

Abstract
Tibial muscular dystrophy (TMD) is a late onset, autosomal dominant distal myopathy that results from mutations in the two last domains of titin. The cascade of molecular events leading from the causative Titin mutations to the preterm death of muscle cells in TMD is largely unknown. In this study we examined the mRNA and protein changes associated with the myopathology of TMD. To identify these components we performed gene expression profiling using muscle biopsies from TMD patients and healthy controls. The profiling results were confirmed through quantitative real-time PCR and protein level analysis. One of the pathways identified was activation of endoplasmic reticulum (ER) stress response. ER stress activates the unfolded protein response (UPR) pathway. UPR activation was supported by elevation of the marker genes HSPA5, ERN1 and the UPR specific XBP1 splice form. However, UPR activation appears to be insufficient to correct the protein abnormalities causing its activation because degenerative TMD muscle fibres show an increase in ubiquitinated protein inclusions. Abnormalities of VCP-associated degradation pathways are also suggested by the presence of proteolytic VCP fragments in western blotting, and VCP's accumulation within rimmed vacuoles in TMD muscle fibres together with p62 and LC3B positive autophagosomes. Thus, pathways controlling turnover and degradation, including autophagy, are distorted and lead to degeneration and loss of muscle fibres.
Introduction
Tibial muscular dystrophy (TMD, OMIM: #600334, Udd myopathy) is an autosomal dominant distal myopathy with a particularly high prevalence in the Finnish population [1]–[3]. The disease is caused by heterozygous mutations in the last two exons (Mex5-6) of the Titin (TTN) gene [4]. Five other phenotypes have been reported to be associated with C-terminal titin mutations: recessive limb-girdle muscular dystrophy type 2J (LGMD2J) [2], dominant hereditary myopathy with early respiratory failure [5], [6], recessive early-onset myopathy with fatal cardiomyopathy [7], core myopathy with heart disease [8] and centronuclear myopathy [9]. All Finnish TMD patients reported so far share the 11 bp deletion/insertion FINmaj founder mutation [1], [2], which results in C-terminal defects in the M-line region of the sarcomeric titin protein [2]. Several other mutations in the last two exons of TTN have been shown to cause TMD in patients of other European populations [4], [10], [11].
TMD is clinically characterised by atrophy and weakness in the muscles of the anterior compartment of the lower leg (tibialis anterior, extensor hallucis longus and extensor digitorum longus) [3], with typical onset between 35–55 years [12]. Homozygous FINmaj mutations cause the manifestation of the completely different, and much more severe, early onset LGMD2J (OMIM: #608807) [2], [3], [13]. TMD and LGMD2J patients do not have facial muscle weakness, dysphagia or clinically manifest cardiomyopathy. TMD muscle biopsies show myopathic–dystrophic morphology with rare fibre necrosis and frequent rimmed vacuolated fibres in affected muscles [3], [12], [14].
Titin molecules are integral to striated muscle structure and function by forming the backbone of the continuous third filament system of the myofibrils. The titin molecule stretches from the Z-disc to the M-line of the sarcomere. One of titin's core functions is to provide muscle elasticity by returning thin and thick filaments to their positions after contractions [15], [16]. The last Ig-domain (M10) containing the FINmaj mutation is adjacent to the differentially spliced [17] is7 domain that contains a calpain-3 (CAPN3) binding site [13]. Immunofluorescent (IF) microscopy of homozygous mutant muscles has revealed an absence of C-terminal titin epitopes from at least the three last domains M9, is7 and M10 [2]. Together with the loss of M-line CAPN3 and mislocalization of the C-terminal ligand obscurin, this suggests that the FINmaj mutation leads to a proteolytic cleavage of the titin C-terminus [13], [18], [19].
As the downstream molecular pathology caused by the FINmaj mutation is still largely unknown, the objective of this study was to identify relevant changes through global expression profiling of affected distal muscles from TMD patients. Significantly changed pathways were identified and confirmed through quantitative real-time PCR and protein level analysis of marker molecules.
Materials and Methods
Ethics statement
All patient biopsies were obtained with written informed consent and according to the Helsinki declaration. The study was approved by the institutional review board of Helsinki University Hospital.
Muscle biopsies
All patients were diagnosed based on DNA mutation testing. Control and patient biopsies were obtained from the distal muscles of male and female patients in a range of ages (37–92; Table 1).
Table 1. Summary of patient and control muscle biopsies.
| Expression array | qPCR | WB/IF/IHC | Biopsy | Diagnosis | Sex | Age at biopsy | Pathology |
| T1 | rT1 | EHL | TMD | M | 53 | Rimmed vacuoles | |
| T2 | GL | TMD | M | 62 | No rimmed vacuoles | ||
| T3 | TA | TMD | M | 48 | Rimmed vacuoles | ||
| T4 | rT2 | TMD 5 | EHL | TMD | M | 65 | Rimmed vacuoles |
| T5 | SOL | TMD | M | 52 | Rimmed vacuoles | ||
| T6 | TP | TMD | M | 52 | No major pathology | ||
| T7 | TA | TMD | M | 52 | Rimmed vacuoles | ||
| rT3 | EHL | TMD | M | 92 | Not known | ||
| rT4 | TA | TMD | M | 73 | Rimmed vacuoles | ||
| rT5 | TA | TMD | M | 78 | Rimmed vacuoles | ||
| TMD 1 | EHL | TMD | M | 62 | Rimmed vacuoles | ||
| TMD 2 | TA | TMD | M | 55 | Rimmed vacuoles | ||
| TMD 3 | GM | TMD | M | 67 | Rimmed vacuoles | ||
| TMD 4 | EDL | TMD | F | 50–55 | Rimmed vacuoles | ||
| TMD 6 | TA | TMD | M | 44 | Rimmed vacuoles | ||
| C1 | rC1 | TP | Control | M | 76 | Normal | |
| C2 | EHL | Control | M | 76 | Normal | ||
| C3 | rC2 | TA | Control | M | 80 | Normal | |
| C4 | rC3 | EHL | Control | M | 80 | Normal | |
| C5 | SOL | Control | M | 76 | Normal | ||
| Ctrl 1 | TA | Control | M | 37 | Normal | ||
| Ctrl 2 | TA | Control | F | 48 | Normal | ||
| Ctrl 3 | THP | Control | F | 91 | Normal |
Tibialis anterior (TA), tibialis posterior (TP), gastrocnemius lateralis (GL), gastrocnemius medialis (GM), soleus (SOL), extensor hallucis longus (EHL), extensor digitorum longus (EDL), thigh posterior (THP), immunofluorescence microscopy (IF), western blotting (WB) and immunohistochemistry (IHC). The expression array controls (C1, C2 & C5) and (C3 & C4) were collected from amputation material from the distal lower limb muscles of two individuals.
RNA preparation
RNA was extracted from frozen muscles samples using RNAeasy mini kit's (Qiagen, USA) recommended protocol. The RNA quality was assessed using a 2100 Bioanalyzer and the Eukaryote total RNA Nano series II program (Agilent technologies, USA).
Microarray analysis
3 µg of total RNA was used to generate double stranded cDNA. The cRNA was generated using either one- or two-cycle eukaryote target labelling along with controls supplied with the kit (Affymetrix, USA). The samples were biotin labelled and the cRNA randomly fragmented before hybridisation to Affymetrix U133plus2 microarrays following the manufacturer's instructions. The microarray chips were washed and stained using Affymetrix Fluidics Station and scanned using a GeneChip Scanner 3000.
Expression values were determined using Affymetrix Microarray Suite 5.0 and GeneSpring GX version 11.0 (Agilent Technologies, USA) was used for pre-processing the data using the MAS5 algorithm. All probe sets with absent flags were removed from the data. The data was baseline transformed to the median of all samples. Differentially expressed genes were detected using an unpaired t-test with unequal variance (Welch correction). P-values were corrected using a Benjamini-Hochberg adjustment for multiple testing. Probe sets having a P<0.05 and at least a two-fold change were considered to be differentially expressed. The microarray data is available from the GEO website (http://www.ncbi.nlm.nih.gov/geo/) with accession number GSE42806.
Pathway Analysis
Pathway analysis was done using Ingenuity Pathway Analysis software (www.ingenuity.com). The significance of each canonical pathway having more affected genes than expected by chance in the data set was determined by the Fisher's exact t-test (P<0.05).
Quantitative real-time PCR
RNA was extracted from tissue biopsies using a Trizol (Invitrogen, USA) based protocol according to the manufacturer's suggestions. 1 µg of total RNA was reverse-transcribed to cDNA using SuperScript II Reverse Transcriptase (Invitrogen, USA) following the manufacturer's protocol. Quantification of the cDNA was performed using TaqMan based quantitative real-time PCR. The primers and probes used were the spliced form of XBP1 (Hs00231936_m1), HSPA5 (Hs00607129_gH), ERN1 (Hs00980093_m1), JUN (Hs99999141_s1), HSPB1 (Hs03044127_g1) and GAPDH (4333764F). 10 µl TaqMan master mix (Applied Biosystems, USA), 0.5 µl 1∶10 diluted cDNA and 2 µl of primer and probe set were used in a 20 µl total reaction volume. Amplification and detection were performed using the ABI 7500 system (Applied Biosystems, USA). The PCR thermal conditions were 50°C for 2 min, 95°C for 10 s and 60°C for 1 min. Each sample was performed in triplicate and the expression was normalised to GAPDH using standard curves for each gene on the same plate.
Statistical analysis
Statistical significance of the quantitative real-time PCR and western blotting results were calculated using an unpaired t-test (P<0.05).
Western blotting
Muscle biopsies were homogenized with 19 volumes of sample buffer containing 4 M urea and 4% SDS at 100°C for 5 min. Samples were resolved using 12% SDS-PAGE gels with 4% stacking gels and transferred to PVDF membranes (Bio-Rad Laboratories, USA). Membranes were blocked in TBST 5% milk for 1 h at +4°C. Primary and secondary HPR-conjugated antibodies were incubated at +4°C for 1 h each, followed by 6× TBST (5 min) washes. Antibodies were detected using the SuperSignal West Femto ECL substrate (Pierce, Thermo Scientific, USA) and captured using photographic film. Protein loading was assessed by staining of the post-blotting gel using Bio-Safe Coomassie Stain (Bio-Rad Laboratories, USA). Antibodies used are listed in Table S1. Quantitative analysis of western blotting bands was performed using ImageJ version 1.46f (http://rsb.info.nih.gov/ij/).
Immunohistochemistry and immunofluorescence
Muscle biopsies were snap frozen in liquid nitrogen cooled isopentane to make 6 µm cryosections on SuperFrost (Kindler GmbH, Germany). The sections were fixed in 4% PFA for 10 min, permeabilized in 0.2% Triton X-100 for 10 min, and blocked in 5% BSA for 30 min at room temperature. The DAB immunohistochemistry (IHC) (Universal DAB detection, Ventana, USA) was performed using the BenchMark automated immunostainer following manufacturer's instructions. LAMP2 IHC signal was enhanced using an amplification kit (cat. 760-080, Ventana, USA). Herovici staining (a modified van Gieson stain) on frozen muscle sections was used for identification of the rimmed vacuoles. The immunofluorescence microscopy was performed as described earlier [20]. Antibodies are listed in Table S1.
Results
Expression profiling and pathway analysis
The expression profiles of seven TMD and five control biopsy samples were hierarchically clustered (Fig. 1). Significantly (P<0.05) changed probe sets in the TMD samples representing all differentially expressed transcripts are given in Table S2. Ingenuity Pathway Analysis (IPA) showed that endoplasmic reticulum (ER) stress response-, NRF2 mediated oxidative stress-, PTEN-, integrin- and EIF2- signalling pathways were among the most clearly affected pathways in TMD (Fig. 2A, 2B & 2C). There were also a range of pathway changes that have been identified in other myopathies with rimmed vacuoles such as; SAPK/JNK apoptosis- [21], p70S6K- [22], [23], protein ubiquitination- [23], [24] and mitochondrial dysfunction- [24], [25] signalling (Fig. 2B, 2C, & 2D).
Figure 1. Hierarchal clustering of TMD and control expression profiles.
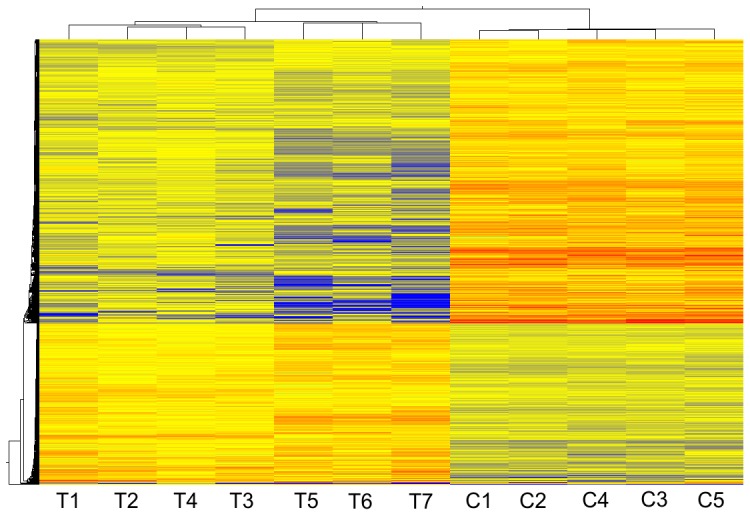
Clustering of differentially expressed probes using centroid linkage and euclidean similarity in GeneSpring GX v11.0. The expression profiles of TMD (T1–T5) and controls (C1–C5) are represented as a heatmap (blue = low expression, red = high expression). A list of significantly changed probes is in Table S2.
Figure 2. Ingenuity Pathway Analysis of TMD expression profiles.

(A) Cell stress pathways affected in TMD also showing the ratio of significantly changed genes versus all genes in the given pathway (oxidative stress response; abbreviated to OSR). (B) Significantly altered cell stress and toxicity pathways, (C) apoptosis pathways, and (D) selected other pathways. The dotted line indicates the statistical significance threshold from a Fisher's exact t-test (P<0.05).
Verification of selected expression changes by quantitative real-time PCR
We chose to investigate ER stress response in TMD muscle since this pathway had the highest fraction of genes with significantly changed expression compared to controls. Unfolded protein response (UPR) is the main mechanism used to alleviate the effects of ER stress. HSPB1 proteins are involved in the ER stress response [26], whilst HSPA5 (BIP) [27], ERN1 (IRE1) [28] and the spliced form of XBP1 [29] are involved in the UPR pathway. Significant (P<0.05) up-regulation was observed in the expression of HSPA5 (Fig. 3A), HSPB1 (Fig. 3B), ERN1 (Fig. 3C) and the previously described UPR specific XBP1 splice isoform (Fig. 3D) in TMD samples versus controls.
Figure 3. Quantitative real-time PCR analysis of key components of selected pathways.

Expression of (A) HSPA5, (B) HSPB1, (C) ERN1, (D) spliced (s) XBP1, and (E) JUN normalised to GAPDH by quantitative real-time PCR from muscle biopsy total RNA (arbitrary units). *Indicates statistical significance in an unpaired t-test (P<0.05) comparison to the control group. The average and standard deviation of five TMD and three control biopsies each performed in triplicate are shown.
The SAPK/JNK apoptotic signalling pathway was significantly (P<0.05) changed in the TMD samples compared to controls in IPA analysis. We analysed JUN (CJUN) expression by quantitative real-time PCR, as it is a final stage component of this pathway, and found significantly elevated expression levels of JUN in TMD samples (Fig. 3E).
Analyses of UPR pathway components on the protein level
Numerous rimmed vacuoles were identified in TMD biopsy sections by Herovici staining (Fig. 4A). We observed abnormalities in the ubiquitin-proteasome system (UPS) such as cytoplasmic ubiquitin containing inclusions (Fig. 4B) in the rimmed vacuolated fibres in IHC. We also observed the presence of HSPA5 granular cytoplasmic dots in non-vacuolated fibres by IF microscopy in TMD muscle biopsies (Fig. 5A), which were not observed in controls (Fig. 5B). This was in line with the up-regulation of HSPA5 we found by quantitative real-time PCR analysis (Fig. 3A). By western blotting HSPA5 levels were increased in two out of five biopsies (Fig. 6A).
Figure 4. Histology and immunohistochemical analysis of TMD muscle biopsies.

(A) Herovici histology staining of TMD muscle sections showing rimmed vacuolar regions within the fibres. (B) Immunohistochemical (IHC) analysis of TMD muscle sections stained for ubiquitin showing localised cytoplasmic ubiquitin containing inclusions in atrophic and rimmed vacuolated fibres and immunoreactivity at the edge of rimmed vacuoles. (C) Autophagosome marker LC3B staining showed strong immunoreactivity within the rimmed vacuolar regions. (D) VCP staining showed immunoreactivity in bodies within a large subset of rimmed vacuoles. (E) p62 staining showed immunoreactivity in rimmed vacuoles and granular immunoreactive dots (shown by an arrow) in an atrophic fibre. (F) Lysosomal protein LAMP2 staining showed no overall increase in rimmed vacuolated fibres. Two separate TMD fibres (A–C and D–F) with rimmed vacuoles in serial sections are shown. Controls of (G) Herovici staining, (H) ubiquitin, (I) LC3B, (J) VCP, (K) p62 and (L) LAMP2 IHC are included for comparison. Sections are from samples TMD 6 and Ctrl 3 in Table 1. Scale bar 50 µm.
Figure 5. Immunofluorescent microscopy of TMD and control biopsies.

(A) HSPA5 staining showed the presence of cytoplasmic granular dots in non-vacuolated TMD muscle fibres. The LC3B channel showed the area has no vacuolated fibres and no autofluorescent material present. (B) A control of HSPA5/LC3B double staining was included for comparison. (C) LAMP2 and LC3B double staining of a TMD muscle fibre showed a representative rimmed vacuole with massive accumulation of LC3B inside and negligible signal from LAMP2 around the edge of the fibre. The top right corner of the LC3B image showed a rimmed vacuole in bright field. (D) A control of LAMP2/LC3B double staining was included for comparison. All images are nuclear counterstained with DAPI (blue signal). Sections are from samples TMD 6 and Ctrl 3 in Table 1. Scale bar 50 µm.
Figure 6. Protein level analysis of UPR and ERAD in TMD biopsies.

(A) Western blotting showed HSPA5 increases in two in five samples when compared to controls. (B) Immunoblotting VCP showed an increase in full length VCP in two TMD samples and an extra band (∼70 kDa) just below the full length VCP in a third sample. Bands at approx. 25 kDa (red boxes) are observed in three of the TMD samples. (C) Samples were also blotted with p97D179 antiserum specific for VCP cleaved at D179 and show bands at approx. 25 kDa in three of the same samples. Total protein content is shown by the myosin band in Coomassie blue staining and showed equal sample loading. A graph of the quantification of the VCP cleavage product (25 kDa) signals normalised to the Coomassie myosin band showed a trend towards being increased in TMD in a t-test (P = 0.077).
Analysis of the ERAD pathway at the protein level
Abnormal or misfolded proteins within the ER are directed into the ER-associated protein degradation (ERAD) pathway. VCP is involved in proteasome-autophagy crosstalk [30], [31] and is a core component of the ERAD pathway [32]. In all 5 TMD biopsies tested by IHC, we observed a variable frequency of VCP-positive material connected with rimmed vacuoles (Fig. 4D). Abnormal VCP immunoreactivity was present as bodies in over half of the rimmed vacuoles. VCP labelling was also found in a subset of nuclei in control and TMD sections but did not differ between disease and controls samples. Western blotting (Fig. 6B) showed an increase in full length VCP in two TMD samples and an extra band (70 kDa) below the full length VCP in a third sample. Intriguingly, an extra 25-kDa band was observed in three out of five TMD biopsies and not in the controls. These bands were identified as D179-cleaved VCP by staining with a specific antiserum [33] (Fig. 6C). However, by quantification the increased levels of the 25-kDa VCP cleavage product in TMD did not reach statistical significance (P = 0.077).
Analyses of autophagy pathway components on the protein level
If a protein abnormality cannot be correctly handled and processed through UPR activation using the UPS for degradation, the proteins may be redirected to the autophagic pathway and degraded in the lysosomal system. We observed strong immunoreactivity of the autophagosome marker LC3B in the rimmed vacuoles and in the cytoplasm of very atrophic fibres (Fig. 4C). Accumulation of p62 and the autophagosome marker LC3B have been used to indicate abnormalities in the autophagic degradation pathways [34]–[36]. We observed p62 immunoreactivity in rimmed vacuoles and granular immunoreactive dots in many atrophic fibres (Fig. 4E). There was no overall increase in immunoreactivity in the lysosomal marker LAMP2 in TMD fibres or within the rimmed vacuoles (Fig. 4F & 5C) when compared to controls (Fig. 4L & 5D).
Discussion
The expression profiling results showed that the samples clustered according to the disease diagnosis as expected. IPA showed that there were differences in distinct molecular pathways between the TMD and the control samples. We were able to confirm selected changes in the studied pathways through quantitative real-time PCR and protein level analysis in the TMD samples.
Pathway analysis indicated a change in the unfolded protein response (UPR) pathway in the TMD samples. This was also suggested by the induction of ER stress response gene HSPB1 and UPR genes HSPA5, ERN1 and the splice form of XBP1 in quantitative real-time PCR. The HSPA5 protein also showed restricted areas of increased expression in IF microscopy. Many of the proteins studied showed large variations between samples, and these differences may be due to the patient's biopsy site, disease severity or age at muscle biopsy. UPR activation has been reported in other rimmed vacuolar myopathies with abnormal UPS or lysosomal degradation pathways [37]–[40], and mildly in a mouse model of desminopathy [41]. This suggests UPR may act as a common protective mechanism during myopathic protein based stress. FINmaj mutated titin appears to cause the activation of UPR and lead to accumulation of ubiquitinated proteins. The increase of ubiquitinated protein components in TMD muscle is unlikely to be related to CAPN3 dysfunction. A secondary CAPN3 defect is present in homozygous LGMD2J, but in heterozygous TMD patients CAPN3 expression, although variable, is typically within the normal range [13].
Excess of misfolded proteins in the ER may also activate the ERAD system [42]. A key component of the ERAD pathway is VCP which, among others, retro-translocates unfolded proteins from the ER into the cytosol for degradation by the UPS [43]. Primary mutations in VCP cause a muscle disease with rimmed vacuolar pathology [44], [45]. VCP has also been reported to regulate ubiquitin-containing autophagosome maturation during myopathic proteomic stress [30], [31], and may therefore be involved in autophagosomal activation in TMD muscle. VCP accumulates inside of rimmed vacuoles suggesting that it may have a role in TMD myopathology with similarities to other neurodegenerative proteinopathies such as VCP-mutated myopathy [44], [45]. Western blotting showed an increase in two out of five biopsies and an extra band beneath the full length form in a third biopsy. In addition, we observed unconventional anti-VCP positive bands of approx. 25 kDa in size in 3 of 5 TMD samples. These ∼25 kDa bands were subsequently confirmed to be VCP cleavage products by specific antiserum [33]. The expression of these VCP cleavage fragments has been reported to impair degradation by the UPS system in neuroblastoma cells [33], but further studies are required before this pathway can be linked to the pathomechanism of TMD.
The p62 protein acts as an auxiliary autophagy factor by directly binding ubiquitinated proteins and LC3, to facilitate their degradation by autophagy [35]. However, p62 also shuttles ubiquitinated proteins to the proteasome for degradation [46] and has been found in protein aggregates in other neuromuscular and neurodegenerative disorders [47]–[49]. The accumulation of LC3B positive autophagosome material and p62 within the rimmed vacuolated regions of the degenerative muscle fibres suggests massive activation of compensatory autophagy mechanisms in TMD. Cytoplasmic increase of LAMP2 has been reported in atrophic fibres in other rimmed vacuolar myopathies [50]. However, there was no consistent increase of mature lysosomes based on LAMP2 staining's in atrophic rimmed vacuolated TMD muscle fibres. Apparently the increased induction of autophagy is not paralleled by increased autophagic flux and further processing, which may cause the observed massive increase of LC3B compartments.
JUN, a final stage component of SAPK/JNK apoptosis signalling pathway was significantly increased in quantitative real-time PCR analysis. In distal myopathy with rimmed vacuoles (DMRV) JUN has been shown to be increased in vacuolated fibres [21]. Nevertheless, whether increased apoptosis leads to a loss of muscle fibres in TMD remains unsettled. A previous study [13] has indicated that apoptosis may not be consistently increased in TMD muscle, although apoptosis is increased in the homozygotic LGMD2J [13] muscle. However, the muscle pathology of LGMD2J is different to TMD and does not show rimmed vacuolar changes.
This study, based on expression profiling and molecular pathology, has identified changes in several subcellular molecular pathways that are apparently involved in the TMD pathomechanism. These include activation of UPR accompanied by increased amounts of ubiquitinated proteins and altered autophagic degradation leading to atrophic and rimmed vacuolated degenerated fibres. Apparently a variety of entries, i.e. different mutant proteins, can result in similar changes in catabolic pathways that control protein turnover. The changes identified in this study are downstream secondary effects of the primary FINmaj mutation in titin. The exact molecular mechanisms that trigger UPR, ERAD and autophagic dysregulation as well as the other abnormally regulated pathways identified in this study need further characterization in order to identify steps that could be therapeutically useful.
Supporting Information
A list of antibodies used in western blotting, immunohistochemistry and immunofluorescence.
(DOC)
A list of significantly changed expression array probes in TMD versus controls.
(XLS)
Acknowledgments
We are grateful to all the participating patients for their cooperation in this study and to Helena Luque and Hanna-Liisa Kojo for technical assistance and for the gift of p97D179 antiserum from Andréa LeBlanc (McGill University). Matti Kankainen is also acknowledged for his support in bioformatics.
Funding Statement
The study received funding from the Folkhälsan Research Foundation, the University of Helsinki, the National Doctoral Program of Musculoskeletal Disorders and Biomaterials. Research grants from the Liv och Hälsa Foundation and Vaasa Central Hospital District Medical Research funds are also acknowledged. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Udd B, Vihola A, Sarparanta J, Richard I, Hackman P (2005) Titinopathies and extension of the M-line mutation phenotype beyond distal myopathy and LGMD2J. Neurology 64: 636–642. [DOI] [PubMed] [Google Scholar]
- 2. Hackman P, Vihola A, Haravuori H, Marchand S, Sarparanta J, et al. (2002) Tibial muscular dystrophy is a titinopathy caused by mutations in TTN, the gene encoding the giant skeletal-muscle protein titin. Am J Hum Genet 71: 492–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Udd B (1992) Limb-girdle type muscular dystrophy in a large family with distal myopathy: Homozygous manifestation of a dominant gene? J Med Genet 29: 383–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hackman P, Marchand S, Sarparanta J, Vihola A, Pénisson-Besnier I, et al. (2008) Truncating mutations in C-terminal titin may cause more severe tibial muscular dystrophy (TMD). Neuromuscul Disord 18: 922–928. [DOI] [PubMed] [Google Scholar]
- 5. Lange S, Xiang F, Yakovenko A, Vihola A, Hackman P, et al. (2005) The kinase domain of titin controls muscle gene expression and protein turnover. Science 308: 1599–1603. [DOI] [PubMed] [Google Scholar]
- 6. Palmio J, Evila A, Chapon F, Tasca G, Xiang F, et al. (2013) Hereditary myopathy with early respiratory failure: Occurrence in various populations. J Neurol Neurosurg Psychiatry In press. [DOI] [PubMed] [Google Scholar]
- 7. Carmignac V, Salih MA, Quijano-Roy S, Marchand S, Al Rayess MM, et al. (2007) C-terminal titin deletions cause a novel early-onset myopathy with fatal cardiomyopathy. Ann Neurol 61: 340–351. [DOI] [PubMed] [Google Scholar]
- 8. Chauveau C, Bonnemann CG, Julien C, Kho AL, Marks H, et al. (2013) Recessive TTN truncating mutations define novel forms of core myopathy with heart disease. Hum Mol Genet In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ceyhan-Birsoy O, Agrawal PB, Hidalgo C, Schmitz-Abe K, DeChene ET, et al. (2013) Recessive truncating titin gene, TTN, mutations presenting as centronuclear myopathy. Neurology 81: 1205–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Pollazzon M, Suominen T, Penttilä S, Malandrini A, Carluccio MA, et al. (2010) The first italian family with tibial muscular dystrophy caused by a novel titin mutation. J Neurol 257: 575–579. [DOI] [PubMed] [Google Scholar]
- 11. Van den Bergh PY, Bouquiaux O, Verellen C, Marchand S, Richard I, et al. (2003) Tibial muscular dystrophy in a belgian family. Ann Neurol 54: 248–251. [DOI] [PubMed] [Google Scholar]
- 12. Udd B, Partanen J, Halonen P, Falck B, Hakamies L, et al. (1993) Tibial muscular dystrophy. late adult-onset distal myopathy in 66 finnish patients. Arch Neurol 50: 604–608. [DOI] [PubMed] [Google Scholar]
- 13. Haravuori H, Vihola A, Straub V, Auranen M, Richard I, et al. (2001) Secondary calpain3 deficiency in 2q-linked muscular dystrophy: Titin is the candidate gene. Neurology 56: 869–877. [DOI] [PubMed] [Google Scholar]
- 14. Udd B, Haravuori H, Kalimo H, Partanen J, Pulkkinen L, et al. (1998) Tibial muscular dystrophy-from clinical description to linkage on chromosome 2q31. Neuromuscul Disord 8: 327–332. [DOI] [PubMed] [Google Scholar]
- 15. Gautel M, Mues A, Young P (1999) Control of sarcomeric assembly: The flow of information on titin. Ergeb Physiol 138: 97–137. [DOI] [PubMed] [Google Scholar]
- 16. Labeit S, Kolmerer B (1995) Titins: Giant proteins in charge of muscle ultrastructure and elasticity. Science 270: 293–296. [DOI] [PubMed] [Google Scholar]
- 17. Kolmerer B, Olivieri N, Witt CC, Herrmann BG, Labeit S (1996) Genomic organization of M line titin and its tissue-specific expression in two distinct isoforms. J Mol Biol 256: 556–563. [DOI] [PubMed] [Google Scholar]
- 18. Charton K, Danièle N, Vihola A, Roudaut C, Gicquel E, et al. (2010) Removal of the calpain 3 protease reverses the myopathology in a mouse model for titinopathies. Hum Mol Genet 19: 4608–4624. [DOI] [PubMed] [Google Scholar]
- 19. Fukuzawa A, Lange S, Holt M, Vihola A, Carmignac V, et al. (2008) Interactions with titin and myomesin target obscurin and obscurin-like 1 to the M-band: Implications for hereditary myopathies. J Cell Sci 121: 1841–1851. [DOI] [PubMed] [Google Scholar]
- 20. Raheem O, Olufemi S, Bachinski LL, Vihola A, Sirito M, et al. (2010) Mutant (CCTG)n expansion causes abnormal expression of zinc finger protein 9 (ZNF9) in myotonic dystrophy type 2. Am J Pathol 177: 3025–3036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Yan C, Ikezoe K, Nonaka I (2001) Apoptotic muscle fiber degeneration in distal myopathy with rimmed vacuoles. Acta Neuropathol 101: 9–16. [DOI] [PubMed] [Google Scholar]
- 22. Nogalska A, D'Agostino C, Terracciano C, Engel WK, Askanas V (2010) Impaired autophagy in sporadic inclusion-body myositis and in endoplasmic reticulum stress-provoked cultured human muscle fibers. Am J Pathol 177: 1377–1387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ching JK, Elizabeth SV, Ju J, Lusk C, Pittman SK, et al. (2013) mTOR dysfunction contributes to vacuolar pathology and weakness in valosin-containing protein associated inclusion body myopathy. Hum Mol Genet 22: 1167–1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Askanas V, Engel WK (2008) Inclusion-body myositis: Muscle-fiber molecular pathology and possible pathogenic significance of its similarity to alzheimer's and parkinson's disease brains. Acta Neuropathol 116: 583–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Eisenberg I, Novershtern N, Itzhaki Z, Becker-Cohen M, Sadeh M, et al. (2008) Mitochondrial processes are impaired in hereditary inclusion body myopathy. Hum Mol Genet 17: 3663–3674. [DOI] [PubMed] [Google Scholar]
- 26. Ito H, Iwamoto I, Inaguma Y, Takizawa T, Nagata K, et al. (2005) Endoplasmic reticulum stress induces the phosphorylation of small heat shock protein, Hsp27. J Cell Biochem 95: 932–941. [DOI] [PubMed] [Google Scholar]
- 27. Lin H, Masso-Welch P, Di Y, Cai J, Shen J, et al. (1993) The 170-kDa glucose-regulated stress protein is an endoplasmic reticulum protein that binds immunoglobulin. Mol Biol Cell 4: 1109–1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Chen Y, Brandizzi F (2013) IRE1: ER stress sensor and cell fate executor. Trends Cell Biol 23, 11: 547–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Yoshida H, Matsui T, Yamamoto A, Okada T, Mori K (2001) XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell 107: 881–891. [DOI] [PubMed] [Google Scholar]
- 30. Ju JS, Fuentealba RA, Miller SE, Jackson E, Piwnica-Worms D, et al. (2009) Valosin-containing protein (VCP) is required for autophagy and is disrupted in VCP disease. J Cell Biol 187: 875–888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Tresse E, Salomons FA, Vesa J, Bott LC, Kimonis V, et al. (2010) VCP/p97 is essential for maturation of ubiquitin-containing autophagosomes and this function is impaired by mutations that cause IBMPFD. Autophagy 6: 217–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Meyer H, Bug M, Bremer S (2012) Emerging functions of the VCP/p97 AAA-ATPase in the ubiquitin system. Nat Cell Biol 14: 117–123. [DOI] [PubMed] [Google Scholar]
- 33. Halawani D, Tessier S, Anzellotti D, Bennett DA, Latterich M, et al. (2010) Identification of caspase-6-mediated processing of the valosin containing protein (p97) in alzheimer's disease: A novel link to dysfunction in ubiquitin proteasome system-mediated protein degradation. J Neurosci 30: 6132–6142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Weihl CC, Pestronk A (2010) Sporadic inclusion body myositis: Possible pathogenesis inferred from biomarkers. Curr Opin Neurol 23: 482–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Pankiv S, Clausen TH, Lamark T, Brech A, Bruun JA, et al. (2007) p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem 282: 24131–24145. [DOI] [PubMed] [Google Scholar]
- 36. Lee HS, Daniels BH, Salas E, Bollen AW, Debnath J, et al. (2012) Clinical utility of LC3 and p62 immunohistochemistry in diagnosis of drug-induced autophagic vacuolar myopathies: A case-control study. PLoS One 7: e36221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Liewluck T, Hayashi YK, Ohsawa M, Kurokawa R, Fujita M, et al. (2007) Unfolded protein response and aggresome formation in hereditary reducing-body myopathy. Muscle Nerve 35: 322–326. [DOI] [PubMed] [Google Scholar]
- 38. Vattemi G, Engel WK, McFerrin J, Askanas V (2004) Endoplasmic reticulum stress and unfolded protein response in inclusion body myositis muscle. Am J Pathol 164: 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Boito CA, Fanin M, Gavassini BF, Cenacchi G, Angelini C, et al. (2007) Biochemical and ultrastructural evidence of endoplasmic reticulum stress in LGMD2I. Virchows Archiv 451: 1047–1055. [DOI] [PubMed] [Google Scholar]
- 40. Kuga A, Ohsawa Y, Okada T, Kanda F, Kanagawa M, et al. (2011) Endoplasmic reticulum stress response in P104L mutant caveolin-3 transgenic mice. Hum Mol Genet 20: 2975–2983. [DOI] [PubMed] [Google Scholar]
- 41. Lynch JM, Maillet M, Vanhoutte D, Schloemer A, Sargent MA, et al. (2012) A thrombospondin-dependent pathway for a protective ER stress response. Cell 149: 1257–1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Meusser B, Hirsch C, Jarosch E, Sommer T (2005) ERAD: The long road to destruction. Nat Cell Biol 7: 766–772. [DOI] [PubMed] [Google Scholar]
- 43. Halawani D, Latterich M (2006) p97: The cell's molecular purgatory? Mol Cell 22: 713–717. [DOI] [PubMed] [Google Scholar]
- 44. Watts GDJ, Wymer J, Kovach MJ, Mehta SG, Mumm S, et al. (2004) Inclusion body myopathy associated with paget disease of bone and frontotemporal dementia is caused by mutant valosin-containing protein. Nat Genet 36: 377–381. [DOI] [PubMed] [Google Scholar]
- 45. Palmio J, Sandell S, Suominen T, Penttilä S, Raheem O, et al. (2011) Distinct distal myopathy phenotype caused by VCP gene mutation in a finnish family. Neuromuscul Disord 21: 551–555. [DOI] [PubMed] [Google Scholar]
- 46. Seibenhener ML, Babu JR, Geetha T, Wong HC, Krishna NR, et al. (2004) Sequestosome 1/p62 is a polyubiquitin chain binding protein involved in ubiquitin proteasome degradation. Mol Cell Biol 24: 8055–8068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Olive M, van Leeuwen FW, Janue A, Moreno D, Torrejon-Escribano B, et al. (2008) Expression of mutant ubiquitin (UBB+1) and p62 in myotilinopathies and desminopathies. Neuropathol Appl Neurobiol 34: 76–87. [DOI] [PubMed] [Google Scholar]
- 48. Nogalska A, Terracciano C, D'Agostino C, King Engel W, Askanas V (2009) p62/SQSTM1 is overexpressed and prominently accumulated in inclusions of sporadic inclusion-body myositis muscle fibers, and can help differentiating it from polymyositis and dermatomyositis. Acta Neuropathol 118: 407–413. [DOI] [PubMed] [Google Scholar]
- 49. Zatloukal K, Stumptner C, Fuchsbichler A, Heid H, Schnoelzer M, et al. (2002) p62 is a common component of cytoplasmic inclusions in protein aggregation diseases. Am J Pathol 160: 255–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Tsuruta Y, Furuta A, Furuta K, Yamada T, Kira J, et al. (2001) Expression of the lysosome-associated membrane proteins in myopathies with rimmed vacuoles. Acta Neuropathol 101: 579–584. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
A list of antibodies used in western blotting, immunohistochemistry and immunofluorescence.
(DOC)
A list of significantly changed expression array probes in TMD versus controls.
(XLS)