

Abstract
Background and Purpose
Intestinal nutrient transporters may mediate the uptake of drugs. The aim of this study was to investigate whether sertraline interacts with the intestinal proton-coupled amino acid transporter 1 PAT1 (SLC36A1).
Experimental Approach
In vitro investigations of interactions between sertraline and human (h)PAT1, hSGLT1 (sodium-glucose linked transporter 1) and hPepT1 (proton-coupled di-/tri-peptide transporter 1) were conducted in Caco-2 cells using radiolabelled substrates. In vivo pharmacokinetic investigations were conducted in male Sprague–Dawley rats using gaboxadol (10 mg·kg−1, p.o.) as a PAT1 substrate and sertraline (0–30.6 mg·kg−1). Gaboxadol was quantified by hydrophilic interaction chromatography followed by MS/MS detection.
Key Results
Sertraline inhibited hPAT1-mediated L-[3H]-Pro uptake in Caco-2 cells. This interaction between sertraline and PAT1 appeared to be non-competitive. The uptake of the hSGLT1 substrate [14C]-α–methyl-D-glycopyranoside and the hPepT1 substrate [14C]-Gly-Sar in Caco-2 cells was also decreased in the presence of 0.3 mM sertraline. In rats, the administration of sertraline (0.1–10 mM, corresponding to 0.3–30.6 mg·kg−1, p.o.) significantly reduced the maximal gaboxadol plasma concentration and AUC after its administration p.o.
Conclusions and Implications
Sertraline is an apparent non-competitive inhibitor of hPAT1-mediated transport in vitro. This inhibitory effect of sertraline is not specific to hPAT1 as substrate transport via hPepT1 and hSGLT1 was also reduced in the presence of sertraline. In vivo, sertraline reduced the amount of gaboxadol absorbed, suggesting that the inhibitory effect of sertraline on PAT1 occurs both in vitro and in vivo. Hence, sertraline could alter the bioavailability of drugs absorbed via PAT1.
Keywords: PAT1 (SLC36A1), gaboxadol, sertraline, small intestinal absorption, in vivo transporter, transporter-mediated pharmacokinetics
Introduction
The proton-coupled amino acid transporter PAT1 (SLC36A1; see Alexander et al., 2011 for correct nomenclature) is expressed in the luminal membrane of enterocytes from rat and man, where it transports amino acids (proline, glycine and alanine) and osmolytes (betaine and taurine) from the intestinal lumen into the enterocyte (Thwaites and Anderson, 2006). The human intestinal cell line Caco-2 has been extensively used for investigations of human (h)PAT1 activity (Chen et al., 2003; Anderson et al., 2004). Based on data from in vitro studies, PAT1 functions as a drug transporter of vigabatrin, δ-aminolevulinic acid and gaboxadol (Abbot et al., 2005; Larsen et al., 2009;2010; Frolund et al., 2010a; Broberg et al., 2012). Furthermore, some proline derivatives and creatine analogues with pharmacological activity have been identified as substrates (Metzner et al., 2004;2009). In the case of gaboxadol, in vivo investigations have confirmed that its intestinal absorption is mediated by PAT1 (Larsen et al., 2009;2010; Broberg et al., 2012). The translation from in vitro substrate identification to in vivo relevance of the PAT1 transporter is challenging. Animals devoid of the slc36a1 gene are currently not available, therefore, in vivo investigations rely on the use of inhibitors of PAT1-mediated transport. The inhibitors identified so far fall into two very different categories, that is, dipeptides and indole derivatives (Metzner et al., 2005; Frolund et al., 2012). The dipeptides have a relatively poor affinity for hPAT1, whereas indole-containing amino acids and amines, such as 5-hydroxy-tryptamine (5-HT), L-tryptophan, 5-hydroxy-L-tryptophan (5-HTP) and tryptamine, have high affinities for hPAT1 of approximately 1–6 mM (Metzner et al., 2005). For in vivo investigations the inhibitors must, therefore, be administered in high doses in order to achieve a sufficient degree of PAT1 inhibition. Accordingly, this introduces the possibility of adverse effects, as previously demonstrated in rat experiments where gaboxadol was co-administered, p.o., with 5-HTP, which resulted in a decreased clearance of gaboxadol as compared to when gaboxadol was administered alone (Larsen et al., 2010).
5-HT is biosynthesized from L-tryptophan. L-tryptophan is an essential amino acid, which is hydroxylated to 5-HTP via tryptophan hydroxylase. 5-HTP is then converted to 5-HT by the enzyme aromatic amino acid decarboxylase. Tryptophan, 5-HTP and 5-HT are all inhibitors of hPAT1 (Metzner et al., 2005). We therefore hypothesized that drugs belonging to the selective 5-HT re-uptake inhibitor (SSRI) group could interact with PAT1, and that a high-affinity inhibitor could be identified within this group of compounds. Hence, the aim of the present study was to investigate if SSRI-related compounds are able to interact with hPAT1 and to assess whether sertraline has an effect on their in vivo absorption via PAT1 using gaboxadol as a prototypical PAT1 substrate.
Methods
Caco-2 cell culture
Caco-2 cells were cultured as previously described (Larsen et al., 2008). In brief, Caco-2 cells (DSMZ, Braunschweig, Germany) were seeded at a density of 1.7 × 105 cells per well for a 24-well plate and 6.0 × 104 cells per well for a 96-well plate, where the cells were grown for 6 or 13 days. For trans-epithelial electrical resistance (TEER) measurements, Caco-2 cells were seeded onto polycarbonate membranes (0.42 cm2, 0.4 μm pore size) of Transwell™ (Corning Life Sciences, New York, NY, USA) inserts at a density of 8.9 × 104 cells·cm−2. Caco-2 cell monolayers grown on Transwell inserts were used for experiments 21 days after seeding. All experiments were performed 24 h after change of media. Caco-2 cells were used in passages 8–20.
In vitro uptake studies in Caco-2 cell monolayers
The uptake studies were performed on cells grown at the bottom of 24-well plates for 6 or 13 days. Uptake and transport studies were performed in HBSS buffer (in mM: CaCl2, 1.26; MgCl2, 0.49; MgSO4, 0.41; KCl, 5.33; KH2PO4, 0.44; NaCl, 138; Na2HPO4, 0.34; D-glucose, 5.56; NaHCO3, 4.17) supplemented with 0.05% BSA. In some studies, the HBSS buffers were not supplemented with BSA, as stated in the figure legends. Compounds used were dissolved directly in HBSS, except for sertraline, which was dissolved in water and then diluted in 2x HBSS. The cells were equilibrated with HBSS, pH 7.4 (±0.05% BSA), and 10 mM HEPES 37°C on an orbital shaker (90 r.p.m.) for 15 min. The buffer was then aspirated and 300 μL of the test solutions [HBSS, pH 6.0 (±0.05% BSA), and 10 mM 2-(N-morpholino)ethanesulfonic acid (MES), isotope and investigated compound] were added. The test solutions were adjusted to pH 6.0 before use. After 5 min incubation with the Caco-2 cells, the test solutions were removed and the cells were washed three times with ice-cold HBSS buffer. The cells were detached using 200 μL 0.1% Triton X-100 in H2O and incubated at 37°C for at least 15 min. The cell homogenate was transferred to a scintillation vial and 2 mL scintillation fluid was added. The radioactivity was counted by liquid scintillation spectrometry (Packard TriCab 2100TR liquid scintillation counter; Meriden, CT, USA). All isotopes were used at an activity of 1 μCi·mL−1. For uptake of α-methyl-D-glycopyranoside (α-MDG), experiments were conducted in HBSS buffer, pH 6.0 without glucose. Non-hPAT1-specific uptake was estimated from uptake of L-[3H]-Pro in the presence of a surplus of Pro by measuring the radioactivity in the sample, which was then used to correct the uptake data for non-specific cellular uptake.
Expression of hPAT1mRNA and protein in Caco-2 cells
RNA was isolated from Caco-2 cells using Nucleospin® RNA/protein isolation kit according to the protocol provided by the manufacturer (Macherey-Nagel GmbH and Co., Düren, Germany). The isolated RNA was purified from genomic DNA by treatment with DNAse using DNAse I amplification grade according to the protocol provided by the manufacturer (Sigma-Aldrich, Steinheim, Germany). Reverse transcriptase was performed with moloney murine leukemia virus high-performance reverse transcriptase according to manufacturer's protocol (Epicentre, Maddison, WI, USA). The PCR was performed using HotStarTaq Plus DNA Polymerase according to manufacturer's protocol (Qiagen, Copenhagen, Denmark). The primers were designed to match hPAT1 and human β-actin and were purchased from Invitrogen (Hellerup, Denmark). The primers against hPAT1 were antisense: ACTTTAAACAGGTGATAGAAGCGGCCAATG and sense: TGAGGGTTATGCTGCCTTGGATATTAGCTC giving a product of 480 bp. The primers against β-actin were antisense: AGC ACT GTG TTG GC and sense: GGA CTT CGA GCA AGA GAT GG giving a reaction product of 234 bp. The initial activation of the polymerase was run for 5 min at 95°C followed by 28 cycles of denaturation for 30 s at 94°C, annealing for 1 min at 69°C and extension at 72°C for 1 min. The final extension was 10 min. The resulting reaction products were run on 1% agarose gel using 50 bp DNA-ladder (Invitrogen) for identification of the product. As a negative control, the primers were tested against the purified RNA, which resulted in no visual bands (not shown). The products were visualized with 0.5 μg·mL−1 ethidium bromide in a fluorchemQ image station (Alpha Innotech/Cell Biosciences, Santa Clara, CA, USA).
Protein was isolated from Caco-2 cells using Nucleospin RNA/protein isolation kit (Macherey-Nagel, Düren, Germany) according to the protocol provided. Protein concentration was determined using Bradford reagent following the protocol provided by the manufacturer (Sigma-Aldrich, St. Louis, MO, USA). A total of 10 μg of protein from Caco-2 cells was loaded onto a gradient gel (4–15%) (Mini-PROTEAN® TGX™ Precast Gel, Bio-Rad Laboratories, Hercules, CA, USA) together with 5 μL Precision Dual Color Plus (Bio-Rad Laboratories) to identify the protein size. Electrophoresis was run at 150 V until the protein reached the edge of the gel. The proteins were transferred to a PVDF membrane at 50 V (350 mA) for 40 min. The membrane was stained with a custom-made rabbit polyclonal anti-PAT1 raised against the human N-terminal (N-PAT1) peptide sequence MSTQRLRNEEYHDYSSTDSS (21st Century Biochemicals, Marlboro, MA, USA) and GAPDH (FL-335) (Santa Cruz Biotechnology, Dallas, TX, USA). Secondary antibody used for Western blot analysis was polyclonal goat anti-rabbit IgG (Invitrogen, Carlsbad, CA, USA). As a negative control, the membrane was blotted with the secondary antibody alone, which showed no visible bands after development. Chemiluminescent detection of the proteins was performed using ECL Plus Western Blotting Detection Reagents (Amersham Biosciences, Amersham, UK).
LDH release from Caco-2 cells
LDH release (LDH-release assay) from Caco-2 cells was measured according to the instructions provided by the manufacturer (Roche, Mannheim, Germany). Caco-2 cells were seeded in a 96-well plate and cultured for 6 days. The cells were equilibrated with HBSS, pH 7.4, supplemented with 0.05% BSA and 10 mM HEPES for 15 min on an orbital shaker (90 r.p.m.) at 37°C, and 150 μL of test solutions containing sertraline were added. A total of 10% Triton X-100 was used as a positive control resulting in 100% LDH release, LDH release in HBSS buffer was taken as baseline release and 10 mM L-Pro was used as a negative control. A total of 100 μL sample solutions were withdrawn from the cell after 5 min incubation with sertraline, control HBSS buffer or L-Pro, whereas the cells were incubated for 30 min with Triton X-100. The LDH content was assayed with an LDH-kit [Cat. no. 04 744 926 001 (Roche)] according to the manufacturer's instructions.
TEER measurements
The TEER at 37°C was used as a parameter for integrity of the Caco-2 cell monolayers after addition of sertraline. The TEER was measured by the CellZscope device (nanoAnalytics, Münster, Germany) kindly borrowed from POLYGEN Denmark ApS (Horsens, Denmark). The module used was suitable for 24 Transwell-filter systems, grown with Caco-2 cell monolayers. Buffers used were as described above.
Xenopus laevis oocytes
X. laevis oocytes were obtained and treated as previously described (Frolund et al., 2012). L-[3H]-Pro uptake (40 μM, 3 μCi·mL−1) was measured in Ringer's solution (in mM: NaCl, 115; KCl, 2.5; CaCl2, 1.8; MgCl2, 0.1; MES, 10), pH adjusted to 6.0, in the absence and presence of 10 mM L-Pro or 1–1000 μM sertraline. Before commencement of the uptake experiments, the oocytes were washed in room temperature Ringer's solution. Uptake was initiated by transferring the oocytes to 24-well plates containing 500 μL of the appropriate uptake solution, and measured at room temperature for 60 min, while the oocytes were maintained on an orbital shaker (60 r.p.m.). Uptake was terminated by transferring the oocytes to 2 mL ice-cold Ringer's solution and washing them three times with 1 mL ice-cold Ringer's solution. Individual oocytes were transferred to scintillation vials containing 200 μL 10% (w/v) SDS and vortexed briefly in order to homogenize the oocytes. A total of 2 mL scintillation fluid was added and the radioactivity was counted by liquid scintillation spectrometry (Packard TriCab 2100TR liquid scintillation counter; Meriden, CT, USA).
Animal studies
All animal studies were approved by the Animal Welfare Committee, appointed by the Danish Ministry of Justice, and were carried out in compliance with EC Directive 86/609/EEC, the Danish law regulating experiments on animals and NIH Guidelines for the Care and Use of Laboratory Animals. In addition, all studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (Kilkenny et al., 2010; McGrath et al., 2010). Male Sprague–Dawley rats (Charles River Laboratories, Sulzfeld, Germany) were acclimatized and maintained on standard feed with free access to water for a minimum of 5 days prior to the experiment. Before entry to the pharmacokinetic experiment, the animals were fasted for 16–20 h. The weight of the animals at the day of administration was 330–390 g.
In vivo absorption of gaboxadol in Sprague–Dawley rats
Male Sprague–Dawley rats were randomly assigned to receive a pre-dose of sertraline, p.o., to achieve the following concentrations of sertraline (in mM): 0, 0.1, 0.25, 0.5, 1.0, 2.5, 5.0, 10.0, corresponding to the following doses (in mg·kg−1): 0.3, 0.8, 1.5, 3.1, 15.3, 30.6. The seven dose groups consisted of 6–12 animals each. Thirty minutes after administration of the pre-dose, the animals received gaboxadol (10 mg·kg−1, p.o.). Both doses were administered by oral gavage (pre-dose, 10.0 mL·kg−1; gaboxadol, 1.0 mL·kg−1). All oral solutions were adjusted to a pH of 4.7. The osmolality was adjusted to iso-osmolality using mannitol or NaCl. Blood samples (0.2 mL) were taken from the lateral tail vein by individual vein puncture and collected into microvettes coated with lithium-heparin (Sarstedt, Nürnberg, Germany). Blood samples were collected 5, 15, 30, 45, 60 min and 1.5, 2, 3, 4, 6 h after gaboxadol dosing as previously done by Broberg et al. (2012). The plasma was harvested immediately by centrifuging 3600× g for 10 min at 5°C and stored at −80°C until further analysis. The animals were killed by an i.v. injection of carbon dioxide after the study.
Quantification of gaboxadol in plasma
Fifty microlitres of plasma samples were spiked with internal standard (d4-gaboxadol) and gaboxadol was extracted from the plasma samples by solid phase extraction using the Oasis MCX μElution Plate system (Waters, Milford, MA, USA) according to the protocol supplied by the manufacturer. The effluent was evaporated to dryness and reconstituted in 200 μL 5 mM ammonium acetate pH 4.2 : acetonitrile (10:90), shaken for 10 min at 900 r.p.m. and centrifuged for 10 min at 5200× g (10°C). Standards were added to control (blind) plasma and prepared similar to the plasma samples. Gaboxadol was separated by hydrophilic interaction chromatography and quantified by MS/MS detection using a protocol modified from Kall et al. (2007). The LC-MS/MS system was obtained from Waters and consisted of a Quattro Ultima Pt mass spectrometer (ACQUITY UPLC) with acquisition software Masslynx version 4.1 and an ACQUITY UPLC BEH Amide column (1.7 μm, 2.1 × 100 mm). A gradient mobile phase method with two mobile phases was used. Mobile phase A consisted of 5 mM ammonium acetate pH 4.2 : acetonitrile : water (5:90:5) and mobile phase B consisted of 5 mM ammonium acetate pH 4.2 : acetonitrile : water (5:10:85). The gradient range was mobile phase A : B (92:8–50:50). The flow was 0.6 mL·min−1 and the injection volume was 7.5 μL. The column temperature was kept at 60°C. The total run time was 3.8 min and the elution time of gaboxadol was approximately 1.7 min. The detection was performed in positive ionization mode where gaboxadol (precursor 140.9 Da, product 123.9 Da) and d4-gaboxadol (precursor 144.9, product 127.9 Da) were measured by multiple-reaction-monitoring. The signals were linear over the concentration range of 25–10 000 ng·mL−1 and the limit of quantification by this procedure was 25 ng·mL−1. The gaboxadol recovery was 80–120%.
Data analysis
The initial uptake rate (U, nmol·min−1·cm−2) of L-Pro, glycyl-sarcosine (Gly-Sar) or α-MDG in Caco-2 cells was calculated as the amount of compound (Q, nmol) accumulated in the cells in a 5 min period (t, min) across the well area (A, cm2). The accumulation of L-Pro within the X. laevis oocytes was calculated based on the assumption that the intercellular volume of an oocyte is 1 μL (Ferrell, 1999).
Cellular uptake of L-Pro in the presence of increasing concentrations of sertraline was fitted to a four-parameter logistic equation (Equation 2005) to obtain an IC50 value:
| 1 |
U is % inhibition of the L-Pro uptake at inhibitor concentration I, Umax is the initial uptake of L-Pro ([I] = 0 mM) ∼100%, Umin is the lowest measured uptake of L-Pro at maximum inhibitor concentration, I is the concentration of inhibitor (mM) and nH is the Hill coefficient.
The concentration-dependent hPAT1-specific uptake of L-Pro in Caco-2 cells in the absence or presence of sertraline was fitted to a Michaelis–Menten type equation (Equation 2004):
| 2 |
where Umax (nmol·min−1·cm−2) is the maximal uptake rate, Km (mM) is the Michaelis constant and [S] (mM) is the L-Pro concentration.
The pharmacokinetic parameters were determined using Phoenix™ WinNonlin® version 5.2 (Pharsight Corporation, Mountain View, CA, USA). The plasma concentration data were fitted to a non-compartment model. Cmax and tmax were found as mean or median values of the animals' plasma profiles within each group. The AUClast was calculated using the linear trapezoidal rule from time zero to Cmax and log linear from Cmax to the last measured plasma concentration. Linear regression of 3–8 (with a median of 6) data points was used to obtain the elimination rate constant, ke. Results are expressed as mean ± SEM or median [Q25%; Q75%] of 6–12 animals per dosing group.
Statistical analysis
Statistical analysis was performed by use of GraphPad Prism (GraphPad Prism Software, version 5.04, San Diego, CA, USA). Parametric values are expressed as mean ± SEM and were analysed for statistical differences using a one sample t-test, Student's t-test or one-way anova followed by either Dunnett's or Tukey's multiple comparison test. Non-parametric values are expressed as median [Q25%; Q75%] and were analysed for statistical differences using Kruskal–Wallis followed by Dunn's multiple comparison test. The following levels of significance were assigned: *P < 0.05, **P < 0.01, ***P < 0.001 and ****P < 0.0001.
Chemicals and reagents
The chemicals used were of analytical grade unless otherwise stated. Caco-2 cells were obtained from Deutsche Sammlung von Mikroorganismen und Zellkulturen (German Collection of Cell Cultures) (DSMZ GmbH). Penicillin, streptomycin, L-glutamine, DMEM, magnesium sulfate, magnesium gluconate, L-proline, 5-HTP, GABA, MES, N-acetyltryptophan, sertraline hydrochloride, DMSO, Gly-Sar, phlorizin dehydrate, α-lobelin hydrochloride, tyramine, α-methyl5-HT maleate salt, 5-HT hydrochloride, 5-methoxy-N,N-dimethyltryptamine, N,N-dimethyl-2(3-ethyl-5-methyl-4-(isoxazolyl)-β-hydroxytryptamine, 1-methylindole-2-carboxylic acid and glycyl-L-proline (Gly-Pro) were from Sigma (St. Louis, MO, USA). 2-Methyl5-HT was from Santa Cruz Biotechnology. Citalopram hydrobromide and d4-gaboxadol were donated by Lundbeck A/S (Valby, Denmark). BSA was from Biotech Line (Slangerup, Denmark). HEPES was obtained from AppleChem GmbH (Darmstadt, Germany). HBSS was purchased from Invitrogen (Taastrup, Denmark). Phosphoric acid 85% was from JT Baker (Deventer, Netherlands). Potassium dihydrogen phosphate, sodium hydrogen carbonate, D-glucose and potassium dihydrogen phosphate were from Merck (Darmstadt, Germany). Acetonitril was from Lab-Scan Analytical Sciences (Sowinskiego, Poland). Triton X-100 was from MP-Biomedicals (Eschwege, Germany). Ultima Gold scintillation liquid, L-[3H]-Pro (75 Ci·mmol−1), [3H]-GABA (35 Ci·mmol−1) and [14C]-α-MDG (310 mCi·mmol−1) were purchased from Perkin Elmer (Boston, MA, USA). [14C]-Gly-Sar (56 mCi·mmol−1) was from GE-Healthcare (Freiburg, Germany). [14C]-gaboxadol (25 mCi·mmol−1) was from H. Lundbeck A/S. LDH kit (Cat. No. 04 744 926 001) was from Roche.
Results
Inhibition of hPAT1-mediated L-proline uptake by 5-HT-related compounds
The uptake of the hPAT1 substrate L-[3H]-Pro (13 nM, 1 μCi·mL−1) was measured in Caco-2 cells in the presence of various 5-HT-related compounds as well as some known PAT1 ligands (Figure 1A). The compounds were investigated at 10 mM, except for lobeline, α-methyl 5-HT, 5-HT and sertraline due to insufficient solubility in the buffers used. The SSRI sertraline (at 0.86 mM) was the most effective inhibitor of L-[3H]Pro uptake among the compounds investigated (Figure 1A). In contrast, another SSRI, citalopram, up to a concentration of 10 mM, showed no inhibition of L-[3H]-Pro uptake. In agreement with previous studies, tryptophan, 5-HTP and 5-HT inhibited L-Pro uptake. Likewise, α-lobeline, α-methyl 5-HT, N,N-dimethyl-2(3-ethyl-5-methyl)-4-(isoxazolyl)-β-hydroxytryptamine and 2-methyl 5-HT inhibited L-Pro uptake, whereas tyramine, 5-methoxy-N,N-dimethyltryptamine and N-acetyltryptophan did not show any inhibition of L-Pro uptake at the concentrations investigated.
Figure 1.
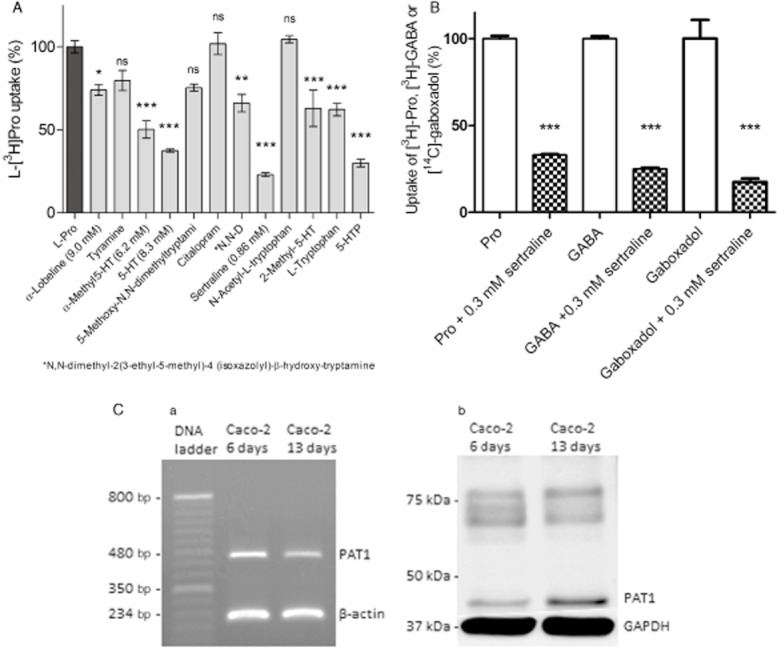
Inhibition of uptake of PAT1 substrates in Caco-2 cells. All compounds were dissolved in HBSS supplemented with 0.05% BSA and 10 mM MES; pH adjusted to 6.0 and added to Caco-2 cells grown for 7 days on the bottom of 24-well plates. Uptake was measured for 5 min. (A) The uptake of the hPAT1 substrate L-[3H]-Pro (13 nM, 1 μCi·mL−1) was measured in the absence or presence of 10 mM test compound unless otherwise stated in parentheses. The uptake measured in the absence of inhibitor was taken as 100% (control). Each bar represents the mean ± SEM of at least three different wells (n = 3). The one-way anova showed significant difference between the means at a level of P < 0.0001; *P < 0.05, **P < 0.01, ***P < 0.001 significantly different from control, analysed by Dunnett's multiple comparison test. (B) The uptake of the hPAT1 substrates L-[3H]-Pro (13 nM, 1 μCi·mL−1), [3H]-GABA (14 nM, 1 μCi·mL−1), [14C]-gaboxadol (40 μM, 1 μCi·mL−1) was measured in the absence or presence of 0.3 mM sertraline. The uptake measured in the absence of sertraline was taken as 100%. Uptake of gaboxadol was measured in the absence of BSA. Each bar represents the mean ± SEM of three different Caco-2 cell passages (n = 3). Student's t-test showed a significant difference between the means measured in the absence and presence of sertraline. ****P < 0.0001 significantly different from the uptake measured in the absence of sertraline. (C) Expression of hPAT1 in Caco-2 cells cultured for 6 or 13 days. (a) The expression of hPAT1 mRNA after RT-PCR. Amplicons were separated on a 1% agarose gel using a 50 bp DNA ladder. The products were visualized with 0.5 μg·mL−1 ethidium bromide [hPAT1 (480 bp) and β-actin (234 bp)]. (b) The level of expression of hPAT1 protein was visualized by SDS-PAGE and Western blotting.
In order to investigate the ability of sertraline to inhibit uptake of hPAT1 substrates further, the effect of a lower sertraline concentration on the uptake of [3H]-GABA (14 nM, 1 μCi·mL−1) and [14C]-gaboxadol (40 μM, 1 μCi·mL−1) was determined (Figure 1B). In the presence of 0.3 mM sertraline, the uptake of L-Pro, GABA and gaboxadol was reduced significantly, suggesting that hPAT1-mediated uptake of these substrates into Caco-2 cells is inhibited by sertraline. The presence of hPAT1 in the Caco-2 cells used was confirmed by RT-PCR and Western blotting as shown in Figure 1C.
Sertraline is a high-affinity, apparently non-competitive, inhibitor of L-proline uptake
The ability of sertraline to concentration-dependently inhibit the uptake of L-[3H]-Pro (13 nM, 1 μCi·mL−1) in Caco-2 cells was investigated. As shown in Figure 2A, sertraline inhibited the uptake of L-Pro in a concentration-dependent manner with an IC50 value of 241 μM (logIC50 2.38 ± 0.03) in the presence of 0.05% BSA. This is the highest inhibition constant reported for an hPAT1 inhibitor so far. As sertraline is known to bind efficiently to plasma protein, the effect of the BSA present in the uptake buffer was investigated to access the accuracy of the estimated IC50 value. Removing BSA from the uptake buffer had no significant effect on the uptake rate of L-[3H]-Pro (13 nM, 1 μCi·mL−1) or the ability of 0.3 mM sertraline to inhibit the uptake of L-Pro (Figure 2B). The affinity of sertraline measured in the absence of BSA had an IC50 value of 177 μM (logIC50 2.25 ± 0.05) (Figure 2A). The IC50 values obtained in the absence or presence of BSA were not significantly different (P = 0.09, n = 3), suggesting that the presence of BSA does not affect the estimated IC50 value. To investigate the nature of the sertraline-induced inhibition of L-Pro uptake via hPAT1, the kinetic parameters for concentration-dependent L-Pro uptake were determined in the presence of different sertraline concentrations (Figure 2C). The uptake of L-Pro without sertraline was concentration-dependent and could be described by a Km of 5.5 ± 1.8 mM and a Vmax of 2.2 ± 0.4 nmol·cm−2·min−1. In the presence of 0.1 or 0.3 mM sertraline, the concentration-dependent uptake of L-Pro resulted in Km values of 2.7 ± 1.4 mM and 6.8 ± 7.9 mM respectively. The corresponding Vmax values were 1.1 ± 0.2 nmol·cm−2·min−1 and 0.7 ± 0.4 nmol·cm−2·min−1. The uptake of L-Pro was totally abolished in the presence of 0.5 mM sertraline, as shown in Figure 2C. The effect of sertraline on L-Pro uptake was thus to decrease the maximal uptake capacity, which was significant at 0.3 mM (P < 0.05, n = 3), without having a significant effect on the Michaelis constant. This was further confirmed by the Dixon plot shown in Figure 2D, where the straight lines intercept each other on the X-axis, yielding a Ki value of 145 ± 21 μM. This indicates that sertraline acts as a non-competitive inhibitor of hPAT1-mediated L-Pro uptake in Caco-2 cells. The concentration-dependent uptake of gaboxadol was measured as well (Figure 2E), and could be described by a Km of 7.4 ± 1.3 mM and a Vmax of 1.5 ± 0.08 nmol·cm−2·min−1. In the presence of 0.3 mM sertraline, the uptake of gaboxadol was decreased (Figure 2E), and the Km was estimated to be 9.1 ± 5.5 mM and Vmax 0.4 ± 0.13 nmol·cm−2·min−1.
Figure 2.

Inhibition of L-[3H]-Pro uptake by sertraline in Caco-2 cells. All compounds were dissolved in HBSS supplemented with 0.05% BSA and 10 mM MES; pH adjusted to 6.0 and added to Caco-2 cells grown for 7 days on the bottom of 24-well plates. Apical uptake was measured for 5 min. (A) The apical uptake of L-[3H]-Pro (13 nM, 1 μCi·mL−1) was measured in the presence of increasing concentrations of sertraline in the absence or presence of BSA. The uptake measured in the absence of sertraline was taken as 100%. The IC50 value in the presence of BSA (circles) was estimated using Eq. 2005 to 241 μM (LogIC50 of 2.38 ± 0.03), and to 177 μM (LogIC50 of 2.25 ± 0.05) in the absence of 0.05% BSA (open triangles). Each data point represents the mean ± SEM of three different passages (n = 3). (B) The uptake of L-[3H]-Pro (13 nM, 1 μCi·mL−1) was measured in the absence or presence of 0.05% BSA and 0.3 mM sertraline (Sert). Each bar represents the mean ± SEM of three different cell passages (n = 3). Student's t-test showed no significant difference between uptake measured in the absence or presence of BSA. (C) Concentration-dependent uptake of Pro [L-[3H]-Pro (13 nM, 1 μCi·mL−1)] in the absence (circles) or presence of 0.1 mM (squares), 0.3 mM (triangles) or 0.5 mM (diamonds) sertraline. Results are mean ± SEM of three different passages (n = 3). The solid lines represent the non-linear regression of the results fitted to Eq. 2004. (D) A Dixon plot of the data presented in Figure 2C. (E) Concentration-dependent uptake of gaboxadol [[14C]-gaboxadol (40 nM, 1 μCi·mL−1)] in the absence (circles) or presence of 0.3 mM (triangles) sertraline. Results are mean ± SEM of three different passages (n = 3). The solid lines represent the non-linear regression of the results fitted to Eq. 2004. (F) Cytotoxicity of sertraline, evaluated by a LDH-release assay, at different concentrations. L-Pro 10 mM was a negative control and 10% Triton X-100 was a positive control. The statistical analysis was performed by a one-way anova followed by a Tukey's multiple comparison test (n = 3). The responses evoked by sertraline were at all concentrations significantly different from the positive control (10% Triton X-100; P < 0.01), but not significantly different from the negative control (10 mM L-Pro). (G) TEER as a function of time in the presence or absence of different sertraline concentrations. The arrow indicates the addition of buffer containing sertraline. The data are a representative example from one Caco-2 cell passage. Caco-2 cells were cultured for 21 days on permeable Transwell filters. The pattern was confirmed in three different cell passages. The TEER was followed in a CellZscope.
To determine whether sertraline has an effect on the barrier and biochemical properties of the Caco-2 cells, the LDH released from the cells during the 5 min incubation period was measured and the changes in TEER across confluent Caco-2 cell monolayers were monitored. No significant change in LDH release was observed for 10 mM L-Pro, or 0.1, 0.3 or 1.0 mM sertraline (Figure 2F). However, the TEER across Caco-2 cell monolayers was affected by the presence of sertraline in a manner that was dependent on the concentration applied (Figure 2G). The TEER value of Caco-2 cell monolayers exposed to 0.1, 0.2 and 0.3 mM sertraline for 15 min sertraline was not different from that of cell monolayers incubated with HBSS buffer. However, the TEER in the presence of 0.1 and 0.2 mM sertraline increased over time compared with the TEER measured in HBSS buffer. The TEER in the presence of 0.3 mM sertraline was comparable to that in control buffer for approximately 45 min, after which it decreased. Upon application of buffers containing 0.65 and 1.0 mM sertraline, the TEER dropped rapidly and reached that of the filter value, which was approximately 22 Ω·cm2. This suggests a concentration-dependent effect of sertraline on the TEER of Caco-2 cell monolayers.
Sertraline also inhibits transport via other membrane transporters
To determine whether the observed effect of sertraline was specific for hPAT1-mediated uptake, its effect on the uptake of substrates for two other membrane transporters expressed at the apical membrane of Caco-2 cells was investigated (Figure 3A and B). The uptake of 3.2 μM of the hSGLT1 substrate α-MDG (1 μCi·mL−1 [14C]-α-MDG) was decreased by 49% in the presence of 0.3 mM sertraline (P < 0.001, n = 3), and by approximately 80% in the presence of 1 mM of the SGLT1 inhibitor phlorizin (P < 0.001, n = 3) (Figure 3A). Likewise, the uptake of 18 μM of the hPepT1 substrate Gly-Sar (1 μCi·mL−1 [14C]-Gly-Sar) was significantly decreased by 57% in the presence of 0.3 mM sertraline (P < 0.001, n = 5), and 91% in the presence of 10 mM Gly-Pro (P < 0.001, n = 4) (Figure 3B). These results suggest that sertraline also inhibits other nutrient transporters in Caco-2 cells.
Figure 3.
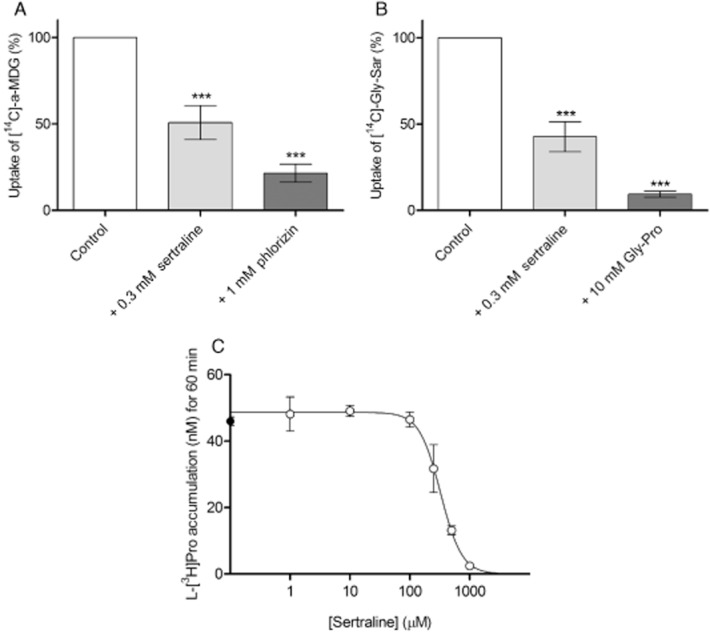
(A) Uptake of 3.2 μM (1 μCi·mL−1) [14C]-α-MDG in Caco-2 cells in the absence (control) or presence of 0.3 mM sertraline or 1.0 mM phlorizin. The uptake was measured for 5 min in HBSS supplemented with 0.05% BSA and 10 mM MES; pH adjusted to 6.0, in three individual cell monolayers. The data represent mean ± SEM (n = 3). The data were analysed by a one-sample t-test; ** and **** denote P < 0.01 and P < 0.0001 significantly different from 100% respectively. (B) Uptake of 18 μM (1 μCi·mL−1) [14C]-Gly-Sar in Caco-2 cells in the absence (control) or presence of 0.3 mM sertraline or 10 mM Gly-Pro. The uptake was measured for 5 min in HBSS supplemented with 0.05% BSA and 10 mM MES; pH adjusted to 6.0 in three individual cell monolayers. The data represent mean ± SEM (n = 3). The data were analysed by a one-sample t-test; ** and **** denote P < 0.01 and P < 0.0001 significantly different from 100% respectively. (C) L-[3H]-Pro (13 nM, 1 μCi·mL−1) accumulation (nM; assuming an intercellular volume of 1 μL) in naked X. laevis oocytes. Uptake was measured for 60 min, and shown as a function of the sertraline concentration. Each data point represents the mean ± SEM of five to six oocytes. Sertraline concentration-dependently decreased L-[3H]-Pro uptake in oocytes with an IC50 value of 327 μM (LogIC50 of 2.5 ± 0.1) and a Hill slope of −2.5 ± 0.9.
To determine whether the effect of sertraline on membrane transporters was specific for the in vitro method employed, that is, Caco-2 cells, the uptake of L-[3H]-Pro (40 μM, 3 μCi·mL−1) was measured in naked X. laevis oocytes (Figure 3C). Sertraline concentration-dependently decreases L-[3H]-Pro uptake in oocytes to the same degree as 10 mM cold L-Pro (4.8 ± 0.4 nM, data not shown), with an IC50 value of 327 μM (logIC50 2.5 ± 0.1) and a Hill slope of −2.5 ± 0.9, indicating that sertraline has effects that are not only evident in Caco-2 cells.
Sertraline affects the pharmacokinetic profile of gaboxadol after p.o. administration in rats
To determine whether the effect of sertraline on PAT1 in vitro could also be demonstrated in vivo, the plasma-concentration time profile for 10 mg·kg−1 gaboxadol, a PAT1 substrate, was obtained in rats pretreated p.o. with a solution containing 0–10 mM sertraline (corresponding to 0–30.6 mg·kg−1) (Figure 4). The maximal plasma concentration of gaboxadol was reached relatively fast regardless of the concentration of sertraline in the p.o. solution (Table 2005). The gaboxadol plasma concentration at the first sampling time, that is, at 5 min, is shown as an inset in Figure 4. Here it is evident that the initial absorption of gaboxadol is highly affected by the presence of sertraline. Sertraline also decreased the maximal plasma concentration of gaboxadol and reduced the AUC compared with the control group without sertraline (Table 2005). However, within the different sertraline groups, no concentration-dependent effects on the Cmax or AUC were observed. The elimination rate constant stayed the same in all the groups. The relative bioavailability of gaboxadol was thus reduced between 20 and 41% in the presence of sertraline. Moreover, the maximal gaboxadol plasma concentration was reduced by between 37 and 53% in the presence of sertraline. Collectively, these results indicate that sertraline has an effect on the initial absorption of gaboxadol, hence, the maximal plasma concentration and the overall exposure to gaboxadol were reduced in the presence of sertraline but with no clear concentration-dependent effect. This effect of sertraline was evident at the lowest concentration investigated (0.1 mM corresponding to 0.3 mg·kg−1).
Figure 4.
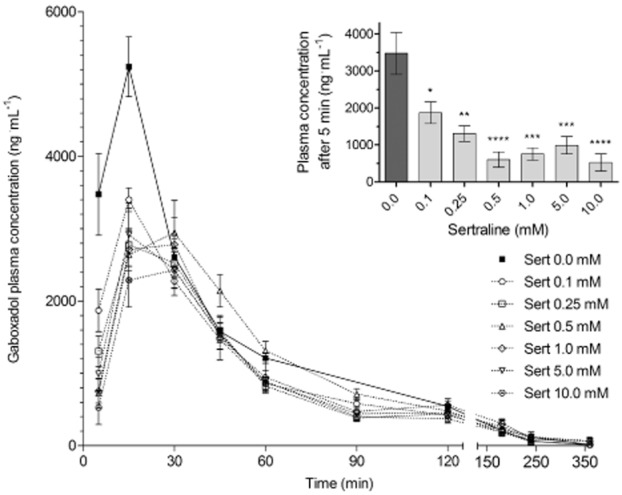
Gaboxadol plasma concentration versus time profile following its administration p.o. Sprague–Dawley rats were pretreated with 0–10 mM (0–30.6 mg·kg−1) sertraline (Sert) p.o. 30 min before gaboxadol administration p.o. The gaboxadol dose was 10 mg·kg−1. Data are presented as mean ± SEM of measurements obtained from 6 to 12 rats per dosing group. Inset: Gaboxadol plasma concentrations measured in the first sampling time at 5 min as a function of sertraline concentration (pre-treatment). The one-way anova showed significant difference between the means at a level of P < 0.0001; *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001 significantly different from 0.0 mM sertraline, analysed by Dunnett's multiple comparison test.
Pharmacokinetic parameters of gaboxadol after its administration p.o. to rats
| Sertraline | tmax | Cmax | AUClast | ke | |
|---|---|---|---|---|---|
| (mM) | (mg·kg−1) | (min) | (ng·mL−1) | (h·ng·mL−1) | (h−1) |
| 0 | 0 | 15.0 [15.0; 15.0] | 5524 ± 387 | 4227 ± 175 | 0.88 ± 0.05 |
| 0.1 | 0.3 | 15.0 [15.0; 15.0] | 3402 ± 165** | 3074 ± 134** | 1.02 ± 0.09 |
| 0.25 | 0.8 | 15.0 [15.0; 18.8] | 3033 ± 309*** | 2753 ± 155**** | 1.09 ± 0.14 |
| 0.5 | 1.5 | 30.0 [26.3; 30.0]** | 3078 ± 281*** | 3383 ± 308* | 1.21 ± 0.14 |
| 1.0 | 3.1 | 22.5 [15.0; 30.0] | 3154 ± 575*** | 3056 ± 326** | 1.19 ± 0.15 |
| 5.0 | 15.3 | 22.5 [15.0; 30.0] | 3008 ± 404*** | 2795 ± 231*** | 1.05 ± 0.11 |
| 10.0 | 30.6 | 30.0 [15.0; 30.0]* | 2609 ± 344**** | 2506 ± 213**** | 1.04 ± 0.16 |
Sprague–Dawley (gaboxadol, Gbx dose of 10 mg·kg−1) rats were pretreated with sertraline (0–10 mM) p.o. 30 min before gaboxadol administration. Cmax, AUClast and ke are expressed as mean ± SEM, and tmax is expressed as median [Q25%; Q75%] of measurement from 6 to 12 rats per dosing group. Significant difference from control (0 mM sertraline) was investigated by parametric testing using a one-way anova followed by Dunnett's multiple comparison test or non-parametric testing using Kruskal–Wallis followed by Dunn's multiple comparison test;
P < 0.05,
P < 0.01,
P < 0.001, and
P < 0.0001.
Discussion and conclusions
Sertraline is a 5-HT re-uptake inhibitor with nM binding affinity for the 5-HT transporter SERT (SLC6A4). However, sertraline is also an inhibitor of the dopamine transporter DAT (SLC6A3). Sertraline is used clinically to treat depression at an initial dose of 50 mg, which may be increased to a maximal daily dose of 200 mg. Assuming these doses are taken with the standard 250 mL water, the initial gastrointestinal sertraline concentration would be 0.65–2.6 mM. Even with an intestinal dilution, these concentrations are quite comparable with the concentration investigated in the present study.
Sertraline is an apparent non-competitive non-specific inhibitor of PAT1-mediated transport in vitro
In this study, we determined whether 5-HT mimetics are able to affect hPAT1-mediated transport in Caco-2 cells. Sertraline, as opposed to citalopram, reduced hPAT1-mediated transport, with, to our knowledge, the highest apparent affinity (241 μM) reported for PAT1 so far. Normally, PAT1 substrates have affinities in the range of 1–20 mM (Thwaites and Anderson, 2011). Inhibitors containing an indole group have affinities in the range of 1–6 mM (Metzner et al., 2005; Larsen et al., 2009), whereas dipeptide inhibitors have lower affinities (Frolund et al., 2012). Our investigation of the mechanism of sertraline-induced inhibition of hPAT1-mediated transport in Caco-2 cells revealed a non-competitive type of inhibition. To our knowledge, non-competitive inhibitors have not been described for PAT1, whereas such inhibitors have been described for the intestinal proton-coupled di-/tri-peptide transporter PepT1 (SLC15A1) (Sawada et al., 1999; Terada et al., 2000; Zhu et al., 2000; Knutter et al., 2008; Omkvist et al., 2010). Sertraline was recently suggested to be a non-competitive inhibitor of the organic cation transporter OCT3 (SLC22A3) in stably transfected HEK293 cells (IC50 around 7.4 μM) (Zhu et al., 2012), and has also been shown to inhibit substrate transport via the human plasma membrane monoamine transporter (SLC29A4) (IC50 around 5 μM) (Haenisch and Bonisch, 2010). An apparent non-competitive inhibition of PAT1-mediated transport could be induced either by direct binding to the transport protein or via a general effect on the cell system expressing the transporter in question. In Caco-2 cells, sertraline was also able to reduce hSGLT1-mediated transport of α-MDG and hPepT1-mediated uptake of Gly-Sar. It should be noted that even though Gly-Sar is also a substrate of hPAT1 (Frolund et al., 2010a,b2010b), with the Gly-Sar concentration used here only hPepT1 was involved in its cellular uptake. Similar to PAT1, PepT1 is a proton-coupled transporter whereas SGLT1 is mainly sodium coupled; hence, sertraline appears to affect membrane transporters in Caco-2 cells in a general manner with a resulting decrease in transport activity. Similar results were obtained in naked oocytes, where sertraline decreased L-Pro uptake with an IC50 value quite comparable to the one obtained in Caco-2 cells (327 and 241 μM respectively). Because sertraline affects L-Pro uptake in naked oocytes, it was not possible to use the X. laevis oocyte expression system to investigate if sertraline actually binds to or is transported by hPAT1. The effect of sertraline on membrane transporters could be a secondary result of sertraline affecting a cellular process. The most obvious explanation for the effects of sertraline on a variety of solute carriers would be a toxic effect, which could either abolish the electro-chemical gradients or destroy the integrity of the cell membrane. However, when the cytotoxicity of sertraline was investigated in Caco-2 cells using the LDH-release assay, no toxic effects were detected up to 1 mM, which is the highest concentration used in the in vitro studies. Using the MTT assay, Zhu et al. (2012) found that incubating HEK293 cells with 0.1 mM sertraline for 20 min at 37°C did not affect their viability. In contrast, Gil-Ad et al. (2008) found that in human colonic HT-29 cells incubated for 24 h with 0–30 μM sertraline, sertraline concentration-dependently reduced cell viability and inhibited cell proliferation. In our study, the cytotoxicity was evaluated over a 5 min period, comparable to the time period of the uptake experiments. In contrast, the TEER across Caco-2 cells was measured in the presence of various sertraline concentrations over a longer time period. Importantly, sertraline concentrations of 0.1, 0.2 and 0.3 mM did not cause an immediate change in TEER over the experimental time of the uptake studies. Taken together, the results from the LDH-release assay and TEER measurements suggest that the membrane of the Caco-2 cells exposed to up to 0.3 mM sertraline was not disrupted and that the Caco-2 cell layer remained tight over the 5 min time period that the uptake studies lasted.
Surprisingly, over time the two lower sertraline concentrations, 0.1 and 0.2 mM, increased the TEER measured relative to control cells, whereas 0.3 mM gradually decreased the TEER and 0.65 and 1.0 mM sertraline rapidly abolished TEER shortly after their addition to the Caco-2 cell monolayer. These changes in TEER indicate that sertraline has a concentration-dependent effect on the functions determining the TEER of the Caco-2 cell monolayer. Previously, sertraline was found to inhibit cardiac Na+ and Ca2+ channels in spontaneously beating isolated guinea pig atria (Pousti et al., 2009), the inwardly rectifying K+-channel KIR4.1 when expressed in HEK293 cells (Ohno et al., 2007), the volume-regulated anion channels in cultured pulmonary artery endothelial cells (Maertens et al., 2002), and the L-type Ca2+ and transient outward K+ currents in rat ventricular myocytes (Park et al., 1999). In epithelial cells (MDCK), sertraline has been shown to increase intracellular Ca2+ concentrations concentration-dependently in the range 1–100 μM (Huang et al., 2009). This increase in intracellular Ca2+ occurred rapidly, reaching a maximal response within 1 min, after which it gradually decreased. This increased intracellular Ca2+ concentration was shown to result from the ability of sertraline to deplete intracellular calcium stores and also to increase the influx of Ca2+ (Huang et al., 2009). Therefore, the effects of sertraline on the Caco-2 cell monolayer TEER may be related to changes in intracellular Ca2+ concentration, known to affect both activity of proteins and cause changes in ion gradients across the cell membrane. Alternatively, an inhibition of K+ channels could lead to a depolarization, which would inhibit the driving force for substrate uptake via the electrogenic transporters hPAT1, hSGLT1 and hPepT. In conclusion, sertraline produces a number of cellular effects in vitro of which decreased cellular transport via hPAT1 is just one.
Sertraline affects the pharmacokinetic profile of the PAT1 substrate gaboxadol in rats
To investigate whether the effect observed with sertraline in vitro is also evident in vivo, the effect of sertraline, administered p.o., on the pharmacokinetic profile of gaboxadol (administered p.o.) was studied in rats. Gaboxadol was chosen as a protypical substrate of PAT1-mediated absorption as earlier studies in Caco-2 cells, rats and dogs have shown that the majority of intestinal transport and hence intestinal absorption is mediated via PAT1 (Larsen et al., 2009; Broberg et al., 2012; Frolund et al., 2012). Previously, we observed that p.o. administration of gaboxadol along with other substrates or inhibitors of PAT1 leads to a reduction in the plasma concentration of gaboxadol measured after 5 min, and that this is normally not followed by a reduction in total AUC (Larsen et al., 2010; Broberg et al., 2012). Furthermore, depending on the animal model employed, an effect may be seen on Cmax and/or tmax (Larsen et al., 2009; Broberg et al., 2012). In the present study sertraline reduced the plasma concentration of gaboxadol measured after 5 min and the Cmax at all doses investigated. Not only did sertraline reduce the Cmax of gaboxadol and it also reduced the total gaboxadol exposure as estimated by the total area under the plasma concentration time profile. However, sertraline did not affect the time to reach maximal plasma concentration or the elimination rate constant of gaboxadol. The effects of sertraline on gaboxadol Cmax and AUC were evident at the lowest dose of sertraline administered as a 30 min pre-dose, that is, 0.3 mg·kg−1 corresponding to a concentration of 100 μM in the oral solution given to the animals. Increasing the sertraline dose did not produce any further reduction in the AUC or Cmax. Previous investigations have shown that when 5 mg·kg−1 sertraline was administered i.p. to mice once daily for 6 days, a minor reduction in AUC of bupropion and hydroxybupropion was observed after i.p. administration of bupropion (Molnari et al., 2012). This was not associated with any alterations in other pharmacokinetic parameters, and the reduction in AUC induced by the sertraline pretreatment was proposed to be due to a drug-drug interaction at the metabolic stage; sertraline and bupropion are both substrates of cytochrome P450 2B6. However, in the present study it is unlikely that the effect of sertraline on the pharmacokinetic parameters of gaboxadol occurs via a metabolic drug-drug interaction. Gaboxadol is mainly excreted unchanged with a minor form being O-glucorunidated by UGT1A9 (Chu et al., 2009), whereas sertraline is metabolized by UGT2B7-mediated N-carbamoyl glucuronidation (Obach et al., 2005). As it has also been suggested that sertraline is a P-gp substrate, its effect on on the pharmacokinetics of fexofenadine administered p.o. has also been investigated in healthy human volunteers. About 50 mg of sertraline was given p.o. for 7 days, after which 60 mg of fexofenadine was given p.o. With this dosage regimen, sertraline did not alter the pharmacokinetic parameters of fexofenadine, although a slight reduction in fexofenadine AUC was observed (Saruwatari et al., 2012). Fexofenadine is a substrate of efflux transporters such as P-gp, but part of its intestinal absorption is also mediated via OATP2B1 (Ming et al., 2011). One could speculate that if sertraline inhibits P-gp-mediated transport of fexofenadine, it would lead to increased plasma exposure, but if it also causes a general decrease in the activity of the absorptive membrane transporter, the reduction in OATP2B1-mediated fexofenadine uptake would negate this increase with the resulting absorption being unaffected by the sertraline treatment.
In conclusion, sertraline affects the pharmacokinetic parameters of gaboxadol in vivo, thereby confirming the results obtained in vitro. Sertraline can also affect the absorption of other compounds absorbed mainly via transport proteins and further studies are needed to assess this, as well as to investigate the exact mechanism whereby sertraline induces an inhibitory effect on membrane transporters such as PAT1. However, our results do provide convincing evidence that, although sertraline can be considered a high-affinity inhibitor of PAT1, it is not a specific for PAT1 and hence is not suitable as an investigational tool for experimental pharmacology investigating PAT1-mediated drug transport.
Acknowledgments
The authors wish to acknowledge the staff at the animal facilities and Jette Pedersen for support with the bioanalysis (H. Lundbeck A/S). We thank Birgitte Eltong and Maria Læssøe Pedersen for technical guidance and culturing of cells (University of Copenhagen). Carlsberg Foundation is acknowledged for financial support. POLYGEN Denmark ApS (Horsens, Denmark) is acknowledged for lending us the CellZcope. Dr Mie Larsen Broberg is acknowledged for help with the TEER measurements, and Marie Darting is acknowledged for help with the oocyte experiments.
Glossary
- 5-HTP
5-hydroxy-L-tryptophan
- α-MDG
α-methyl-D-glycopyranoside
- Cmax
maximal plasma concentration
- Gly-Pro
glycyl-L-proline
- Gly-Sar
glycyl-sarcosine
- ke
elimination rate constant
- Km
Michaelis constant
- L-Pro
L-proline
- MES
2-(N-morpholino)ethanesulfonic acid
- PAT1
proton-coupled amino acid transporter 1 (SLC36A1)
- PepT1
proton-coupled di-/tri-peptide transporter 1 (SLC15A1)
- Q25%
25% percentile
- Q75%
75% percentile
- Sert
sertraline
- SGLT1
sodium-glucose linked transporter 1 (SLC5A1)
- SSRI
selective 5-HT re-uptake inhibitor
- TEER
trans-epithelial electrical resistance
- tmax
time of maximal plasma concentration
Conflict of interest
None.
References
- Abbot EL, Grenade DS, Kennedy DJ, Gatfield KM, Thwaites DT. Vigabatrin transport across the human intestinal epithelial (Caco-2) brush-border membrane is via the H(+)-coupled amino-acid transporter hPAT1. Br J Pharmacol. 2005;147:298–306. doi: 10.1038/sj.bjp.0706557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Mathie A, Peters JA. Guide to receptors and channels (GRAC) 5th edition. Br J Pharmacol. 2011;164:S1–S324. doi: 10.1111/j.1476-5381.2011.01649_1.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson CM, Grenade DS, Boll M, Foltz M, Wake KA, Kennedy DJ, et al. H+/amino acid transporter 1 (PAT1) is the imino acid carrier: an intestinal nutrient/drug transporter in human and rat. Gastroenterology. 2004;127:1410–1422. doi: 10.1053/j.gastro.2004.08.017. [DOI] [PubMed] [Google Scholar]
- Broberg M, Holm R, Tonsberg H, Frolund S, Ewon KB, Nielsen A, et al. Function and expression of the proton-coupled amino acid transporter PAT1 along the rat gastrointestinal tract: implications for intestinal absorption of gaboxadol. Br J Pharmacol. 2012;167:654–665. doi: 10.1111/j.1476-5381.2012.02030.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, Fei YJ, Anderson CM, Wake KA, Miyauchi S, Huang W, et al. Structure, function and immunolocalization of a proton-coupled amino acid transporter (hPAT1) in the human intestinal cell line Caco-2. J Physiol. 2003;546:349–361. doi: 10.1113/jphysiol.2002.026500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu XY, Liang Y, Cai X, Cuevas-Licea K, Rippley RK, Kassahun K, et al. Metabolism and renal elimination of gaboxadol in humans: role of UDP-glucuronosyltransferases and transporters. Pharm Res. 2009;26:459–468. doi: 10.1007/s11095-008-9799-5. [DOI] [PubMed] [Google Scholar]
- Ferrell JE., Jr Xenopus oocyte maturation: new lessons from a good egg. Bioessays. 1999;21:833–842. doi: 10.1002/(SICI)1521-1878(199910)21:10<833::AID-BIES5>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- Frolund S, Cutillas O, Larsen M, Brodin B, Nielsen CU. Delta-aminolevulinic acid is a substrate for SLC36A1 (hPAT1) Br J Pharmacol. 2010a;159:1339–1353. doi: 10.1111/j.1476-5381.2009.00620.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frolund S, Holm R, Brodin B, Nielsen CU. The proton-coupled amino acid transporter, SLC36A1 (hPAT1), transports Gly-Gly, Gly-Sar and other Gly-Gly mimetics. Br J Pharmacol. 2010b;161:589–600. doi: 10.1111/j.1476-5381.2010.00888.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frolund S, Langthaler L, Kall MA, Holm R, Nielsen CU. Intestinal drug transport via the proton-coupled amino acid transporter PAT1 (SLC36A1) is inhibited by Gly-X(aa) dipeptides. Mol Pharm. 2012;9:2761–2769. doi: 10.1021/mp300345e. [DOI] [PubMed] [Google Scholar]
- Gil-Ad I, Zolokov A, Lomnitski L, Taler M, Bar M, Luria D, et al. Evaluation of the potential anti-cancer activity of the antidepressant sertraline in human colon cancer cell lines and in colorectal cancer-xenografted mice. Int J Oncol. 2008;33:277–286. [PubMed] [Google Scholar]
- Haenisch B, Bonisch H. Interaction of the human plasma membrane monoamine transporter (hPMAT) with antidepressants and antipsychotics. Naunyn Schmiedebergs Arch Pharmacol. 2010;381:33–39. doi: 10.1007/s00210-009-0479-8. [DOI] [PubMed] [Google Scholar]
- Huang CJ, Kuo DH, Chang KH, Shieh P, Chen FA, Fang YC, et al. Effect of the antidepressant sertraline on Ca2+ fluxes in Madin-Darby canine renal tubular cells. J Recept Signal Transduct Res. 2009;29:342–348. doi: 10.3109/10799890903295135. [DOI] [PubMed] [Google Scholar]
- Kall MA, Fu I, Dige T, Vallano P, Woolf E, Jorgensen M. Development and validation of a selective and sensitive bioanalytical procedure for the quantitative determination of gaboxadol in human plasma employing mixed mode solid phase extraction and hydrophilic interaction liquid chromatography with tandem mass spectroscopic detection. J Chromatogr B Analyt Technol Biomed Life Sci. 2007;858:168–176. doi: 10.1016/j.jchromb.2007.08.029. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG. Animal research: Reporting in vivo experiments: The ARRIVE guidelines. Br J Pharmacol. 2010;160:1577–1579. doi: 10.1111/j.1476-5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knutter I, Wollesky C, Kottra G, Hahn MG, Fischer W, Zebisch K, et al. Transport of angiotensin-converting enzyme inhibitors by H+/peptide transporters revisited. J Pharmacol Exp Ther. 2008;327:432–441. doi: 10.1124/jpet.108.143339. [DOI] [PubMed] [Google Scholar]
- Larsen M, Larsen BB, Frolund B, Nielsen CU. Transport of amino acids and GABA analogues via the human proton-coupled amino acid transporter, hPAT1: characterization of conditions for affinity and transport experiments in Caco-2 cells. Eur J Pharm Sci. 2008;35:86–95. doi: 10.1016/j.ejps.2008.06.007. [DOI] [PubMed] [Google Scholar]
- Larsen M, Holm R, Jensen KG, Brodin B, Nielsen CU. Intestinal gaboxadol absorption via PAT1(SLC36A1): modified absorption in vivo following co-administration of L-tryptophan. Br J Pharmacol. 2009;157:1380–1389. doi: 10.1111/j.1476-5381.2009.00253.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen M, Holm R, Jensen KG, Sveigaard C, Brodin B, Nielsen CU. 5-Hydroxy-L-tryptophan alters gaboxadol pharmacokinetics in rats: involvement of PAT1 and rOat1 in gaboxadol absorption and elimination. Eur J Pharm Sci. 2010;39:68–75. doi: 10.1016/j.ejps.2009.10.013. [DOI] [PubMed] [Google Scholar]
- Maertens C, Droogmans G, Verbesselt R, Nilius B. Block of volume-regulated anion channels by selective serotonin reuptake inhibitors. Naunyn Schmiedebergs Arch Pharmacol. 2002;366:158–165. doi: 10.1007/s00210-002-0567-5. [DOI] [PubMed] [Google Scholar]
- McGrath J, Drummond G, McLachlan E, Kilkenny C, Wainwright C. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metzner L, Kalbitz J, Brandsch M. Transport of pharmacologically active proline derivatives by the human proton-coupled amino acid transporter hPAT1. J Pharmacol Exp Ther. 2004;309:28–35. doi: 10.1124/jpet.103.059014. [DOI] [PubMed] [Google Scholar]
- Metzner L, Kottra G, Neubert K, Daniel H, Brandsch M. Serotonin, L-tryptophan, and tryptamine are effective inhibitors of the amino acid transport system PAT1. FASEB J. 2005;19:1468–1473. doi: 10.1096/fj.05-3683com. [DOI] [PubMed] [Google Scholar]
- Metzner L, Dorn M, Markwardt F, Brandsch M. The orally active antihyperglycemic drug beta-guanidinopropionic acid is transported by the human proton-coupled amino acid transporter hPAT1. Mol Pharm. 2009;6:1006–1011. doi: 10.1021/mp9000684. [DOI] [PubMed] [Google Scholar]
- Ming X, Knight BM, Thakker DR. Vectorial transport of fexofenadine across Caco-2 cells: involvement of apical uptake and basolateral efflux transporters. Mol Pharm. 2011;8:1677–1686. doi: 10.1021/mp200026v. [DOI] [PubMed] [Google Scholar]
- Molnari JC, Hassan HE, Myers AL. Effects of sertraline on the pharmacokinetics of bupropion and its major metabolite, hydroxybupropion, in mice. Eur J Drug Metab Pharmacokinet. 2012;37:57–63. doi: 10.1007/s13318-011-0065-6. [DOI] [PubMed] [Google Scholar]
- Obach RS, Cox LM, Tremaine LM. Sertraline is metabolized by multiple cytochrome P450 enzymes, monoamine oxidases, and glucuronyl transferases in human: an in vitro study. Drug Metab Dispos. 2005;33:262–270. doi: 10.1124/dmd.104.002428. [DOI] [PubMed] [Google Scholar]
- Ohno Y, Hibino H, Lossin C, Inanobe A, Kurachi Y. Inhibition of astroglial Kir4.1 channels by selective serotonin reuptake inhibitors. Brain Res. 2007;1178:44–51. doi: 10.1016/j.brainres.2007.08.018. [DOI] [PubMed] [Google Scholar]
- Omkvist DH, Brodin B, Nielsen CU. Ibuprofen is a non-competitive inhibitor of the peptide transporter hPEPT1 (SLC15A1): possible interactions between hPEPT1 substrates and ibuprofen. Br J Pharmacol. 2010;161:1793–1805. doi: 10.1111/j.1476-5381.2010.01000.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park KS, Kong ID, Park KC, Lee JW. Fluoxetine inhibits L-type Ca2+ and transient outward K+ currents in rat ventricular myocytes. Yonsei Med J. 1999;40:144–151. doi: 10.3349/ymj.1999.40.2.144. [DOI] [PubMed] [Google Scholar]
- Pousti A, Bakhtiarian A, Najafi R, Deemyad T, Brumand K, Hosseini MJ. Effect of sertraline on ouabain-induced arrhythmia in isolated guinea-pig atria. Depress Anxiety. 2009;26:E106–E110. doi: 10.1002/da.20407. [DOI] [PubMed] [Google Scholar]
- Saruwatari J, Yasui-Furukori N, Niioka T, Akamine Y, Takashima A, Kaneko S, et al. Different effects of the selective serotonin reuptake inhibitors fluvoxamine, paroxetine, and sertraline on the pharmacokinetics of fexofenadine in healthy volunteers. J Clin Psychopharmacol. 2012;32:195–199. doi: 10.1097/JCP.0b013e318248ddb9. [DOI] [PubMed] [Google Scholar]
- Sawada K, Terada T, Saito H, Hashimoto Y, Inui K. Effects of glibenclamide on glycylsarcosine transport by the rat peptide transporters PEPT1 and PEPT2. Br J Pharmacol. 1999;128:1159–1164. doi: 10.1038/sj.bjp.0702895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terada T, Sawada K, Saito H, Hashimoto Y, Inui K. Inhibitory effect of novel oral hypoglycemic agent nateglinide (AY4166) on peptide transporters PEPT1 and PEPT2. Eur J Pharmacol. 2000;392:11–17. doi: 10.1016/s0014-2999(00)00119-9. [DOI] [PubMed] [Google Scholar]
- Thwaites DT, Anderson CM. Deciphering the mechanisms of intestinal imino (and amino) acid transport: the redemption of SLC36A1. Biochim Biophys Acta. 2006;1768:179–197. doi: 10.1016/j.bbamem.2006.10.001. [DOI] [PubMed] [Google Scholar]
- Thwaites DT, Anderson CM. The SLC36 family of proton-coupled amino acid transporters and their potential role in drug transport. Br J Pharmacol. 2011;164:1802–1816. doi: 10.1111/j.1476-5381.2011.01438.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu HJ, Appel DI, Grundemann D, Richelson E, Markowitz JS. Evaluation of organic cation transporter 3 (SLC22A3) inhibition as a potential mechanism of antidepressant action. Pharmacol Res. 2012;65:491–496. doi: 10.1016/j.phrs.2012.01.008. [DOI] [PubMed] [Google Scholar]
- Zhu T, Chen XZ, Steel A, Hediger MA, Smith DE. Differential recognition of ACE inhibitors in Xenopus laevis oocytes expressing rat PEPT1 and PEPT2. Pharm Res. 2000;17:526–532. doi: 10.1023/a:1007556630189. [DOI] [PubMed] [Google Scholar]