

Abstract
Altered metabolism is fundamental to the growth and survival of cancer cells. Pyruvate kinase M2 (PKM2), a key enzyme in cancer metabolism, has been demonstrated to play a central role not only in metabolic reprogramming but also in direct regulation of gene expression and subsequent cell cycle progression. This review outlines the current understanding of PKM2 protein kinase activity and regulatory mechanisms underlying PKM2 expression, enzymatic activity, and nuclear localization, thus highlighting PKM2 as a potential therapeutic target.
Keywords: PKM2, the Warburg effect, gene expression
1. Introduction
In the 1920s, Otto Warburg observed that despite the availability of oxygen, most cancer cells produce energy predominantly by a high rate of glycolysis followed by lactic acid fermentation in the cytosol rather than by a comparatively low rate of glycolysis followed by pyruvate oxidation in the mitochondria (as is the case in most normal cells) [1; 2]. This phenomenon is termed the Warburg effect or aerobic glycolysis. Warburg found the ratio of glycolysis to respiration to be a fundamental difference between normal and cancer cells. He hypothesized that aerobic glycolysis arose from respiratory injury (mitochondrial injury) [3]. However, later studies show that mitochondrial defects are infrequent in cancer cells [4; 5]. Moreover, cancer cells can switch back to mitochondrial respiration under certain conditions [6]. It has been established that sustained aerobic glycolysis in certain cancer cells is linked to activation of oncogenes or to suppression of tumor suppressor functions [7].
Pyruvate kinase (PK) regulates the final rate-limiting step of glycolysis and catalyzes the transfer of a phosphate group from phosphoenolpyruvate (PEP) to adenosine diphosphate. This transfer yields one molecule each of pyruvate and adenosine triphosphate [8; 9]. PKM1, PKM2, PKL, and PKR are pyruvate kinase isoforms expressed in different types of mammalian cells and tissues. Alternate splicing of PKM pre-mRNA by hnRNP A1/2 and polypyrimidine-tract binding (PTB) protein splicing factors leads to PKM2 generation by including exon 10 and excluding exon 9 [10; 11]. PKM2 is expressed in some differentiated tissues, such as lung, fat, retina, and pancreatic islets, as well as in all cells with a high rate of nucleic synthesis, such as normal proliferating cells and embryonic cells [12; 13; 14; 15; 16; 17]. Regardless of tissue origin, most cancer cells have increased expression of PKM2, which positions PKM2 as an attractive target for cancer therapy [18]. This review highlights findings on PKM2 regulation and functioning in cancer cells, focusing on regulation of PKM2 expression in tumorigenesis, regulation of PKM2 enzyme activity, protein kinase activity of PKM2, and regulation of nuclear localization of PKM2.
2. Regulation of PKM2 expression in tumorigenesis
Regulation of PKM transcription
The PKM gene promoter consists of three cis-acting regions and three GC boxes [19; 20]. A mutation in a GC box resulted in a 50% decrease in promoter activity [21]. Five putative binding sites for SP1 and SP3 were found in the PKM promoter [22]. Glucose promotes the dephosphorylation of SP1, which increases SP1’s DNA binding activity and enhances PKM2 expression. The latter is a prerequisite for tumor cell proliferation (Fig.1A, left panel) [23]. Conversely, overexpression of SP3 represses PKM promoter activity. Hypoxia downregulates SP3, and thereby removes the associated transcriptional repression and activates the promoter activity (Fig.1A, right panel) [24]. In proliferating rat thymocytes, the reduced reactive peroxide anion generation observed during the S phase increases the binding of SP1 to its consensus sequence in the PKM promoter and subsequently enhances PKM2 expression [25]. Insulin also induces PKM promoter activation (independent of glucose and glucosamine) [26]. In phosphatase and tensin homolog (PTEN)-null fatty liver, peroxisome proliferator-activated receptor gamma (PPARγ) binds to hexokinase 2 and PKM promoters to activate transcription [27]. PKM2 gene expression can be regulated by microRNAs (miRNAs), which are noncoding RNAs that bind onto specific mRNA molecules and promote degradation of target mRNA and/or hinder the translation process. Both miR-133a and miR-133b target PKM transcript; these miRNAs were significantly reduced in tongue squamous cell carcinoma cells relative to paired normal epithelial cells [28].
Fig.1.
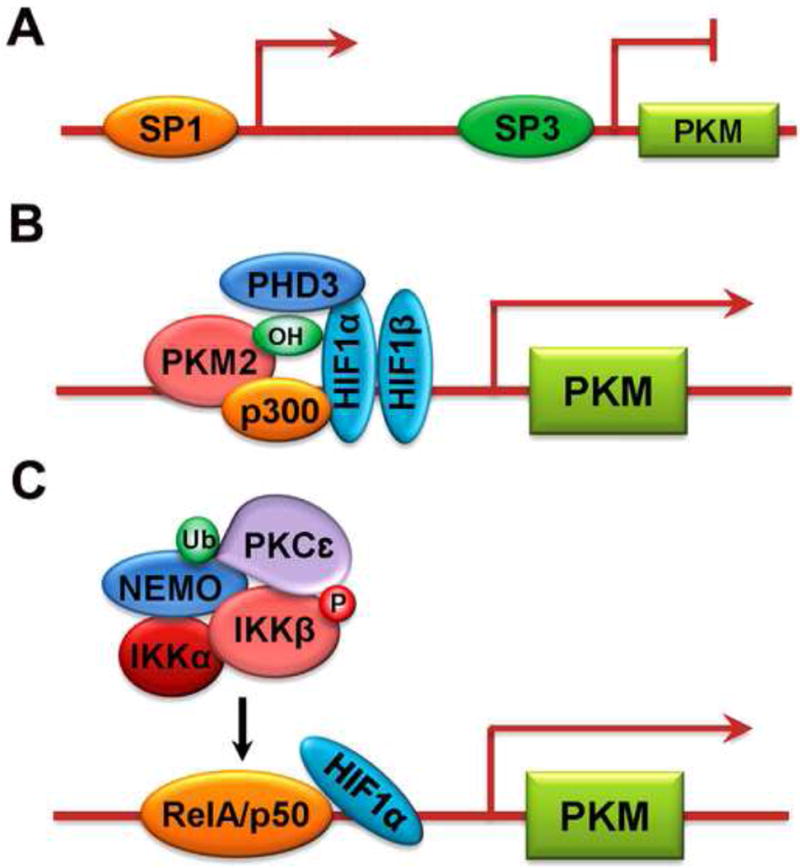
Regulation of PKM transcription.
(A) PKM promoter activity is activated by transcription factor SP1 and inhibited by SP3.
(B) PKM2 hydroxylated by PHD3 interacts directly with HIF1, which increases transcriptional activity of HIF1 and promotes recruitments of p300 to the PKM promoter.
(C) Monoubiquitylated PKC interacts with NEMO and recruits the IKK complex, leading to phosphorylation of IKK. IKK -activated RelA/p50, which is associated with HIF1, binds to the PKM promoter and initiates the transcription of PKM2.
Mammalian target of rapamycin (mTOR) signaling is frequently deregulated during multistep oncogenic processes. mTOR increases PKM2 expression by binding hypoxia-inducible factor 1 (HIF1) to the promoter region immediately upstream of exon 1 of PKM [29]. Under hypoxic conditions, PKM2, acting in a feedback loop, interacts directly with HIF1 and promotes transactivation of HIF1 downstream genes by enhancing HIF1 DNA-binding activity and recruiting p300 to hypoxia response elements (HREs). The interaction between PKM2 and HIF1 is mediated by the prolyl hydroxylase 3 (PHD3)-dependent hydroxylation of PKM2 at proline 403/408 (Fig.1B) [30]. Because both PKM and EGLN3 (encoding PHD3) [31] are HIF1 target genes, the positive feedback loop that promotes HIF1 activity maintains high expression of PKM2 and other glycolytic genes, which accelerates metabolic reprogramming of cancer cell metabolism.
Yang et al. have recently shown how PKM2 is transcriptionally upregulated in normoxic conditions by growth factors [32]. Activation of epidermal growth factor receptor (EGFR), which occurs in many types of cancer, results in the binding of the SH2 domain of phospholipase C (PLC) 1 to autophosphorylated EGFR and in activation of PLC 1. Diacylglycerol generated by PLC 1 activates PKC, which results in RINCK1-dependent monoubiquitylation of PKC at K321 at the plasma membrane. Monoubiquitylated PKC interacts with a ubiquitin-binding domain in the NEMO zinc finger region and recruits the cytosolic IKK complex composed of NEMO, IKK, and IKK, to the plasma membrane, where PKC phosphorylates IKK at serine 177 and activates IKK. Activated RelA interacts with HIF1, which is required for RelA to bind the PKM promoter. PKC - and NF-κB-dependent PKM2 upregulation is required for the EGFR-promoted Warburg effect and tumorigenesis. In addition, PKM2 expression correlates with EGFR and IKK activity in human glioblastoma specimens and with glioma malignancy grade. These results highlight the distinct regulation of NF-κB activation between EGFR and well-studied inflammation responses and cytokine stimulation. These results also reveal the importance of metabolic cooperation between EGFR and NF-κB pathways in PKM2 upregulation and tumorigenesis (Fig.1C).
Regulation of alternative splicing of PKM pre-mRNA
PKM2 expression is regulated not only at the transcriptional level but also by alternative splicing of PKM pre-mRNA. PKM1 and PKM2 are produced from the mutually exclusive alternative splicing of PKM pre-mRNA, reflecting the inclusion of either exon 9 (PKM1) or exon 10 (PKM2). In adult tissues, low expression of three heterogeneous, unclear ribonucleoproteins (hnRNPs)—PTB (also known as hnRNP1), hnRNPA1, and hnRNPA2—allows for recognition of exon 9 by splicing machinery and disrupts intronic structures favorable for exon 10 inclusion. However, in rapidly proliferating cells—such as embryonic and cancer cells—PTB, hnRNPA1 and hnRNPA2 are upregulated by oncogenic transcription factor c-Myc, which is overexpressed in many human tumors. PTB, hnRNPA1 and hnRNPA2 then bind to splicing signals flanking exon 9 and represses exon 9’s inclusion [11; 29]. c-Myc, which is downstream from β–catenin activation, increases the expression of glucose transporter 1 (GLUT1), lactate dehydrogenase A (LDHA), PTB, and PTB-dependent PKM2, thereby promoting the Warburg effect [33; 34].
3. Regulation of PKM2 enzyme activity
Effects of metabolic intermediates on glycolytic activity of PKM2
PKM2 activity can be regulated by different mechanisms (Fig.2) and PKM2 may exist in both tetrameric and dimeric forms. Kinetic analysis has revealed that the tetrameric form has high affinity (Km, 0.03 mM) to its substrate PEP, whereas the dimeric form has low affinity to PEP (Km, 0.46) [18; 35; 36]. Fructose-1,6-biphosphate (FBP), a glycolysis intermediate product upstream of PKM2, is an allosteric activator of PKM2. It is proposed that the inter-conversion between dimeric and tetrameric PKM2 is dynamic. Increased levels of dimeric PKM2 that is less active than tetrameric lower the rate of glycolysis and increase the accumulation of FBP, and the latter promotes the tetramerization of PKM2. Tetrameric PKM2 in turn increases the rate of glycolysis and decreases FBP concentration, leading to dimerization of PKM2 [37].
Fig.2.
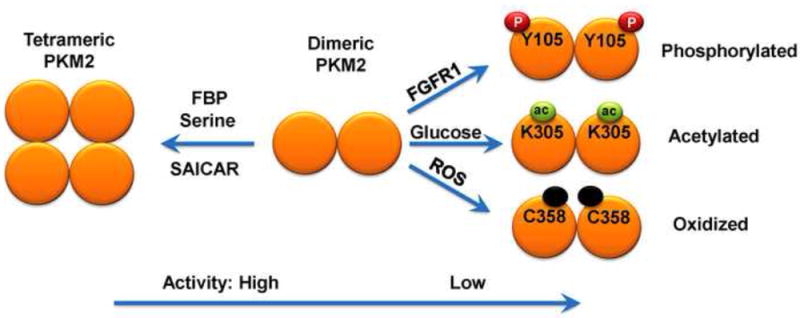
Regulation of glycolytic activity of PKM2.
PKM2 glycolytic activity can be regulated by not only metabolic intermediates, but also post-translational modification of PKM2 induced by different stimuli.
PKM2 is a phosphotyrosine-binding protein [38], as evidenced by the observation that nuclear PKM2 binds to tyrosine-phosphorylated -catenin and activates -catenin [39]. The interaction between PKM2 and phosphotyrosine releases FBP and reduces PKM2 activity. In tumor cells, PKM2 has been found mainly in its less active, dimeric form, which initially appears paradoxical given the high rate of lactate production in these cells. However, the exchange between dimeric and tetrameric forms of PKM2 can be very dynamic, and dimeric PKM2 may allow all glycolytic intermediates above the PKM2 reaction to accumulate, thereby providing a high level of metabolic precursors for synthetic processes [35].
Like the well-known FBP, serine has been reported to be an allosteric activator of PKM2 [40]. Serine can bind to and activate human PKM2 in a manner similar to but independent of FBP. Serine biosynthesis is an anabolic pathway required for growth and proliferation [41]. When serine is abundant, PKM2 is fully active, enabling maximal use of glucose through glycolysis. However, when serine is limited, PKM2 activity is immediately curtailed, enabling rapid diversion of glucose-derived carbon to serine biosynthesis and thus compensating for the serine shortfall. In addition to creating a build-up of glycolytic intermediates for serine biosynthesis, blocking PKM2 activity shifts glucose metabolism away from lactate production in the cytosol to citrate production in the mitochondria. This shift provides the energy required for the rapid proliferation. Intriguingly, succinylaminoimidazolecarboxamideribose-5’-phosphate (SAICAR), an intermediate of the de novo purine nucleotide synthesis pathway, has also been shown to specifically regulate PKM2 activity allosterically and independently of FBP. SAICAR binding sites on PKM2 are close to the FBP binding pocket [42]. Upon glucose starvation, cellular SAICAR concentration has been shown to increase in an oscillatory manner. Increased amount of SAICAR stimulates PKM2 activity, increases glucose and glutamine consumption rates, and promotes cancer cell survival. This allosteric regulation may explain how cancer cells coordinate different metabolic pathways to optimize their growth in the often nutrient-limited tumor microenvironment [42].
Regulation of PKM2 glycolytic activity by post-translational modification
Protein post-translational modifications (PTMs) increase the functional diversity of the proteome by the covalent addition of functional groups or proteins, proteolytic cleavage of regulatory subunits, or degradation of entire proteins. These modifications, which include phosphorylation, glycosylation, ubiquitination, nitrosylation, methylation, acetylation, lipidation, and proteolysis, influence almost all aspects of normal cell biology and pathogenesis [43]. A variety of PTMs can regulate the catalytic activity of PKM2 (Fig.2). PKM2 can be directly phosphorylated by fibroblast growth factor receptor type 1 at tyrosine 105. PKM2 Y105 phosphorylation inhibits the formation of active, tetrameric PKM2 by disrupting the binding of cofactor FBP and thus suppressing PKM2 activity. Expression of PKM2 Y105F mutant resistant to phosphorylation in cancer cells impairs cell proliferation and tumorigenesis [44]. Protein acetylation is another modification commonly used in regulating many cellular processes, including metabolic pathway. Most intermediate metabolic enzymes are acetylated and regulated by acetylation [45]. PKM2 is acetylated at lysine 305 under high glucose concentration [46]. Acetylated PKM2 with reduced activity interacts with HSC70 (a chaperone for chaperone-mediated autophagy), which leads to its lysosomal-dependent degradation. Overexpression of acetylation-mimetic PKM2 K305Q mutant increases levels of glycolytic intermediates and promotes cell proliferation and tumorigenesis.
PKM2 is also involved in the removal of excessive intracellular reactive oxygen species (ROS) [47]. High concentrations of ROS can damage cellular components and compromise cell viability [48]. Controlling intracellular ROS concentrations is therefore critical for cell proliferation and survival. Acute increases in intracellular ROS inhibit PKM2 activity through oxidation of PKM2 cysteine 358, which diverts glucose flux to the pentose phosphate pathway and thus generates reducing potential sufficient to detoxify ROS. Lung cancer cells in which oxidation-resistant PKM2 C358S mutant is expressed exhibit increased sensitivity to oxidative stress and impaired tumor formation [47].
4. Protein kinase activity of PKM2
In addition to functioning cytosolically as a glycolytic enzyme, PKM2 functions as a protein kinase. Upon EGFR activation, nuclear PKM2 binds to c-Src -phosphorylated -catenin at Y333 and enhances transcriptional activity of -catenin, thereby promoting expression of its downstream gene CCND1 (encoding cyclin D1) and MYC [39]. Intriguingly, a catalytically inactive PKM2 mutant translocates to the nucleus and binds to -catenin but fails to induce cyclin D1 expression. A recent study by Yang et al.[49] has demonstrated that PKM2 directly binds to histone H3 and phosphorylates it at threonine 11 upon EGFR activation. This phosphorylation, which uses PEP as a phosphate donor, is required for the dissociation of HDAC3 from CCND1 and MYC promoters and for subsequent acetylation of histone H3 at lysine 9. PKM2-dependent histone H3 modifications are essential for EGF-promoted gene expression as well as for cell proliferation and tumorigenesis [49; 50]. Like histone H3, STAT3 is a substrate for PKM2 kinase activity. Nuclear PKM2 phosphorylates STAT3 at tyrosine 705 and activates MEK5 transcription [51]. These results suggest PKM2 can function as a dual-specificity kinase like MEK/MAP2K and GSK3, which belong to Ser/Thr kinase groups, but can also phosphorylate on tyrosine. Kinases use many different mechanisms to phosphorylate their proper substrates selectively to maintain the specificity that is observed in signaling pathways. Ser/Thr or Tyr specificity is determined by the structure of the catalytic cleft of the kinase and local interactions between the kinase cleft and the substrate phosphorylation site [52]. In vitro kinase assay of these studies showed that the bacterial purified PKM2 most as tetramer can phosphorylate Histone H3 at Thr 11, whereas PKM2 R399E mutant constantly as dimer presents much higher phosphorylation of Stat3 at Tyr705 than PKM2 WT [49; 51]. This suggests that PKM2 confirmation may contribute to its specificity. Of course, different structure of substrate phosphorylation sites may also be an important factor that affects its specificity. The gel filtration chromatography suggested that nuclear PKM2 was completely dimer, while the cytoplasmic PKM2 existed in both dimer and tetramer [51]. Given the fact that tetrameric PKM2 has protein kinase activity towards Histone H3 at least in vitro assay and high abundance of PEP in cytosol, it is reasonable to hypothesize that PKM2 might also function as a protein kinase outside of nucleus, which remains to be further identified. Together, these reports highlight the role of PKM2 as a protein kinase in the regulation of gene expression and tumorigenesis.
5. Regulation of nuclear localization of PKM2
As a glycolytic enzyme, PKM2 predominantly localizes in cytosol. However, PKM2 has also been shown to translocate to the nucleus under certain circumstances. In Cos-7 cells, nuclear translocation of PKM2 can be triggered by different apoptotic agents, such as UV and H2O2. Expression of the fusion protein of PKM2 with SV40 large T-antigen nuclear localization signal (NLS) induces caspase-independent cell death [53]. Conversely, interleukin-3-induced nuclear translocation of PKM2 is required for Jak2-dependent cell proliferation [54]. Furthermore, PKM2 was reported to interact with SUMO-E3 ligase PIAS3, the interaction is required for sumoylation and nuclear translocation of PKM2 [55]. These evidences indicate that PKM2 can translocate into the nucleus. However, the mechanisms of nuclear translocation of PKM2 and whether this translocation is unique to PKM2 were not answered until recently in a report by Yang et al. (Fig.3) [34]. Upon EGFR activation, the docking groove of extracellular signal-regulated kinase 2 (ERK2) binds to PKM2 in a region encoded by exon 10. PKM2 (not PKM1) is phosphorylated at serine 37 by ERK2. Phosphorylated PKM2 recruits peptidyl-prolylcis-trans isomerase NIMA-interacting 1 (PIN1) for cis-trans isomerization of PKM2. Isomerization of PKM2 exposes the NLS sequence located in a region encoded by exon 10 of PKM. This exposure promotes the association of importin 5 with PKM2 and nuclear translocation of PKM2. In the nucleus, PKM2 coactivates -catenin to induce c-Myc expression, resulting in upregulation of GLUT1 and LDHA and, in a positive feedback loop, PTB-dependent PKM2 expression. Expression of PKM2 S37A, which fails to translocate to the nucleus, impairs EGFR-promoted Warburg effect and brain tumor development. These findings explain why PKM2, but not its alternate splicing form PKM1, is instrumental for growth factor-induced Warburg effect, gene expression, and tumorigenesis.
Fig.3.
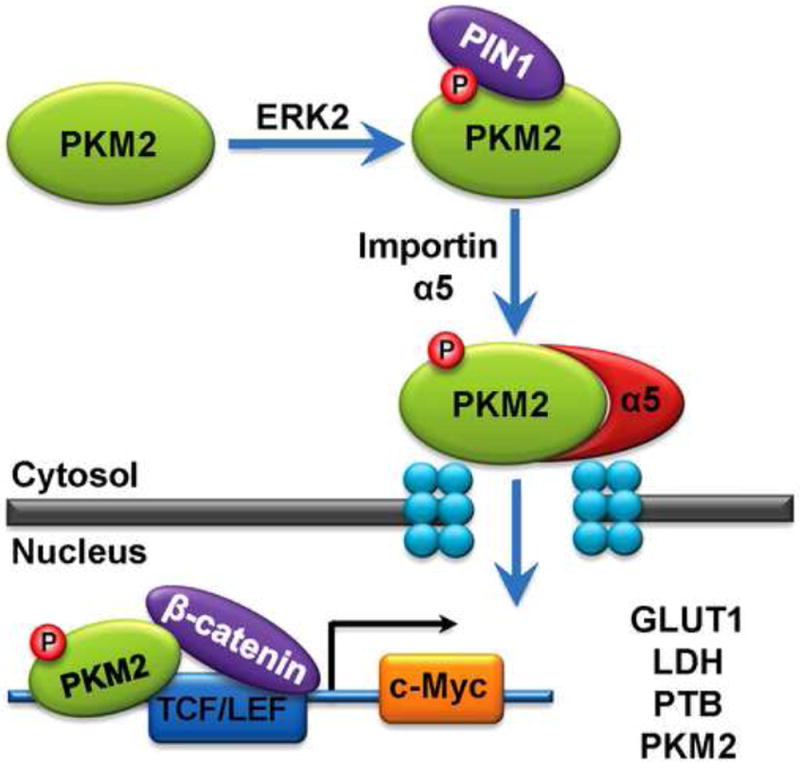
A model of EGF-induced nuclear translocation of PKM2 and PKM2-promoted Warburg effect.
Upon EGF receptor activation, PKM2 is phosphorylated by ERK2 at S37. PIN1 binds to and isomerizes phosphorylated PKM2, resulting in the recruitment of importin 5 and translocation of PKM2 into the nucleus. Nuclear PKM2 binds to -catenin and enhances c-Myc expression, which subsequently increases the expression of GLUT1, LDHA, PTB and PKM2 itself, thereby promoting the Warburg effect and tumorigenesis.
6. Therapeutic effects of PKM2 modulation
High expression of PKM2 in most of cancers made it an attractive target for cancer therapy. Depletion of PKM2 leads to apoptosis or sensitize tumor cells to chemotherapy [56; 57]. Hence, it is reasonable to propose that inhibition of PKM2 slows down tumor growth or even kills tumor cells. This was supported by the use of specific PKM2 inhibitors, which showed inhibitory effects on cancer cell growth and survival [28; 58]. Shikonin, a component of a Chinese herbal medicine was recently reported to inhibit PKM2 and induce cell death. [59]. A clinical trial with a potential PKM2 inhibitor was even initiated to evaluate the clinical significance of PKM2 inhibition [60].
However, this strategy became controversial recently since the finding that inhibition of PKM2 by many post-translational modifications could support cancer cell proliferation [44; 46]. Furthermore, replacing less active PKM2 with constitutively active PKM1 effectively attenuates cancer cell proliferation in vivo and in vitro [61]. These results suggest that high pyruvate kinase activity may suppress tumor growth by diverting glycolytic intermediates from anabolic processes to energy production. This was further supported by the finding that exposure to small-molecule PKM2 activators inhibits the growth of xenograft tumors. Afterwards, several PKM2 –specific activators were developed. Like FBP, the natural activator of PKM2, these molecules increase the affinity of PKM2 for PEP [62; 63; 64].
More recently, discovery of the nuclear functions of PKM2, especially its protein kinase activity, made the strategies targeting PKM2 more diverse. PKM2 nuclear functions contribute to cell proliferation. These activities can be separate or complementary to the well-described cytoplasmic role of PKM2 as the gatekeeper of glycolysis and metabolic flux distribution [65]. Therefore, inhibition of PKM2 nuclear functions provides a new approach to treat cancer. Protein kinase activity of PKM2 is the major nuclear function of PKM2 by far. It has been recently reported that the pyruvate kinase and protein kinase activities of PKM2 could be reciprocally regulated by growth signals [66]. Growth factor stimulations significantly increase the dimer/tetramer PKM2 ratio in cells and consequently activate the protein kinase activity of PKM2 and promote tumor growth [51]. They found PKM2 dimer has higher protein kinase activity than the tetramer, whereas the tetramer has higher pyruvate kinase activity. Based on this study, only those activators of PKM2, who can force PKM2 tetramer formation, could inhibit PKM2 protein kinase activity as well as activate its pyruvate kinase activity, and thereby inhibit tumor cell proliferation by two-pronged ways. However, one critical question needs to be answered before that. Is the catalytic domain of PKM2 protein kinase activity the same as that of its pyruvate kinase activity? If they share the same catalytic domain, the previously developed PKM2 inhibitors may also inhibit its protein kinase activity. If not, new inhibitors targeting PKM2 protein kinase activity need to be further developed in future. The exact catalytic domain of PKM2 protein kinase activity remains to be clarified. It is also important to identify the conditions in which inhibiting or activating PKM2 would inhibit tumor growth.
7. Conclusions
Mutations in oncogenes and tumor suppressor genes cause alterations to multiple intracellular signaling pathways. These alterations reengineer tumor cell metabolism to allow enhanced survival and growth. PKM2, a key enzyme controlling the rate-limiting step of glycolysis, has been demonstrated to play a central role in metabolic reprogramming during cancer progression. Tumor cells have multiple ways to regulate PKM2 that are favorable to cell growth and survival, including PKM2 expression, localization, post-translational modification, and allosteric regulation. PKM2’s newly characterized non-metabolic functions as a transcriptional coactivator and protein kinase (especially as a histone kinase globally regulating histone phosphorylation and acetylation) endow it with the ability to regulate gene expression, cell cycle progression, and metabolism in a feedback loop. All of these findings reflect the great complexity of PKM2 regulation and PKM2’s role as a gatekeeper for both metabolism and cell cycle progression in cancer cells. This gatekeeper role makes PKM2 an attractive target for cancer treatment.
Acknowledgments
This work was supported by National Cancer Institute grants 2R01CA109035 (Z.L.) and CA16672 (Cancer Center Support Grant); a research grant (RP110252 and RP130389; Z. L.) from the Cancer Prevention and Research Institute of Texas (CPRIT); an American Cancer Society Research Scholar Award RSG-09-277-01-CSM (Z.L.); the James S. McDonnell Foundation 21st Century Science Initiative in Brain Cancer Research Award (220020318; Z.L.); a Sister Institution Network Fund from The University of Texas MD Anderson Cancer Center (Z.L.); and the Odyssey fellowship from The University of Texas MD Anderson Cancer Center (W.Y.).
Footnotes
Conflict of Interest Statement
None
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Gatenby RA, Gillies RJ. Why do cancers have high aerobic glycolysis? Nat Rev Cancer. 2004;4:891–899. doi: 10.1038/nrc1478. [DOI] [PubMed] [Google Scholar]
- 2.Kim JW, Dang CV. Cancer’s molecular sweet tooth and the Warburg effect. Cancer Res. 2006;66:8927–8930. doi: 10.1158/0008-5472.CAN-06-1501. [DOI] [PubMed] [Google Scholar]
- 3.Warburg O. On the origin of cancer cells. Science. 1956;123:309–314. doi: 10.1126/science.123.3191.309. [DOI] [PubMed] [Google Scholar]
- 4.Koppenol WH, Bounds PL, Dang CV. Otto Warburg’s contributions to current concepts of cancer metabolism. Nat Rev Cancer. 2011;11:325–337. doi: 10.1038/nrc3038. [DOI] [PubMed] [Google Scholar]
- 5.Ward PS, Thompson CB. Metabolic reprogramming: a cancer hallmark even warburg did not anticipate. Cancer Cell. 2012;21:297–308. doi: 10.1016/j.ccr.2012.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mazurek S. Pyruvate kinase type M2: a key regulator of the metabolic budget system in tumor cells. Int J Biochem Cell Biol. 2011;43:969–980. doi: 10.1016/j.biocel.2010.02.005. [DOI] [PubMed] [Google Scholar]
- 7.Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324:1029–1033. doi: 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Altenberg B, Greulich KO. Genes of glycolysis are ubiquitously overexpressed in 24 cancer classes. Genomics. 2004;84:1014–1020. doi: 10.1016/j.ygeno.2004.08.010. [DOI] [PubMed] [Google Scholar]
- 9.Majumder PK, Febbo PG, Bikoff R, Berger R, Xue Q, McMahon LM, Manola J, Brugarolas J, McDonnell TJ, Golub TR, Loda M, Lane HA, Sellers WR. mTOR inhibition reverses Akt-dependent prostate intraepithelial neoplasia through regulation of apoptotic and HIF-1-dependent pathways. Nat Med. 2004;10:594–601. doi: 10.1038/nm1052. [DOI] [PubMed] [Google Scholar]
- 10.Noguchi T, Inoue H, Tanaka T. The M1- and M2-type isozymes of rat pyruvate kinase are produced from the same gene by alternative RNA splicing. The Journal of biological chemistry. 1986;261:13807–13812. [PubMed] [Google Scholar]
- 11.David CJ, Chen M, Assanah M, Canoll P, Manley JL. HnRNP proteins controlled by c-Myc deregulate pyruvate kinase mRNA splicing in cancer. Nature. 2010;463:364–368. doi: 10.1038/nature08697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Corcoran E, Phelan JJ, Fottrell PF. Purification and properties of pyruvate kinase from human lung. Biochim Biophys Acta. 1976;446:96–104. doi: 10.1016/0005-2795(76)90101-x. [DOI] [PubMed] [Google Scholar]
- 13.Tolle SW, Dyson RD, Newburgh RW, Cardenas JM. Pyruvate kinase isozymes in neurons, glia, neuroblastoma, and glioblastoma. J Neurochem. 1976;27:1355–1360. doi: 10.1111/j.1471-4159.1976.tb02615.x. [DOI] [PubMed] [Google Scholar]
- 14.Reinacher M, Eigenbrodt E. Immunohistological demonstration of the same type of pyruvate kinase isoenzyme (M2-Pk) in tumors of chicken and rat. Virchows Arch B Cell Pathol Incl Mol Pathol. 1981;37:79–88. doi: 10.1007/BF02892557. [DOI] [PubMed] [Google Scholar]
- 15.Schering B, Eigenbrodt E, Linder D, Schoner W. Purification and properties of pyruvate kinase type M2 from rat lung. Biochim Biophys Acta. 1982;717:337–347. doi: 10.1016/0304-4165(82)90188-x. [DOI] [PubMed] [Google Scholar]
- 16.MacDonald MJ, Chang CM. Pancreatic islets contain the M2 isoenzyme of pyruvate kinase. Its phosphorylation has no effect on enzyme activity. Mol Cell Biochem. 1985;68:115–120. doi: 10.1007/BF00219375. [DOI] [PubMed] [Google Scholar]
- 17.Brinck U, Eigenbrodt E, Oehmke M, Mazurek S, Fischer G. L- and M2-pyruvate kinase expression in renal cell carcinomas and their metastases. Virchows Arch. 1994;424:177–185. doi: 10.1007/BF00193498. [DOI] [PubMed] [Google Scholar]
- 18.Mazurek S, Boschek CB, Hugo F, Eigenbrodt E. Pyruvate kinase type M2 and its role in tumor growth and spreading. Semin Cancer Biol. 2005;15:300–308. doi: 10.1016/j.semcancer.2005.04.009. [DOI] [PubMed] [Google Scholar]
- 19.Noguchi T, Yamada K, Inoue H, Matsuda T, Tanaka T. The L- and R-type isozymes of rat pyruvate kinase are produced from a single gene by use of different promoters. The Journal of biological chemistry. 1987;262:14366–14371. [PubMed] [Google Scholar]
- 20.Wang Z, Takenaka M, Imai E, Yamada K, Tanaka T, Noguchi T. Transcriptional regulatory regions for expression of the rat pyruvate kinase M gene. Eur J Biochem. 1994;220:301–307. doi: 10.1111/j.1432-1033.1994.tb18626.x. [DOI] [PubMed] [Google Scholar]
- 21.Yamada K, Noguchi T. Nutrient and hormonal regulation of pyruvate kinase gene expression. Biochem J. 1999;337(Pt 1):1–11. [PMC free article] [PubMed] [Google Scholar]
- 22.Netzker R, Weigert C, Brand K. Role of the stimulatory proteins Sp1 and Sp3 in the regulation of transcription of the rat pyruvate kinase M gene. Eur J Biochem. 1997;245:174–181. doi: 10.1111/j.1432-1033.1997.00174.x. [DOI] [PubMed] [Google Scholar]
- 23.Schafer D, Hamm-Kunzelmann B, Brand K. Glucose regulates the promoter activity of aldolase A and pyruvate kinase M2 via dephosphorylation of Sp1. FEBS Lett. 1997;417:325–328. doi: 10.1016/s0014-5793(97)01314-8. [DOI] [PubMed] [Google Scholar]
- 24.Discher DJ, Bishopric NH, Wu X, Peterson CA, Webster KA. Hypoxia regulates beta-enolase and pyruvate kinase-M promoters by modulating Sp1/Sp3 binding to a conserved GC element. The Journal of biological chemistry. 1998;273:26087–26093. doi: 10.1074/jbc.273.40.26087. [DOI] [PubMed] [Google Scholar]
- 25.Schafer D, Hamm-Kunzelmann B, Hermfisse U, Brand K. Differences in DNA-binding efficiency of Sp1 to aldolase and pyruvate kinase promoter correlate with altered redox states in resting and proliferating rat thymocytes. FEBS Lett. 1996;391:35–38. doi: 10.1016/0014-5793(96)00701-6. [DOI] [PubMed] [Google Scholar]
- 26.Marshall S, Bacote V, Traxinger RR. Discovery of a metabolic pathway mediating glucose-induced desensitization of the glucose transport system. Role of hexosamine biosynthesis in the induction of insulin resistance. The Journal of biological chemistry. 1991;266:4706–4712. [PubMed] [Google Scholar]
- 27.Panasyuk G, Espeillac C, Chauvin C, Pradelli LA, Horie Y, Suzuki A, Annicotte JS, Fajas L, Foretz M, Verdeguer F, Pontoglio M, Ferre P, Scoazec JY, Birnbaum MJ, Ricci JE, Pende M. PPARgamma contributes to PKM2 and HK2 expression in fatty liver. Nat Commun. 2012;3:672. doi: 10.1038/ncomms1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wong TS, Liu XB, Chung-Wai Ho A, Po-Wing Yuen A, Wai-Man Ng R, Ignace Wei W. Identification of pyruvate kinase type M2 as potential oncoprotein in squamous cell carcinoma of tongue through microRNA profiling. Int J Cancer. 2008;123:251–257. doi: 10.1002/ijc.23583. [DOI] [PubMed] [Google Scholar]
- 29.Sun Q, Chen X, Ma J, Peng H, Wang F, Zha X, Wang Y, Jing Y, Yang H, Chen R, Chang L, Zhang Y, Goto J, Onda H, Chen T, Wang MR, Lu Y, You H, Kwiatkowski D, Zhang H. Mammalian target of rapamycin up-regulation of pyruvate kinase isoenzyme type M2 is critical for aerobic glycolysis and tumor growth. Proc Natl Acad Sci U S A. 2011;108:4129–4134. doi: 10.1073/pnas.1014769108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Luo W, Hu H, Chang R, Zhong J, Knabel M, O’Meally R, Cole RN, Pandey A, Semenza GL. Pyruvate kinase M2 is a PHD3-stimulated coactivator for hypoxia-inducible factor 1. Cell. 2011;145:732–744. doi: 10.1016/j.cell.2011.03.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pescador N, Cuevas Y, Naranjo S, Alcaide M, Villar D, Landazuri MO, Del Peso L. Identification of a functional hypoxia-responsive element that regulates the expression of the egl nine homologue 3 (egln3/phd3) gene. Biochem J. 2005;390:189–197. doi: 10.1042/BJ20042121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yang W, Xia Y, Cao Y, Zheng Y, Bu W, Zhang L, You MJ, Koh MY, Cote G, Aldape K, Li Y, Verma IM, Chiao PJ, Lu Z. EGFR-Induced and PKCepsilon Monoubiquitylation-Dependent NF-kappaB Activation Upregulates PKM2 Expression and Promotes Tumorigenesis. Mol Cell. 2012;48:771–784. doi: 10.1016/j.molcel.2012.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lu Z. Nonmetabolic functions of pyruvate kinase isoform M2 in controlling cell cycle progression and tumorigenesis. Chinese journal of cancer. 2012;31:5–7. doi: 10.5732/cjc.011.10446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yang W, Zheng Y, Xia Y, Ji H, Chen X, Guo F, Lyssiotis CA, Aldape K, Cantley LC, Lu Z. ERK1/2-dependent phosphorylation and nuclear translocation of PKM2 promotes the Warburg effect. Nat Cell Biol. 2012;14:1295–1304. doi: 10.1038/ncb2629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mazurek S. Pyruvate kinase type M2: a key regulator within the tumour metabolome and a tool for metabolic profiling of tumours. Ernst Schering Found Symp Proc. 2007:99–124. doi: 10.1007/2789_2008_091. [DOI] [PubMed] [Google Scholar]
- 36.Eigenbrodt E, Reinacher M, Scheefers-Borchel U, Scheefers H, Friis R. Double role for pyruvate kinase type M2 in the expansion of phosphometabolite pools found in tumor cells. Crit Rev Oncog. 1992;3:91–115. [PubMed] [Google Scholar]
- 37.Dombrauckas JD, Santarsiero BD, Mesecar AD. Structural basis for tumor pyruvate kinase M2 allosteric regulation and catalysis. Biochemistry. 2005;44:9417–9429. doi: 10.1021/bi0474923. [DOI] [PubMed] [Google Scholar]
- 38.Christofk HR, Vander Heiden MG, Wu N, Asara JM, Cantley LC. Pyruvate kinase M2 is a phosphotyrosine-binding protein. Nature. 2008;452:181–186. doi: 10.1038/nature06667. [DOI] [PubMed] [Google Scholar]
- 39.Yang W, Xia Y, Ji H, Zheng Y, Liang J, Huang W, Gao X, Aldape K, Lu Z. Nuclear PKM2 regulates beta-catenin transactivation upon EGFR activation. Nature. 2011;480:118–122. doi: 10.1038/nature10598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.de Koning TJ, Snell K, Duran M, Berger R, Poll-The BT, Surtees R. L-serine in disease and development. Biochem J. 2003;371:653–661. doi: 10.1042/BJ20021785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chaneton B, Hillmann P, Zheng L, Martin AC, Maddocks OD, Chokkathukalam A, Coyle JE, Jankevics A, Holding FP, Vousden KH, Frezza C, O’Reilly M, Gottlieb E. Serine is a natural ligand and allosteric activator of pyruvate kinase M2. Nature. 2012;491:458–462. doi: 10.1038/nature11540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Keller KE, Tan IS, Lee YS. SAICAR stimulates pyruvate kinase isoform M2 and promotes cancer cell survival in glucose-limited conditions. Science. 2012;338:1069–1072. doi: 10.1126/science.1224409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lu Z, Hunter T. Degradation of activated protein kinases by ubiquitination. Annu Rev Biochem. 2009;78:435–475. doi: 10.1146/annurev.biochem.013008.092711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hitosugi T, Kang S, Vander Heiden MG, Chung TW, Elf S, Lythgoe K, Dong S, Lonial S, Wang X, Chen GZ, Xie J, Gu TL, Polakiewicz RD, Roesel JL, Boggon TJ, Khuri FR, Gilliland DG, Cantley LC, Kaufman J, Chen J. Tyrosine phosphorylation inhibits PKM2 to promote the Warburg effect and tumor growth. Sci Signal. 2009;2:ra73. doi: 10.1126/scisignal.2000431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhao S, Xu W, Jiang W, Yu W, Lin Y, Zhang T, Yao J, Zhou L, Zeng Y, Li H, Li H, Shi J, An W, Hancock SM, He F, Qin L, Chin J, Yang P, Chen X, Lei Q, Xiong Y, Guan KL. Regulation of cellular metabolism by protein lysine acetylation. Science. 2010;327:1000–1004. doi: 10.1126/science.1179689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lv L, Li D, Zhao D, Lin R, Chu Y, Zhang H, Zha Z, Liu Y, Li Z, Xu Y, Wang G, Huang Y, Xiong Y, Guan KL, Lei QY. Acetylation targets the M2 isoform of pyruvate kinase for degradation through chaperone-mediated autophagy and promotes tumor growth. Mol Cell. 2011;42:719–730. doi: 10.1016/j.molcel.2011.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Anastasiou D, Poulogiannis G, Asara JM, Boxer MB, Jiang JK, Shen M, Bellinger G, Sasaki AT, Locasale JW, Auld DS, Thomas CJ, Vander Heiden MG, Cantley LC. Inhibition of pyruvate kinase M2 by reactive oxygen species contributes to cellular antioxidant responses. Science. 2011;334:1278–1283. doi: 10.1126/science.1211485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wellen KE, Thompson CB. Cellular metabolic stress: considering how cells respond to nutrient excess. Mol Cell. 2010;40:323–332. doi: 10.1016/j.molcel.2010.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yang W, Xia Y, Hawke D, Li X, Liang J, Xing D, Aldape K, Hunter T, Alfred Yung WK, Lu Z. PKM2 phosphorylates histone H3 and promotes gene transcription and tumorigenesis. Cell. 2012;150:685–696. doi: 10.1016/j.cell.2012.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lu Z. PKM2 functions as a histone kinase. Cell Cycle. 2012;11:4101–4102. doi: 10.4161/cc.22325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gao X, Wang H, Yang JJ, Liu X, Liu ZR. Pyruvate kinase M2 regulates gene transcription by acting as a protein kinase. Mol Cell. 2012;45:598–609. doi: 10.1016/j.molcel.2012.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ubersax JA, Ferrell JE., Jr Mechanisms of specificity in protein phosphorylation. Nature reviews Molecular cell biology. 2007;8:530–541. doi: 10.1038/nrm2203. [DOI] [PubMed] [Google Scholar]
- 53.Stetak A, Veress R, Ovadi J, Csermely P, Keri G, Ullrich A. Nuclear translocation of the tumor marker pyruvate kinase M2 induces programmed cell death. Cancer Res. 2007;67:1602–1608. doi: 10.1158/0008-5472.CAN-06-2870. [DOI] [PubMed] [Google Scholar]
- 54.Hoshino A, Hirst JA, Fujii H. Regulation of cell proliferation by interleukin-3-induced nuclear translocation of pyruvate kinase. The Journal of biological chemistry. 2007;282:17706–17711. doi: 10.1074/jbc.M700094200. [DOI] [PubMed] [Google Scholar]
- 55.Spoden GA, Morandell D, Ehehalt D, Fiedler M, Jansen-Durr P, Hermann M, Zwerschke W. The SUMO-E3 ligase PIAS3 targets pyruvate kinase M2. J Cell Biochem. 2009;107:293–302. doi: 10.1002/jcb.22125. [DOI] [PubMed] [Google Scholar]
- 56.Christofk HR, Vander Heiden MG, Harris MH, Ramanathan A, Gerszten RE, Wei R, Fleming MD, Schreiber SL, Cantley LC. The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth. Nature. 2008;452:230–233. doi: 10.1038/nature06734. [DOI] [PubMed] [Google Scholar]
- 57.Kefas B, Comeau L, Erdle N, Montgomery E, Amos S, Purow B. Pyruvate kinase M2 is a target of the tumor-suppressive microRNA-326 and regulates the survival of glioma cells. Neuro-oncology. 2010;12:1102–1112. doi: 10.1093/neuonc/noq080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Vander Heiden MG, Christofk HR, Schuman E, Subtelny AO, Sharfi H, Harlow EE, Xian J, Cantley LC. Identification of small molecule inhibitors of pyruvate kinase M2. Biochemical pharmacology. 2010;79:1118–1124. doi: 10.1016/j.bcp.2009.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chen J, Xie J, Jiang Z, Wang B, Wang Y, Hu X. Shikonin and its analogs inhibit cancer cell glycolysis by targeting tumor pyruvate kinase-M2. Oncogene. 2011;30:4297–4306. doi: 10.1038/onc.2011.137. [DOI] [PubMed] [Google Scholar]
- 60.Porporato PE, Dhup S, Dadhich RK, Copetti T, Sonveaux P. Anticancer targets in the glycolytic metabolism of tumors: a comprehensive review. Frontiers in pharmacology. 2011;2:49. doi: 10.3389/fphar.2011.00049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Anastasiou D, Yu Y, Israelsen WJ, Jiang JK, Boxer MB, Hong BS, Tempel W, Dimov S, Shen M, Jha A, Yang H, Mattaini KR, Metallo CM, Fiske BP, Courtney KD, Malstrom S, Khan TM, Kung C, Skoumbourdis AP, Veith H, Southall N, Walsh MJ, Brimacombe KR, Leister W, Lunt SY, Johnson ZR, Yen KE, Kunii K, Davidson SM, Christofk HR, Austin CP, Inglese J, Harris MH, Asara JM, Stephanopoulos G, Salituro FG, Jin S, Dang L, Auld DS, Park HW, Cantley LC, Thomas CJ, Vander Heiden MG. Pyruvate kinase M2 activators promote tetramer formation and suppress tumorigenesis. Nature chemical biology. 2012;8:839–847. doi: 10.1038/nchembio.1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Boxer MB, Jiang JK, Vander Heiden MG, Shen M, Skoumbourdis AP, Southall N, Veith H, Leister W, Austin CP, Park HW, Inglese J, Cantley LC, Auld DS, Thomas CJ. Evaluation of substituted N,N’-diarylsulfonamides as activators of the tumor cell specific M2 isoform of pyruvate kinase. Journal of medicinal chemistry. 2010;53:1048–1055. doi: 10.1021/jm901577g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Jiang JK, Boxer MB, Vander Heiden MG, Shen M, Skoumbourdis AP, Southall N, Veith H, Leister W, Austin CP, Park HW, Inglese J, Cantley LC, Auld DS, Thomas CJ. Evaluation of thieno[3,2-b] pyrrole[3,2-d] pyridazinones as activators of the tumor cell specific M2 isoform of pyruvate kinase. Bioorganic & medicinal chemistry letters. 2010;20:3387–3393. doi: 10.1016/j.bmcl.2010.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Walsh MJ, Brimacombe KR, Veith H, Bougie JM, Daniel T, Leister W, Cantley LC, Israelsen WJ, Vander Heiden MG, Shen M, Auld DS, Thomas CJ, Boxer MB. 2-Oxo-N-aryl-1,2,3,4-tetrahydroquinoline-6-sulfonamides as activators of the tumor cell specific M2 isoform of pyruvate kinase. Bioorganic & medicinal chemistry letters. 2011;21:6322–6327. doi: 10.1016/j.bmcl.2011.08.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chaneton B, Gottlieb E. Rocking cell metabolism: revised functions of the key glycolytic regulator PKM2 in cancer. Trends in biochemical sciences. 2012;37:309–316. doi: 10.1016/j.tibs.2012.04.003. [DOI] [PubMed] [Google Scholar]
- 66.Gao X, Wang H, Yang JJ, Chen J, Jie J, Li L, Zhang Y, Liu ZR. Reciprocal Regulation of Protein Kinase and Pyruvate Kinase Activities of Pyruvate Kinase M2 by Growth Signals. The Journal of biological chemistry. 2013 doi: 10.1074/jbc.M112.448753. [DOI] [PMC free article] [PubMed] [Google Scholar]