0.32 M). For MAO-B activity determination, homogenates were pre-incubated with clorgyline (0.15 μM) for 60 min then with 14C-PEA for 20 min at 37°C. For MAO-A activity determination, homogenates were pre-incubated with selegiline (0.15 μM) for 60 min, then were incubated with 14C-5-HT for 30 min at 37°C. The deaminated products were extracted into toluene/ethyl acetate (1:1, vol/vol), to which was added a solution of 2,5-diphenyl oxazole to a final concentration of 0.4% w·v−1 before determination by liquid scintillation counting (LKB-Wallac Rakbeta 1211). MAO activity is expressed as the amount of radioactive deaminated product per microgram of protein in a 20 or 30 min incubation period as appropriate.
Gliosis quantitation
Because an increase in MAO-B activity was found in some of the lesioned rats, and because MAO-B is the isoform associated with glial cells, we quantitated the degree of gliosis induced by single-and double-lesion procedures by determination of the glial marker protein glial fibrillary acidic protein (GFAP) in striatal tissue homogenates, and by GFAP-positive cell counting in striatal tissue sections.
GFAP quantification was carried out on the striatal sucrose homogenate used for determination of MAO activity. Aliquots of the sucrose homogenate were diluted in lysis buffer (15 mM Tris, 250 mM sucrose, 1 mM EDTA, 0.05% NP-40), sonicated and then centrifuged for 10 min at 4°C, 1000 g. The supernatant was assayed with sandwich ELISA. Briefly, a 96-well plate was coated with mouse anti-GFAP (dilution 1:500) overnight at 4°C, homogenates (300 ng protein each well) were added and incubated for 1 h at room temperature. Non-specific binding was blocked with 1% BSA followed by incubation for 1 h with rabbit anti-GFAP (dilution 1:500). Finally, alkaline phosphatase conjugated anti-rabbit antibody and its substrate p-nitrophenyl phosphate were used to detect bound primary antibody. A linear standard curve was obtained correlating the optical density of the product with the amount of protein in the homogenate.
Additional groups of lesioned rats were prepared for GFAP-positive cell counts: sham lesion (n = 4), single lesion (n = 6) and double lesion (n = 8). Four weeks after the induction of the lesions and 1 week after the apomorphine screening test, rats were anaesthetized with ketamine/xylazine and perfused via the left cardiac ventricle with PBS (pH = 7.4, 10 mL) followed by 4% paraformaldehyde in PBS. Brains were removed and post-fixed for 4 days. After dehydration in alcohols (70, 80, 96% ethanol, then 100% isopropanol) and clearing in organic solvent (chloroform) brains were embedded in paraffin. Coronal sections (5 μm) of striatum from levels 1.6, 1.2, 0.8, 0.5 and 0.2 mm anterior to bregma were deparaffinized in xylene, rehydrated in decreasing concentrations of ethanol and submitted to antigen retrieving procedure by microwaving in phosphate-citrate buffer pH = 6.8 (0.017 M citric acid and 0.066 M dibasic sodium phosphate in double-distilled water). After permeabilization in 0.5%Triton X100 in PBS and quenching of endogenous peroxidase in hydrogen peroxide (0.3% in absolute ethanol), sections were blocked in PBS containing 5% BSA and 5% FBS with 0.1% Triton X100 and incubated overnight in mouse anti-GFAP antibody 1:500 (Sigma). For antibody visualization sections were treated with biotinylated secondary antibody to mouse, followed by HRP-streptavidin complex, then exposed to 3-amino 9-ethylcarbazole (AEC) chromogen reagent and counterstained with haematoxylin (Histostain-SP Kit, Zymed Laboratories Inc., Invitrogen). For GFAP-positive cell counting, 25 pictures were taken from left striatum at each level from bregma as shown earlier. Only GFAP-positive glial cells sectioned through the central part of the nucleus were counted (125 pictures for every rat) and data were statistically analysed by one-way anova followed by Bonferoni multiple comparison test.
Protein determination
Protein content was obtained using the Lowry assay (Lowry et al., 1951).
Microdialysis experiments
One day before the microdialysis study, rats were anaesthetized with ketamine/xylazine (70:7 mg·kg−1 i.p.) and a silicon guide cannula (CMA/12) was implanted in the skull with its end directed to the upper limit of the left striatum (0.2 mm anterior, 2.5 mm left, 3.5 mm ventral to bregma, according to a rat brain stereotaxic atlas (Paxinos and Watson, 1982). Rats were left to recover for 24 h. On the next day, rats were anaesthetized using isoflurane inhalation and maintained on isoflurane anaesthesia administered via a nose cone while the rat's body temperature was maintained at 37°C with a thermostatically controlled heating pad. A dialysis probe (CMA12, 4 mm membrane length) was inserted into the guide cannula and perfused continuously with artificial CSF (composition in mM: NaCl 14.7, KCl 0.27, CaCl2 0.12, MgCl2 0.085) at a flow rate of 2 μL·min−1. After a 1 h equilibration period, carbidopa (6 mg·kg−1) was given i.p., and then baseline dialysate collection was commenced each 20 min for 1 h in tubes containing 10 μL of a preservative solution containing 0.1 M perchloric acid and 0.05 mM disodium EDTA. L-DOPA (25 mg·kg−1) was then given i.p., and sample collection was continued for 5 h. At the end of the experiment, rats were killed and striata were dissected for MAO activity measurement as well as DA and 5-HIAA content determination, and the position of the probe was determined by direct observation of the probe track.
Rats bearing either single or double lesions were randomly assigned for daily MAO inhibitor treatment 2 weeks after the apomorphine test. Saline, rasagiline 0.05 mg·kg−1 or clorgyline 0.2 mg·kg−1 were administered s.c. daily for 14 days. On day 14, the microdialysis experiment was carried out as described earlier, and on this day, MAO inhibitors were given 2 h before L-DOPA injection.
In a separate experiment designed to test the effect of partial MAO-A inhibition, rats bearing the double lesion were treated daily with either clorgyline 0.0033 mg·kg−1 s.c. or saline for 14 days. On day 14, the microdialysis experiment was carried out as described earlier, and on this day, clorgyline was given 2 h before L-DOPA injection. The doses of carbidopa and L-DOPA were as mentioned earlier.
Microdialysis following inhibition of the OCT-3 in single-and double-lesion rats was carried out as described earlier 7 days after screening with apomorphine, but microdialysis tubings were filled with either artificial CSF or 50 μM decynium-22 (D-22; OCT-3 inhibitor). Carbidopa and L-DOPA were administered as mentioned earlier.
Dialysate samples were analysed for DA, DOPAC and HVA by HPLC with electrochemical detection. Separation of DA and its metabolites was achieved using an Inertsil ODS-2 column (GL Sciences, Tokyo, Japan) with a mobile phase composed of 100 mM sodium dihydrogen phosphate, 1 mM octanesulfonic acid and 269 μM disodium EDTA, with 2.5% v/v methanol and 4.5% v/v acetonitrile, in HPLC grade deionized water, pH 2.75. Detection of compounds was enabled by an ESA Coulochem II (model 5200, ESA, Chelmsford, MA, USA) electrochemical unit operated in redox mode and coupled to an ESA guard cell at a potential of +300 mV placed before the analytical cell (ESA model 5010). Column eluates were reduced to +100 mV at detector 1 of the analytical cell, and measured at −400 mV at detector 2. The limit of detection for DA, DOPAC and HVA was 0.01 pmol.
Immunofluorescence determination of MAO-A and MAO-B localization in brain sections
Brain sections (5 μm) were prepared as described for GFAP-positive cell counts and permeabilized with 0.5% Triton in PBS for 10 min at room temperature. Afterwards, a blocking solution (for MAO-A staining, 5% goat serum and 1% BSA, for MAO-B only 5% BSA in PBS) was added, and sections were incubated for 2 h at room temperature. Incubation with primary antibodies [rabbit anti-MAO A 1:100, goat anti-MAO B 1:150, anti-GFAP 1:1000, rabbit anti-OCT-3 1:100, mouse anti-tyrosine hydroxylase 1:1000, mouse anti-DARPP-32 (DA-and cAMP-regulated phosphoprotein, Mr 32 kDa)1:500, rabbit anti-DARPP-32 1:500] was carried out overnight at 4°C and incubation with secondary fluorescent antibodies (1:1000) for 1 h at room temperature. Sections were observed with a laser confocal microscope (LSM 510 Meta).
Immunohistochemical determination of tyrosine hydroxylase-and tryptophan hydroxylase-positive neurons
This experiment was carried out to verify the degrees of dopaminergic and serotonergic lesioning produced by the procedures mentioned earlier. An additional group of 11 double-lesioned and 7 control rats were prepared for in vivo fixation as described for GFAP-positive cell counts. Coronal 25 μm sections were cut at topographically-identified levels (for substantia nigra from −4.8 to −6.0 and for raphe nucleus from −7.0 to −9.2 mm from bregma according to stereotactic atlas of Paxinos and Watson, 1982). For calculation of raphe nucleus cells, sections were taken every 200 μm between the bregma levels mentioned earlier and only cells with visible nucleus were counted. Immunohistochemical detection of tyrosine hydroxylase and tryptophan hydroxylase was carried out as described for GFAP-positive cell counts. Secondary antibodies were visualized using AEC or 3-3′-diaminobenzidine tetrahydrochloride (DAB).
Statistical analysis
All data are presented as mean ± SEM or mean + SEM for graphical data. One way anova was applied on the results of MAO activity and GFAP expression followed by Bonferroni multiple comparison test. Statistical analysis of microdialysis data was done using two-way anova followed by Bonferroni multiple comparisons test post hoc for difference between treatments at each time point. For comparison of maximum peak levels unpaired t-test was used. All statistical analyses were performed on original data, not on percentages.
Results
Striatal MAO activity, amine depletion and GFAP expression, following single and double lesions
Initially, we determined the alteration in MAO activity in striatal tissue in rats given single and double lesions. The extent of lesioning of dopaminergic and serotonergic striatal inputs was determined as described in Methods. In rats given a 6-OHDA lesion, and which showed the stated rotation criteria, striatal DA tissue levels were reduced by 99.6 ± 0.087% control (n = 7 single-and 7 double-lesioned striata, mean ± SEM). The 5,7-DHT lesion produced 92.4 ± 1.05% (mean ± SEM, n = 54) reduction in striatal 5-HIAA content compared with intact striata indicating denervation of most 5-HT terminals innervating the striatum (Hall et al., 1999; Tanaka et al., 1999). The nearly complete absence of tyrosine hydroxylase-positive neurons in SNpc in an identically prepared group of double-lesioned rats was confirmed by immunohistochemistry (Figure 1), and cresyl violet staining confirmed the absence of cells with the typical morphology of dopaminergic neurons in lesioned SNpc. Numbers of tryptophan hydroxylase-positive neurons in dorsal part of dorsal raphe nucleus were reduced by 86.4 ± 2.9% in this additional group of lesioned rats (representative section shown in Figure 1). In view of the very high dopaminergic lesion extent used in this study, in which the extensive lesion develops rapidly (within a few days) and recovery of innervation does not occur (Schwarting and Huston, 1996), the exact timing of lesion extent determination in relation to microdialysis and immunohistochemical determinations is not critical, and was carried out either 4 or 7 weeks after administration of the neurotoxins. Gliosis extent was also not expected to alter substantially over this time period, as 4 to 7 weeks is after the reactive period when gliosis commences.
Figure 1.
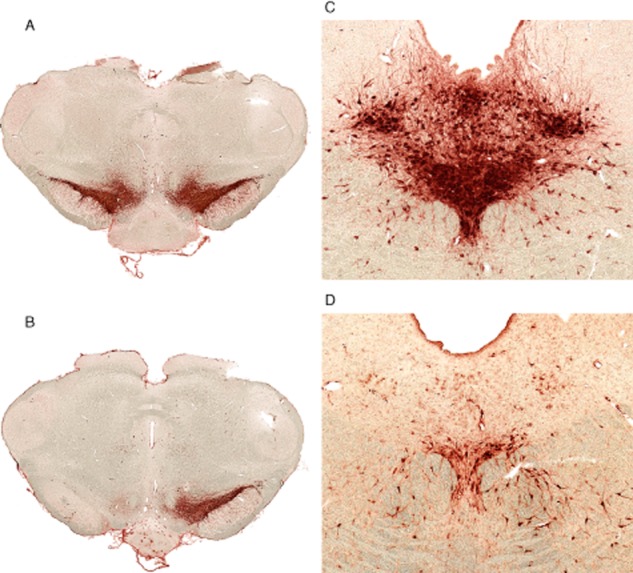
Immunohistochemical demonstration of lesion extent by 6-OHDA and 5,7-DHT. (A, B) tyrosine hydroxylase-positive neurons in coronal midbrain sections of normal rat brain (A) and rat with double lesion (B), 4 weeks after lesioning. (C, D) tryptophan hydroxylase-positive neurons in dorsal raphe nucleus area of normal (C) and double-lesioned (D) brain 4 weeks after lesioning. Ruler, 1 mm.
Striatal MAO-A activity was not affected by either single or double lesion (Figure 2). On the other hand, there was a significant increase in MAO-B activity in the double lesioned striata (137.5 ± 0.06%, P < 0.01) compared with that of sham-operated rats four weeks after lesioning. A significant increase was seen also when compared with single-lesioned striata (P < 0.05, Figure 2A). Expression of the glial marker GFAP was measured in the same homogenates of the striata (the lesioned side) used for the MAO activity assay and significantly increased (P < 0.001) in the double-lesioned striata compared with sham and to single-lesioned striata, while there was no significant change in its expression in the single-lesioned striata 4 weeks after lesioning (Figure 3A). Glial cell counts were significantly increased (P < 0.001) in both single-and double-lesioned striata immunostained for GFAP expression compared with sham-lesioned striata, but a larger increase was seen in the double-lesioned compared with the single-lesioned striata (Figure 3B). The greater fold increase in the glial marker GFAP as shown by elisa compared with GFAP-positive cell counts is probably the result of increased astrocytic processes in each astrocyte of the newly formed glial cells, as GFAP is expressed throughout the astrocytic cell. The fact that double-lesioned rats showed a greater degree of gliosis than single-lesioned rats is possibly due to the higher exposure of the target striatal tissue to 5,7-DHT than to 6-OHDA, the latter having been injected to the tissue surrounding the medial forebrain bundle while the former was injected intra-cerebroventricularly.
Figure 2.
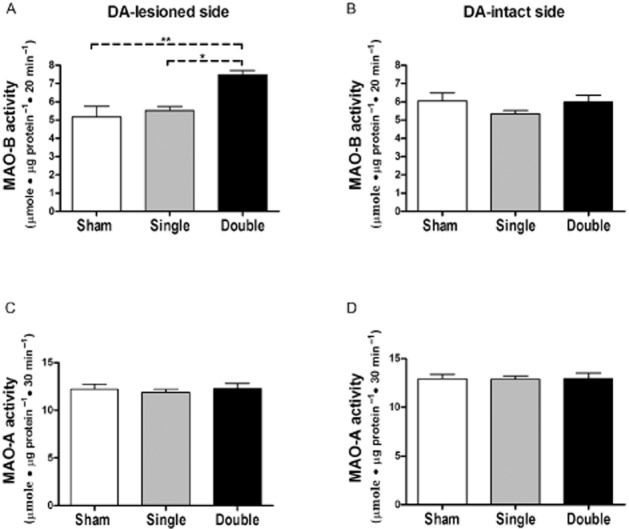
Striatal MAO activity in lesioned rats. MAO-B (A and B) and MAO-A (C and D) activities were determined in DA-intact and DA-lesioned side of striatum from rats bearing either unilateral sham (n = 4), unilateral single (dopaminergic; n = 7) or double (unilateral dopaminergic and bilateral serotonergic; n = 6) lesions 4 weeks after lesioning. Data are expressed as mean + SEM of μmole metabolite·μg protein−1·30 min−1 for MAO-A, and μmole metabolite·μg protein−1·20 min−1 for MAO-B. *P < 0.05, **P < 0.01 by one way anova followed by Bonferroni multiple comparison test.
Figure 3.
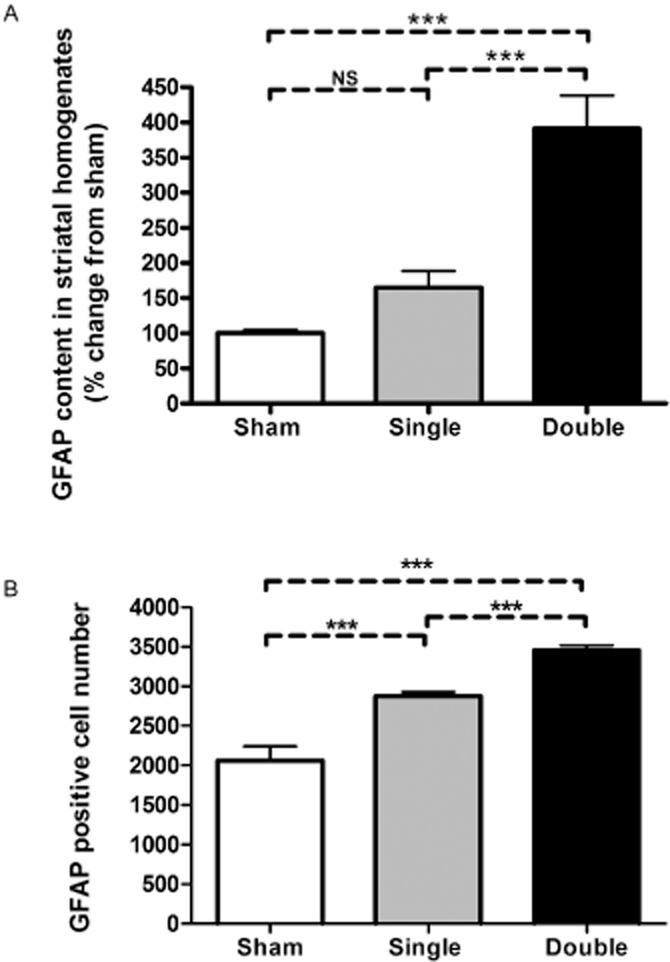
(A) GFAP quantitation in homogenates of the lesioned striata of rats bearing either unilateral sham, unilateral single (dopaminergic) or double (unilateral dopaminergic and bilateral serotonergic) lesions. GFAP content in the striatal homogenates was measured by sandwich ELISA 4 weeks after the induction of lesion (n = 4 for sham, n = 7 for single-and n = 6 for double-lesion rats), and is expressed as mean % change from sham-lesioned striata + SEM. *** P < 0.001 by one-way anova followed by Bonferroni multiple comparison test. NS, not significant. (B) GFAP positive cell counts in 5 μm sections of DA-lesioned striata of rats bearing either unilateral sham (n = 4), single (dopaminergic, n = 6) or double (dopaminergic and serotonergic, n = 8) lesion. For each rat, 25 coronal photomicrographs were taken from left striatum at each of the levels1.6, 1.2, 0.8, 0.5 and 0.2 mm anterior to bregma. Data shown represent the average counts + SEM for each group. ***P < 0.001 by one-way anova followed by Bonferroni multiple comparison test.
Striatal MAO activity following clorgyline and rasagiline treatment
The dose of clorgyline used for selective inhibition of MAO-A (0.2 mg·kg−1 s.c. daily for 14 days) caused 94.9 ± 2.4% inhibition of MAO-A and 22.5 ± 12.4% inhibition of MAO-B. The dose of rasagiline used for selective inhibition of MAO-B (0.05 mg·kg−1 s.c. daily for 14 days) caused 92.9 ± 1.5% inhibition of this enzyme form, and 14.9 ± 9.2% (P > 0.05) inhibition of MAO-A. A similar extent of inhibition of MAO-A as that associated with this dose of rasagiline was caused by 0.0033 mg·kg−1 s.c. clorgyline daily for 14 days (18.7 ± 5.8%, P < 0.05) with insignificant inhibition of MAO-B.
MAO inhibition effects on microdialysate levels of DA and metabolites following L-DOPA administration
Systemic administration of L-DOPA causes a marked increase in striatal extracellular DA levels following lesion of the nigro-striatal pathway, which is reduced by additional lesion of the serotonergic input (Tanaka et al., 1999). Following on this finding, we probed the role of MAO-A and MAO-B in metabolism of DA produced from L-DOPA in single-and double-lesioned rats, by use of isoform-selective MAO inhibitors. Microdialysate DA levels before L-DOPA administration in saline-treated single and double-lesion rats were very low and close to detection limit (Figure 4A and B). We confirmed the findings of Tanaka et al. (1999), that following L-DOPA administration, DA levels in saline-treated rats were higher in those given a single lesion than in double lesion rats (P < 0.001 by two-way anova over the time period 20–200 min post L-DOPA). In both lesion models, the levels of the L-DOPA metabolite, 3-O-methyl DOPA, were similar in all treatment groups (not shown), indicating a consistency of L-DOPA dosing and absorption following the i.p. administration.
Figure 4.
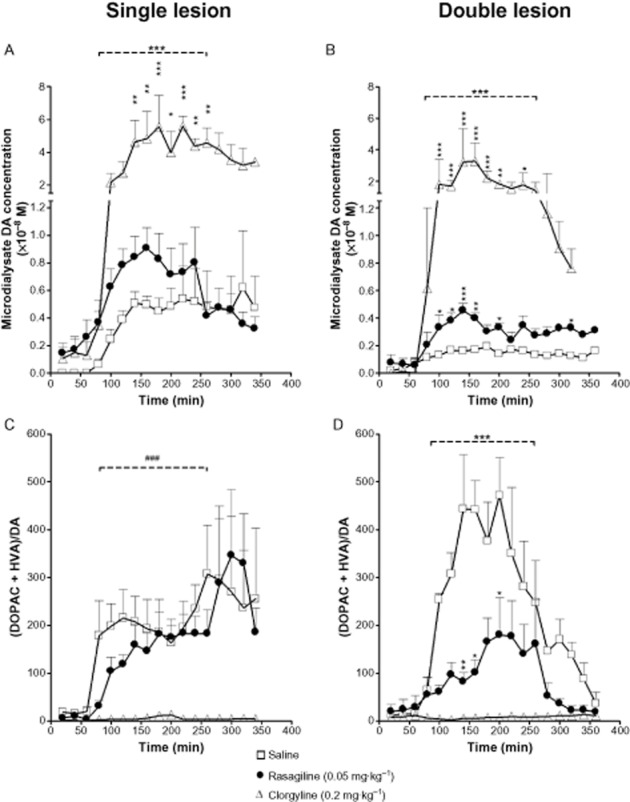
Microdialysis study of L-DOPA-derived DA and DA metabolite levels in DA-lesioned striatum of single (unilateral dopaminergic only) or double (unilateral dopaminergic plus bilateral serotonergic) lesion rats. Rats bearing either single or double lesion were treated daily for two weeks with either saline, rasagiline (0.05 mg·kg−1) or clorgyline (0.2 mg·kg−1), by s.c. injection. Rasagiline increased L–DOPA-derived DA levels in both lesion models when compared with saline treatment; however, the magnitude of fold increase over saline control in peak DA levels was larger in the double lesion (B) compared with the single lesion rats (A) (2.8-vs. 1.8–fold, respectively) and duration of increased DA level was also increased in the double lesion group. This effect was accompanied by a decrease in DA oxidative metabolism (expressed as [DOPAC + HVA]/DA ratio) in the double lesion model only (D). Clorgyline treatment caused a significant difference from saline in all post-L-DOPA DA and metabolite levels. Rats were anaesthetized with isoflurane on day 14, microdialysis probes were inserted into lesioned striatum via a guide cannula implanted 24 h previously and MAO inhibitor was administered. One hour later carbidopa (6 mg·kg−1 i.p.) was administered and microdialysis collections were started (time 0). L-DOPA methyl ester (25 mg·kg−1 i.p.) was administered 60 min later. Data are expressed as mean DA or metabolite dialysate concentration + SEM for n = 4–6 rats per group. For horizontal bars in A, B and D, ***P < 0.001 over the time period between 20 and 200 min post L-DOPA for difference between both rasagiline and clorgyline curves from saline curve by two way anova. Bonferroni post hoc test was performed for comparison between treatments at each time point (*P < 0.05, **P < 0.01, ***P < 0.001). In C, ###P < 0.001 for statistical significance over the time period between 20 and 200 min post L-DOPA for difference between clorgyline curve only from saline curve by two-way anova.
Prior treatment with clorgyline increased significantly DA levels in both lesion models (P < 0.001 by two-way anova over the time period of 20–200 min post L-DOPA; Figure 4A and B) leading to an increase in peak DA levels following L-DOPA of more than 10 fold in both single (55.4 ± 19.1 vs. 4.5 ± 0.9 nM, ratio 12.3, unpaired t-test at 180 min P < 0.05) and double (32.1 ± 12.2 vs. 1.6 ± 0.3 nM, ratio 19.7, unpaired t-test at 140 min P < 0.05) lesion groups. Rasagiline treatment also significantly increased DA levels following L-DOPA in both lesion models (P < 0.001 by two-way anova over the time period 20–200 min post L-DOPA), but to a much smaller extent than clorgyline. It should be noted, however, that the magnitude of effect of rasagiline compared with saline pretreatment on peak DA levels post L-DOPA in double-lesion rats (4.5 ± 0.55 vs. 1.6 ± 0.2 nM, ratio 2.8, unpaired t-test at 140 min P < 0.01) was greater than that in single-lesion rats (9.1 ± 1.5 vs. 5.0 ± 0.7 nM, ratio 1.8, unpaired t-test at 160 min P < 0.05). Moreover, higher DA levels were maintained in rasagiline-treated rats for a longer period of time following L-DOPA administration (Figure 4B) in the double lesion model. The ratio of (DOPAC + HVA)/DA microdialysate concentrations, an index of oxidative metabolism of DA by MAO, was significantly reduced by rasagiline in double-lesioned, but not in single-lesioned rats by comparison with saline pretreatment (P < 0.001 by two-way anova over the time period 20–200 min post–L-DOPA; Figure 4C and D). The ratio (DOPAC + HVA)/DA was reduced to a much greater extent by clorgyline than by rasagiline in both lesion models and the increase in this ratio following L-DOPA was completely suppressed by clorgyline (Figure 4C and D).
In view of the marked effect of clorgyline to increase DA microdialysate levels, an additional experiment was carried out in a group of double-lesioned rats to examine the effect of inhibition of MAO-A by low-dose clorgyline (0.0033 mg·kg−1) to the same extent as was seen in the rasagiline-treated animals (i.e. about 20% inhibition of enzyme activity). In this group of double-lesioned rats, pretreatment with this low dose of clorgyline did not increase the post L-DOPA DA microdialysate levels (data not shown).
Effect of inhibition of low-affinity uptake transporters on microdialysate levels of DA and metabolites following L-DOPA administration
The organic transporter OCT-3 and the plasma-membrane transporter (PMAT) mediate low-affinity transport of catecholamines and both are expressed in neurons of rat striatum (Vialou et al., 2007; Cui et al., 2009; Kilkenny et al., 2012). In Parkinsonian striatum, where the DAT is absent, and the SERT may be reduced, the low-affinity transporters will play a more important role in uptake of extracellular DA to sites of metabolism within striatal neurons and glia. We therefore investigated the role of the low-affinity uptake pathway in clearance of L–DOPA-derived DA microdialysate levels in striatum of single-and double-lesion rats using D-22, an inhibitor of OCT-3 and PMAT (Hayer-Zillgen et al., 2002; Engel and Wang, 2005). In this experiment, the treatment group received 50 μM D-22 by infusion through the dialysis probe into the striatum while the control group received only artificial CSF. Following L-DOPA administration, as previously, DA levels in CSF-treated rats were higher in those given a single-lesion than in double-lesion rats (P < 0.001 by two-way anova over the period 20–240 min post L-DOPA; Figure 5A and B). DA levels post L-DOPA injection in both lesion models were significantly higher in the D–22-treated group compared with the control group throughout the whole time of sampling (P < 0.001) As can be seen in Figure 5B, the peak increase in DA levels in the double lesion group occurred very soon after the L-DOPA injection, whereas in the single lesion group it was delayed, and it should be noted that in the double-lesioned striata the levels of DA in the D-22 treated rats were significantly higher when compared at each time point to the CSF-treated rats (time 20–140 min post L-DOPA), while in the single-lesioned striata the difference was significant only for later time points (200–220 min post L-DOPA). The ratio (DOPAC + HVA)/DA was significantly decreased following D-22 treatment in both models (Figure 5C and D), mainly as a result of reduction in HVA levels. Levels of 3-OMD were similar in both treatment groups in each lesion model, indicating consistency of L-DOPA administration (not shown).
Figure 5.
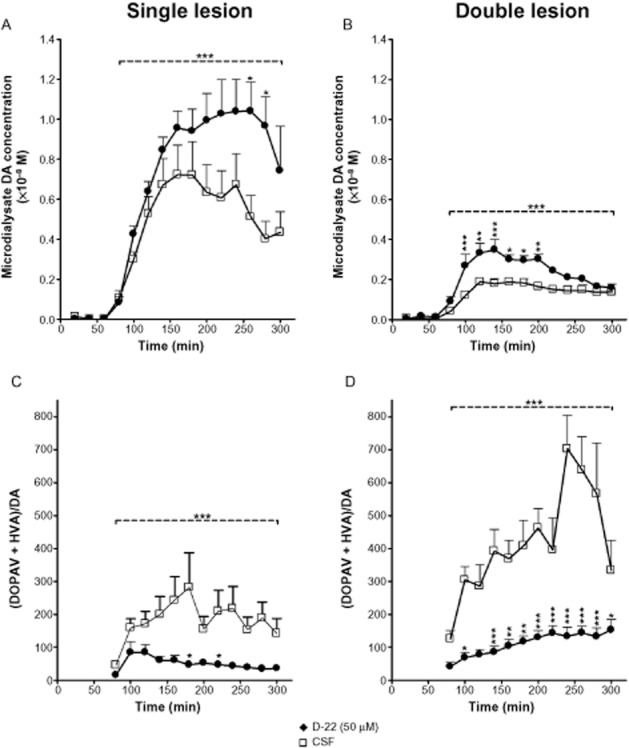
The effect of D-22 on L-DOPA-derived DA levels in DA-lesioned striatum of both single (dopaminergic) and double (dopaminergic and serotonergic) lesion models. D-22 increased L-DOPA-derived DA levels in both lesion models when compared with artificial cerebrospinal fluid (CSF) infusion. This increase in DA levels was accompanied by a decrease in DA oxidative metabolism (expressed as [DOPAC + HVA] / DA ratio) in both lesion models (C and D). Rats bearing either type of lesion were infused via the dialysis probe with either 50 μM D-22 at 2 μL·min−1 or artificial CSF. Infusion started 1 h before dialysate collection to achieve stable tissue levels of the drug. Carbidopa (6 mg·kg−1 i.p.) was administered at the start of microdialysis collections (time 0), and L-DOPA (25 mg·kg−1) was administered 60 min later. Data are expressed as mean + SEM for n = 7–9 rats per group. ***P < 0.001 for time periods indicated by horizontal bars (20–240 min post L-DOPA) by two-way anova for comparison between treatments (saline and D-22). Bonferroni post hoc test was performed for comparison between treatments at each time point (*P < 0.05, **P < 0.01, ***P < 0.001).
MAO subtypes localization in striatal sections
In order to correlate the results of MAO activity determination in lesioned striata with the effects of MAO inhibitors on microdialysate DA levels, we conducted immunofluorescence experiments to localize MAO-A and MAO-B in striatal tissue. MAO-A antibody was validated by positive staining of noradrenergic cells in locus coeruleus (Figure 6A) and MAO-B antibody positively stained GFAP-positive cells in the striatum (Figure 7B). Interestingly, MAO-A was clearly localized in MSNs expressing DARPP-32 in intact striatum (Figure 6B). Furthermore, MAO-A was still expressed in MSNs even following the application of the different lesions (Figure 6B). However, no MAO-A staining was seen in glial cells (Figure 6C). MAO-B was not solely localized in glial cells, weaker staining of MAO-B was also detected in MSN's in both lesion models (Figure 7A); however, confirmation of the presence or absence of MAO-B in MSN requires application of additional techniques, and will be carried out in a future study. Structures identified morphologically as fibre bundles stained positively for MAO-A (Figures 6 and 7). The staining of DARPP-32 in striatal MSN occurs to a variable extent, because of the different degrees of DA activity at striatal dopaminergic receptors on these cells, which varies the expression level of DARPP-32.
Figure 6.
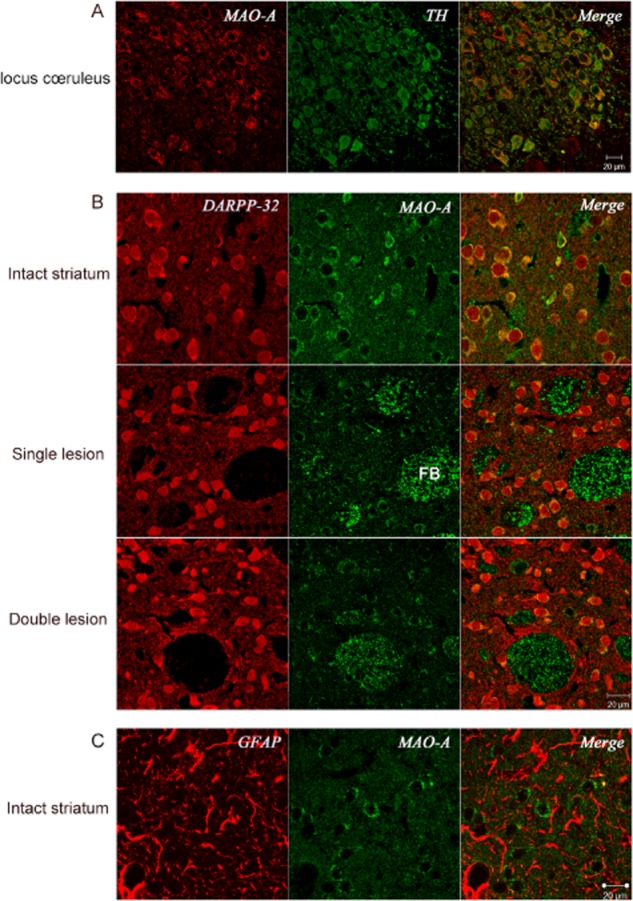
MAO-A localization in brain sections (5 μm) using polyclonal anti-MAO-A antibody. (A) Section from intact locus coeruleus, additionally stained for tyrosine hydroxylase using monoclonal anti-tyrosine hydroxylase antibody, confirming known localization of MAO-A to noradrenergic neurons of this brain area. (B) Striatal sections from intact, single-(ipsilateral dopaminergic) and double-(ipsilateral dopaminergic plus bilateral serotonergic) lesioned rats, additionally stained with monoclonal anti-DARPP-32 as indicator of MSN, showing localization of MAO-A in MSN and fibre bundles (FB), and similar distribution of MAO-A in intact, single-and double-lesioned rats (representative sections shown from n = 3, 6 and 5, respectively, rats with similar results in each). (C) Striatal section from normal control rat stained for glial cells using monoclonal anti-GFAP antibody, showing absence of MAO-A from glial cells. Images were obtained using confocal microscope.
Figure 7.
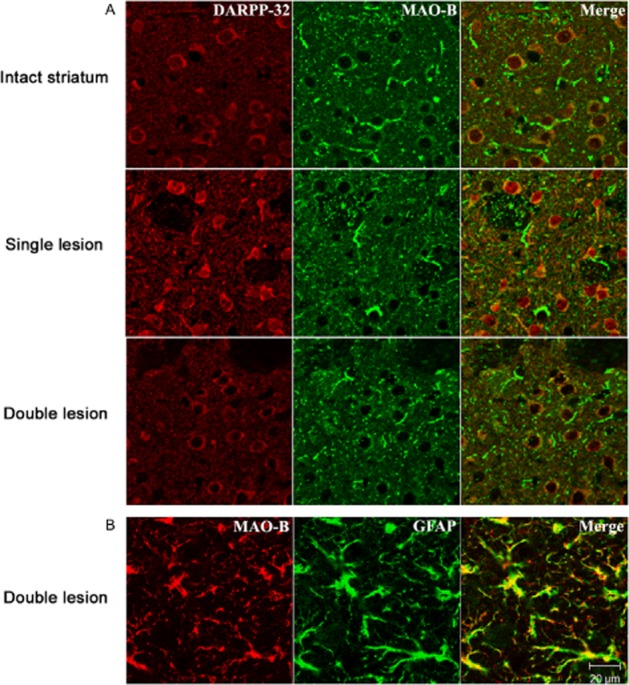
MAO-B localization in brain sections (5 μm) using polyclonal anti-MAO-B antibody. (A) Striatal sections from intact, single (ipsilateral dopaminergic) and double (ipsilateral dopaminergic plus bilateral serotonergic) lesioned rats, additionally stained for MSN using polyclonal anti DARPP-32. Most MAO-B containing structures are not co-localised with DARPP-32. Representative images are shown from n = 3, 4 and 4 intact, single-and double-lesioned rats respectively. (B) Striatal section from double-lesion rat additionally stained for glial cells using monoclonal anti-GFAP antibody. MAO-B colocalizes with GFAP in most glial cell processes. Images were obtained using confocal microscope.
Discussion
The activity of MAO is critically important in determining effective concentrations of DA at receptor sites in the striatum following administration of L-DOPA. Many techniques have been applied to the question of the localization of the enzyme subtypes in neurons and glia of the brain. Although several studies have shown an enrichment of MAO-A in tyrosine hydroxylase-positive neurons of the locus coeruleus and other catecholaminergic neuronal groups including SNpc neurons (Westlund et al., 1993), not all agree with the finding of MAO-A localization in dopaminergic neurons of the SNpc (Konradi et al., 1988; Willoughby et al., 1988; Arai et al., 1998; Hida et al., 1999). Experiments using human brain synaptosomes showed that DA nerve endings in caudate nucleus contain exclusively MAO-A (O'Carroll et al., 1987), but the subtype of the enzyme in synaptic vesicles may differ from that found in cell bodies of SNpc. Although conclusive direct evidence for the expression of MAO-A by nigro-striatal dopaminergic neurons is still lacking, indirect evidence points to a role of MAO-A in deamination of DA released from axon terminals of these neurons. Thus, inhibition of MAO-A by clorgyline, but not selective inhibition of MAO-B by selegiline or rasagiline increases striatal tissue steady-state DA levels and reduces DOPAC in intact rat brain (Waldmeier et al., 1976; Waldmeier et al., 1981; Finberg and Youdim, 2002).
Our findings of no detectable change in MAO-A content of whole striatal tissue after destruction of SNpc are at variance with those of other researchers (Agid et al., 1973; Demarest et al., 1980) who carried out a similar study, but found significant reduction in MAO-A levels following partial destruction of SNpc. It should be pointed out, however, that in these previous studies, the extent of DA depletion would have been much less than in the present study, because we selected rats for a high degree of striatal DA depletion (>99%) by using the functional index of contralateral turning. Under these conditions, serotonergic neurons hyperinnervate the striatum, which could increase MAO-A activity of the tissue, assuming the presence of this enzyme form in serotonergic axonal varicosities, and such an increase may offset any reduction caused by the loss of dopaminergic neurons. Our finding of lack of change of striatal MAO-A activity following DA as well as DA plus 5-HT lesion can be compared with the study of Carlsson et al. (1981) who saw no change or a modest increase in striatal MAO-A following unilateral brain hemitransection at the level of the caudal hypothalamus. Similarly, (Van der Krogt et al., 1983) found no change in striatal MAO-A content following 6-OHDA-induced lesion of SNpc. Our finding of no change in striatal MAO-A activity following the double lesion indicates that most of the MAO-A enzyme in striatum is not located in dopaminergic or serotonergic neurons. In this respect, it should also be remembered that axonal varicosities comprise only a very small percent by volume of the tissue even though within axonal varicosities the enzyme is highly concentrated. When whole tissue enzyme activity is determined, the contribution of this small compartment to the enzyme activity of whole striatal tissue may be minor, unless the specific activity of the enzyme in the dopaminergic axonal varicosities is many fold that of the other neuronal elements in the tissue. Our findings of increased MAO-B content of striatum following both dopaminergic and serotonergic deafferentation are consistent with the gliosis caused by this procedure, because MAO-B is the enzyme form that has been detected in astrocytes in the brain of both rat and man (Levitt et al., 1982; Westlund et al., 1985; Ekblom et al., 1993) as also in our current findings. The increase in number of GFAP-positive cells and GFAP tissue content (Figure 3) was greater than the increase in MAO-B activity (Figure 2) caused by the lesions, which may be related to the much less profound immunohistochemical staining of MAO-B in glia than that of GFAP (Figure 7). Increased striatal expression of GFAP has been described following striatal and nigral injection of 6-OHDA (Nomura et al., 2000; Rodrigues et al., 2001).
Rasagiline is a selective inhibitor of MAO-B, which is currently in clinical use in Pd. In contrast to selegiline, it is not metabolized to amphetamines, and does not possess amphetamine-like effects (Glezer and Finberg, 2003). Previous studies on DA oxidative metabolism have revealed that both endogenously released-and L–DOPA-derived DA is largely metabolized by MAO-A in intact and in hemi-Parkinsonian rats (Butcher et al., 1990; Paterson et al., 1991; Scarr et al., 1994; Wachtel and Abercrombie, 1994; Finberg et al., 1995). In the study of Wachtel and Abercrombie (1994) only a minor effect of clorgyline on DA microdialysate was seen in unilateral 6–OHDA-lesioned rats, but the degree of lesioning in their study was probably much lower than in the present and our previous one (Finberg et al., 1995). The present study is the first to investigate the effect of subacute MAO-B inhibition in 6-OHDA lesioned rats on the metabolism of DA produced from L-DOPA. The doses of rasagiline and clorgyline were chosen to be selective according to previous studies (Waldmeier et al., 1981; Finberg et al., 1995; Lamensdorf et al., 1996) and achieved 95% inhibition of the target enzyme with less than 20% inhibition of the other form. The activity of MAO in catecholaminergic neurons is considered to be in great excess, as it is necessary to inhibit the enzyme by more than 80% in order to see functional changes in behaviour (Youdim and Finberg, 1983). However, because of the marked effect of MAO-A inhibition on DA levels, we carried out an additional experiment that showed that 20% reduction in MAO-A activity by clorgyline did not cause an increase in DA levels in double lesion rats, and so the effect of rasagiline on DA levels is due to MAO-B inhibition, and the 15% inhibition of MAO-A produced by rasagiline does not contribute to this effect.
Because 5-HT neurons are a major site for DA production from L-DOPA, a larger dose of L-DOPA is needed to initiate turning behaviour and induce detectable DA levels in the microdialysate in the double lesion model compared with the single lesion. We used 25 mg·kg−1 of L-DOPA as in a previous study (Tanaka et al., 1999), in which it was shown that this dose could induce turning behaviour in double lesion rats. The need for increasing the dose of L-DOPA with time is a well-known phenomenon in treating human Pd. This might be explained by the increasing loss of 5-HT as well as DA innervation in the striatum along with the progress of the disease (see introduction), thus supporting the validity of the double lesion model for investigating the metabolism of DA from L-DOPA in advanced stage PD.
In agreement with our previous studies (Finberg et al., 1995), MAO-A inhibition produced a larger increase in DA extracellular levels and decrease in oxidative metabolites than did inhibition of MAO-B in the single lesion model, and in the present study, we found that MAO-A inhibition also caused a greater effect on DA metabolism than did MAO-B inhibition in the double-lesioned rats. This finding leads to the interesting question as to where is MAO-A located in the double lesioned striatum, and how does it still contribute to DA metabolism following L-DOPA administration? Our present immunohistochemical study showed that MAO-A is expressed in GABA-ergic MSNs, which occupy more than 90% of the striatal neuropil, but not in astrocytes. The role of these neurons in DA metabolism might become more significant in the absence of the dopaminergic and serotonergic axon terminals in the striatum.
The effect of rasagiline on DA levels following L-DOPA was of a greater relative magnitude in the double than in the single-lesioned striatum. This might be explained by the absence of the serotonergic terminals, which presumably express MAO-A and that play a major role in the uptake and metabolism of DA in the case of the single dopaminergic lesion. Consequently, the contribution of other cells, such as astrocytes expressing MAO-B, in the production and metabolism of DA becomes more significant in the double-lesion rats. Our results support this assumption, because the double denervation of dopaminergic and serotonergic terminals was accompanied by a marked increase in GFAP expression. The recent demonstration of occurrence of the amine transporter OCT-3 in striatal astrocytes (Cui et al., 2009) is in accord with a role of astrocytes in metabolism of DA produced from L-DOPA in the double-lesioned striatum. The expression of OCT-3 and PMAT in non-aminergic striatal neurons indicates the possibility that these neurons, which express MAO-A, play a major role in uptake and metabolism of DA in the absence of DA and 5-HT axonal varicosities, which express the high-affinity transporters DAT and SERT respectively (Vialou et al., 2007).
In conclusion, when both DA and 5-HT terminals are eliminated, the MAO-B enzyme plays a larger part in DA metabolism, but MAO-A remains the major enzyme. The precise part played by MSN and glial cells in DA metabolism depends on the degree of expression of the low-affinity transporters in each cell type together with the expression degree and affinities of the two MAO isoforms for DA, and cannot be stated in a quantitative way in the current research. In striatum devoid of DA and 5-HT neuronal varicosities, however, our present data indicate an important role of low-affinity transporters, and MAO-A expressed in MSN, in metabolism of DA derived from L-DOPA. These results have clinical relevance as they explain the efficacy of MAO-B inhibition in advanced stages of PD. In such patients, administration of rasagiline together with L-DOPA increases ‘on’ period duration and reduces adverse symptoms in ‘off’ period (Parkinson Study Group, 2005), which correlates with the increased duration of elevated striatal DA levels following L-DOPA seen in double lesion rats. In addition, the results of our study indicate that inhibitors of OCT-3 could be useful in treatment of Pd, by enhancing and possibly prolonging the elevation of extracellular levels of DA in striatum following L-DOPA administration.
Glossary
- 5,7-DHT
5,7-dihydroxytryptamine
- 5-HIAA
5-hydroxyindoleacetic acid
- 5-HT
5-hydroxytryptamine
- 6-OHDA
6-hydroxydopamine
- AAAD
L-aromatic amino acid decarboxylase
- DA
dopamine
- DARPP-32
dopamine and cAMP-regulated phosphoprotein, Mr 32 kDa
- DAT
dopamine transporter
- DOPAC
3,4-dihydroxyphenylacetic acid
- HVA
homovanillic acid
- L-DOPA
L-3,4-dihydroxyphenylalanine
- MSN
medium spiny neuron
- OCT-3
organic cation transporter-3
- Pd
Parkinson's disease
- PEA
phenylethylamine
- SERT
serotonin transporter
- SNpc
substantia nigra, pars compacta
Conflict of interest
John Finberg is a co-inventor of rasagiline and receives royalties from sale of the drug; however, this fact had no impact on the ideas and concepts expressed in this paper.
References
- Agid Y, Javoy F, Youdim MB. Monoamine oxidase and aldehyde dehydrogenase activity in the striatum of rats after 6-hydroxydopamine lesion of the nigrostriatal pathway. Br J Pharmacol. 1973;48:175–178. doi: 10.1111/j.1476-5381.1973.tb08238.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arai R, Horiike K, Hasegawa Y. Dopamine-degrading activity of monoamine oxidase is not detected by histochemistry in neurons of the substantia nigra pars compacta of the rat. Brain Res. 1998;812:275–278. doi: 10.1016/s0006-8993(98)00983-4. [DOI] [PubMed] [Google Scholar]
- Butcher SP, Fairbrother IS, Kelly JS, Arbuthnott GW. Effects of selective monoamine oxidase inhibitors on the in vivo release and metabolism of dopamine in the rat striatum. J Neurochem. 1990;55:981–988. doi: 10.1111/j.1471-4159.1990.tb04587.x. [DOI] [PubMed] [Google Scholar]
- Carlsson A, Fowler CJ, Magnusson T, Oreland L, Wiberg A. The activities of monoamine oxidase-A and-B, succinate dehydrogenase and acid phosphatase in the rat brain after hemitransection. Naunyn Schmiedebergs Arch Pharmacol. 1981;316:51–55. doi: 10.1007/BF00507227. [DOI] [PubMed] [Google Scholar]
- Carta M, Carlsson T, Kirik D, Bjorklund A. Dopamine released from 5-HT terminals is the cause of L-DOPA-induced dyskinesia in Parkinsonian rats. Brain. 2007;130(Pt 7):1819–1833. doi: 10.1093/brain/awm082. [DOI] [PubMed] [Google Scholar]
- Cui M, Aras R, Christian WV, Rappold PM, Hatwar M, Panza J, et al. The organic cation transporter-3 is a pivotal modulator of neurodegeneration in the nigrostriatal dopaminergic pathway. Proc Natl Acad Sci U S A. 2009;106:8043–8048. doi: 10.1073/pnas.0900358106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demarest KT, Smith DJ, Azzaro AJ. The presence of the type A form of monoamine oxidase within nigrostriatal dopamine-containing neurons. J Pharmacol Exp Ther. 1980;215:461–468. [PubMed] [Google Scholar]
- Ekblom J, Jossan SS, Bergstrom M, Oreland L, Walum E, Aquilonius SM. Monoamine oxidase-B in astrocytes. Glia. 1993;8:122–132. doi: 10.1002/glia.440080208. [DOI] [PubMed] [Google Scholar]
- Engel K, Wang J. Interaction of organic cations with a newly identified plasma membrane monoamine transporter. Mol Pharmacol. 2005;68:1397–1407. doi: 10.1124/mol.105.016832. [DOI] [PubMed] [Google Scholar]
- Finberg JP, Youdim MB. Pharmacological properties of the anti-Parkinson drug rasagiline; modification of endogenous brain amines, reserpine reversal, serotonergic and dopaminergic behaviours. Neuropharmacology. 2002;43:1110–1118. doi: 10.1016/s0028-3908(02)00216-2. [DOI] [PubMed] [Google Scholar]
- Finberg JP, Wang J, Goldstein DS, Kopin IJ, Bankiewicz KS. Influence of selective inhibition of monoamine oxidase A or B on striatal metabolism of L-DOPA in hemiparkinsonian rats. J Neurochem. 1995;65:1213–1220. doi: 10.1046/j.1471-4159.1995.65031213.x. [DOI] [PubMed] [Google Scholar]
- Glezer S, Finberg JP. Pharmacological comparison between the actions of methamphetamine and 1-aminoindan stereoisomers on sympathetic nervous function in rat vas deferens. Eur J Pharmacol. 2003;472:173–177. doi: 10.1016/s0014-2999(03)01906-x. [DOI] [PubMed] [Google Scholar]
- Green AR, Youdim MB. Effects of monoamine oxidase inhibition by clorgyline, deprenil or tranylcypromine on 5-hydroxytryptamine concentrations in rat brain and hyperactivity following subsequent tryptophan administration. Br J Pharmacol. 1975;55:415–422. doi: 10.1111/j.1476-5381.1975.tb06946.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall FS, Devries AC, Fong GW, Huang S, Pert A. Effects of 5,7-dihydroxytryptamine depletion of tissue serotonin levels on extracellular serotonin in the striatum assessed with in vivo microdialysis: relationship to behavior. Synapse. 1999;33:16–25. doi: 10.1002/(SICI)1098-2396(199907)33:1<16::AID-SYN2>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- Hayer-Zillgen M, Bruss M, Bonisch H. Expression and pharmacological profile of the human organic cation transporters hOCT1, hOCT2 and hOCT3. Br J Pharmacol. 2002;136:829–836. doi: 10.1038/sj.bjp.0704785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hida T, Hasegawa Y, Arai R. Histochemical study of dopamine-degrading monoamine oxidase activity in dopaminergic neurons of rat brain. Brain Res. 1999;842:491–495. doi: 10.1016/s0006-8993(99)01873-9. [DOI] [PubMed] [Google Scholar]
- Inazu M, Takeda H, Matsumiya T. Functional expression of the norepinephrine transporter in cultured rat astrocytes. J Neurochem. 2003;84:136–144. doi: 10.1046/j.1471-4159.2003.01514.x. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG. Animal research: reporting in vivo experiments: the ARRIVE guidelines. J Gene Med. 2010;12:561–563. doi: 10.1002/jgm.1473. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne WJ, Cuthill IC, Emerson M, Altman DG. Improving bioscience research reporting: the ARRIVE guidelines for reporting animal research. Osteoarthritis Cartilage. 2012;20:256–260. doi: 10.1016/j.joca.2012.02.010. [DOI] [PubMed] [Google Scholar]
- Konradi C, Svoma E, Jellinger K, Riederer P, Denney R, Thibault J. Topographic immunocytochemical mapping of monoamine oxidase-A, monoamine oxidase-B and tyrosine hydroxylase in human post mortem brain stem. Neuroscience. 1988;26:791–802. doi: 10.1016/0306-4522(88)90099-1. [DOI] [PubMed] [Google Scholar]
- Lamensdorf I, Youdim MB, Finberg JP. Effect of long-term treatment with selective monoamine oxidase A and B inhibitors on dopamine release from rat striatum in vivo. J Neurochem. 1996;67:1532–1539. doi: 10.1046/j.1471-4159.1996.67041532.x. [DOI] [PubMed] [Google Scholar]
- Levitt P, Pintar JE, Breakefield XO. Immunocytochemical demonstration of monoamine oxidase B in brain astrocytes and serotonergic neurons. Proc Natl Acad Sci U S A. 1982;79:6385–6389. doi: 10.1073/pnas.79.20.6385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Real A, Rodriguez-Pallares J, Guerra MJ, Labandeira-Garcia JL. Localization and functional significance of striatal neurons immunoreactive to aromatic L-amino acid decarboxylase or tyrosine hydroxylase in rat Parkinsonian models. Brain Res. 2003;969:135–146. doi: 10.1016/s0006-8993(03)02291-1. [DOI] [PubMed] [Google Scholar]
- Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. Protein measurement with the Folin phenol reagent. J Biol Chem. 1951;193:265–275. [PubMed] [Google Scholar]
- McGrath JC, Drummond GB, McLachlan EM, Kilkenny C, Wainwright CL. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda T, Kannari K, Shen H, Arai A, Tomiyama M, Matsunaga M, et al. Rapid induction of serotonergic hyperinnervation in the adult rat striatum with extensive dopaminergic denervation. Neurosci Lett. 2003;343:17–20. doi: 10.1016/s0304-3940(03)00295-7. [DOI] [PubMed] [Google Scholar]
- Miller DW, Abercrombie ED. Role of high-affinity dopamine uptake and impulse activity in the appearance of extracellular dopamine in striatum after administration of exogenous L-DOPA: studies in intact and 6-hydroxydopamine-treated rats. J Neurochem. 1999;72:1516–1522. doi: 10.1046/j.1471-4159.1999.721516.x. [DOI] [PubMed] [Google Scholar]
- Mura A, Jackson D, Manley MS, Young SJ, Groves PM. Aromatic L-amino acid decarboxylase immunoreactive cells in the rat striatum: a possible site for the conversion of exogenous L-DOPA to dopamine. Brain Res. 1995;704:51–60. doi: 10.1016/0006-8993(95)01104-8. [DOI] [PubMed] [Google Scholar]
- Navailles S, Bioulac B, Gross C, De Deurwaerdere P. Serotonergic neurons mediate ectopic release of dopamine induced by L-DOPA in a rat model of Parkinson's disease. Neurobiol Dis. 2010;38:136–143. doi: 10.1016/j.nbd.2010.01.012. [DOI] [PubMed] [Google Scholar]
- Nomura T, Yabe T, Rosenthal ES, Krzan M, Schwartz JP. PSA-NCAM distinguishes reactive astrocytes in 6-OHDA-lesioned substantia nigra from those in the striatal terminal fields. J Neurosci Res. 2000;61:588–596. doi: 10.1002/1097-4547(20000915)61:6<588::AID-JNR2>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- O'Carroll AM, Fowler CJ, Phillips JP, Tobbia I, Tipton KF. The deamination of dopamine by human brain monoamine oxidase. Specificity for the two enzyme forms in seven brain regions. Naunyn Schmiedebergs Arch Pharmacol. 1983;322:198–202. doi: 10.1007/BF00500765. [DOI] [PubMed] [Google Scholar]
- O'Carroll AM, Tipton KF, Sullivan JP, Fowler CJ, Ross SB. Intra-and extra-neuronal deamination of dopamine and noradrenaline by the two forms of human brain monoamine oxidase. Implications for the neurotoxicity of N-methyl-4-phenyl-1,2,3,6-tetrahydropyridine. Biog Amines. 1987;4:165–178. [Google Scholar]
- Otsuka S, Kobayashi Y. Radioisotopic assay for monoamine oxidase determinations in human plasma. Biochem Pharmacol. 1964;13:995–1006. doi: 10.1016/0006-2952(64)90096-6. [DOI] [PubMed] [Google Scholar]
- Parkinson Study Group. A randomized placebo-controlled trial of rasagiline in levodopa-treated patients with Parkinson disease and motor fluctuations: the PRESTO study. Arch Neurol. 2005;62:241–248. doi: 10.1001/archneur.62.2.241. [DOI] [PubMed] [Google Scholar]
- Paterson IA, Juorio AV, Berry MD, Zhu MY. Inhibition of monoamine oxidase-B by (–)-deprenyl potentiates neuronal responses to dopamine agonists but does not inhibit dopamine catabolism in the rat striatum. J Pharmacol Exp Ther. 1991;258:1019–1026. [PubMed] [Google Scholar]
- Paxinos G, Watson C. The Rat Brain in Stereotaxic Coordinates. Sydney: Academic Press; 1982. [Google Scholar]
- Rodrigues RW, Gomide VC, Chadi G. Astroglial and microglial reaction after a partial nigrostriatal degeneration induced by the striatal injection of different doses of 6-hydroxydopamine. Int J Neurosci. 2001;109:91–126. doi: 10.3109/00207450108986528. [DOI] [PubMed] [Google Scholar]
- Scarr E, Wingerchuk DM, Juorio AV, Paterson IA. The effects of monoamine oxidase B inhibition on dopamine metabolism in rats with nigro-striatal lesions. Neurochem Res. 1994;19:153–159. doi: 10.1007/BF00966810. [DOI] [PubMed] [Google Scholar]
- Schwarting RK, Huston JP. The unilateral 6-hydroxydopamine lesion model in behavioral brain research. Analysis of functional deficits, recovery and treatments. Prog Neurobiol. 1996;50:275–331. doi: 10.1016/s0301-0082(96)00040-8. [DOI] [PubMed] [Google Scholar]
- Tanaka H, Kannari K, Maeda T, Tomiyama M, Suda T, Matsunaga M. Role of serotonergic neurons in L-DOPA-derived extracellular dopamine in the striatum of 6-OHDA-lesioned rats. Neuroreport. 1999;10:631–634. doi: 10.1097/00001756-199902250-00034. [DOI] [PubMed] [Google Scholar]
- Tashiro Y, Kaneko T, Sugimoto T, Nagatsu I, Kikuchi H, Mizuno N. Striatal neurons with aromatic L-amino acid decarboxylase-like immunoreactivity in the rat. Neurosci Lett. 1989;100:29–34. doi: 10.1016/0304-3940(89)90655-1. [DOI] [PubMed] [Google Scholar]
- Van der Krogt JA, Koot-Gronsveld E, Van den Berg CJ. Localization of rat striatal monoamine oxidase activities towards dopamine, serotonin and kynuramine by gradient centrifugation and nigro-striatal lesions. Life Sci. 1983;33:615–623. doi: 10.1016/0024-3205(83)90249-7. [DOI] [PubMed] [Google Scholar]
- Vialou V, Balasse L, Dumas S, Giros B, Gautron S. Neurochemical characterization of pathways expressing plasma membrane monoamine transporter in the rat brain. Neuroscience. 2007;144:616–622. doi: 10.1016/j.neuroscience.2006.09.058. [DOI] [PubMed] [Google Scholar]
- Wachtel SR, Abercrombie ED. L-3,4-dihydroxyphenylalanine-induced dopamine release in the striatum of intact and 6-hydroxydopamine-treated rats: differential effects of monoamine oxidase A and B inhibitors. J Neurochem. 1994;63:108–117. doi: 10.1046/j.1471-4159.1994.63010108.x. [DOI] [PubMed] [Google Scholar]
- Waldmeier PC, Delini-Stula A, Maitre L. Preferential deamination of dopamine by an A type monoamine oxidase in rat brain. Naunyn Schmiedebergs Arch Pharmacol. 1976;292:9–14. doi: 10.1007/BF00506483. [DOI] [PubMed] [Google Scholar]
- Waldmeier PC, Felner AE, Maitre L. Long term effects of selective MAO inhibitors on MAO activity and amine metabolism. In: Youdim MB, Paykel ES, editors. Monoamine Oxidase Inhibitors. New York: Wiley; 1981. pp. 87–102. [Google Scholar]
- Westlund KN, Denney RM, Kochersperger LM, Rose RM, Abell CW. Distinct monoamine oxidase A and B populations in primate brain. Science. 1985;230:181–183. doi: 10.1126/science.3875898. [DOI] [PubMed] [Google Scholar]
- Westlund KN, Krakower TJ, Kwan SW, Abell CW. Intracellular distribution of monoamine oxidase A in selected regions of rat and monkey brain and spinal cord. Brain Res. 1993;612:221–230. doi: 10.1016/0006-8993(93)91664-e. [DOI] [PubMed] [Google Scholar]
- Willoughby J, Glover V, Sandler M. Histochemical localisation of monoamine oxidase A and B in rat brain. J Neural Transm. 1988;74:29–42. doi: 10.1007/BF01243573. [DOI] [PubMed] [Google Scholar]
- Yamada H, Aimi Y, Nagatsu I, Taki K, Kudo M, Arai R. Immunohistochemical detection of L-DOPA-derived dopamine within serotonergic fibers in the striatum and the substantia nigra pars reticulata in Parkinsonian model rats. Neurosci Res. 2007;59:1–7. doi: 10.1016/j.neures.2007.05.002. [DOI] [PubMed] [Google Scholar]
- Youdim MBH, Finberg JPM. Monoamine oxidase inhibitor antidepressants. In: Grahame-Smith DG, Cowen PJ, editors. Psychopharmacology. Vol. 1. Amsterdam: Excerpta Medica; 1983. pp. 38–70. [Google Scholar]