

Abstract
Background and Purpose
Available medications for chronic pain provide only partial relief and often cause unacceptable side effects. There is therefore a need for novel molecular targets to develop new therapeutics with improved efficacy and tolerability. Despite encouraging efficacy data in rodents with inhibitors of the neuronal glycine transporter-2 (GlyT2), there are also some reports of toxicity and their development was discontinued.
Experimental Approach
In order to clarify the possibility of targeting GlyT2 for the treatment of pain, we have used an integrated approach comprising in vitro pharmacology, selectivity, bioavailability, in vivo efficacy and safety assessment to analyse the properties and efficacy of ALX-1393 and Org-25543, the two published GlyT2 inhibitors from which in vivo data are available.
Key Results
We report that these compounds have a different set of undesirable properties that limit their usefulness as pharmacological tools. Importantly, we discover that inhibitors of GlyT2 can exert an apparent reversible or irreversible inhibition of the transporter and describe a new class of reversible GlyT2 inhibitors that preserves efficacy while avoiding acute toxicity.
Conclusions and Implications
Our pharmacological comparison of two closely related GlyT2 inhibitors with different modes of inhibition provides important insights into their safety and efficacy profiles, uncovering that in the presence of a GlyT2 mechanism-based toxicity, reversible inhibitors might allow a tolerable balance between efficacy and toxicity. These findings shed light into the drawbacks associated with the early GlyT2 inhibitors and describe a new mechanism that might serve as the starting point for new drug development.
Keywords: glycine, pain, neuropathic pain, reversible inhibitor
Introduction
Chronic pain conditions affect more than a billion people worldwide, a number exceeding those of diabetes, cardiovascular disease and cancer combined (National Research Council, 2011). Available medications, including non-steroidal anti-inflammatory drugs, anticonvulsants, antidepressants, ion channel modulators and opioid drugs, provide only partial relief and often cause adverse effects. There is therefore a need for novel molecular targets for the development of new pain treatments of improved efficacy and tolerability.
Chronic nerve pain (neuropathic pain) arises as a consequence of neurological damage that produces disabling, long-lasting increased response to noxious stimuli (hyperalgesia) and painful response to normally innocuous stimuli (allodynia; Woolf, 1983; Zimmermann and Herdegen, 1996; Scholz and Woolf, 2002). Among the changes resulting from nerve damage that will lead to chronic pain is a reduction in the inhibitory tone in the spinal cord (Basbaum et al., 2009; Latremoliere and Woolf, 2009). Restoring the inhibitory tone therefore appears as a reasonable therapeutic approach for chronic pain.
Glycine is one of the main inhibitory neurotransmitters in the brainstem and spinal cord and its action is regulated via reuptake by the presynaptic glycine transporter-2 (GlyT2), the expression of which is largely restricted to these areas (Jursky and Nelson, 1995; Zafra et al., 1995; Eulenburg et al., 2005; Zafra and Gimenez, 2008). A structurally related glycine transporter, GlyT1, is expressed by glial cells in forebrain regions where glycine plays an excitatory role as a co-agonist required for NMDA receptor activation (Jursky and Nelson, 1995; Zafra et al., 1995; Eulenburg et al., 2005; Zafra and Gimenez, 2008). Following the discovery of GlyT2 as a key regulator of the extracellular levels of glycine in the spinal cord, a surge of attention was devoted to the study of this transporter as a target for neuropathic pain (Dohi et al., 2009). Indeed, intrathecal injection of glycine is anti-nociceptive in rodent models of pain (Huang and Simpson, 2000; Tanabe et al., 2008; Dohi et al., 2009) and an endogenous GlyT2 inhibitor, N-arachidonyl glycine, reduces allodynia and hyperalgesia in rat models of inflammatory and neuropathic pain (Succar et al., 2007; Vuong et al., 2008; Dohi et al., 2009; Edington et al., 2009). Spinal knockdown of GlyT2 also produces a profound anti-allodynic effect in mouse chronic pain models (Morita et al., 2008). Despite this body of evidence and the encouraging initial reports following the generation of specific GlyT2 inhibitors by pharmaceutical companies in the early 2000s, the development of these inhibitors was discontinued and only a limited amount of data about them were ever published (Dohi et al., 2009). Once these compounds were made available to the scientific community, a large number of reports on their efficacy in a variety of chronic pain models have been published (Hermanns et al., 2008; Morita et al., 2008; Dohi et al., 2009; Haranishi et al., 2010), reviving the interest on this target.
Interestingly, the development of compounds against GlyT1 ran into an initial stop following the description of a lethal knockout phenotype and the toxicity of the first generation of inhibitors (Lindsley, 2010). The field would later experience a successful rebound with the discovery of different chemotypes, culminating with the clinical confirmation of the therapeutic benefit of GlyT1 inhibition for schizophrenia (Lindsley, 2010). Because of the lethality of the GlyT2 knockout (Gomeza et al., 2003) and the discontinuation of the GlyT2 programmes, we have performed an evaluation of the properties and efficacy of the two published inhibitors from which in vivo data are available. Here we confirm the efficacy of the brain-penetrant GlyT2 inhibitor Org-25543 in a rodent model of persistent pain, but also uncover a toxicity that closely mimics the GlyT2 knockout phenotype at dose levels compatible with an on-target effect. Importantly, we show that this GlyT2 inhibitor is a tight binder, behaving as an irreversible inhibitor, and report on a closely related reversible compound that avoids acute toxicity while preserving efficacy. Our findings shed light into the drawbacks associated with the early GlyT2 inhibitors and describe how on-target toxicity might be avoided by developing reversible GlyT2 inhibitors, thus opening a new avenue to re-evaluate the potential of this promising target for the treatment of chronic pain.
Methods
All experiments involving animals were approved by the ethical committee for animal experimentation of UCB, in accordance with the European Directive 2010/63/EU on the protection of animals used for scientific purpose and with the Belgian law on the use of laboratory animals. All studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (Kilkenny et al., 1995; McGrath et al., 2010).
Cloning, cell line generation and determination of [3H] glycine uptake
Full length human GlyT1 (NM_201649), human GlyT2 (NM_004211), mouse GlyT1 (NM_008135) and mouse GlyT2 (NM_001146013) were synthesized at Genscript and cloned into pcDNA3.1(+) (Life Technologies, Gent, Belgium). Final constructs were verified by sequencing. The transporters were expressed as transient transfections in FreeStyle™ 293-F suspension cells using 293fectin™ following the manufacturers instruction (all from Life Technologies).
Transfected cells were pelleted and gently re-suspended in uptake buffer containing HEPES 10 mM, NaCl 150 mM, CaCl2 1 mM, KCl 5 mM, MgCl2 1 mM, glucose 10 mM, pH 7.4. Cells were then incubated with test compounds or dimethyl sulfoxide (DMSO) at 37°C for 30 min. The uptake was started by the addition of 10 nM [3H]glycine, and allowed to continue for 30 min, after which the assay was terminated by a rapid filtration on glass fibre filters type A/C (Pall) that had been presoaked for 2 h in 0.1% Polyethyleneimine (Fluka Biochemika, Buchs, Switzerland). The filters were rinsed four times and transferred to plastic vials. Scintillation cocktail (Ultima Gold MV, Perkin Elmer, Zaventem, Belgium) was added and the radioactivity trapped on the filters was measured using a Liquid Scintillation analyser (Tri-Carb, Perkin Elmer). For IC50 value determinations initial experiments were performed by diluting compounds by log steps in DMSO from 1 pM to 100 μM. Accurate IC50 values were then determined by performing 10-point concentration curves over a suitable range using half log dilutions in DMSO. IC50 values were determined using a non-linear curve fitting method (Graphpad Prism® software, version 5.0, Graphpad, San Diego, CA, USA).
Selectivity profile
The selectivity of Org-25543 and compound 1 for GlyT2 was assessed compared with a broad panel of receptors, enzymes and ion channels. The compounds were tested at 10 μM for binding against a panel of more than 40 targets either internally (n = 3 with data in duplicate) or externally (CEREP, Celle l'Evescault, France, study 9140414, n = 1 with data in duplicate). For some of those targets, an IC50 curve was later done internally. Target nomenclature conforms to the British Journal of Pharmacology's Guide to Receptors and Channels (Alexander et al., 2011).
Oocyte glycine current assay
Xenopus leavis oocytes stage V–VI (purchased from EcoCyte Bioscience, Germany) were plated into conical NUNC 96-well plates in Barth's solution and microinjected with the automated screening system Roboocyte® (Multi Channel Systems, Reutlingen, Germany). Injections consisted of 50 nL of GlyT1 or GlyT2 mRNA dissolved in RNAse free water at 0.5 μg·μL−1. Oocytes were then kept at 17°C for 3–6 days before the functional analysis of expressed glycine transporters.
Two electrode voltage clamp recordings were performed with the automated Roboocyte® system using standard recording heads (electrode resistance 300–800 kΩ; current and voltage electrodes filled with potassium acetate at 1.5 M + potassium chloride 1.5 M, Multi-Channel System). Oocytes were impaled and voltage clamped at a holding potential of −60 mV, then rinsed with normal frog ringer buffer. Drugs were applied via a liquid dispenser (Gilson GX271; Gilson, Middleton, WI, USA) coupled to a peristaltic pump (MINIPULS 3, Gilson). The perfusion rate of the oocytes was ∼3 mL·min−1 and all solutions were freshly prepared before each experiment. Glycine application during 20 s evoked an inward current that was fully reversed after a 60-s washout. For compound testing, compounds were applied to the oocytes during 4 min before co-application with 15 μM glycine. Glycine-evoked inward currents were analysed using a Roboocyte Software version 2.2. Prism Graphpad software was then used to generate dose-response curves using non-linear regression analysis. Data were normalized to the first glycine response.
Additional methods included in the Supporting Information.
Results
Properties of the published GlyT2 inhibitors
Published preclinical evidence supporting GlyT2 inhibition as a viable approach for treating pain has been obtained using the non-selective endogenous GlyT2 inhibitor N-arachidonyl glycine (NAGly; Succar et al., 2007; Vuong et al., 2008), and two compounds belonging to different classes that were under development by Organon (Org-25543) and Allelix Pharma (ALX-1393) (Caulfield et al., 2001; Xu et al., 2005). While the published data using these compounds in animal pain models are largely positive, there are some conflicting reports of lack of efficacy or toxicity. To clarify these findings, we analysed the selectivity of Org-25543 and ALX-1393 for GlyT2 over GlyT1, as well as their ability to enter the brain following systemic administration (Figure 1).
Figure 1.
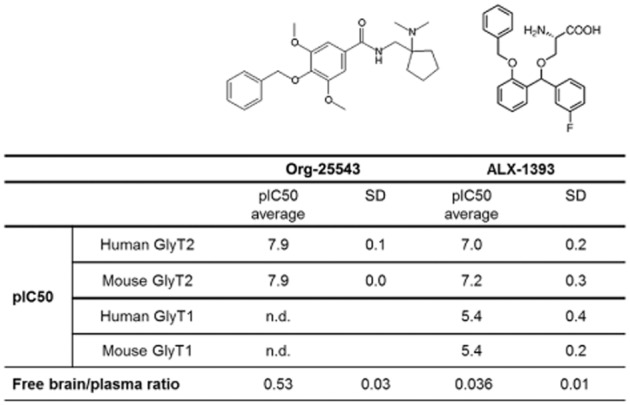
Affinity, selectivity and brain penetration properties of the published GlyT2 inhibitors Org-25543 and ALX-1393. Both compounds inhibited [3H]glycine uptake into HEK293 cells expressing GlyT2 with IC50 in the nanomolar range. ALX-1393 also inhibited [3H]glycine uptake into HEK293 cells expressing GlyT1 at low micromolar concentrations. Free brain/plasma concentration ratio for Org-25543 was determined 35 min after i.v. administration to mice at 2 and 20 mg·kg−1. For ALX-1393, compound tissue concentration was determined 60 min after i.v. administration to mice at 1 and 10 mg·kg−1.
We first evaluated the efficacy and glycine transporter selectivity of Org-25543 and ALX-1393 by measuring [3H]glycine uptake into HEK293 cells recombinantly expressing human or mouse GlyT2 and GlyT1 (Figure 1). Both inhibitors blocked [3H]glycine uptake with nanomolar potency in cells transfected with GlyT2 with no difference in affinity between the human and the mouse variants (12 nM and 100 nM IC50 respectively). Contrarily to Org-25543, ALX-1393 also inhibited [3H]glycine uptake in GlyT1-expressing cells (IC50 value of 4 μM).
To estimate whether systemic administration of Org-25543 and ALX-1393 resulted in sufficient target exposure in the CNS, we measured their concentration in brain tissue at 35 and 60 min, respectively, following i.v. administration. While the lipophilic Org-25543 readily crossed the blood–brain barrier with a free brain/plasma ratio of approximately 0.5, the amino acidic ALX-1393 showed minimal brain penetration, with a ratio of less than 0.05 after 60 min (Figure 1). At 10 mg·kg−1 i.v., the maximal dose of ALX-1393 that could be administered due to limited compound solubility, the calculated free concentration of compound in the brain was predicted to be threefold below its in vitro GlyT2 IC50 in the uptake assay. Because of these limitations, together with poor selectivity versus GlyT1, the subsequent experiments were carried out with Org-25543 only.
Pharmacological inhibition of GlyT2 by Org-25543 reduces formalin-evoked pain
In the formalin model of pain, intraplantar injection of formalin in mice results in a biphasic pain response; a first phase of acute pain due to direct nociceptor stimulation followed by a second phase that involves inflammation and central sensitization in the dorsal horn (Dubuisson and Dennis, 1977; Tjolsen et al., 1992). Efficacy of a test compound during the late phase in this model is regarded as predictive of efficacy in neuropathic pain (Dubuisson and Dennis, 1977; Tjolsen et al., 1992; Saddi and Abbott, 2000). In order to validate the sensitivity of our test, we confirmed the ability of gabapentin, the clinical standard of care, to reduce nociception in the formalin model (Supporting Information Figure S1). To determine the ability of GlyT2 inhibition to reduce pain hypersensitivity, we administered Org-25543 to mice 5 min before formalin injection and monitored their formalin-evoked pain behaviour by measuring paw licking during the 30 min following formalin injection (Figure 2 and Supporting Information Figure S2). Intravenous administration of Org-25543 preceding formalin injection decreased paw licking time during the late phase in a dose-dependent manner (Figure 2) with only a mild effect during the acute phase (Supporting Information Figure S2). The efficacy of Org-25543 to reduce pain behaviour during the late phase was seen at doses as low as 0.06 mg·kg−1 (36% reduction). The maximal effect observed without side effects corresponded to 2 mg·kg−1 (48% reduction), whereas at doses of 20 mg·kg−1, Org-25543 was highly toxic leading to convulsions and lethality within the 30 min time of the experiment.
Figure 2.
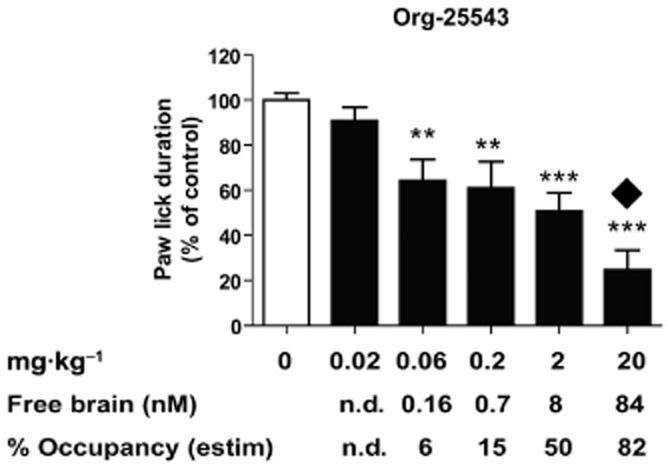
Pharmacological inhibition of GlyT2 by Org-25543 reduces formalin-evoked pain behaviour. Quantification of paw lick duration during the chronic phase in animals treated with formalin. Animals were pretreated with vehicle (n = 20) or Org-25543 0.02 mg·kg−1 (n = 10), 0.06 (n = 10), 0.2 (n = 10), 2 (n = 20) and 20 mg·kg−1 (n = 10, only six included in analysis). No significant effect was seen with Org-25543 at dose levels of 0.02 mg·kg−1. In contrast, doses ≥0.06 mg·kg−1 reduced the paw lick duration during the late phase of the response to formalin injection. At 20 mg·kg−1, Org-25543 induced convulsions and mortality in 4 out of 10 mice (black diamond). Bars represent mean ± SEM. **P < 0.01; ***P < 0.001; one-way anova, followed by a Dunnett's multicomparison test. Free brain concentration of Org-25543 at the end of the experiment (35 min after compound injection) estimated as 2% of total brain concentration based on protein binding data. The percentage of target occupancy at the end of the experiment was calculated based on compound brain concentration and in vitro IC50. Abbreviations: n.d., non-detectable.
Org-25543 displayed very high plasma and brain tissue protein binding, with only ∼2% of the compound being unbound. As a result, the free brain concentration of Org-25543 at the minimal active dose of 0.06 mg·kg−1 was estimated to be around 160 pM (Figure 2). With an in vitro IC50 of 12 nM, such active dose is predicted to achieve less than 10% of target inhibition. Similarly, the toxic dose of 20 mg·kg−1 was estimated to be still below full target occupancy (calculated 82% of glycine transport inhibition, Figure 2). Therefore, both the minimal active dose and the toxic dose required to reproduce the knockout phenotype (Gomeza et al., 2003) were lower than anticipated by the compound affinity for GlyT2.
The lack of agreement between exposure and pharmacodynamic effects of Org-25543 led us to assess its ability to interact with unrelated targets that could explain the observations. A selectivity profiling of Org-25543 against a panel of common and biologically relevant unrelated targets uncovered only partial activity on a limited number of targets with an IC50 above the maximal free compound concentration achieved at the highest dose tested in the formalin experiments (Supporting Information Table S1). Thus, Org-25543 is likely to be selective for GlyT2 at the range of doses used in the pain experiments.
Org-25543 is a biologically irreversible GlyT2 inhibitor
The observed shift between Org-25543 pharmacokinetics and pharmacodynamics is compatible with the profile of a tight binder or irreversible inhibitor. To assess this possibility, we used an oocyte assay that has been previously used to functionally express and characterize GlyT1 and GlyT2 (Wiles et al., 2006; Mezler et al., 2008). Application of glycine to Xenopus laevis oocytes expressing GlyT2 generates concentration-dependent inward currents measured by two electrode voltage clamp (Figure 3). For both transporters, glycine evoked inward currents with an EC50 of 15 μM, which were dependent on extracellular Na+ and Cl− ion levels (Supporting Information Figure S3). Glycine transport by GlyT1, but not GlyT2, was also modulated by external Zn2+ and H+ levels, also in agreement with published observations (Supporting Information Figure S3; Aubrey et al., 2000; Ju et al., 2004). To study the inhibitory properties of Org-25543 on GlyT2 activity, we used a protocol in which we applied three pulses of glycine to determine the compound potency and reversibility (Figure 3A). A first application of 15 μM glycine, which generates an inward current, is followed by a second application in the presence of a test compound that has been pre-incubated for 4 min, which in the example of Figure 3A corresponds to the known GlyT2 inhibitor NAGly (Wiles et al., 2006). After a co-application of compound and glycine and a washout period of 10 min, 15 μM of glycine is again applied in the absence of the inhibitor. While reversible compounds are washed out and the third current is similar in magnitude to the first one, in the case of irreversible or tight-binder inhibitors, a reduction in glycine transport can still be detected following the 10 min washout. Using this protocol, NAGly proved to be an effective and reversible inhibitor of electrogenic glycine transport by GlyT2, fully dissociating from the transporter after a washout period of 10 min (Figure 3A and B).
Figure 3.

Org-25543 is a biologically irreversible GlyT2 inhibitor. Glycine-evoked currents in oocytes expressing human GlyT2. (A, C, E) representative reversibility experiments displaying currents induced by three pulses of glycine in the presence of NAGly (A), Org-25543 (C) or ALX-1393 (E). (B, D, F) Concentration–response curves showing the % of glycine-induced current of the second peak (black) or the third peak (white, following washout) as compared with the first glycine current. Org-25543, but not NAGly or ALX-1393 was irreversible in this assay. Each point represents the average response of five to eight oocytes ± SEM.
In a first experiment, we verified that Org-25543 inhibited GlyT2-mediated glycine currents in Xenopus laevis oocytes with a potency in agreement with the IC50 obtained from [3H]glycine uptake into GlyT2-expressing HEK293 (IC50 = ∼20 nM, Figure 3C and D closed circles). We then determined the apparent reversibility of Org-25543 by measuring the ability of glycine to induce currents following a 10 min washout. In this assay, Org-25543 behaved as an irreversible inhibitor, and a third application of glycine following compound washout induced currents of the same magnitude to those obtained in the presence of the compound during the second application (Figure 3C and D open circles). This indicates that Org-25543 has a very slow off-rate and might behave as a ‘biologically irreversible’ inhibitor of glycine transport, as once the compound is bound to GlyT2, the activity of the transporter is effectively shut down for a significant time period.
We also evaluated whether the amino acid inhibitor ALX-1393 is a reversible or irreversible blocker of GlyT2. During co-application with glycine, ALX-1393 inhibited GlyT2-mediated glycine currents with similar potency to that observed in the radioactive uptake assay (IC50 = ∼25 nM, Figure 3F closed circles). Similarly, we reproduced our observation that ALX-1393 also inhibits GlyT1-mediated glycine transport in Xenopus laevis oocytes with 30 times lower potency when compared with inhibition of GlyT2. Unlike Org-25543, application of glycine following a washout period of ALX-1393 was able to evoke currents, albeit with some remaining partial inhibition (Figure 3E and F open circles). Therefore both available test compounds have a different set of undesirable properties that limit their usefulness as pharmacological tools, either poor brain penetration and activity on GlyT1 as in the case of ALX-1393, or a remarkable tight-binding mode for Org-25543 that might lead to biological irreversibility and potential on-target toxicity.
Generation of a reversible and brain-penetrant GlyT2 inhibitor
In order to understand the toxicity and efficacy observed with Org-25543 and to determine the feasibility of targeting GlyT2 for the treatment of pain we set to identify a reversible, selective and brain-penetrant GlyT2 inhibitor that could be used in vivo. With this aim, we used Org-25543 as a starting point and screened several analogues for activity on GlyT2 and selectivity versus GlyT1 using the radioactive uptake assay on HEK293 cells. This led to the identification of compound 1 (Figure 4A); a compound with an IC50 value of 100 nM for mouse GlyT2, high selectivity over GlyT1, and a selectivity profile against unrelated targets at 10 μM in line with that observed for Org-25543 (Figure 4B). Similarly to Org-25543, the physicochemical parameters of compound 1 appeared to be consistent with CNS penetration, obtaining a 0.4 brain/plasma free concentration ratio 35 min after intraperitoneal injection (Figure 4B). More importantly, the inhibition of GlyT2 by compound 1 was nearly completely reversible in the oocyte reversibility assay, with most of the glycine current recovered following washout even after complete inhibition of GlyT2 (Figure 4C and D). These properties made compound 1 the ideal pharmacological tool to evaluate in animal models whether the potential on-target toxicity of GlyT2 inhibitors might be avoided by developing reversible compounds.
Figure 4.

Compound 1 is a reversible and brain-penetrant GlyT2 inhibitor that reduces formalin-evoked pain behaviour. (A) Compound 1 structure. (B) Table summarizing compound 1 affinity for human and mouse GlyT1 and GlyT2 as well as brain/plasma ratio 35 min after i.p. administration to mice at 10–100 mg·kg−1. (C) Representative trace of a reversibility experiments and (D) concentration–response curves of glycine-induced currents in oocytes expressing human GlyT2. Compound 1 was reversible in this assay. Each point represents the average response of five to eight oocytes ± SEM. (E) Efficacy of compound 1 in the mouse formalin model. Quantification of paw lick duration during the chronic phase in animals treated with formalin. Animals were pretreated with vehicle (n = 20) or compound 1 at 1 mg·kg−1 (n = 10), 3 (n = 10), 10 (n = 10), 25 (n = 20) and 100 mg·kg−1 (n = 10). Doses ≥25 mg·kg−1 reduced the paw lick duration during the late phase of the response to formalin injection. At 100 mg·kg−1, compound 1 produced a profound reduction of pain behaviour with no convulsions or mortality. Bars represent mean ± SEM. ***P < 0.001; one-way anova, followed by a Dunnett's multicomparison test. Free brain concentration of compound 1 at the end of the experiment (35 min after compound injection) estimated as 2% of total brain concentration based on protein binding data. Percentage of target occupancy at the end of the experiment calculated based on compound brain concentration and in vitro IC50.
Reversible GlyT2 inhibitors reduce nociception in the formalin model
We then determined the minimal active dose and target exposure levels of the reversible GlyT2 inhibitor compound 1 in the formalin pain model (Figure 4E and Supporting Information Figure S4). We treated mice with compound 1 via intraperitoneal administration 5 min before formalin injection and monitored their pain behaviour during the second phase of the formalin model. Because of the difference in in vitro potency between Org-25543 and compound 1 (12 nM vs. 100 nM IC50 for mouse GlyT2, respectively) we adapted the dose range administered so that we could compare their efficacy at very low predicted target exposure levels (<10%) as well as at high predicted target occupancy (>50%). The highest dose of compound 1 that could be administered due to solubility limitations was 100 mg·kg−1. This dose was predicted to be sufficient to achieve over 50% estimated target occupancy. Similarly to Org-25543, compound 1 had good brain penetration and very high in vitro binding to brain homogenates, resulting in ∼2% of the compound estimated as being unbound. Based on this finding, the estimated free brain concentration at the minimal dose of compound 1 that produced a statistically significant analgesic effect (33% reduction at 25 mg·kg−1) was 42 nM, which corresponded to a calculated target occupancy of 36% (Figure 4E). Unlike Org-25543, which showed efficacy at very low predicted target exposure levels (<10%), low exposure levels of compound 1 failed to show any significant reduction in the development of hyperalgesia. In contrast, an estimated free brain concentration of 119 nM, achieving over 50% estimated target occupancy, produced a dramatic reduction in pain as seen by 90% reduction in paw licking time in the late phase of the formalin pain model (100 mg·kg−1, Figure 4E), suggesting a good correlation between compound 1 pharmacokinetic and pharmacodynamic properties. These data are consistent with our findings in the oocyte assay and support the reversible profile of compound 1 in vivo.
We did not see any evident serious side effect or mortality in mice treated with the reversible inhibitor compound 1, even at a dose that was able to achieve a profound reduction in nociceptive behaviour during the late phase of the formalin model. This was still true 3 h after the end of the experiment, when animals were killed. In contrast with these findings, the dose of Org-25543 able to produce a 75% reduction in hyperalgesia was highly toxic, inducing spasms, convulsions and the death of four mice out of 10 during the 35 min time of the experiment. Therefore we performed additional experiments in order to better understand the safety profile of both compounds.
Reversible and irreversible GlyT2 inhibitors have a different safety profiles
We determined the overall safety profile of Org-25543 and compound 1 using a range of inactive and active doses (Figure 5) using an Irwin neurobehavioural observation battery consisting of a series of 53 parameters, including central activity and reactivity, neurovegetative reflexes, neuromotor tone and autonomic signs (Irwin, 1968; De Ron et al., 2008). We also evaluated the profile of gabapentin as a positive control (not shown). Our results showed that Org-25543 was characterized by an overall excitatory profile, with dose-dependent increase in the incidence of excitatory side effects including tremors and stereotypies. We already detected other signs of excitability, such as enhanced vigilance and touch response, at the lowest dose. Because of ethical reasons, we were unable to administer the maximal active dose of 20 mg·kg−1 of Org-25543 to animals, a dose for which we already had observed spasms, convulsions and mortality during the formalin test. Compound 1, instead, showed an overall sedative profile comparable with that of gabapentin and characterized by a decrease in vigilance and spontaneous activity associated with a decrease in muscular tone. None of the GlyT2 inhibitors showed signs of general locomotion activity impairment (gate or limb position) predicted to interfere with the pharmacokinetic readout of the formalin model at any of the active doses. Therefore these findings support the hypothesis that the acute toxicity observed with some GlyT2 inhibitors is likely to result from their biologically irreversible profile and that reversible compounds might avoid this toxicity while preserving efficacy.
Figure 5.

Different side effect profile of Org-25543 and compound 1 in the Irwin test. Animals were pretreated with vehicle, 0.02, 0.2 or 2 mg·kg−1 of Org-25543 and 3, 25 and 100 g·kg−1 of compound 1 (n = 3 mice per group). The two intermediate active doses of Org-25543 (0.2 and 2 mg·kg−1) were characterized by tremors and stereotypies, with an incidence one mouse out of three or three mice out of three respectively. The 20 mg·kg−1 dose of Org-25543 was not repeated for the Irwin test and the toxicity signs already observed during the formalin test have been included in the table. No sedation was observed for Org-25543. Compound 1 had a sedative profile starting at the dose of 100 mg·kg−1.
Discussion and conclusions
Because of the elevated incidence and the lack of adequate treatments for chronic pain, there is a need to find new molecular targets that will enable the development of new therapeutics. We have now characterized the properties of two published GlyT2 inhibitors, originating from different companies, for which in vivo reports are available. Our data provide evidence that these compounds have limited usefulness as pharmacological tools, either because of poor brain penetration and activity on GlyT1 or a remarkable tight-binding mode that might lead to on-target toxicity. Our finding that on-target toxicity might be avoided by developing reversible GlyT2 inhibitors opens a new avenue to re-evaluate the potential of this promising target for chronic pain.
The efficacy of GlyT2 inhibitors to reduce nociception in a number of animal models of neuropathic and inflammatory pain has been reported over the years. However, there are some conflicting reports on the efficacy and toxicity of these compounds that can now be re-evaluated in the light of our findings. In one study by Morita et al. (2008), Org-25543 reduced nociceptive behaviour after partial sciatic nerve ligation and in a diabetic neuropathic pain model in mice with an ED50 value of 0.07–0.16 mg·kg−1 when administered i.v. and a minimal active dose of 0.01 mg·kg−1 (Morita et al., 2008). While brain levels of the compound at this minimal active dose fell below detection levels in our study, these findings are consistent with our observed minimal active dose and ED50, both much lower than what would be expected by the in vitro compound affinity for GlyT2. This reported efficacy can be attributed to an on-target effect in the light of our finding that Org-25543 behaves as a biologically irreversible inhibitor of GlyT2. The same cannot be said about the reported efficacy of ALX-1393 when administered peripherally at 0.01 mg·kg−1 in the partial sciatic nerve ligation model and at 0.1 mg·kg−1 in the streptozotocin diabetic neuropathy model (Morita et al., 2008), a phenotype not consistent with an on-target effect given the inability of ALX-1393 to cross the blood brain barrier.
This second compound has been the most extensively characterized one in animal models, and albeit with variations among studies, seems to have a narrow efficacy window characterized by toxicity at the highest doses. When delivered intrathecally, ALX-1393 has been reported to have efficacy with a minimal active dose of 10 ng in three neuropathic and inflammatory models in mice (Morita et al., 2008) or 20 μg in the formalin pain model in rat without any associated motor or respiratory function disturbance (Haranishi et al., 2010). A third study by Hermanns and collaborators failed to detect antinociception at doses lower than 100 μg when delivered intrathecally in the chronic constriction injury model in rats (Hermanns et al., 2008). This minimal active dose was accompanied by serious side effects, including major respiratory depression (Hermanns et al., 2008), a toxic phenotype also observed by Haranishi et al. (2010) at a dose of 60 μg. The toxic phenotype reported for ALX-1393 closely resembles that described for the first generations of GlyT1 inhibitors. This observation would be consistent with the elevated local concentration created by intrathecal delivery of ALX-1393 and the limited selectivity on GlyT2 versus GlyT1 that we report here. Collectively, these data and our findings suggest that the two unrelated GlyT2 inhibitors developed by pharmaceutical companies had a number of undesirable effects that seriously restricted their therapeutic window and questioned the viability of targeting GlyT2 as a therapeutic approach.
Several approved drugs act through an irreversible mode of action, either by covalent binding or by having a very slow dissociation rate from their targets (Copeland et al., 2006; Singh et al., 2011). This property can bring in significant pharmacodynamic and selectivity advantages when no on-target toxicity is present, as in the case of the irreversible monoamine oxidase inhibitors rasagiline and selegiline (Riederer and Laux, 2011). In other cases, such as in the irreversible inhibition of platelet COX by aspirin, this mode of action might be undesirable and has been circumvented by developing reversible COX inhibitors (Bunimov and Laneuville, 2008). Here, we describe for the first time that inhibitors of GlyT2 can exert an apparent reversible or irreversible inhibition of the transporter, leading to a different safety profile. While chronic dosing with compound 1 would be needed to rule out any toxicity associated with reversible GlyT2 inhibition, we show that acute administration of the irreversible Org-25543 had an excitatory profile leading to tremor, seizures and mortality, while the closely related, but reversible compound 1 had a sedative profile instead, comparable with that of gabapentin. Therefore by characterizing two very close analogues with different binding modes to GlyT2, we have uncovered a scenario where a potential mechanism-based acute toxicity can be circumvented using reversible inhibitors.
This new insight is reminiscent of the early days of GlyT1 inhibitors. Despite proven efficacy in animal models of psychosis, the original GlyT1 inhibitors were associated with serious side effects characterized by decreased respiratory activity and lethality, which led to the discontinuation of these programmes (Lindsley, 2010). It was only after the discovery of different chemotypes with different mode of action that efficacy and toxicity could be separated and the development of GlyT1 inhibitors successfully progressed into the clinic (Harsing et al., 2006; Javitt, 2009). In the case of GlyT1, the toxic hyperglycinergic phenotype resulted from sustained increase in glycine levels leading to excessive inhibition of postsynaptic targets via glycine receptors. In the case of GlyT2, sustained inhibition is likely to result in a hypoglycinergic phenotype instead (Rousseau et al., 2008), characterized by hyperekplexia and convulsions in rodents and humans carrying mutations (Gomeza et al., 2003; Rees et al., 2006). Although both transporters could have been regarded as non-viable targets based on initial limited pharmacological data, the discovery that compounds with different modes of action are able to circumvent the initial safety drawbacks remind us of the importance of understanding the mode of inhibition before ruling out a novel target as a viable one.
Taken together, our pharmacological comparison of two closely related GlyT2 inhibitors with different modes of inhibition provides important insights into their safety and efficacy profiles, uncovering that in the presence of a GlyT2 mechanism-based toxicity, reversible inhibitors might allow a tolerable balance between efficacy and toxicity. Our findings also shed light into the drawbacks associated with the early GlyT2 inhibitors and describe a new mechanism that might serve as the starting point for new drug development. These findings have the potential to revive the development of GlyT2 inhibitors and address an unmet medical need for chronic pain patients.
Acknowledgments
The authors would like to thank Maggi Burton, Alvaro Cardenas, Patrik Foerch, Javier Garcia-Ladona, Michel Gillard, Marie Ledecq, Karen Miller, Florence Moureau, Virgile Richard and Christian Wolf for their valuable suggestions on this work. We are also grateful to Eric Schenkel and Rosana Chirico for help with compound formulation, Marie Laetitia Mushikiwabo for support with the formalin studies, David Boucaut for in-life support, Chiara Rospo for bioanalysis and Nathalie Latour for determination of protein binding.
Conflict of interest
All authors are employees of UCB. None of the compounds mentioned in this paper is sold by UCB.
Supporting information
Additional Supporting Information may be found in the online version of this article at the publisher's web-site: http://dx.doi.org/10.1111/bph.12343
Figure S1 Gabapentin reduces formalin-evoked acute pain. Quantification of paw lick duration during the early (acute) and chronic phase in animals treated with formalin. Animals were pretreated with vehicle (n = 10) or gabapentin 10 (n = 10), 30 (n = 10) and 100 mg·kg−1 (n = 10). Bars represent mean ± SEM. *P < 0.05; **P < 0.01; ***P < 0.001; one-way ANOVA, followed by a Dunnett's multicomparison test.
Figure S2 Pharmacological inhibition of GlyT2 by Org-25543 reduces formalin-evoked acute pain. Quantification of paw lick duration during the early (acute) phase in animals treated with formalin. Animals were pretreated with vehicle (n = 20) or Org-25543 0.02 mg·kg−1 (n = 10), 0.06 (n = 10), 0.2 (n = 10), 2 (n = 20) and 20 mg·kg−1 (n = 10, only six included in analysis). No significant effect was seen with Org-25543 at dose levels of 0.02 mg·kg−1. Doses 0.06-2.0 mg·kg−1 induced a small, although significant, decrease in the paw lick duration during the initial 10 min after formalin injection. At 20 mg·kg−1, Org-25543 reduced formalin-evoked acute pain but was accompanied by convulsions and mortality in 4 out of 10 mice (black diamond). Bars represent mean ± SEM. **P < 0.01; ***P < 0.001; one-way ANOVA, followed by a Dunnett's multicomparison test. Free brain concentration and percentage of target occupancy at the end of the experiment was calculated as described for Figure 1. Abbreviations: n.d., non detectable.
Figure S3 Characterization of GlyT1 and 2 transport activity in Xenopus oocytes. (A) Typical current trace evoked by application of glycine to oocyte expressing GlyT2 transporter. (B) Transport of glycine by GlyT1 and GlyT2 generates a concentration-response curve with EC50 values consistent with those previously reported. (C-D) GlyT2 transport activity, measured as glycine-induced current, is dependent on the presence of extracellular sodium and chloride. (E-F) Transport activity of GlyT1, but not GlyT2, is sensitive to inhibition by extracellular Zinc (E) and pH (F), confirming the specificity for each transporter type. Each point represents the average response of four to nine oocytes for B and three to five oocytes for E and F ±SEM.
Figure S4 Reversible GlyT2 inhibition has no effect during the acute phase of the formalin model. Quantification of paw lick duration during the early (acute) phase in animals treated with formalin. Animals were pretreated with vehicle (n = 20) or compound 1 at 1 mg·kg−1 (n = 10), 3 (n = 10), 10 (n = 10), 25 (n = 20) and 100 mg·kg−1 (n = 10). Some limited reduction in paw-licking time was observed in the 25 mg·kg−1, but not at higher dose. Bars represent mean ± SEM. ***P < 0.001; one-way ANOVA, followed by a Dunnett's multicomparison test.
Table S1 Activity of Org-25543 against a panel of common and biologically relevant targets. The pharmacological specificity of Org-25543 was confirmed by assessment in radioligand binding assays in a broad CEREP screen (Paris, France; http://www.cerep.fr) and a collection of in-house targets.
Appendix S1 Supplemental methods.
References
- Alexander SP, Mathie A, Peters JA. Guide to receptors and channels (GRAC), 5th edition. Br J Pharmacol. 2011;164(Suppl 1):S1–324. doi: 10.1111/j.1476-5381.2011.01649_1.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aubrey KR, Mitrovic AD, Vandenberg RJ. Molecular basis for proton regulation of glycine transport by glycine transporter subtype 1b. Mol Pharmacol. 2000;58:129–135. doi: 10.1124/mol.58.1.129. [DOI] [PubMed] [Google Scholar]
- Basbaum AI, Bautista DM, Scherrer G, Julius D. Cellular and molecular mechanisms of pain. Cell. 2009;139:267–284. doi: 10.1016/j.cell.2009.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunimov N, Laneuville O. Cyclooxygenase inhibitors: instrumental drugs to understand cardiovascular homeostasis and arterial thrombosis. Cardiovasc Hematol Disord Drug Targets. 2008;8:268–277. doi: 10.2174/187152908786786250. [DOI] [PubMed] [Google Scholar]
- Caulfield WL, Collie IT, Dickins RS, Epemolu O, McGuire R, Hill DR, et al. The first potent and selective inhibitors of the glycine transporter type 2. J Med Chem. 2001;44:2679–2682. doi: 10.1021/jm0011272. [DOI] [PubMed] [Google Scholar]
- Copeland RA, Pompliano DL, Meek TD. Drug-target residence time and its implications for lead optimization. Nat Rev Drug Discov. 2006;5:730–739. doi: 10.1038/nrd2082. [DOI] [PubMed] [Google Scholar]
- De Ron P, Delaunois A, Hanon E, Lamberty Y, Guyaux M. An original method to interpret neurobehavioral data generated by the Irwin test in the mouse. In: Spink AJ, Ballintijn MR, Bogers ND, Grieco F, Loijens LWS, Noldus LPJJ, Smit G, Zimmerman PH, editors. Proceedings of Measuring Behavior 2008 (Maastricht, The Netherlands, August 26-29, 2008) Wageningen: Noldus Information Technology; 2008. p. 326. [Google Scholar]
- Dohi T, Morita K, Kitayama T, Motoyama N, Morioka N. Glycine transporter inhibitors as a novel drug discovery strategy for neuropathic pain. Pharmacol Ther. 2009;123:54–79. doi: 10.1016/j.pharmthera.2009.03.018. [DOI] [PubMed] [Google Scholar]
- Dubuisson D, Dennis SG. The formalin test: a quantitative study of the analgesic effects of morphine, meperidine, and brain stem stimulation in rats and cats. Pain. 1977;4:161–174. doi: 10.1016/0304-3959(77)90130-0. [DOI] [PubMed] [Google Scholar]
- Edington AR, McKinzie AA, Reynolds AJ, Kassiou M, Ryan RM, Vandenberg RJ. Extracellular loops 2 and 4 of GLYT2 are required for N-arachidonylglycine inhibition of glycine transport. J Biol Chem. 2009;284:36424–36430. doi: 10.1074/jbc.M109.017509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eulenburg V, Armsen W, Betz H, Gomeza J. Glycine transporters: essential regulators of neurotransmission. Trends Biochem Sci. 2005;30:325–333. doi: 10.1016/j.tibs.2005.04.004. [DOI] [PubMed] [Google Scholar]
- Gomeza J, Ohno K, Hulsmann S, Armsen W, Eulenburg V, Richter DW, et al. Deletion of the mouse glycine transporter 2 results in a hyperekplexia phenotype and postnatal lethality. Neuron. 2003;40:797–806. doi: 10.1016/s0896-6273(03)00673-1. [DOI] [PubMed] [Google Scholar]
- Haranishi Y, Hara K, Terada T, Nakamura S, Sata T. The antinociceptive effect of intrathecal administration of glycine transporter-2 inhibitor ALX1393 in a rat acute pain model. Anesth Analg. 2010;110:615–621. doi: 10.1213/ANE.0b013e3181c7ebbb. [DOI] [PubMed] [Google Scholar]
- Harsing LG, Jr, Juranyi Z, Gacsalyi I, Tapolcsanyi P, Czompa A, Matyus P. Glycine transporter type-1 and its inhibitors. Curr Med Chem. 2006;13:1017–1044. doi: 10.2174/092986706776360932. [DOI] [PubMed] [Google Scholar]
- Hermanns H, Muth-Selbach U, Williams R, Krug S, Lipfert P, Werdehausen R, et al. Differential effects of spinally applied glycine transporter inhibitors on nociception in a rat model of neuropathic pain. Neurosci Lett. 2008;445:214–219. doi: 10.1016/j.neulet.2008.09.012. [DOI] [PubMed] [Google Scholar]
- Huang W, Simpson RK. Long-term intrathecal administration of glycine prevents mechanical hyperalgesia in a rat model of neuropathic pain. Neurol Res. 2000;22:160–164. doi: 10.1080/01616412.2000.11741054. [DOI] [PubMed] [Google Scholar]
- Irwin S. Comprehensive observational assessment: Ia. A systematic, quantitative procedure for assessing the behavioral and physiologic state of the mouse. Psychopharmacologia. 1968;13:222–257. doi: 10.1007/BF00401402. [DOI] [PubMed] [Google Scholar]
- Javitt DC. Glycine transport inhibitors for the treatment of schizophrenia: symptom and disease modification. Curr Opin Drug Discov Devel. 2009;12:468–478. [PubMed] [Google Scholar]
- Ju P, Aubrey KR, Vandenberg RJ. Zn2+ inhibits glycine transport by glycine transporter subtype 1b. J Biol Chem. 2004;279:22983–22991. doi: 10.1074/jbc.M312484200. [DOI] [PubMed] [Google Scholar]
- Jursky F, Nelson N. Localization of glycine neurotransmitter transporter (GLYT2) reveals correlation with the distribution of glycine receptor. J Neurochem. 64:1026–1033. doi: 10.1046/j.1471-4159.1995.64031026.x. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG. Animal research: Reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol. 1995;160:1577–1579. doi: 10.1111/j.1476-5381.2010.00872.x. 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latremoliere A, Woolf CJ. Central sensitization: a generator of pain hypersensitivity by central neural plasticity. J Pain. 2009;10:895–926. doi: 10.1016/j.jpain.2009.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindsley C. GlyT1 – up from the ashes. The importance of not condemning a mechanism based on a single chemotype. ACS Chem Neurosci. 1:165–166. doi: 10.1021/cn100017a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath J, Drummond G, McLachlan E, Kilkenny C, Wainwright C. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mezler M, Hornberger W, Mueller R, Schmidt M, Amberg W, Braje W, et al. Inhibitors of GlyT1 affect glycine transport via discrete binding sites. Mol Pharmacol. 2008;74:1705–1715. doi: 10.1124/mol.108.049312. [DOI] [PubMed] [Google Scholar]
- Morita K, Motoyama N, Kitayama T, Morioka N, Kifune K, Dohi T. Spinal antiallodynia action of glycine transporter inhibitors in neuropathic pain models in mice. J Pharmacol Exp Ther. 2008;326:633–645. doi: 10.1124/jpet.108.136267. [DOI] [PubMed] [Google Scholar]
- National Research Council. Relieving Pain in America: A Blueprint for Transforming Prevention, Care, Education, and Research. Washington: The National Academy Press; 2011. [PubMed] [Google Scholar]
- Rees MI, Harvey K, Pearce BR, Chung SK, Duguid IC, Thomas P, et al. Mutations in the gene encoding GlyT2 (SLC6A5) define a presynaptic component of human startle disease. Nat Genet. 2006;38:801–806. doi: 10.1038/ng1814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riederer P, Laux G. MAO-inhibitors in Parkinson's disease. Exp Neurobiol. 2011;20:1–17. doi: 10.5607/en.2011.20.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rousseau F, Aubrey KR, Supplisson S. The glycine transporter GlyT2 controls the dynamics of synaptic vesicle refilling in inhibitory spinal cord neurons. J Neurosci. 2008;28:9755–9768. doi: 10.1523/JNEUROSCI.0509-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saddi G, Abbott FV. The formalin test in the mouse: a parametric analysis of scoring properties. Pain. 2000;89:53–63. doi: 10.1016/S0304-3959(00)00348-1. [DOI] [PubMed] [Google Scholar]
- Scholz J, Woolf CJ. Can we conquer pain? Nat Neurosci. 2002;5(Suppl):1062–1067. doi: 10.1038/nn942. [DOI] [PubMed] [Google Scholar]
- Singh J, Petter RC, Baillie TA, Whitty A. The resurgence of covalent drugs. Nat Rev Drug Discov. 2011;10:307–317. doi: 10.1038/nrd3410. [DOI] [PubMed] [Google Scholar]
- Succar R, Mitchell VA, Vaughan CW. Actions of N-arachidonyl-glycine in a rat inflammatory pain model. Mol Pain. 2007;3:24. doi: 10.1186/1744-8069-3-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanabe M, Takasu K, Yamaguchi S, Kodama D, Ono H. Glycine transporter inhibitors as a potential therapeutic strategy for chronic pain with memory impairment. Anesthesiology. 2008;108:929–937. doi: 10.1097/ALN.0b013e31816c9044. [DOI] [PubMed] [Google Scholar]
- Tjolsen A, Berge OG, Hunskaar S, Rosland JH, Hole K. The formalin test: an evaluation of the method. Pain. 1992;51:5–17. doi: 10.1016/0304-3959(92)90003-T. [DOI] [PubMed] [Google Scholar]
- Vuong LA, Mitchell VA, Vaughan CW. Actions of N-arachidonyl-glycine in a rat neuropathic pain model. Neuropharmacology. 2008;54:189–193. doi: 10.1016/j.neuropharm.2007.05.004. [DOI] [PubMed] [Google Scholar]
- Wiles AL, Pearlman RJ, Rosvall M, Aubrey KR, Vandenberg RJ. N-arachidonyl-glycine inhibits the glycine transporter, GLYT2a. J Neurochem. 2006;99:781–786. doi: 10.1111/j.1471-4159.2006.04107.x. [DOI] [PubMed] [Google Scholar]
- Woolf CJ. Evidence for a central component of post-injury pain hypersensitivity. Nature. 1983;306:686–688. doi: 10.1038/306686a0. [DOI] [PubMed] [Google Scholar]
- Xu TX, Gong N, Xu TL. Inhibitors of GlyT1 and GlyT2 differentially modulate inhibitory transmission. Neuroreport. 2005;16:1227–1231. doi: 10.1097/00001756-200508010-00019. [DOI] [PubMed] [Google Scholar]
- Zafra F, Gimenez C. Glycine transporters and synaptic function. IUBMB Life. 2008;60:810–817. doi: 10.1002/iub.128. [DOI] [PubMed] [Google Scholar]
- Zafra F, Aragon C, Olivares L, Danbolt NC, Gimenez C, Storm-Mathisen J. Glycine transporters are differentially expressed among CNS cells. J Neurosci. 1995;15(5 Pt 2):3952–3969. doi: 10.1523/JNEUROSCI.15-05-03952.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmermann M, Herdegen T. Plasticity of the nervous system at the systematic, cellular and molecular levels: a mechanism of chronic pain and hyperalgesia. Prog Brain Res. 1996;110:233–259. doi: 10.1016/s0079-6123(08)62578-9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Gabapentin reduces formalin-evoked acute pain. Quantification of paw lick duration during the early (acute) and chronic phase in animals treated with formalin. Animals were pretreated with vehicle (n = 10) or gabapentin 10 (n = 10), 30 (n = 10) and 100 mg·kg−1 (n = 10). Bars represent mean ± SEM. *P < 0.05; **P < 0.01; ***P < 0.001; one-way ANOVA, followed by a Dunnett's multicomparison test.
Figure S2 Pharmacological inhibition of GlyT2 by Org-25543 reduces formalin-evoked acute pain. Quantification of paw lick duration during the early (acute) phase in animals treated with formalin. Animals were pretreated with vehicle (n = 20) or Org-25543 0.02 mg·kg−1 (n = 10), 0.06 (n = 10), 0.2 (n = 10), 2 (n = 20) and 20 mg·kg−1 (n = 10, only six included in analysis). No significant effect was seen with Org-25543 at dose levels of 0.02 mg·kg−1. Doses 0.06-2.0 mg·kg−1 induced a small, although significant, decrease in the paw lick duration during the initial 10 min after formalin injection. At 20 mg·kg−1, Org-25543 reduced formalin-evoked acute pain but was accompanied by convulsions and mortality in 4 out of 10 mice (black diamond). Bars represent mean ± SEM. **P < 0.01; ***P < 0.001; one-way ANOVA, followed by a Dunnett's multicomparison test. Free brain concentration and percentage of target occupancy at the end of the experiment was calculated as described for Figure 1. Abbreviations: n.d., non detectable.
Figure S3 Characterization of GlyT1 and 2 transport activity in Xenopus oocytes. (A) Typical current trace evoked by application of glycine to oocyte expressing GlyT2 transporter. (B) Transport of glycine by GlyT1 and GlyT2 generates a concentration-response curve with EC50 values consistent with those previously reported. (C-D) GlyT2 transport activity, measured as glycine-induced current, is dependent on the presence of extracellular sodium and chloride. (E-F) Transport activity of GlyT1, but not GlyT2, is sensitive to inhibition by extracellular Zinc (E) and pH (F), confirming the specificity for each transporter type. Each point represents the average response of four to nine oocytes for B and three to five oocytes for E and F ±SEM.
Figure S4 Reversible GlyT2 inhibition has no effect during the acute phase of the formalin model. Quantification of paw lick duration during the early (acute) phase in animals treated with formalin. Animals were pretreated with vehicle (n = 20) or compound 1 at 1 mg·kg−1 (n = 10), 3 (n = 10), 10 (n = 10), 25 (n = 20) and 100 mg·kg−1 (n = 10). Some limited reduction in paw-licking time was observed in the 25 mg·kg−1, but not at higher dose. Bars represent mean ± SEM. ***P < 0.001; one-way ANOVA, followed by a Dunnett's multicomparison test.
Table S1 Activity of Org-25543 against a panel of common and biologically relevant targets. The pharmacological specificity of Org-25543 was confirmed by assessment in radioligand binding assays in a broad CEREP screen (Paris, France; http://www.cerep.fr) and a collection of in-house targets.
Appendix S1 Supplemental methods.