

Abstract
Background and Purpose
The gallnut of Rhus chinensis MILL and its main constituent penta-O-galloyl-β-D-glucose (PGG) inhibited NF-κB activation in LPS-stimulated peritoneal and colonic macrophages. Here we have investigated PGG mechanisms underlying anti-inflammatory effects of PGG in vitro and in vivo.
Experimental Approach
Male C57BL/6 mice (18–22 g, 6 weeks old) were used to prepare peritoneal and colonic macrophages and for the induction of colitis by intrarectal administration of 2,3,4-trinitrobenzene sulphonic acid (TNBS). A range of inflammatory markers and transcription factors were evaluated by elisa, immunoblotting, flow cytometry and confocal microscopy.
Key Results
Expression of Toll-like receptor (TLR)-4 or Lipopolysaccharide (LPS) binding to TLR-4 in LPS-stimulated peritoneal macrophages was not affected by PGG. However PGG inhibited binding of an anti-MyD88 antibody to peritoneal macrophages, but did not reduce binding of anti–IL-1 receptor-associated kinase (IRAK1) and IRAK4 antibodies to the macrophages with or without transfection with MyD88 siRNA. PGG potently reduced the activation of IRAK1, NF-κB, and MAPKs in LPS- or pepetidoglycan-stimulated peritoneal and colonic macrophages. PGG suppressed IL-1β, TNF-α and IL-6 in LPS-stimulated peritoneal macrophages, while increasing expression of the anti-inflammatorycytokine IL-10. Oral administration of PGG inhibited colon shortening and myeloperoxidase activity in mice with TNBS-induced colitis, along with reducing NF-κB activation and IL-1β, TNF-α, and IL-6 levels, whereas it increased IL-10.
Conclusions and Implications
PGG reduced activation of NF-κB and MAPK signalling pathways by directly interacting with the MyD88 adaptor protein. PGG may ameliorate inflammatory diseases such as colitis.
Keywords: penta-O-galloyl-β-D-glucose, inflammation, MyD88, NF-κB, macrophage
Introduction
Inflammatory bowel disease (IBD), including ulcerative colitis and Crohn's disease, consists of chronically relapsing disorders of the intestine (Shanahan, 2002; Binder, 2004). The pathogenic mechanism was proposed to be dys-regulation of the intestinal immune response to environmental antigens in the gastrointestinal tract, such as intestinal microbiota that mainly reside in the ileum, caecum and colon (Rafii et al., 1999; Atreya et al., 2000; Joh et al., 2011). Normal intestinal microbiota consist of more than 1000 bacterial species, which are directly influenced by endogenous and exogenous factors such as diet, drugs, stress and hormones (Peris-Bondia et al., 2011; Kim et al., 2012; Pendyala et al., 2012). For example, a high-fat diet induces LPS, a known inflammatory mediator, in the intestinal contents of humans and animals, supporting that intestinal microbiota initiate and perpetuate colonic inflammation. More specifically, intestinal bacterial endotoxins penetrate the epithelial barrier and stimulate immune reactions in the mucosa (Radema et al., 1991; Rafii et al., 1999). Subsequently, these toxins induce production of pro-inflammatory cytokines such as IL-1β, IL-6 and TNF-α, and other inflammatory mediators, leading to inflammatory activation via distinct signalling pathways through Toll-like receptors (TLRs) and/or cytokine receptors (Jung et al., 1995; Chow et al., 1999; Cario and Podolsky, 2000; Joh and Kim, 2011; receptor nomenclature follows Alexander et al., 2011). Suppressing the expression of these inflammatory mediators therefore decreases the incidence of inflammatory diseases. The assessment of natural products that may lessen inflammatory diseases has recently become a topic of much interest (Paradkar et al., 2004; Joh and Kim, 2011).
The gallnut of Rhus chinensis MILL (GRC; family Anacardiaceae), which contains 1,2,3,4,6-penta-O-galloyl-β-d-glucose (PGG) as a major constituent (Park et al., 2008), is used for treating diarrhoea, excessive sweating, bleeding, and chronic coughs as a traditional Chinese medicine in China, Japan and Korea (Djakpo and Yao, 2010; Shim et al., 2003; Kim et al., 2005; Ren et al., 2008). It is reported to inhibit α-glucosidase (Shim et al., 2003) and to display anti-anaphylactic (Kim et al., 2005), anti-viral (Nakano et al., 1998) and anti-ischaemic activities (Lee et al., 2012). PGG inhibits TNF-α and IL-6 secretion in LPS-stimulated peripheral blood mononuclear cells (Genfa et al., 2005), and also reduces the TNF-α level in rats after systemic administration of LPS. Oh et al. (2004) reported that PGG blocks IL-8 mRNA expression and secretion in human monocytic U937 cells stimulated with phorbol 12-myristate 13-acetate (PMA) or TNF-α. Finally, PGG significantly inhibits COX-2 and inducible NOS (iNOS) expression, as well as NF-κB activation via inhibition of IκB kinase (IKK) activity in LPS-activated RAW264.7 cells (Pan et al., 2000; Lee et al., 2003).
Our long-standing search for anti-inflammatory agents from natural products showed that GRC potently inhibited the phosphorylation of IL-1 receptor-associated kinase 1 (IRAK1) phosphorylation and NF-κB activation in murine primary peritoneal macrophages, stimulated by LPS- or peptidoglycan (PGN). Recently, we additionally found that PGG, a major constituent of GRC, inhibited the activation of PI3K and MAPKs in LPS-stimulated peritoneal macrophages, which was not investigated in a previous similar report (Pan et al., 2000). Therefore, in this study we attempted to clarify the anti-inflammatory mechanism of PGG by thoroughly investigating the molecular basis of its anti-inflammatory effect, using LPS-induced peritoneal and colonic macrophages, in a model of systemic inflammation induced by LPS and a model of colitis induced by 2,3,4-trinitrobenzene sulphonic acid (TNBS) in mice.
Methods
Animals
All animal care and experimental procedures were in accordance with the NIH and Kyung Hee University guidelines for Laboratory Animals Care and Use, and were approved by the Committee for the Care and Use of Laboratory Animals in the College of Pharmacy, Kyung Hee University (KHP-2012-11-01-R1). All studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (Kilkenny et al., 2010; McGrath et al., 2010). A total of 95 animals were used in the experiments described here. Male C57BL/6 mice (18–22 g, 6 weeks) were supplied from the Orient Animal Breeding Center (Sungnam, Korea). All animals were housed in wire cages at 20–22°C and 50 ± 10% humidity, fed standard laboratory chow (Samyang Co., Seoul, Korea) and allowed water ad libitum.
Isolation and culture of peritoneal and colonic macrophages
To isolate peritoneal macrophages, mice were intraperitoneally injected with 2 mL of 4% sodium thioglycolate. Mice were killed 4 days after injection, and the peritoneal cavities were flushed with 10 mL RPMI 1640. The peritoneal lavage fluids were centrifuged at 200× g for 10 min, and then the cells were resuspended with RPMI 1640 and plated. After incubation for 1 h at 37°C, the cells were washed three times and non-adherent cells were removed by aspiration. Cells were cultured in 24-well plates (5 × 105 cells per well) at 37°C in RPMI 1640 with 10% fetal bovine serum (FBS). Peritoneal macrophages were separated from colonic cells using a biotin-labeled anti-mouse F4/80 antibody (Invitrogen, Carlsbad, CA, USA) and streptavidin magnetic beads (Invitrogen).
To isolate macrophages from the intestine, Peyer's patches were isolated from mouse intestines and digested in DMEM containing 1 mg·mL−1 dispase, 0.25 mg·mL−1 collagenase A and 25 U·mL−1 DNAase (Roche Diagnostic, Indianapolis, IN, USA) at 37°C for 20 min with shaking, and then passed through a 70 μm cell strainer (BD Biosciences, San Diego, CA, USA). The cells were collected by centrifugation and then washed with PBS containing 4% FBS. The cells were resuspended in cold IMag buffer (0.05% BSA and 2 mM EDTA in PBS), and viable cells were counted by Trypan blue exclusion. Colonic macrophages were separated from colonic cells using a biotin-labelled anti-mouse F4/80 antibody and streptavidin magnetic beads (Invitrogen). To examine the anti-inflammatory effect of PGG, peritoneal or colonic macrophages were incubated in the absence or presence of PGG with 50 ng·mL−1 LPS or PGN.
Confocal and fluorescent microscopy
For the p65 assay, peritoneal macrophages were stimulated with LPS (100 ng·mL−1) in the presence or absence of PGG for 60 min. The cells were then fixed with 4% formaldehyde and permeabilized with 0.2% Triton X-100. Then, the cells were stained with a goat anti-p65 polyclonal antibody for 2 h at 4°C, followed by incubation with Alexa 488-conjugated secondary antibody propidium iodide (10 μg·mL−1; Calbiochem Co., San Diego, CA, USA) for 1 h. Images were obtained using confocal microscopy.
For the LPS-TLR4 complex assay, peritoneal macrophages plated on glass slides were incubated at 37°C overnight. Macrophages were stimulated with Alexa Fluor 594-conjugated LPS (10 μg·mL−1; Invitrogen) for 20 min in the presence or absence of PGG. The cells were then fixed with 4% formaldehyde and 3% sucrose for 20 min (Baldwin, 1996; Joh et al., 2012). Then, the cells were stained with a rabbit anti-TLR4 polyclonal antibody for 2 h at 4°C, followed by Alexa Fluor 488-conjugated secondary antibodies for 1 h (Park et al., 2008). The stained cells were then observed by confocal microscopy.
For MyD88 and IRAK assays, peritoneal macrophages were stimulated with LPS (100 ng·mL−1) in the presence or absence of PGG for 30 min. The cells were then fixed with 4% formaldehyde and permeabilized with 0.2% Triton X-100. Then, the cells were stained with goat anti-MyD88, anti-IRAK1, or anti-p-IRAK1 polyclonal antibody for 2 h at 4°C, followed by Alexa Fluor 488-conjugated secondary antibody for 1 h (Park et al., 2008). The stained cells were observed by confocal microscopy.
Flow cytometry
Mouse peritoneal macrophages were incubated with Alexa Fluor 488-conjugated LPS (10 μg·mL−1) for 10 min at room temperature. The cells were then fixed in PBS containing 4% paraformaldehyde and 3% sucrose for 20 min. After washing with PBS, the macrophages were incubated with propidium iodide (10 μg·mL−1) for 10 min and then analysed by flow cytometry.
Transient transfection with siRNA
Cells (3 × 105 cells per well) were seeded in 24-well plates and allowed to rest for 24 h. Then the cells were transfected with 100 nM siRNA for TLR4 or MyD88 using Lipofectamine™ 2000 (Invitrogen) according to the manufacturer's instruction. At 48 h after transfections, cells were treated with or without PGG (5 μM) and/or LPS (50 ng·mL−1).
Model of colitis in mice
Mice were randomly divided into five groups: normal (control) and TNBS-induced colitis groups treated with or without PGG or mesalazine. Each group consisted of seven mice. Colitis was induced by slow intrarectal administration of a 2.5% (w/v) TNBS solution (100 μL) in 50% ethanol into the colon of anaesthetized mice via a thin round-tip needle attached to a 1 mL syringe (Joh et al., 2011). The normal (control) group was treated with the vehicle alone. The needle was inserted so that the tip was 3.5–4.0 cm proximal to the anal verge. To distribute the agents within the entire colon and caecum, mice were held in a vertical position for 30 s after the injection. Using this procedure, >95% of the mice retained the TNBS enema. If an animal quickly excreted the TNBS-ethanol solution, it was excluded from the remainder of the study. PGG (5 or 10 mg·kg−1) [based on the ethnopharmacological dose (2–10 g GRC/day/person) and the preliminary study] or mesalazine (10 mg·kg−1) dissolved in 2% Tween 80 was orally administered once a day for 3 days after TNBS treatment. The mice were killed the day after the final administration of PGG or mesalazine. The colon was quickly removed, opened longitudinally and gently cleared of stool by washing with PBS. Macroscopic assessment of the disease grade was scored according to a previously reported scoring system (0, no ulceration and no inflammation; 1, no ulceration and local hyperaemia; 2, ulceration with hyperaemia; 3, ulceration and inflammation at one site only; 4, two or more sites of ulceration and inflammation; 5, ulceration extending more than 2 cm), and the colon tissue was divided longitudinally for immunoblotting, elisa, myeloperoxidase (MPO) and histological examination.
For the assays of immunoblotting, elisa, and MPO activity, the colon was homogenized in 1 mL ice-cold RIPA containing 1% protease inhibitor cocktail and 1% phosphatase inhibitor cocktail. The homogenates were centrifuged (15 000× g, 4°C) for 15 min, and then the supernatants were collected and used.
For the histological studies, one longitudinal part of each colon was fixed overnight in 50 mM phosphate buffer (pH 7.4) containing 4% paraformaldehyde, and then immersed in a 30% sucrose solution (in 50 mM phosphate buffered saline) and stored at 4°C until sectioning. Frozen colons were sectioned along the coronal plane (10 μm) using a cryostat (Leica Microsystems, AG, Germany), and stained with haematoxylin–eosin.
MPO activity assay
An aliquot (50 μL) of the colon supernatant was added to a reaction mixture of 1.6 mM tetramethyl benzidine and 0.1 mM H2O2, and then incubated at 37°C. The absorbance was obtained at 650 nm over time. MPO activity was defined as the quantity of enzyme degrading 1 μmol·mL−1 of peroxide at 37°C, and expressed in unit·(mg protein)−1 (Joh et al., 2011).
elisa and immunoblotting
For the elisas of IL-1β, IL-6, IL-10 and TNF-α, colons or cell culture supernatants were transferred to 96-well elisa plates. IL-1β, IL-6, IL-10 and TNF-α concentrations were determined using commercial elisa kits (Pierce Biotechnology, Inc., Rockford, IL, USA).
For immunoblot analyses of p-IRAK1, p-IKKβ, p-p65 and β-actin, colon tissue homogenates or collected cells were resuspended in 1 mL RIPA containing 1% protease inhibitor cocktail and 1% phosphatase inhibitor cocktail. After centrifugation, the supernatant was used for the immunoblot assay. The proteins from collected cells were separated by electrophoresis on an 8–10% SDS–polyacrylamide gel, and then transferred to a nitrocellulose membrane and immunoblotted using standard procedures (Joh et al., 2011).
Data analysis
All data are expressed as the mean ± SD. Statistical significance was assessed using one-way anova followed by the Student's t Newman–Keuls test.
Materials
LPS was purified from Escherichia coli O111:B4, PGN was purified from Staphylococcus aureus cell wall component. TNBS, DMEM, RPMI 1640 and mesalazine were purchased from Sigma Co. (St. Louis, MO, USA). Antibodies for MyD88 (sc-8197), IRAK1 (sc-7883), p-IRAK1 (sc-130197), IRAK2 (sc-23652), IRAK4 (sc-34470), IκBα (sc-371), p65 (sc-372), COX-2 (sc-1747-R), iNOS (sc-650), β-actin (sc-47778) and small–interfering RNA (siRNA) for MyD88 and for TLR4 were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Antibodies for TLR4 (2219S), p-p65 (3033 L), p-IκBα (2859S), p-IKKβ (2078S), transforming growth factor-β-activated kinase-1 (TAK1; 4505S), p-TAK1 (9339S), ERK (4695S), p-ERK (9101 L), JNK (9252S), p-JNK (9251S), p38 (9211 L), p38 (9212) were purchased from Cell Signaling Technology (Beverly, MA, USA). All antibodies were used at a dilution of 1:1000. elisa kits for cytokines and PGE2 were purchased from R&D Systems (Minneapolis, MN, USA). PGG (purity, >95%) was isolated from 80% EtOH extract of GRC as previously reported (Park et al., 2008).
Results
PGG reduces the phosphorylation of IRAK1 and p65 under LPS- and PGN-stimulated conditions in macrophages
As PGG is known to be a major active constituent of GRC, we purified PGG and tested it in LPS- or PGN-stimulated peritoneal macrophages (Figure 1A) and colonic macrophages (Figure 1B). In both LPS- or PGN-stimulated macrophage populations, PGG (5 μM) strongly decreased the phosphorylation of IRAK1 and p65, an indicator of NF-κB activation. These results are consistent with our preliminary data using ethanol extracts of GRC (Supporting Information Figure S1). We then assessed cytotoxicity of PGG and found that exposure to 20 μM of PGG for 48 h did not affect the viability of macrophages (Figure 1C).
Figure 1.
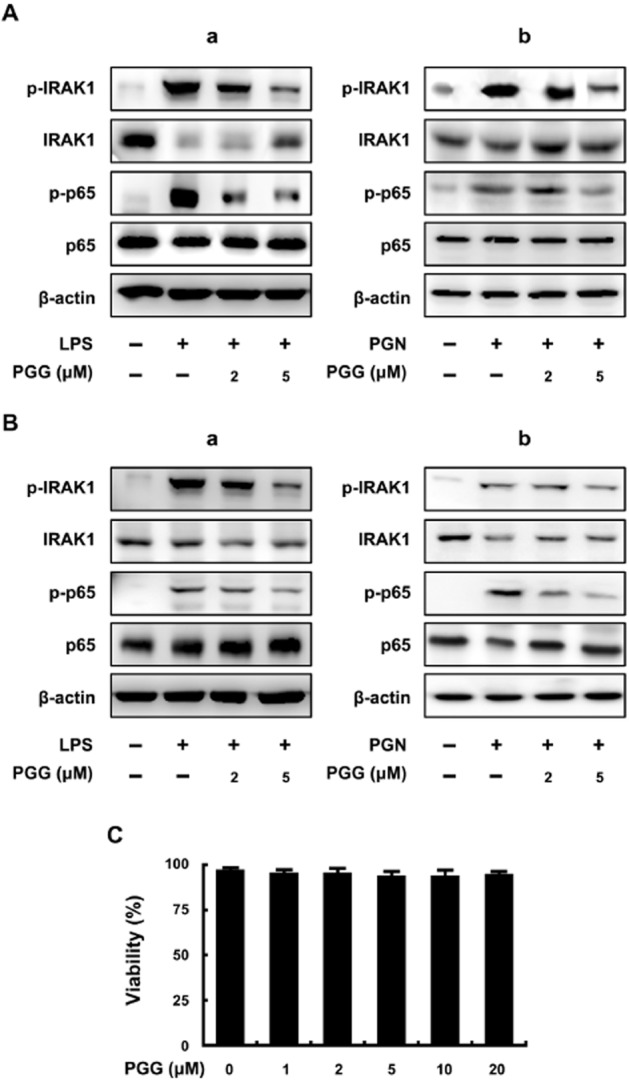
Effect of PGG on IRAK1 phosphorylation and NF-κB activation in LPS- or PGN-stimulated peritoneal and colonic macrophages. (A) Effect in peritoneal macrophages stimulated with LPS (a) or PGN (b). (B) Effect in colonic macrophages stimulated with LPS (a) or PGN (b). Peritoneal macrophages (5 × 105 cells) were treated with 50 ng·mL−1 LPS in the absence or presence of PGG (2 or 5 μM) for 20 h. IRAK1, p-IRAK1, p65, p-p65 and β-actin levels were measured by immunoblotting. (C) Cytotoxicity in peritoneal macrophages. Peritoneal macrophages (5 × 105 cells) were treated with PGG (2, 5 and 20 μM for 48 h.
PGG treatment reduces the phosphorylation of several signalling molecules
Using LPS-stimulated peritoneal macrophages, we investigated the effects of PGG on several intracellular signalling proteins. As shown in Figure 2A, LPS treatment reduced the expression of IRAK2 and IRAK4 expression in peritoneal macrophages and this was prevented by co-treatment with PGG. LPS treatment also increased p-TAK1, p-IKKβ and p-IκBα and these changes were prevented by PGG (5 μM). LPS treatment increased levels of p-ERK, p-JNK and p-p38, components of the MAPK signalling pathway. We found that co-treatment with PGG concentration-dependently reduced p-ERK and p-p38 levels, while only 5 μM of PGG decreased p-JNK. It should be noted that even at the highest PGG concentration, the phosphorylation levels of all three components of the MAPK pathway were still above control levels, suggesting that PGG could not completely prevent this signalling pathway of macrophages from responding to LPS.
Figure 2.
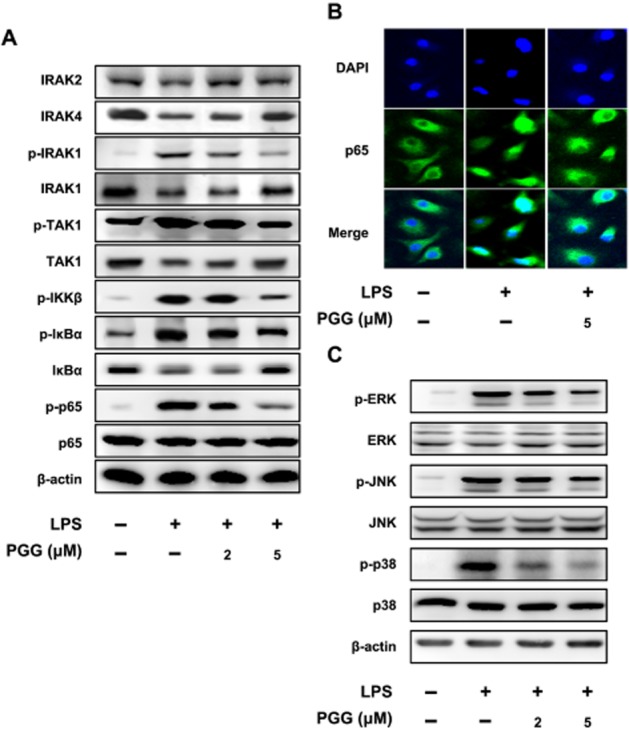
Inhibitory effect of PGG on NF-κB and MAPK signalling pathways in LPS-stimulated peritoneal macrophages. Peritoneal macrophages were treated with 50 ng·mL−1 LPS in the absence or presence of PGG (2 or 5 μM). (A) Effect on IRAK1/NF-κB pathway. (B) Effect on NF-κB nuclear translocation, as detected by confocal microscopy using an antibody against p65 subunit. (C) Effect on MAPK pathway. NF-κB and MAPK phosphorylation as well as nuclear translocation of NF-κB in LPS-stimulated peritoneal macrophages were determined at 90 min after treatment with LPS by immunoblotting and confocal microscopy respectively.
Because PGG inhibited the induction of p-p65, we examined the cellular localization of total p65 during different treatment conditions. Macrophages were left untreated or with LPS alone or with LPS and PGG. Next the cells were fixed and examined for the intracellular location of p65. As showing in Figure 2C, LPS caused p65 to move into the nucleus (DAPI stained). However, PGG decreased the p65 localization in the nucleus of LPS co-treated macrophages. Collectively, these data further support our immunoblot data showing that PGG inhibited activation of NF-κB in macrophages.
To further characterize the effect of PGG on LPS-stimulated macrophages, we assessed a range of cytokines. LPS induced production of the pro-inflammatory cytokines TNF-α, IL-1β and IL-6 (Figure 3A–C). Co-treatment with PGG and LPS produced lower levels of these cytokines, compared with those after to LPS alone. Also, LPS treatment reduced production of IL-10, while PGG treatment restored the production of this cytokine (Figure 3D), as observed earlier for echinocystic acid (Joh et al., 2011). We also measured PGE2, another known mediator of inflammation. PGE2 was significantly increased with LPS treatment and PGG co-treatment reduced this increase (Figure 3E). Production of NO (measured as nitrite) by macrophages was significantly increased by LPS and this production was inhibited by co-treatment with PGG (Figure 3F). Finally, immunoblot analysis showed that COX-2 and iNOS levels were induced by LPS treatment and PGG concentration-dependently reduced these levels. All these data indicate that PGG treatment reduced levels of several pro-inflammatory molecules.
Figure 3.
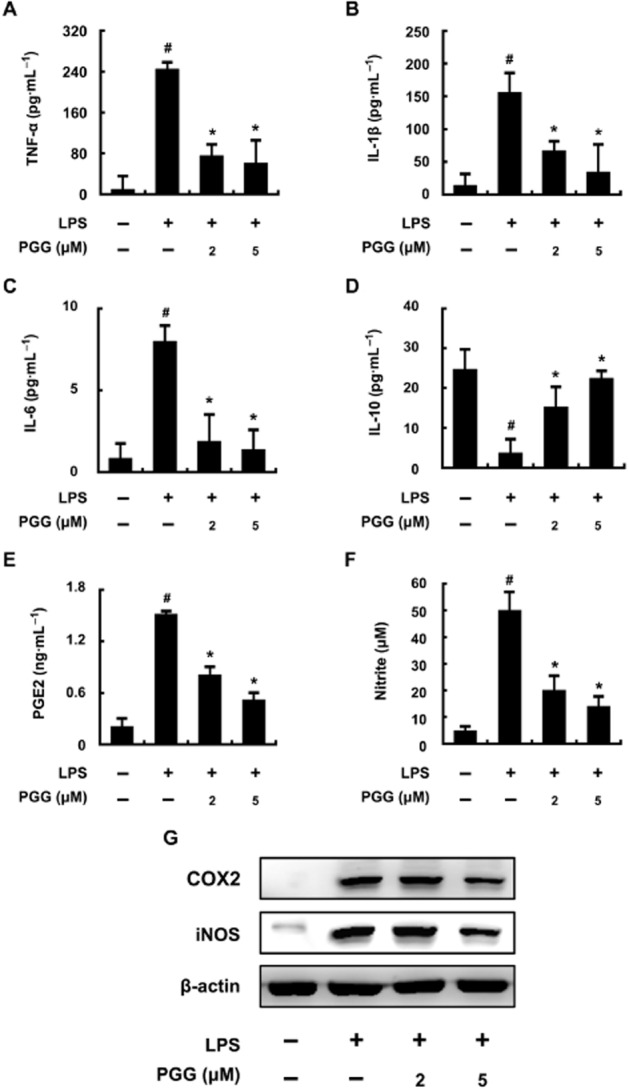
Inhibitory effect of PGG on inflammatory markers in LPS-stimulated peritoneal macrophages. Peritoneal macrophages (5 × 105 cells) were treated with 50 ng·mL−1 LPS in the absence or presence of PGG (2 or 5 μM) for 20 h. The levels of TNF-α (A), IL-1β (B), IL-6 (C), IL-10 (D), PGE2 (E) and NO (F) in culture supernatants were measured by elisa. COX-2 and iNOS levels (G) in the supernatants of lysed cells were measured by immunoblotting. All data are expressed as the mean ± SD (n = 4 in a single experiment). #P < 0.05 significantly different from control group; *P < 0.05, significantly different from LPS alone.
Next, to clarify the mechanism of inhibition by PGG in LPS-stimulated peritoneal macrophages, we examined whether PGG could block the interaction between LPS and TLR4. As shown in Figure 4A, basal autofluorescence was determined in untreated peritoneal macrophages. Other samples were treated with Alexa Fluor 488-conjugated LPS alone or with different concentrations of PGG plus Alexa Fluor 488-conjugated LPS. PGG did not block LPS binding to the macrophage as indicated by a similar shift to the right in fluorescence (percentages show shift from baseline). We further confirmed our FACS assay-based observation by confocal microscopy (Figure 4B) to show that PGG did not block the binding of LPS to TLR4 on the cell surface. We also showed by immunoblot analysis that LPS treatment caused an increase in TLR4 expression and PGG treatment did not diminish this LPS-induced increase (Figure 4C).
Figure 4.
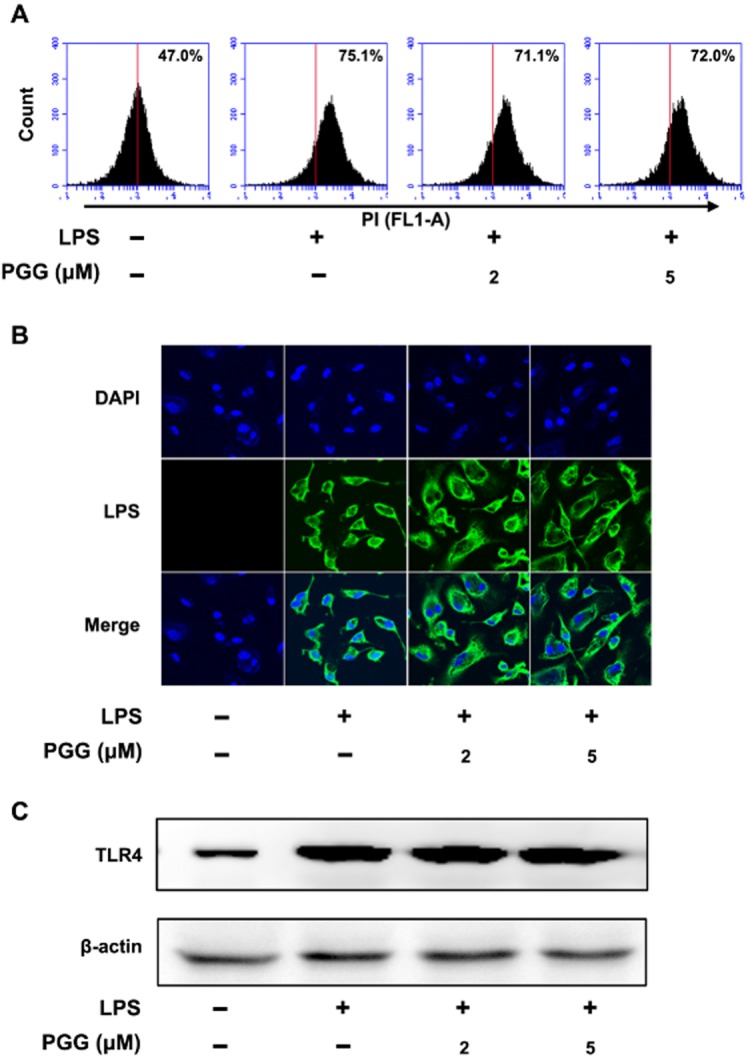
Effect of PGG on the binding of LPS to TLR-4 on peritoneal macrophages. Peritoneal macrophages (5 × 105 cells) isolated from mice were incubated with Alexa Fluor 488-conjugated LPS for 20 min in the absence or presence of PGG (2 or 5 μM), and then analysed by flow cytometry (A), confocal microscopy (B), and immunoblotting (C). TLR4 and β-actin were analysed by immunoblotting.
We next examined some intracellular effectors that are associated with TLR4 and are upstream of NF-κB. Macrophages were treated with siRNA for MyD88, plus LPS and PGG. We then monitored the cellular localization of MyD88, IRAK4, p-IRAK1 and IRAK1 (Figure 5). Treatment with MyD88 siRNA reduced MyD88 levels in the cell (Figure 5A) and PGG plus LPS treatment also decreased MyD88 levels in cells without MyD88 siRNA, a result which may be due to PGG blocking the MyD88 antibody from binding or to an actual decrease in MyD88 expression. Expression of IRAK4 was increased by MyD88 siRNA treatment, regardless of LPS or PGG treatment, compared with controls (Figure 5B). Moreover, LPS treatment also decreased the levels of IRAK4, consistent with data in Figure 2A. The presence of p-IRAK1 was markedly increased in the nucleus of LPS-treated macrophages, an effect reduced by PGG. Cells treated with MyD88 siRNA did not show similar accumulation of p-IRAK1 (Figure 5C). When total IRAK1 was examined (Figure 5D), no significant change in intensity within the cells was observed for the different treatment groups. PGG (5 and 10 μM) did not inhibit IRAK-1 kinase activity in vitro, assayed according to our already described method (Joh et al., 2011; data not shown).
Figure 5.

Inhibitory effect of PGG on IRAK1 and NF-κB activation in LPS-stimulated peritoneal macrophages transfected with MyD88 siRNA. (A) Effect on MyD88 expression. (B) Effect on IRAK4 expression. (C) Effect on p-IRAK1 expression. (D) Effect on IRAK1 expression. Peritoneal macrophages isolated from mice were treated with 50 ng·mL−1 LPS in the absence or presence of PGG (5 μM) for 90 min. Alexa Fluor 488-conjugated IRAK1 and p-IRAK1 were detected in siRNA-transfected peritoneal macrophages. (E) Effect on IRAK1 and IRAK4 degradation and IRAK1 phosphorylation. MyD88, IRAK1, p-IRAK1, IRAK4 and β-actin were analysed by immunoblotting. Peritoneal macrophages were treated with 50 ng·mL−1 LPS in the absence or presence of PGG (5 μM) for 90 min.
To further confirm the results from confocal microscopy, we performed immunoblot analysis for MyD88, IRAK4, p-IRAK1 and IRAK. LPS treatment alone did not influence MyD88 expression. However, IRAK4 expression was decreased and p-IRAK1 expression was increased. PGG plus LPS co-treated macrophages did not significantly influence MyD88 expression, but showed more normalization of IRAK4, p-IRAK1 and IRAK1 levels. MyD88 siRNA macrophages showed no significant change in MyD88 levels for all the groups. Further, we observed that LPS alone or LPS plus PGG treatments did not significantly change the levels of IRAK4, p-IRAK1 and IRAK1. Collectively, these data demonstrate the treatment with PGG may directly influence MyD88 levels in activated macrophages. Alternatively, PGG may bind to the MyD88 adaptor protein and block its interactions with other signalling molecules.
Inhibitory effect of PGG on TNBS-induced colitis in mice
We then investigated the effects of PGG on another in vivo model of inflammation, TNBS-induced colitis in mice. TNBS caused severe colonic inflammation, assayed as colon shortening, increased MPO activity, NF-κB activation and IRAK1 phosphorylation. Treatment with PGG or mesalazine significantly reduced TNBS-induced weight loss (Figure 6A), macroscopic disease (Figure 6A), and shortening of the colon (Figure 6C), and MPO activity (Figure 6D). Macroscopic images of the colons are shown in Figure 6 and microscopic examinations of sections stained with haematoxylin–eosin are displayed below for the different treatment groups. The anti-colitis effects of PGG were comparable to those of that of mesalazine (10 mg·kg-1).
Figure 6.

Effect of PGG on body weight (A), macroscopic disease (B), colon length (C), colonic MPO activity (D), and histology (haematoxylin–eosin staining) (E) in mice with TNBS-induced colitis. TNBS, except in the control group, was intrarectally administered to mice treated with saline, PGG or mesalazine. Test compounds [Cpd; PGG (5 or 10 mg·kg−1), mesalazine (MS; 10 mg·kg−1), or saline] were orally administered for 3 days after TNBS treatment. The mice were killed at 20 h after the final administration of PGG or mesalazine. All values are the mean ± SD (n = 7). #P < 0.05, significantly different from control group; *P < 0.05, significantly different from TNBS alone.
Next we monitored protein expression levels after different treatments. Although TLR4 levels were not changed in the different treatment groups (Figure 7A), TNBS treatment did increase p-IRAK1, p-IKKβ, p-p65, COX-2 and iNOS levels. Both PGG and mesalzine reduced the levels of p-IRAK1, p-IKKβ and p-p65 proteins, and also inhibited the expression of COX-2 and iNOS. Furthermore, PGG (10 mg·kg−1) almost totally blocked the increase in TNF-α, IL-1β and IL-6 expression in mice with TNBS-induced colitis (Figure 7B). However, PGG restored the levels of IL-10, which had been depressed by TNBS, to nearly normal values. Nevertheless, PGG, at a much higher dose (25 mg·kg−1), did not show acute toxicity (body weight, abnormal behaviour and death) when observed for 7 days under the conditions used in the experiment (data not shown).
Figure 7.

Effect of PGG on phosphorylation of IRAK1 and p65 and expression of iNOS, COX-2 (A), and proinflammatory cytokines (B) in mice with TNBS-induced colitis. TNBS, except in the control group, was intrarectally administered to mice treated with saline, PGG or mesalazine. PGG (5 or 10 mg·kg−1), mesalazine (MS, 10 mg·kg−1), or saline was orally administered for 3 days after TNBS treatment. The mice were killed at 20 h after the final administration of test compound (Cpd; PGG or mesalazine). (A) TLR4, IRAK1, p-IRAK1, IKKβ, p-IKKβ, COX-2, iNOS and β-actin were analysed by immunoblotting. (B) TNF-α (a), IL-1β (b), IL-6 (c), IL-10 (d) were analysed by elisa kits. All values are the mean ± SD (n = 7). #P < 0.05, significantly different from control group; *P < 0.05, significantly different from TNBS alone.
Discussion
A number of in vitro and in vivo studies have shown that PGG exhibits a wide range of biological activities in the therapy and prevention of several major human diseases including cancer, diabetes and inflammation (Zhang et al., 2009). PGG inhibits the expression of TNF-α in LPS-stimulated human peripheral blood mononuclear cells (Feldman et al., 2001) and the secretion of TNF-α and IL-6 induced by IL-1β in rat synoviocytes (Wu and Gu, 2009). PGG also inhibits IL-8 mRNA expression and secretion in human monocytic U937 cells stimulated with PMA or TNF-α (Lee et al., 2007). PGG strongly suppresses PMA and calcium ionophore A23187 induced expression of TNF-α, IL-1β and IL-6 in human mast cells (Lee et al., 2007). In addition, PGG significantly inhibits COX-2 and iNOS activity, as well as NO production in LPS-stimulated RAW264.7 cells (Lee et al., 2003). PGG inhibits iNOS and NADPH-diaphorase activities and NO release in LPS-induced peritoneal macrophages (Yokozawa et al., 2000), and significantly reduces serum TNF-α level in LPS-stimulated endotoxaemic mice (Genfa et al., 2005). The anti-inflammatory effect of PGG may be due to inhibition of the phosphorylation of IκB in the cytosol, which inhibits NF-κB activation in LPS-activated RAW264.7 cells (Pan et al., 2000).
In the present study, we observed that PGG exhibited an anti-inflammatory effect in LPS- or PGN-induced peritoneal macrophages and in LPS-induced colonic macrophages. PGG also potently inhibited the expression levels of COX-2 and iNOS as well as their products, PGE2 and NO, in LPS-treated peritoneal macrophages, mechanistically explaining the previously observed inhibitory effect of PGG against COX-2 and iNOS activity (Lee et al., 2003). Furthermore, LPS induced expression of pro-inflammatory cytokines as previously reported (Brightbill et al., 2000; Cao et al., 2005; Joh and Kim, 2011). However, in our experiments, LPS reduced IL-10 expression, in contrast to earlier reports showing that LPS increased IL-10 expression in macrophages (Brightbill et al., 2000; Cao et al., 2005; Soudi et al., 2013). This discrepancy should be resolved by further study. PGG inhibited the LPS-induced expression of pro-inflammatory cytokines TNF-α IL-1β and IL-6, but increased the LPS-reduced IL-10 expression, results similar to those of other anti-inflammatory agents (Dutra et al., 2011; Clemente et al., 2012).
In terms of intracellular signalling, PGG inhibited LPS-induced IRAK1 degradation and phosphorylation, IKKβ phosphorylation, IκBα degradation and translocation of the p65 subunit of NF-κB into the nucleus. Furthermore, PGG inhibited the phosphorylation of MAPKs, JNK, ERK and p38, even though PGG did not inhibit the interaction between LPS and its receptor, TLR4, on the cell membrane of peritoneal macrophages. Interestingly, PGG inhibited binding of a fluorescent MyD88 antibody to peritoneal macrophages and the fluorescent anti-MyD88 antibody was also not bound to peritoneal macrophages treated with MyD88 siRNA. Finally, this study also revealed that PGG did not inhibit the binding of fluorescent anti-IRAK1 and IRAK4 antibodies to peritoneal macrophages, irrespective of transfection with MyD88 siRNA. Therefore, these results suggest that PGG, by binding directly to MyD88, may inhibit the signalling from TLRs activated by LPS or PGN, normally transferred by MyD88 on to IRAK1.
In humans, IBD is a severe form of intestinal inflammation, the pathogenesis of which remains to be clearly understood. It is thought that the disease might be attributed to complex mucosal immune responses to resident enteric bacteria (Duchmann et al., 1995; Jung et al., 1995). The innate immune system recognizes the presence of specific bacterial antigens through pattern recognition receptors (Ingalls et al., 1999). TLRs, which serve as a major link between the innate and adaptive mucosal immune responses, act as transmembrane co-receptors in the cellular response to pathogen antigens. Of the TLRs, TLR2 and TLR4 are pattern recognition receptors that respond to PGN (isolated from Gram-positive bacteria) and LPS (isolated from Gram-negative bacteria) respectively (Chow et al., 1999; Ingalls et al., 1999; Maldonado-Bernal et al., 2005). These TLRs activate the secretion of pro-inflammatory mediators from monocytes, macrophages and dendritic cells, thereby triggering the activation of the acquired immune response. Particularly, TLR4 is potently expressed in intestinal epithelial cells from the colons of patients with IBD (Cario and Podolsky, 2000) and significantly up-regulated in mice with experimental colitis (Pasternak et al., 2010). LPS may potentially activate the TLR4-NF-κB signalling pathway in IBD (Chow et al., 1999; Pasternak et al., 2010). The activation of NF-κB in mucosal macrophages is accompanied by an increased capacity of these cells to produce and secrete IL-1, IL-6 and TNF-α, which activates NF-κB. The adapter protein, MyD88, is used by all TLRs (except TLR3) as a link to the pathway of NF-κB activation (Kawai and Akira, 2007; Cario and Podolsky, 2000). TIRAP (Toll-IL 1 receptor domain-containing adapter protein) is necessary to recruit MyD88 to TLR2 and TLR4, and then MyD88 signals through IRAKs. Thus, binding of LPS or PGN to TLRs induces phosphorylation of IRAK1, PI3K and/or MAPKs, which are involved in host defence mechanisms, either by identification of pathogens or as receptors for pro-inflammatory cytokines.
Various inflammatory diseases, including colitis and systemic inflammation, involve the over-expression of pro-inflammatory cytokines such as TNF-α and IL-1β, and inflammatory mediators, such as NO and PGE2 via NF-κB and MAPK pathways in macrophages (Tak and Firestein, 2001; Moynagh, 2005). Similar to the effects of kalopanaxsaponin A (Joh and Kim, 2011), compound K (Joh et al., 2011) and echinocystic acid, PGG inhibited MAPK, IRAK/ NF-κB and PI3K-AKT pathways. However, the anti-inflammatory mechanism of PGG was different to those of kalopanaxsaponin A, compound K and echinocystic acid. Indeed, PGG inhibited activation of PI3K/AKT, NF-κB and MAPKs in macrophages, by preventing the signals from LPS- or PGN-bound TLRs to be transferred to IRAKs via MyD88.
In the model of colitis induced by TNBS in mice, PGG suppressed the macroscopic features of the colitis such as weight loss and colon shortening, and intracellular signalling events such as the activation of NF-κB and phosphorylation of IKKβ, IκBα and IRAK1. Furthermore, PGG also inhibited the expression of pro-inflammatory cytokines. In our model we used a lower dose of TNBS (2.5 mg per mouse), compared with earlier models (3–10 mg per mouse; Ukil et al., 2006; Okamoto et al., 2013) and found that the anti-colitis effect of PGG (10 mg·kg−1) was comparable to that of mesalazine (10 mg·kg−1). Mesalazine is used clinically at oral doses of 10–70 mg·kg−1 per day in patients with IBD (Das et al., 1973; Sandborn and Hanauer, 2003). In many studies of experimental colitis in mice, mesalazine is given at oral doses of 50 or 100 mg·kg−1 (Malekinejad et al., 2013; Sann et al., 2013). However, in our preliminary studies, when we gave mesalazine at 10, 20 or 50 mg·kg−1 to our model of colitis in mice, we could not find any significant difference of anti-colitis effects such as colon shortening and macroscopic score between these doses (data not shown).
In conclusion, our results suggest, overall, that PGG may inhibit colitis by interfering with the TLR4-MyD88 signalling pathway. Our finding that PGG also reduced mortality, along with reduced expression of pro-inflammatory cytokines, in mice treated with LPS in a model of systemic inflammation (Supporting Information Figure S2), would provide additional support for this suggested mode of action of PGG. Based on these findings, we conclude that PGG may ameliorate inflammation by inhibiting MyD88 signalling pathways to NF-κB and/or MAPK.
Acknowledgments
This study was supported by a grant from the World Class University Program through the National Research Foundation of Korea funded by the Ministry of Education, Science and Technology (R33-2008-000-10018-0).
Glossary
- GRC
Rhus chinensis MILL
- IBD
inflammatory bowel disease
- IKK
IκB kinase
- iNOS
inducible NOS
- IRAK
IL-1 receptor-associated kinase
- MPO
myeloperoxidase
- MyD88
myeloid differentiation primary response gene
- PGG
penta-O-galloyl-β-D-glucose
- TAK1
transforming growth factor-β-activated kinase-1
- TLR
Toll-like receptor
- TNBS
2,3,4-trinitrobenzene sulphonic acid
Conflict of interest
The authors state no conflict of interest.
Supporting information
Additional Supporting Information may be found in the online version of this article at the publisher's web-site:
http://dx.doi.org/10.1111/bph.12333
Figure S1 Effect of GRC on IKKβ phosphorylation, IκB-αdegradation and NF-κB activation in LPS-stimulated peritoneal and colonic macrophages. (A) Effect in peritoneal macrophages stimulated with LPS. (B) Effect in colonic macrophages stimulated with LPS. Peritoneal macrophages (5 × 105 cells) were treated with 50 ng·mL·1 LPS in the absence or presence of GRC (5 or 10 μg·mL·1) for 20 h. p-IKKβ, I, IκB-α, p-IκB-α, p65, p-p65 and β-actin levels were measured by immunoblotting.
Figure S2 Inhibitory effect of PGG on serum IL-1β and TNF-α levels and death of mice injected i.p. with LPS. (A) Effect on IL-1β (a) and TNF-α production (b). Mice were injected i.p. with or without PGG (5 or 10 mg·kg·1) in the presence or absence of LPS (25 mg·kg·1). The normal, control group was treated with vehicle alone instead of LPS and PGG. Mice were killed at 12 h after LPS injection and serum levels of IL-1β and TNF-α were measured using ELISA kits. (B) Effect on mortality. Septic shock was induced by injection of LPS (25 mg·kg·1, i.p.) at 1 h after treatment with PGG (10 mg·kg·1). Survival rates were monitored over 72 h. All data are shown as the mean ± SD (n = 6). #P < 0.05, significantly different from control group; *P < 0.05, significantly different from LPS alone.
References
- Alexander SP, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 5th edition. Br J Pharmacol. 2011;164(Suppl. 1):S1–324. doi: 10.1111/j.1476-5381.2011.01649_1.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atreya R, Multer J, Fintoo S, Müllberg J, Jostock T, Wirtz S, et al. Blockade of interleukin 6 trans signaling suppresses T-cell resistance against apoptosis in chronic intestinal inflammation: evidence in Crohn disease and experimental colitis in vivo. Nat Med. 2000;6:583–588. doi: 10.1038/75068. [DOI] [PubMed] [Google Scholar]
- Baldwin AS., Jr The NF-κB and IκB proteins: new discoveries and insights. Annu Rev Immunol. 1996;14:649–681. doi: 10.1146/annurev.immunol.14.1.649. [DOI] [PubMed] [Google Scholar]
- Binder V. Epidemiology of IBD during the twentieth century: an integrated view. Best Pract Res Clin Gastroenterol. 2004;18:463–479. doi: 10.1016/j.bpg.2003.12.002. [DOI] [PubMed] [Google Scholar]
- Brightbill HD, Plevy SE, Modlin RL, Smale ST. A prominent role for Sp1 during lipopolysaccharide-mediated induction of the IL-10 promoter in macrophages. J Immunol. 2000;164:1940–1951. doi: 10.4049/jimmunol.164.4.1940. [DOI] [PubMed] [Google Scholar]
- Cao S, Liu J, Song L, Ma X. The protooncogene c-Maf is an essential transcription factor for IL-10 gene expression in macrophages. J Immunol. 2005;174:3484–3492. doi: 10.4049/jimmunol.174.6.3484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cario E, Podolsky DK. Differential alteration in intestinal epithelial cell expression of toll-like receptor 3 (TLR3) and TLR4 in inflammatory bowel disease. Infect Immun. 2000;68:7010–7017. doi: 10.1128/iai.68.12.7010-7017.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chow JC, Young DW, Golenbock DT, Christ WJ, Gusovsky F. Toll-like receptor-4 mediates lipolysaccharide-induced signal transduction. J Biol Chem. 1999;274:10689–10692. doi: 10.1074/jbc.274.16.10689. [DOI] [PubMed] [Google Scholar]
- Clemente TR, Dos Santos AN, Sturaro JN, Gotardo EM, Oliveira de CC, Acedo SC, et al. Infliximab modifies mesenteric adipose tissue alterations and intestinal inflammation in rats with TNBS-induced colitis. Scand J Gastroenterol. 2012;47:943–950. doi: 10.3109/00365521.2012.688213. [DOI] [PubMed] [Google Scholar]
- Das KM, Eastwood MA, McManus JP, Sircus W. The metabolism of salicylazosulphapyridine in ulcerative colitis. I. The relationship between metabolites and the response to treatment in inpatients. Gut. 1973;14:631–641. doi: 10.1136/gut.14.8.631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Djakpo O, Yao W. Rhus chinensis and Galla Chinensis – folklore to modern evidence: review. Phytother Res. 2010;24:1739–1747. doi: 10.1002/ptr.3215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duchmann R, Kaiser I, Hermann E, Mayet W, Ewe K, Meyer zum Büschenfelde KH. Tolerance exists towards resident intestinal flora but is broken in active inflammatory bowel disease. Clin Exp Immunol. 1995;102:448–455. doi: 10.1111/j.1365-2249.1995.tb03836.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutra RC, Claudino RF, Bento AF, Marcon R, Schmidt EC, Bouzon ZL, et al. Preventive and therapeutic euphol treatment attenuates experimental colitis in mice. PLoS ONE. 2011;6:e27122. doi: 10.1371/journal.pone.0027122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldman KS, Sahasrabudhe K, Lawlor MD, Wilson SL, Lang CH, Scheuchenzuber WJ. In vitro and in vivo inhibition of LPS-stimulated tumor necrosis factor-alpha secretion by the gallotannin beta-D-pentagalloylglucose. Bioorg Med Chem Lett. 2001;11:1813–1815. doi: 10.1016/s0960-894x(01)00332-8. [DOI] [PubMed] [Google Scholar]
- Genfa L, Jiang Z, Hong Z, Yimin Z, Liangxi W, Guo W, et al. The screening and isolation of an effective anti-endotoxin monomer from Radix Paeoniae Rubra using affinity biosensor technology. Int Immunopharmacol. 2005;5:1007–1017. doi: 10.1016/j.intimp.2005.01.013. [DOI] [PubMed] [Google Scholar]
- Ingalls RR, Heine H, Lien E, Yoshimura A, Golenbock D. Lipopolysaccharide recognition, CD14, and lipopolysaccharide receptors. Infect Dis Clin North Am. 1999;13:341–353. doi: 10.1016/s0891-5520(05)70078-7. [DOI] [PubMed] [Google Scholar]
- Joh EH, Kim DH. Kalopanaxsaponin A ameliorates experimental colitis in mice by inhibiting IRAK-1 activation in the NF-κB and MAPK pathways. Br J Pharmacol. 2011;162:1731–1742. doi: 10.1111/j.1476-5381.2010.01195.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joh EH, Lee IA, Jung IH, Kim DH. Ginsenoside Rb1 and its metabolite compound K inhibit IRAK-1 activation – the key step of inflammation. Biochem Pharmacol. 2011;82:278–286. doi: 10.1016/j.bcp.2011.05.003. [DOI] [PubMed] [Google Scholar]
- Joh EH, Gu W, Kim DH. Echinocystic acid ameliorates lung inflammation in mice and alveolar macrophages by inhibiting the binding of LPS to TLR4 in NF-κB and MAPK pathways. Biochem Pharmacol. 2012;84:331–340. doi: 10.1016/j.bcp.2012.04.020. [DOI] [PubMed] [Google Scholar]
- Jung HC, Eckmann I, Yang SK, Panja A, Fierer J, Morzycka-Wroblewska E, et al. A distinct array of proinflammatory cytokine is expressed in human colon epithelia cells in response to bacterial invasion. J Clin Invest. 1995;95:55–65. doi: 10.1172/JCI117676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawai T, Akira S. Signaling to NF-kappaB by Toll-like receptors. Trends Mol Med. 2007;13:460–469. doi: 10.1016/j.molmed.2007.09.002. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG. Animal research: Reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1577–1579. doi: 10.1111/j.1476-5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim KA, Gu W, Lee IA, Joh EH, Kim DH. High fat diet-induced gut microbiota exacerbates inflammation and obesity in mice via the TLR4 signaling pathway. PLoS ONE. 2012;7:e47713. doi: 10.1371/journal.pone.0047713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SH, Park HH, Lee S, Jun CD, Choi BJ, Kim SY, et al. The anti-anaphylactic effect of the gall of Rhus javanica is mediated through inhibition of histamine release and inflammatory cytokine secretion. Int Immunopharmacol. 2005;5:1820–1829. doi: 10.1016/j.intimp.2005.06.007. [DOI] [PubMed] [Google Scholar]
- Lee K, Kim J, Lee BJ, Park JW, Leem KH, Bu Y. Protective effects of Galla Rhois, the excrescence produced by the sumac aphid, Schlechtendalia chinensis, on transient focal cerebral ischemia in the rat. J Insect Sci. 2012;12:10. doi: 10.1673/031.012.0110. doi: 10.1673/031.012.0110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SH, Park HH, Kim JE, Kim JA, Kim YH, Jun CD, et al. Allose gallates suppress expression of pro-inflammatory cytokines through attenuation of NF-kappaB in human mast cells. Planta Med. 2007;73:769–773. doi: 10.1055/s-2007-981553. [DOI] [PubMed] [Google Scholar]
- Lee SJ, Lee IS, Mar W. Inhibition of inducible nitric oxide synthase and cyclooxygenase-2 activity by 1,2,3,4,6-penta-O-galloyl-beta-D-glucose in murine macrophage cells. Arch Pharm Res. 2003;26:832–839. doi: 10.1007/BF02980029. [DOI] [PubMed] [Google Scholar]
- McGrath J, Drummond G, McLachlan E, Kilkenny C, Wainwright C. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maldonado-Bernal C, Kirschning CJ, Rosenstein Y, Rocha LM, Rios-Sarabia N, Espinosa-Cantellano M, et al. The innate immune response to Entamoeba histolytica lipopeptidophosphoglycan is mediated by toll-like receptors 2 and 4. Parasite Immunol. 2005;27:127–137. doi: 10.1111/j.1365-3024.2005.00754.x. [DOI] [PubMed] [Google Scholar]
- Malekinejad H, Shafie-Irannejad V, Hobbenaghi R, Tabatabaie SH, Moshtaghion SM. Comparative protective effect of hawthorn berry hydroalcoholic extract, atorvastatin, and mesalamine on experimentally induced colitis in rats. J Med Food. 2013;16:593–601. doi: 10.1089/jmf.2012.2672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moynagh PN. The NF-kappaB pathway. J Cell Sci. 2005;118:4589–4592. doi: 10.1242/jcs.02579. [DOI] [PubMed] [Google Scholar]
- Nakano M, Kurokawa M, Hozumi T. Suppression of recurrent genital herpes simplex virus type 2 infection by Rhus javanica in guinea pigs. Antiviral Res. 1998;39:25–33. doi: 10.1016/s0166-3542(98)00023-0. [DOI] [PubMed] [Google Scholar]
- Oh GS, Pae HO, Choi BM, Lee HS, Kim IK, Yun YG, et al. Penta-O-galloyl-beta-D-glucose inhibits phorbol myristate acetateinduced interleukin-8 gene expression in human monocytic U937 cells through its inactivation of nuclear factor-kappaB. Int Immunopharmacol. 2004;4:377–386. doi: 10.1016/j.intimp.2003.10.010. [DOI] [PubMed] [Google Scholar]
- Okamoto Y, Hara T, Ebato T, Fukui T, Masuzawa T. Brazilian propolis ameliorates trinitrobenzene sulfonic acid-induced colitis in mice by inhibiting Th1 differentiation. Int Immunopharmacol. 2013;16:178–183. doi: 10.1016/j.intimp.2013.04.004. [DOI] [PubMed] [Google Scholar]
- Pan MH, Lin-Shiau SY, Ho CT, Lin JH, Lin JK. Suppression of lipopolysaccharide-induced nuclear factor-kappaB activity by theaflavin-3,3′-digallate from black tea and other polyphenols through down-regulation of IkappaB kinase activity in macrophages. Biochem Pharmacol. 2000;59:357–367. doi: 10.1016/s0006-2952(99)00335-4. [DOI] [PubMed] [Google Scholar]
- Paradkar PN, Blum PS, Berhow MA, Baumann H, Kuo SM. Dietary isoflavones suppress endotoxin-induced inflammatory reaction in liver and intestine. Cancer Lett. 2004;215:21–28. doi: 10.1016/j.canlet.2004.05.019. [DOI] [PubMed] [Google Scholar]
- Park EJ, Zhao YZ, An RB, Kim YC, Sohn DH. 1,2,3,4,6-Penta-O-galloyl-beta-D-glucose from Galla Rhois protects primary rat hepatocytes from necrosis and apoptosis. Planta Med. 2008;74:1380–1383. doi: 10.1055/s-2008-1081300. [DOI] [PubMed] [Google Scholar]
- Pasternak BA, D'Mello S, Jurickova II, Han X, Willson T, Flick L, et al. Lipopolysaccharide exposure is linked to activation of the acute phase response and growth failure in pediatric Crohn's disease and murine colitis. Inflamm Bowel Dis. 2010;16:856–869. doi: 10.1002/ibd.21132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pendyala S, Walker JM, Holt PR. A high-fat diet is associated with endotoxemia that originates from the gut. Gastroenterology. 2012;142:1100–1101. doi: 10.1053/j.gastro.2012.01.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peris-Bondia F, Latorre A, Artacho A, Moya A, D'Auria G. The active human gut microbiota differs from the total microbiota. PLoS ONE. 2011;6:e22448. doi: 10.1371/journal.pone.0022448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radema SA, Deventer van SJ, Cerami A. Interleukin 1 beta is expressed predominantly by enterocytes in experimental colitis. Gastroenterology. 1991;100:1180–1186. [PubMed] [Google Scholar]
- Rafii F, Embdin van R, Lieshout van LMC. Changes in bacterial enzymes and PCR profiles of fecal bacteria from a patient with ulcerative colitis before and after antimicrobial treatments. Dig Dis Sci. 1999;44:637–642. doi: 10.1023/a:1026634229934. [DOI] [PubMed] [Google Scholar]
- Ren Z, Zhu B, Wang D, Ma E, Su D, Zhong Y. Comparative population structure of Chinese sumac aphid Schlechtendalia chinensis and its primary host-plant Rhus chinensis. Genetica. 2008;132:103–112. doi: 10.1007/s10709-007-9153-6. [DOI] [PubMed] [Google Scholar]
- Sandborn WJ, Hanauer SB. Systematic review: the pharmacokinetic profiles of oral mesalazine formulations and mesalazine pro-drugs used in the management of ulcerative colitis. Aliment Pharmacol Ther. 2003;17:29–42. doi: 10.1046/j.1365-2036.2003.01408.x. [DOI] [PubMed] [Google Scholar]
- Sann H, Erichsen JV, Hessmann M, Pahl A, Hoffmeyer A. Efficacy of drugs used in the treatment of IBD and combinations thereof in acute DSS-induced colitis in mice. Life Sci. 2013;92:708–718. doi: 10.1016/j.lfs.2013.01.028. [DOI] [PubMed] [Google Scholar]
- Shanahan F. Crohn's disease. Lancet. 2002;359:62–69. doi: 10.1016/S0140-6736(02)07284-7. [DOI] [PubMed] [Google Scholar]
- Shim YJ, Doo HK, Ahn SY, Kim YS, Seong JK, Park IS, et al. Inhibitory effect of aqueous extract from the gall of Rhus chinensis on alpha-glucosidase activity and postprandial blood glucose. J Ethnopharmacol. 2003;85:283–287. doi: 10.1016/s0378-8741(02)00370-7. [DOI] [PubMed] [Google Scholar]
- Soudi S, Zavaran-Hosseini A, Muhammad Hassan Z, Soleimani M, Jamshidi Adegani F, Hashemi SM. Comparative study of the effect of LPS on the function of BALB/c and C57BL/6 peritoneal macrophages. Cell J. 2013;15:45–54. [PMC free article] [PubMed] [Google Scholar]
- Tak PP, Firestein GS. NF-kappaB: a key role in inflammatory diseases. J Clin Invest. 2001;107:7–11. doi: 10.1172/JCI11830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ukil A, Maity S, Das PK. Protection from experimental colitis by theaflavin-3,3'-digallate correlates with inhibition of IKK and NF-kappaB activation. Br J Pharmacol. 2006;149:121–131. doi: 10.1038/sj.bjp.0706847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu M, Gu Z. Screening of bioactive compounds from moutan cortex and their anti-inflammatory activities in rat synoviocytes. Evid Based Complement Alternat Med. 2009;6:57–63. doi: 10.1093/ecam/nem066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokozawa T, Chen CP, Tanaka T, Kitani K. A study on the nitric oxide production-suppressing activity of Sanguisorbae Radix components. Biol Pharm Bull. 2000;23:717–722. doi: 10.1248/bpb.23.717. [DOI] [PubMed] [Google Scholar]
- Zhang J, Li L, Kim SH, Hagerman AE, Lü J. Anti-cancer, anti-diabetic and other pharmacologic and biological activities of penta-galloyl-glucose. Pharm Res. 2009;26:2066–2080. doi: 10.1007/s11095-009-9932-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Effect of GRC on IKKβ phosphorylation, IκB-αdegradation and NF-κB activation in LPS-stimulated peritoneal and colonic macrophages. (A) Effect in peritoneal macrophages stimulated with LPS. (B) Effect in colonic macrophages stimulated with LPS. Peritoneal macrophages (5 × 105 cells) were treated with 50 ng·mL·1 LPS in the absence or presence of GRC (5 or 10 μg·mL·1) for 20 h. p-IKKβ, I, IκB-α, p-IκB-α, p65, p-p65 and β-actin levels were measured by immunoblotting.
Figure S2 Inhibitory effect of PGG on serum IL-1β and TNF-α levels and death of mice injected i.p. with LPS. (A) Effect on IL-1β (a) and TNF-α production (b). Mice were injected i.p. with or without PGG (5 or 10 mg·kg·1) in the presence or absence of LPS (25 mg·kg·1). The normal, control group was treated with vehicle alone instead of LPS and PGG. Mice were killed at 12 h after LPS injection and serum levels of IL-1β and TNF-α were measured using ELISA kits. (B) Effect on mortality. Septic shock was induced by injection of LPS (25 mg·kg·1, i.p.) at 1 h after treatment with PGG (10 mg·kg·1). Survival rates were monitored over 72 h. All data are shown as the mean ± SD (n = 6). #P < 0.05, significantly different from control group; *P < 0.05, significantly different from LPS alone.