

Abstract
Usher syndrome is an autosomal recessive disorder manifesting hearing loss, retinitis pigmentosa and vestibular dysfunction, and having three clinical subtypes. Usher syndrome type 1 is the most severe subtype due to its profound hearing loss, lack of vestibular responses, and retinitis pigmentosa that appears in prepuberty. Six of the corresponding genes have been identified, making early diagnosis through DNA testing possible, with many immediate and several long-term advantages for patients and their families. However, the conventional genetic techniques, such as direct sequence analysis, are both time-consuming and expensive. Targeted exon sequencing of selected genes using the massively parallel DNA sequencing technology will potentially enable us to systematically tackle previously intractable monogenic disorders and improve molecular diagnosis.
Using this technique combined with direct sequence analysis, we screened 17 unrelated Usher syndrome type 1 patients and detected probable pathogenic variants in the 16 of them (94.1%) who carried at least one mutation. Seven patients had the MYO7A mutation (41.2%), which is the most common type in Japanese. Most of the mutations were detected by only the massively parallel DNA sequencing. We report here four patients, who had probable pathogenic mutations in two different Usher syndrome type 1 genes, and one case of MYO7A/PCDH15 digenic inheritance.
This is the first report of Usher syndrome mutation analysis using massively parallel DNA sequencing and the frequency of Usher syndrome type 1 genes in Japanese. Mutation screening using this technique has the power to quickly identify mutations of many causative genes while maintaining cost-benefit performance. In addition, the simultaneous mutation analysis of large numbers of genes is useful for detecting mutations in different genes that are possibly disease modifiers or of digenic inheritance.
Introduction
Usher syndrome (USH) is an autosomal recessive disorder characterized by hearing loss (HL), retinitis pigmentosa (RP) and vestibular dysfunction. Three clinical subtypes can be distinguished. USH type 1 (USH1) is the most severe among them because of profound HL, absent vestibular responses, and prepubertal onset RP. USH type 2 (USH2) is characterized by congenital moderate to severe HL, with a high-frequency sloping configuration. The vestibular function is normal and onset of RP is in the first or second decade. The onset of the visual symptoms such as night blindness in USH usually occurs several years later than in USH1. USH type 3 (USH3) is characterized by variable onset of progressive HL, variable onset of RP, and variable impairment of vestibular function (normal to absent) [1], [2].
To date, nine genetic loci for USH1(USH1B-H, J, and K) have been mapped to chromosomes 11q13.5, 11p15.1, 10q22.1, 21q21, 10q21-q22, 17q24-q25, 15q22-q23 (USH1H and J), and 10p11.21–q21.1 [2], [3], [4]. Six of the corresponding genes have been identified: the actin-based motor protein myosin VIIa (MYO7A, USH1B) [5]; two cadherin-related proteins, cadherin 23 (CDH23, USH1D) [6] and protocadherin 15 (PCDH15, USH1F) [7]; and two scaffold proteins, harmonin (USH1C) [8] and sans (USH1G) [9]; the Ca2+- and integrin-binding protein (CIB2, USH1J) [4]. In Caucasian USH1 patients, previous studies showed that mutations in MYO7A, USH1C, CDH23, PCDH15, and USH1G, were found in 39–55%, 7–14%, 7–35%, 7–11%, and 0–7%, respectively (the frequency of CIB2 is still unknown) [10], [11], [12]. In Japanese, Nakanishi et al. showed that MYO7A and CDH23 mutations are present in USH1 patients [13], however, the frequency is not yet known. In addition, mutations in three corresponding genes (usherin USH2A [14], G protein-coupled receptor 98; GPR98 [15], and deafness, autosomal recessive 31; DFNB31 [16]) have been reported so far in USH2, and USH3 is caused by mutations in the clarin 1 (CLRN1) [17] gene.
Comprehensive molecular diagnosis of USH has been hampered both by genetic heterogeneity and the large number of exons for most of the USH genes. The six USH1 genes collectively contain 180 coding exons [4], [9], [10] the three USH2 genes comprise 175 coding exons [15], [16], [18], and the USH3 gene has five coding exons [17]. In addition some of these genes are alternatively spliced ([4], [7], [8], [16], [17] and NCBI database: http://www.ncbi.nlm.nih.gov/nuccore/). Thus far, large-scale mutation screening has been performed using direct sequence analysis, but that is both time-consuming and expensive. We thought that targeted exon sequencing of selected genes using the Massively Parallel DNA Sequencing (MPS) technology would enable us to systematically tackle previously intractable monogenic disorders and improve molecular diagnosis.
Therefore, in this study, we have conducted genetic analysis using MPS-based genetic screening to find mutations in nine causative USH genes (except CIB2) in Japanese USH1 patients.
Results
Mutation analysis of the nine USH genes in 17 unrelated USH1 patients revealed 19 different probable pathogenic variants, of which 14 were novel (Table 1).
Table 1. Possible pathogenic variants found in this study.
| Gene | Mutation type | Nucleotide change | Amino acid change | exon/intron number | Domain | control (in 384 alleles) | SIFT Score | PolyPhen Score | Reference |
| MYO7A | Frameshift | c.1623dup | p.Lys542GlnfsX5 | Exon 14 | - | N/A | - | - | Le Quesne Stabej et al. (2012) |
| c.4482_4483insTG | p.Trp1495CysfsX55 | Exon 34 | - | N/A | - | - | This study | ||
| c.6205_6206delAT | p.Ile2069ProfsX6 | Exon 45 | - | N/A | - | - | This study | ||
| Nonsense | c.1477C>T | p.Gln493X | Exon 13 | - | N/A | - | - | This study | |
| c.1708C>T | p.Arg570X | Exon 15 | - | N/A | - | - | This study | ||
| c.2115C>A | p.Cys705X | Exon 18 | - | N/A | - | - | This study | ||
| c.6321G>A | p.Trp2107X | Exon 46 | - | N/A | - | - | This study | ||
| Missense | c.2074G>A | p.Val692Met | Exon 17 | Motor domain | 0 | 0.09 | 0.982 | This study | |
| c.2311G>T | p.Ala771Ser | Exon 20 | IQ 2 | 0.0026 | 0.01 | 0.825 | Nakanishi et al. (2010) | ||
| c.6028G>A | p.Asp2010Asn | Exon 44 | FERM 2 | 0 | 0 | 0.925 | Jacobson et al. (2009) | ||
| CDH23 | Frameshift | c.3567delG | p.Arg1189ArgfsX5 | Exon 30 | - | N/A | - | - | This study |
| c.5780_5781delCT | p.Ser1927Cysfs16 | Exon 44 | - | N/A | - | - | This study | ||
| Splicing | c.5821-2A>G | ? | Intron 44 | - | N/A | - | - | This study | |
| Nonsense | c.6319C>T | p.Arg2107X | Exon 48 | - | N/A | - | - | Nakanishi et al. (2010) | |
| PCDH15 | Splicing | c.158-1G>A | ? | Intron 3 | - | N/A | - | - | This study |
| Nonsense | c.1006C>T | p.Arg336X | Exon 10 | - | N/A | - | - | This study | |
| c.2971C>T | p.Arg991X | Exon 22 | - | N/A | - | - | Roux et al. (2006) | ||
| c.3337G>T | p.Glu1113X | Exon 25 | - | N/A | - | - | This study | ||
| Missense | c.3724G>A | p.Val1242Met | Exon 28 | Cadherin 11 | 0 | 0 | 1 | This study |
Computer analysis to predict the effect of missense variants on MYO7A protein function was performed with sorting intolerant from tolerant (SIFT; http://sift.jcvi.org/), and polymorphism phenotyping (PolyPhen2; http://genetics.bwh.harvard.edu/pph2/).
N/A: not applicable.
All mutations were detected in only one patient each and sixteen of the 17 patients (94.1%) carried at least one mutation, while one patient had no mutations. Thirteen of the 16 mutation carriers each had two pathogenic mutations (Table 2).
Table 2. Details of phenotype and genotype of 17 USH1 patients.
| Sample No. | Age | Sex | Allele1 | Allele2 | Hereditary form | Onset of night blindness | Cataract | Hearing Aid | Cochlear Implant |
| MYO7A | |||||||||
| 1 | 37 | M | p.Gln493X | p.Trp1495CysfsX55 | sporadic | 13 | no | unilateral | unilateral |
| 2 | 41 | W | p.l2069fsX6 | p.l2069fsX6 | AR | unknown | both eyes | bilateral | no |
| 5 | 54 | M | p.Val692Met | p.Val692Met | AR | 5 | both eyes | no | no |
| 6 | 54 | W | p.Arg570X | p.Arg570X | sporadic | 6 | no | no | no |
| 8 | 14 | M | p.Lys542GlnfsX5 | p.Lys542GlnfsX5 | sporadic | 6 | no | unilateral | unilateral |
| 11 | 54 | M | p.Asp2010Asn | p.Trp2107X | sporadic | 13 | no | no | no |
| 17 | 56 | W | p.Cys705X | p.Cys705X | sporadic | unknown | no | no | no |
| CDH23 | |||||||||
| 7 | 12 | W | p.Arg1189ArglfsX5 | p.Arg1189ArglfsX5 | sporadic | 12 | both eyes | no | bilateral |
| 9 | 9 | M | p.Ser1927Cysfs16 | c.5821-2A>G | sporadic | 8 | no | unilateral | unilateral |
| 15 | 16 | W | p.Arg2107X | p.Arg2107X | sporadic | unknown | no | no | no |
| PCDH15 | |||||||||
| 3 | 47 | W | p.Glu1113X | p.Glu1113X | sporadic | 5 | both eyes | no | no |
| 16 | 28 | W | p.Arg991X | p.Arg991X | AR | 10 | no | no | no |
| 10 | 62 | M | p.Arg962Cys | unknown | sporadic | 9 | both eyes | no | no |
| 12 | 52 | M | p.Arg336X | unknown | sporadic | 3 | no | no | no |
| 13 | 51 | M | p.Val1242Met | unknown | sporadic | 10 | no | no | no |
| MYO7A * 1 /PCDH15 * 2 | |||||||||
| 4 | 21 | M | p.Ala771Ser* 1 | c.158-1G>A* 2 | sporadic | 10 | no | unilateral | unilateral |
| unknown | |||||||||
| 14 | 64 | W | unknown | unknown | sporadic | 15 | both eyes | unilateral | no |
*All subjects have congenital deafness and RP.
Nonsense, frame shift, and splice site mutations are all classified as pathogenic, whereas missense mutations are presumed to be probable pathogenic variants based on results of prediction software for evaluation of the pathogenicity of missense variants (Table 1).
Of the 19 probable pathogenic mutations that we found, 17 were detected by MPS. The remaining two (p.Lys542GlnfsX5 in MYO7A and c.5821-2A>G in CDH23) were sequenced by direct sequence analysis.
Of our 17 USH patients, seven had MYO7A mutations (41.2%), three had CDH23 mutations (17.6%), and two had PCDH15 mutations (11.8%). We did not find any probable pathogenic mutations in USH1C, USH1G, and USH2/3 genes.
Four USH1 patients (Cases #3, 5, 8, 15) had probable pathogenic mutations in two different USH genes, with one being a biallelic mutation (Table 3). The other heterozygous/homozygous mutations were missense variants. Three of these patients (Cases #3, 5, 8) presented with earlier RP onset (night blindness) than in the other patients with two pathogenic mutations (Cases #1, 6, 7, 9, 11, 16) (p = 0.007) (Fig. 1).
Table 3. The patients with mutations in two different genes.
| Sample | Genes with two pathogenic mutations | Gene with one heterozygous mutation | Nucleotide change | Amino acid change | control | SIFT score | PolyPhen score | Referense |
| 5 | MYO7A | CDH23 | c.C719T | p.P240L* | 0.26 | 0.06 | 0.999 | Wagatsuma et al. (2007) |
| 8 | MYO7A | CDH23 | c.2568C>G | p.Ile856Met | 0 | 0.08 | 1 | This study |
| 15 | CDH15 | USH1C | c.2437T>G | p.Tyr813Asp | 0 | 0.19 | 0.932 | This study |
| 3 | PCDH15 | USH1G | c.28C>T | p.Arg10Trp | 0 | 0.19 | 1 | This study |
*homozygotes.
Figure 1. The number of mutations and the age of RP onset in Usher syndrome type 1 patients.
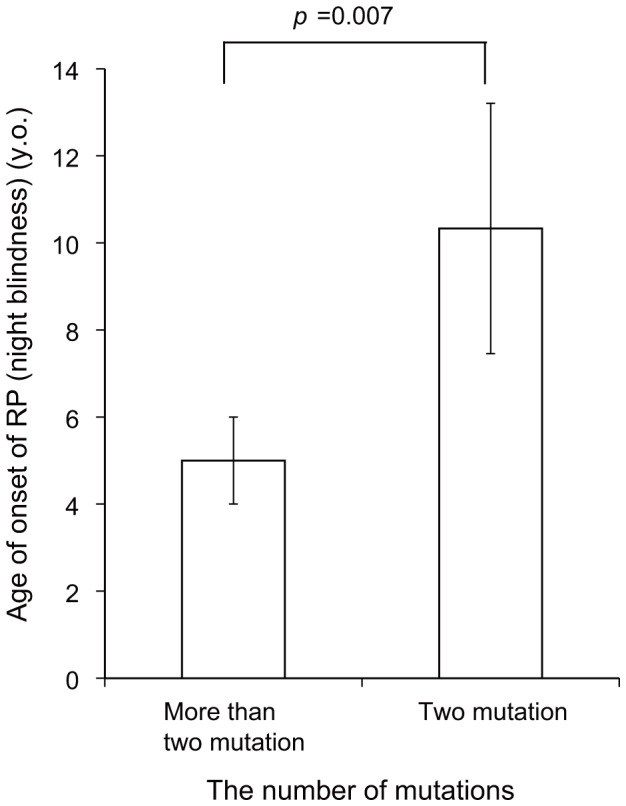
The age of RP onset is earlier in the patients with more than two pathogenic mutations. RP: retinitis pigmentosa.
One patient (Case #4) had heterozygote mutations in two USH1 genes (p.Ala771Ser in MYO7A and c.158-1G>A in PCDH15). His parents and one brother were found to also be carriers for these mutations. Another brother had no variants (Fig. 2).
Figure 2. Pedigree and sequence chromatograms of the patient with the p.Ala771Ser in MYO7A and c.158-1G>A in PCDH15 mutations.

(A) The pedigree and sequence results of the proband and family. (B) Sequence chromatograms from wild-type and mutations. The proband, his mothor and one brother carried a heterozygous 2311G>T transition in exon 20, which results in an alanine to a serine (Ala771Ser) in MYO7A. Another variation, 158-1G>A in intron 3 of PCDH15, was derived from the proband and his father. Another brother had no variants.
Discussion
For USH1, early diagnosis has many immediate and several long-term advantages for patients and their families [1]. However, diagnosis in childhood, based on a clinical phenotype, has been difficult because patients appear to have only non-syndromic HL in childhood and RP develops in later years. Although early diagnosis is now possible through DNA testing, performing large-scale mutation screening for USH genes in all non-syndromic HL children has been both time-consuming and expensive. Therefore, the availability of MPS, which facilitates comprehensive large-scale mutation screening [19] is a very welcome advance.
MPS technology enabled us to detect pathogenic mutations in USH1 patients efficiently, identifying one or two pathogenic/likely pathogenic mutations in 16 of 17 (94.1%) cases. This was comparable to previous direct sequence analysis results such as Bonnet et al. who detected one or two mutations in 24 out of 27 (89%) USH1 patients [11] and Le Quesne Stabej et al. who detected one or two mutations in 41 out of 47 (87.2%) USH1 patients [12].
In addition, MPS assists in the analysis of disease modifiers and digenic inheritance because it simultaneously investigates many causative genes for a specific disease, such as in our case, USH. Previous reports have described several USH cases with pathogenic mutations in two or three different USH genes [11], [12], [20]. In our study, four patients had two pathogenic mutations in one gene and missense variants in a different gene (Table 3). We considered the latter to possibly be a disease modifier. For example, USH1C:p.Tyr813Asp, which occurred in 0/384 control chromosomes and was predicted to be “probably damaging” by the Polyphen program, was found with a homozygous CDH23 nonsense mutation (p.Arg2107X) (Case #15). As for what the variant “modifies”, we speculate that for USH1 patients with a disease modifier, RP symptoms such as night blindness show an earlier onset. However, we think that profound HL and the absence of vestibular function in USH1 patients are not affected by modifiers as they are congenital and therefore not progressive.
Ebermann et al. described a USH2 patient with “digenic inheritance.” a heterozygous truncating mutation in GPR98, and a truncating heterozygous mutation in PDZ domain-containing 7 (PDZD7), which is reported to be a cause of USH [20]. Our USH1 patient (Case #4) had segregated MYO7A:p.Ala771Ser and PCDH15:c.158-1G>A. Molecular analyses in mouse models have shown many interactions among the USH1 proteins [2]. In particular, MYO7A directly binds to PCDH15 and both proteins are expressed in an overlapping pattern in hair bundles in a mouse model [21]. PCDH15:c.158-1G>A, predicted to alter the splice donor site of intron 3, has been classified as pathogenic. MYO7A:p.Ala771Ser is a non-truncating mutation, but was previously reported as disease-causing [13]. So, we consider the patient to be the first reported case of MYO7A/PCDH15 digenic inheritance.
However, we should be aware of two limitations of MPS technology. First, the target region of MPS cannot cover all coding exons of USH genes. Actually, the coverage of the target exons was 97.0% in our study. So, it is impossible to detect a mutation in a region which is not covered using this system (Case #9: c.5821-2A>G). Secondarily, the MPS system used in this study, is not effective for detecting homo-polymer regions, for example poly C stretch [22] (Case #8: p.Lys542GlnfsX5). In addition, concerning pathogenecity of mutations identified, functional analysis will be necessary to draw the final conclusion in the future.
In UK and US Caucasian USH1 patients, USH1B (MYO7A) has been reported as the most common USH1 genetic subtype [11], [12], while USH1F (PCDH15) has been reported as the most common USH1 genetic subtype in North American Ashkenazi Jews [23]. In Japanese, our study revealed that the most common type was MYO7A (41.7%), which was similar to the frequency in the above Caucasian patients (46.8∼55%) [11], [12]. However, the small number of USH1 patients in our study might have biased the frequency and further large cohort study will be needed in the future.
In addition, most of our detected mutations were novel. We have previously reported genes responsible for deafness in Japanese patients and observed differences in mutation spectrum between Japanese (who are probably representative of other Asian populations) and populations with European ancestry [24].
In conclusion, our study was the first report of USH mutation analysis using MPS and the frequency of USH1 genes in Japanese. Mutation screening using MPS has the potential power to quickly identify mutations of many causative genes such as USH while maintaining cost-benefit performance. In addition, the simultaneous mutation analysis of large numbers of genes was useful for detecting mutations in different genes that are possibly disease modifiers or of digenic inheritance.
Materials and Methods
Subjects
We screened 17 Japanese USH1 patients (aged 9 to 64 years): three from autosomal recessive families (non-affected parents and two or more affected siblings), and 14 from sporadic families. There were 9 males and 8 females. None of the subjects had any other noteworthy symptoms. All subjects or next of kin on the behalf of the minors/children gave prior written informed consent for participation in the project, and the Ethical Committee of Shinshu University approved the study and the consent procedure.
Amplicon Library Preparation
An Amplicon library of the target exons was prepared with an Ion AmpliSeq Custom Panel (Applied Biosystems, Life Technologies, Carlsbad, CA) designed with Ion AmpliSeq Designer (https://www.ampliseq.com/browse.action) for nine USH genes by using Ion AmpliSeq Library Kit 2.0 (Applied Biosystems, Life Technologies) and Ion Xpress Barcode Adapter 1–16 Kit (Applied Biosystems, Life Technologies) according to the manufacturers' procedures.
In brief, DNA concentration was measured with Quant-iT dsDNA HS Assay (Invitrogen, Life Technologies) and Qubit Fluorometer (Invitrogen, Life Technologies) and DNA quality was confirmed by agarose gel electrophoresis. 10 ng of each genomic DNA sample was amplified, using Ion AmpliSeq HiFi Master Mix (Applied Biosystems, Life Technologies) and AmpliSeq Custom primer pools, for 2 min at 99°C, followed by 15 two-step cycles of 99°C for 15 sec and 60°C for 4 min, ending with a holding period at 10°C in a PCR thermal cycler (Takara, Shiga, Japan). After the Multiplex PCR amplification, amplified DNA samples were digested with FuPa enzyme at 50°C for 10 min and 55°C for 10 min and the enzyme was successively inactivated for 60°C for 20 min incubation. After digestion, diluted barcode adapter mix including Ion Xpress Barcode Adapter and Ion P1 adaptor were ligated to the end of the digested amplicons with ligase in the kit for 30 min at 22°C and the ligase was successively inactivated at 60°C for 20 min incubation. Adaptor ligated amplicon libraries were purified with the Agencourt AMPure XP system (Beckman Coulter Genomics, Danvers, MA). The amplicon libraries were quantified by using Ion Library Quantitation Kit (Applied Biosystems, Life Technologies) and the StepOne plus realtime PCR system (Applied Biosystems, Life Technologies) according to the manufacturers' procedures. After quantification, each amplicon library was diluted to 20 pM and the same amount of the 12 libraries for 12 patients were pooled for one sequence reaction.
Emulsion PCR and Sequencing
The emulsion PCR was carried out with the Ion OneTouch System and Ion OneTouch 200 Template Kit v2 (Life Technologies) according to the manufacturer's procedure (Publication Part Number 4478371 Rev. B Revision Date 13 June 2012). After the emulsion PCR, template-positive Ion Sphere Particles were enriched with the Dynabeads MyOne Streptavidin C1 Beads (Life Technologies) and washed with Ion OneTouch Wash Solution in the kit. This process were performed using an Ion OneTouch ES system (Life Technologies).
After the Ion Sphere Particle preparation, MPS was performed with an Ion Torrent Personal Genome Machine (PGM) system using the Ion PGM 200 Sequencing Kit and Ion 318 Chip (Life Technologies) according to the manufacturer's procedures.
Base Call and Data Analysis
The sequence data were processed with standard Ion Torrent Suite Software and Torrent Server successively mapped to human genome sequence (build GRCh37/hg19) with Torrent Mapping Alignment Program optimized to Ion Torrent data. The average of 562.33 Mb sequences with about 4,300,000 reads was obtained by one Ion 318 chip. The 98.0% sequences were mapped to the human genome and 94% of them were on the target region. Average coverage of depth in the target region was 314.2 and 93.8% of them were over 20 coverage.
After the sequence mapping, the DNA variant regions were piled up with Torrent Variant Caller plug-in software. Selected variant candidates were filtered with the average base QV (minimum average base quality 25), variant frequency (40–60% for heterozygous mutations and 80–100% for homozygous mutations) and coverage of depth (minimum coverage of depth 10). After the filtrations, variant effects were analyzed with the wANNOVAR web site [25], [26] (http://wannovar.usc.edu) including the functional prediction software for missense variants: Sorting Intolerant from Tolerant (SIFT; http://sift.jcvi.org/), and Polymorphism Phenotyping (PolyPhen2; http://genetics.bwh.harvard.edu/pph2/). The sequencing data was available in the DNA databank of Japan (Accession number: DRA001273).
Algorithm
Missense, nonsense, and splicing variants were selected among the identified variants. Variants were further selected as less than 1% of: 1) the 1000 genome database (http://www.1000genomes.org/), 2) the 5400 exome variants (http://evs.gs.washington.edu/EVS/), and 3) the in-house control. Candidate mutations were confirmed by Sanger sequencing and the responsible mutations were identified by segregation analysis using samples from family members of the patients. In addition, the cases with heterozygous or no causative mutation were fully sequenced by Sanger sequencing for USH1 genes in order to verify the MPS results.
Direct Sequence Analysis
Primers were designed with the Primer 3 plus web server (http://www.bioinformatics.nl/cgi-bin/primer3plus/primer3plus.cgi). Each genomic DNA sample (40 ng) was amplified using Ampli Taq Gold (Life Technologies) for 5 min at 94°C, followed by 30 three-step cycles of 94°C for 30 sec, 60°C for 30 sec, and 72°C for 30 sec, with a final extension at 72°C for 5 min, ending with a holding period at 4°C in a PCR thermal cycler (Takara, Shiga, Japan). The PCR products were treated with ExoSAP-IT (GE Healthcare Bio, Buckinghamshire, UK) and by incubation at 37°C for 60 min, and inactivation at 80°C for 15 min. After the products were purified, we performed standard cycle sequencing reaction with ABI Big Dye terminators in an ABI 3130xl sequencer (Life Technologies).
Accession numbers
MYO7A, [NM_000260.3]; USH1C, [NM_ 153676.3]; CDH23, [NM_ 022124.5]; PCDH15, [NM_ 033056.3]; USH1G, [NM_ 173477.2]; USH2A, [NM_206933.2]; GPR98, [NM_ 032119.3]; DFNB31, [NM_ 015404.3]; CLRN1, [NM_ 174878.2]; PDZD7, [NM_ 001195263.1].
Acknowledgments
We thank A. C. Apple-Mathews for help in preparing the manuscript.
Funding Statement
This study was supported by a Health and Labour Sciences Research Grant for Comprehensive Research on Disability Health and Welfare from the Ministry of Health, Labour and Welfare of Japan (S.U.), and by a Grant-in-Aid for Scientific Research from the (then) Ministry of Education, Science and Culture of Japan (http://www.mext.go.jp/english/) (S.U.). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Kimberling WJ, Hildebrand MS, Shearer AE, Jensen ML, Halder JA, et al. (2010) Frequency of Usher syndrome in two pediatric populations: Implications for genetic screening of deaf and hard of hearing children. Genet Med 12: 512–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Yan D, Liu XZ (2010) Genetics and pathological mechanisms of Usher syndrome. J Hum Genet 55: 327–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Jaworek TJ, Bhatti R, Latief N, Khan SN, Riazuddin S, et al. (2012) USH1K, a novel locus for type I Usher syndrome, maps to chromosome 10p11.21-q21.1. J Hum Genet 57: 633–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Riazuddin S, Belyantseva IA, Giese AP, Lee K, Indzhykulian AA, et al. (2012) Alterations of the CIB2 calcium- and integrin-binding protein cause Usher syndrome type 1J and nonsyndromic deafness DFNB48. Nat Genet 44: 1265–1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Weil D, Blanchard S, Kaplan J, Guilford P, Gibson F, et al. (1995) Defective myosin VIIA gene responsible for Usher syndrome type 1B. Nature 374: 60–61. [DOI] [PubMed] [Google Scholar]
- 6. Bork JM, Peters LM, Riazuddin S, Bernstein SL, Ahmed ZM, et al. (2001) Usher syndrome 1D and nonsyndromic autosomal recessive deafness DFNB12 are caused by allelic mutations of the novel cadherin-like gene CDH23. Am J Hum Genet 68: 26–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ahmed ZM, Riazuddin S, Bernstein SL, Ahmed Z, Khan S, et al. (2001) Mutations of the protocadherin gene PCDH15 cause Usher syndrome type 1F. Am J Hum Genet 69: 25–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Verpy E, Leibovici M, Zwaenepoel I, Liu XZ, Gal A, et al. (2000) A defect in harmonin, a PDZ domain-containing protein expressed in the inner ear sensory hair cells, underlies Usher syndrome type 1C. Nat Genet 26: 51–55. [DOI] [PubMed] [Google Scholar]
- 9. Mustapha M, Chouery E, Torchard-Pagnez D, Nouaille S, Khrais A, et al. (2002) A novel locus for Usher syndrome type I, USH1G, maps to chromosome 17q24-25. Hum Genet 110: 348–350. [DOI] [PubMed] [Google Scholar]
- 10. Ouyang XM, Yan D, Du LL, Hejtmancik JF, Jacobson SG, et al. (2005) Characterization of Usher syndrome type I gene mutations in an Usher syndrome patient population. Hum Genet 116: 292–299. [DOI] [PubMed] [Google Scholar]
- 11. Bonnet C, Grati M, Marlin S, Levilliers J, Hardelin JP, et al. (2011) Complete exon sequencing of all known Usher syndrome genes greatly improves molecular diagnosis. Orphanet J Rare Dis 6: 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Le Quesne Stabej P, Saihan Z, Rangesh N, Steele-Stallard HB, Ambrose J, et al. (2012) Comprehensive sequence analysis of nine Usher syndrome genes in the UK National Collaborative Usher Study. J Med Genet 49: 27–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Nakanishi H, Ohtsubo M, Iwasaki S, Hotta Y, Takizawa Y, et al. (2010) Mutation analysis of the MYO7A and CDH23 genes in Japanese patients with Usher syndrome type 1. J Hum Genet 55: 796–800. [DOI] [PubMed] [Google Scholar]
- 14. Eudy JD, Yao S, Weston MD, Ma-Edmonds M, Talmadge CB, et al. (1998) Isolation of a gene encoding a novel member of the nuclear receptor superfamily from the critical region of Usher syndrome type IIa at 1q41. Genomics 50: 382–384. [DOI] [PubMed] [Google Scholar]
- 15. Weston MD, Luijendijk MW, Humphrey KD, Moller C, Kimberling WJ (2004) Mutations in the VLGR1 gene implicate G-protein signaling in the pathogenesis of Usher syndrome type II. Am J Hum Genet 74: 357–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Aller E, Jaijo T, van Wijk E, Ebermann I, Kersten F, et al. (2010) Sequence variants of the DFNB31 gene among Usher syndrome patients of diverse origin. Mol Vis 16: 495–500. [PMC free article] [PubMed] [Google Scholar]
- 17. Joensuu T, Hamalainen R, Yuan B, Johnson C, Tegelberg S, et al. (2001) Mutations in a novel gene with transmembrane domains underlie Usher syndrome type 3. Am J Hum Genet 69: 673–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Nakanishi H, Ohtsubo M, Iwasaki S, Hotta Y, Usami S, et al. (2011) Novel USH2A mutations in Japanese Usher syndrome type 2 patients: marked differences in the mutation spectrum between the Japanese and other populations. J Hum Genet 56: 484–490. [DOI] [PubMed] [Google Scholar]
- 19. Miyagawa M, Nishio SY, Ikeda T, Fukushima K, Usami S (2013) Massively parallel DNA sequencing successfully identifies new causative mutations in deafness genes in patients with cochlear implantation and EAS. PLoS One [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ebermann I, Phillips JB, Liebau MC, Koenekoop RK, Schermer B, et al. (2010) PDZD7 is a modifier of retinal disease and a contributor to digenic Usher syndrome. J Clin Invest 120: 1812–1823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Senften M, Schwander M, Kazmierczak P, Lillo C, Shin JB, et al. (2006) Physical and functional interaction between protocadherin 15 and myosin VIIa in mechanosensory hair cells. J Neurosci 26: 2060–2071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Loman NJ, Misra RV, Dallman TJ, Constantinidou C, Gharbia SE, et al. (2012) Performance comparison of benchtop high-throughput sequencing platforms. Nat Biotechnol 30: 434–439. [DOI] [PubMed] [Google Scholar]
- 23. Ben-Yosef T, Ness SL, Madeo AC, Bar-Lev A, Wolfman JH, et al. (2003) A mutation of PCDH15 among Ashkenazi Jews with the type 1 Usher syndrome. N Engl J Med 348: 1664–1670. [DOI] [PubMed] [Google Scholar]
- 24. Usami S, Wagatsuma M, Fukuoka H, Suzuki H, Tsukada K, et al. (2008) The responsible genes in Japanese deafness patients and clinical application using Invader assay. Acta Otolaryngol 128: 446–454. [DOI] [PubMed] [Google Scholar]
- 25. Wang K, Li M, Hakonarson H (2010) ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res 38: e164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Chang X, Wang K (2012) wANNOVAR: annotating genetic variants for personal genomes via the web. J Med Genet 49: 433–436. [DOI] [PMC free article] [PubMed] [Google Scholar]