

The Mre11 complex (Mre11, Rad50, and Nbs1) is central to the DNA damage response (DDR). Rad50 contains a conserved Zn2+-dependent homodimerization interface, the Rad50 hook domain. Petrini and colleagues analyze mutants with reduced hook domain function to delineate hook-dependent Mre11 complex functions. A Rad50 mutant partially defective for dimerization was found to hyperactivate DDR signaling. Heterozygous Rad50 mutant mice exhibit severe ATM-dependent hydrocephalus and tumorigenesis defects. The data reveal a previously unrecognized influence of the Rad50 hook domain on DDR signaling, tissue homeostasis, and tumorigenesis.
Keywords: Mre11 complex, double-strand breaks, Rad50, ATM
Abstract
The Mre11 complex (Mre11, Rad50, and Nbs1) is a central component of the DNA damage response (DDR), governing both double-strand break repair and DDR signaling. Rad50 contains a highly conserved Zn2+-dependent homodimerization interface, the Rad50 hook domain. Mutations that inactivate the hook domain produce a null phenotype. In this study, we analyzed mutants with reduced hook domain function in an effort to stratify hook-dependent Mre11 complex functions. One of these alleles, Rad5046, conferred reduced Zn2+ affinity and dimerization efficiency. Homozygous Rad5046/46 mutations were lethal in mice. However, in the presence of wild-type Rad50, Rad5046 exerted a dominant gain-of-function phenotype associated with chronic DDR signaling. At the organismal level, Rad50+/46 exhibited hydrocephalus, liver tumorigenesis, and defects in primitive hematopoietic and gametogenic cells. These outcomes were dependent on ATM, as all phenotypes were mitigated in Rad50+/46 Atm+/− mice. These data reveal that the murine Rad50 hook domain strongly influences Mre11 complex-dependent DDR signaling, tissue homeostasis, and tumorigenesis.
The Mre11 complex is a large multicomponent assembly that comprises two Mre11, two Rad50, and two Nbs1 (Xrs2 in Saccharomyces cerevisiae) molecules that plays a central role in the DNA damage response (DDR). The complex consists of a globular domain in which Mre11 and Nbs1 associate with the Walker A and B domains of Rad50 (Fig. 1A). A large coiled-coil domain of Rad50 extends out from the globular domain and folds back on itself in an anti-parallel manner, with the Rad50 hook domain at the apex. The hook domain is a highly conserved interface that mediates homotypic dimerization via the coordination of a Zn2+ atom by two invariant cysteines from each of two Rad50 protomers (Hopfner et al. 2002).
Figure 1.

Analysis of Rad50 hook dimerization. (A, top panel) Schematic representation of the Mre11 complex in two possible configurations. Rad50 globular domain shown in brown, Mre11 is in blue, and Nbs1 is in gray. (Bottom panel) Sequence of the hook domain from S. cerevisiae, Mus musculus, and the Rad5046 and Rad5047 mutants. (B) Gel filtration profile of Rad50-HK proteins. (C) Fractions from B were resolved on an acrylamide gel and stained with Coomassie. (D) Molecular weight and Zn2+ content of the indicated Rad50-HK proteins determined by SEC-MALS and ICP-MS, respectively. Theoretical molecular weight for a monomeric (P1), dimeric (P2 and Rad50-HKWT), and trimeric/tretrameric (P3) species.
The Mre11 complex is a sensor of DNA double-strand breaks (DSBs) that is required for DDR signaling primarily via the kinase ATM (Stracker and Petrini 2011). Recent data suggest that the complex may also influence the activation of ATR, which is primarily activated in response to DNA replication stress (Stiff et al. 2005; Duursma et al. 2013; Shiotani et al. 2013). These observations underlie the view that the Mre11 complex plays an integral role in the regulation of signaling within the DDR network.
The Mre11 complex plays a key role in DSB repair, promoting both homology-directed DSB repair (HDR) and nonhomologous end-joining (NHEJ). With respect its role in HDR, genetic and physical evidence suggests that the primary function of the Mre11 complex is to promote HDR between sister chromatids via the hook and coiled-coil domains of Rad50 (Bressan et al. 1999; Hartsuiker et al. 2001; Gonzalez-Barrera et al. 2003; Wiltzius et al. 2005; Hohl et al. 2011). Mre11 complex-mediated NHEJ appears to be primarily dependent on DNA-binding sites in the globular domain (Williams et al. 2008, 2009), although we showed that the hook and coiled-coil domains also influence this process, presumably via long-range effects on the disposition of globular domain components (Hohl et al. 2011).
Structural analyses of the complex suggest a model in which the Mre11 complex's effect on DSB repair is attributable to its ability to bridge DNA molecules in trans and in cis (de Jager et al. 2001, 2004; van Noort et al. 2003; Moreno-Herrero et al. 2005). In trans, two complexes bind DNA independently by their globular domains and, via association through the Rad50 hook domain, would bridge sister chromatids and promote HDR between them. Indeed, studies in yeast have shown that the dimerization function of the hook domain underlies the influence of this domain on DSB repair, telomere maintenance, and meiotic DSB formation (Wiltzius et al. 2005). In cis, the globular domain could bridge two DNA ends in a manner that would promote NHEJ (Williams et al. 2008). In this mode, end-to-end DNA bridging is effected by adjacent monomeric components of the dimeric globular domain (Lammens et al. 2011; Lim et al. 2011).
Mutations that target the invariant cysteines of the Rad50 hook domain (Hopfner et al. 2002; Wiltzius et al. 2005; Hohl et al. 2011; He et al. 2012) globally disrupt Mre11 complex functions and thus phenocopy the null Rad50 mutation. Hence, the specific contribution of the Rad50 hook to the repair and signaling functions of the Mre11 complex has not been examined in isolation from the other domains of the complex.
To gain insight into Rad50 hook domain's role, our laboratory targeted the residues adjacent to the invariant cysteine residues in S. cerevisiae in an effort to obtain hypomorphic alleles. The alleles obtained were examined and found to encode hook domains with reduced Zn2+-dependent dimerization and increased DNA damage sensitivity (M Hohl, C Tous, T Kochańczyk, A Krężel, A Aguilera, and JHJ Petrini, in prep.). In this study, two of the yeast alleles (rad50-46 and rad50-47) were modeled in mice.
We found that in the absence of the wild-type Rad50 protein, both Rad5046 and Rad5047 alleles failed to support viability at either the cellular or organismal level, consistent with the view that the Rad50 hook domain is integral to the functions of the Mre11 complex. The Rad5046 and Rad5047 gene products exerted dominant gains of function in the presence of the wild-type protein.
Heterozygous Rad50+/46 and Rad50+/47 cells were relatively proficient in DNA repair but exhibited chronic DDR signaling at the cellular level and severe ATM-dependent phenotypes at the organismal level. The data suggest that the Rad5046 and Rad5047 proteins promote aberrant heterotypic dimerization at the hook domain interface. As the functional defect imparted by Rad5046 and Rad5047 predominantly affected Mre11 complex-dependent DDR signaling, these data reveal a previously unrecognized influence of the Rad50 hook domain on Mre11 complex-dependent regulation of the apical DDR kinases.
Results
Rad50 hook domain mutants
Having previously shown that alteration of the cysteine residues of the Rad50 hook domain was tantamount to complete elimination of the protein (Hopfner et al. 2002), we reasoned that alteration of adjacent residues might result in a partial loss of function that would provide insight regarding the functional importance of the Rad50 hook. Using S. cerevisiae, we identified two mutants, designated rad50-46 and rad50-47, that behaved as hook domain hypomorphs and have in common the presence of charged residues in the vicinity of the cysteines that coordinate zinc (Fig. 1A). As a consequence, both mutants showed reduced hook-mediated dimerization and reduced affinity for Zn2+ (M Hohl, C Tous, T Kochańczyk, A Krężel, A Aguilera, and JHJ Petrini, in prep.).
A 22.7-kDa fragment of bacterially produced mouse Rad50 (Rad50-HK) that contained the hook domain flanked by small segments of the coiled-coil region was purified and examined by size exclusion chromatography and multiangle light scattering (SEC-MALS). Rad50-HKWT eluted as a single peak (Fig. 1B,C) with a molecular weight of 44.6 kDa, indicating a dimeric species. Inductively coupled mass spectrometry (ICP-MS) revealed that the protein to Zn2+ molar ratio of this species was 2.4, approximating the expected value of 2 for a Zn2+-dependent dimer (Fig. 1D). In contrast, Rad50-HK46 eluted in three peaks (Fig. 1B). Peak 2 (P2) was similar to Rad50-HKWT in molecular weight and Zn2+ content. Peak 1 (P1) and peak 3 (P3) contained very little Zn2+ and represent monomers and higher-molecular-weight aggregates, respectively (Fig. 1D). These data suggest that the Rad5046 hook domain is structurally compromised but retains some ability to dimerize and bind Zn2+.
Derivation of Rad5046 and Rad5047 mice
To analyze the function of the Rad50 hook in vivo, mice expressing analogs of the yeast rad50-46 and rad50-47 alleles were derived. The mice initially retained a LoxP-flanked neo cassette within intron 13 of the mutant Rad50 locus (Supplemental Fig. S1A,B). These alleles were designated Rad50+/Neo46 and Rad50+/Neo47. Rad50+/Neo47 chimeric mice were unable to produce viable offspring. No live Rad50+/Neo47 births were noted among 327 pups produced from 27 different chimeras. PCR genotyping of tissues from Rad50+/Neo47 chimeras (including isolated sperm) indicated that the Rad50Neo47 allele was present (Supplemental Fig. S1C). On this basis, we concluded that the Rad50Neo47 allele exerted a dominant lethal effect. In contrast, Rad50+/Neo46 chimeric mice transmitted the Rad50Neo46 allele to viable offspring, and 50% of the embryonic stem cell-derived pups were Rad50+/Neo46.
Rad50Neo46 also exerted a dominant phenotype. The effects observed were more pronounced following deletion of the LoxP-flanked neo cassette (henceforth Rad50+/46 mice) by crossing to CAG-Cre mice. Rad50+/46 mice were born at the expected Mendelian ratios but displayed intense skin hyperpigmentation (Fig. 2A), and 27% of them developed hydrocephalus compared with 4% of Rad50+/Neo46 (Fig. 2B). RT–PCR analysis indicated that the expression of Rad50Neo46 was lower than Rad5046 (Supplemental Fig. S1D). Hence, the extent of dominance was correlated with the dosage of the Rad5046 gene product.
Figure 2.

Rad5046 allele is dominant. (A) Representative image showing hyperpigmentation in tail and paws in a Rad50+/46 mouse compared with a Rad50+/+ littermate. (B) Incidence of hydrocephalus in Rad50+/46, Rad50+/Neo46, and Rad50+/46 Atm+/− mice and representative X-ray images of an 8-wk-old Rad50+/46 mouse with hydrocephalus and a Rad50+/+ littermate. (C) Rad50, Mre11, and Nbs1 protein levels in Rad50+/+ and Rad50+/46 SV40-immortalized MEFs and Nbs1 immunoprecipitation to assess Mre11 complex integrity in mock-treated cells (−) or 1 h after 5 Gy IR. Mre11ATLD1/ATLD1 cells were used as control for cells with reduced Mre11 complex levels (Theunissen et al. 2003). (D) SV40-immortalized MEFs were treated with camptothecin for 24 h, and colonies were assessed 10 d later. Experiments were done three times in triplicate using two independent cell lines per genotype. Error bars depict standard deviation (SD). (E) Primary mouse fibroblasts Rad50+/+ Pim1+/DR-GFP and Rad50+/46 Pim1+/DR-GFP were infected with SceI-IRES-hCD4 retrovirus, and GFP+ cells were assessed 72 h after infection. The efficiency of GFP+ is measured relative to the infected-only (CD4+) cells. Experiments were done in triplicate with two independent cell lines per genotype. P-value was determined by Wilcoxon rank-sum test. (F) SV40-immortalized MEFs of the indicated genotypes were left untreated (t = 0) or irradiated with 2 Gy IR, and the levels of pS1987-ATM and γH2AX were assessed in the indicated time points.
Rad50+/46 males were infertile (discussed further below). This precluded interbreeding of heterozygotes to obtain Rad5046/46 mice. Instead, Rad50+/46 females were crossed with Rad50Δ/+ males (Luo et al. 1999). No Rad5046/Δ mice were obtained from 90 offspring (χ2, P < 10−4), indicating that the Rad5046 allele does not support embryonic viability.
Rad50+/46 and Rad50+/Δ mice were crossed with Rad50ind/ind (in which exons 1 and 2 are flanked by loxP sites) (Adelman et al. 2009) and a tamoxifen-inducible Cre (Ventura et al. 2007). In this setting, Cre-mediated excision of the floxed exons of the Rad50ind allele to generate Rad5046/− and Rad50Δ/− cells in vivo was carried out. The induction of Cre is variable in vivo, and so affected tissues were likely to exhibit mosaicism for the deletion genotype. Tamoxifen administered in the food produced excision in the Rad50ind allele in various tissues but was predominant in the intestine (Supplemental Fig. S2B). Rad5046/− and Rad50Δ/− mosaic mice did not live beyond 2 wk due to intestinal failure. Histological analysis of the gut 9 d after beginning the treatment evidenced a profound alteration of Rad5046/− intestinal morphology (Supplemental Fig. S2C). These data indicate that the Rad5046 allele does not support the viability of proliferating cells in vivo.
DNA repair and checkpoint signaling in Rad50+/46 cells
We sought to determine the molecular basis of the Rad50+/46 phenotype. SV40-immortalized Rad50+/46 and Rad50+/+ mouse embryonic fibroblasts (MEFs) showed similar levels of Mre11 complex proteins, and immunoprecipitations indicated that complex integrity was not overtly compromised (Fig. 2C).
The DNA repair functions of the Mre11 complex were modestly affected in Rad50+/46 cells. Rad50+/46 cells showed increased sensitivity to camptothecin at high doses (Fig. 2D) but were not sensitive to other DNA-damaging agents (Supplemental Fig. S3A). Accordingly, treatment with clastogens failed to increase the yield of chromosomal aberrations relative to wild-type cells (Supplemental Fig. S3B). The DR-GFP assay, in which an I-SceI/GFP reporter is integrated at the Pim1 locus (Kass et al. 2013), was employed to assess HDR proficiency. An I-SceI-IRES-hCD4 retroviral vector, which allowed cell sorting of I-SceI-expressing cells, was used to minimize the confounding effects of poor transfectability and varying transfection efficiencies of the cells examined. We found that Rad50+/46 cells consistently exhibited a trend of lower HDR efficiency, although the difference was not statistically significant (P = 0.17) (Fig. 2E).
The Mre11 complex is required for ATM activation (Stracker and Petrini 2011). Rad50+/46 cells exhibited indices of spontaneous ATM activation, including pS1987-ATM and γH2AX formation (Fig. 2F). IR-induced ATM activation in Rad50+/46 cells was otherwise normal and occurred with wild-type kinetics (Fig. 2F). Rad50+/47 skin fibroblasts generated from chimeric mice phenocopied Rad50+/46 cells in all respects (Supplemental Fig. S4). These results suggested that Rad50+/46 (and Rad50+/47) cells exhibited spontaneous DNA damage. An alternative nonexclusive possibility is that the Rad5046 and Rad5047 alleles are hypermorphic with respect to DNA damage signaling, as observed previously in Rad50S/S mice (and yeast) (Usui et al. 2006), and thereby exhibited indices of ATM activation at levels disproportional to the extent of DNA damage. This latter interpretation predicts that Rad5046 phenotypes would be ATM-dependent.
The Rad50+/46 phenotype is ATM-dependent
Precedent for the hypothesis that Rad50+/46 phenotypes could be ATM-dependent comes from Rad50S/S mice, which at both the cellular and organismal levels exhibited phenotypes reminiscent of Rad50+/46 (Bender et al. 2002; Morales et al. 2005). We found that most aspects of the Rad50+/46 pathology, including hydrocephalus (Fig. 2B), were rescued in the context of Atm heterozygosity, demonstrating the acute ATM dependence of the Rad50+/46 phenotype.
Mre11 complex-dependent ATM signaling affects the viability and proliferative behavior of Lin− Sca1+ c-Kit+ (LSK) cells, which contain primitive hematopoietic components, including hematopoietic stem cells (HSCs) and lineage progenitors (Morales et al. 2008). Rad50+/46 mice showed an altered LSK population characterized by an expansion (average increase of 4.7-fold) of the multipotent precursors (MPPs) (Fig. 3A). This phenotype was also dependent on ATM because it was suppressed in Rad50+/46 Atm+/− mice (Fig. 3A). The increased MPP population was not correlated with elevated levels of its proximal descendants, the common lymphoid progenitor (CLP) and common myeloid progenitors (CMPs) (Fig. 3B). CFU-S assays, which measure the ability of MPPs to differentiate and form colonies in the spleen of lethally irradiated mice, were carried out (Spangrude et al. 1988). We noted a 50% reduction in the colony-forming ability of Rad50+/46 MPP cells (Fig. 3C). These data indicate that the expanded Rad50+/46 MPP population is not fully functional.
Figure 3.

ATM-dependent hematopoietic alterations in Rad50+/46 mice. (A) Quantification of long-term hematopoietic stem cells (LT-HSC), short-term hematopoietic stem cells (ST-HSC), and MPPs in bone marrow from mice of the indicated genotypes. Bars denote the average ± standard error of mean (SEM). Rad50+/+, n = 8; Rad50+/46, n = 8,; Rad50+/46 Atm+/−, n = 2. (B) Quantification of CMP, granulocyte macrophage progenitor (GMP), megakaryocyte erythroid progenitor (MEP), and CLP. Bars denote the average ± SEM. Rad50+/+, n = 5; Rad50+/46, n = 5; Rad50+/46 Atm+/−, n = 2. P-values were determined by unpaired t-test. (C) Representative images and quantification of CFU-S assay at day 13 using bone marrow cells of the indicated genotype. Bars denote the average ± SEM of all the spleens obtained from three independent experiments. Rad50+/+, n = 21; Rad50+/46, n = 20; Rad50+/46 Atm+/−, n = 5. P-values were determined by unpaired t-test. (D) Kaplan-Meier survival curves of Atm−/−, Rad50S/S, Rad50S/S Atm−/− (previously published; Morales et al. 2005), Rad50+/46, and Rad50+/46 Atm−/− mice. Rad50+/46 mouse survival was not assessed beyond 18 mo, and thus the events were censored at that age.
A remarkable outcome in Rad50S/S Atm−/− mice was the suppression of lymphomagenesis associated to ATM deficiency (Morales et al. 2005; Usui et al. 2006). Similarly, the latency of thymic lymphomas typical of the Atm−/− genoytpe was markedly increased in Rad50+/46 Atm−/− mice (Fig. 3D). In light of recent data suggesting a role for the Mre11 complex in ATR activation (Duursma et al. 2013; Shiotani et al. 2013), the suppression of lymphomagenesis in Rad50+/46 Atm−/− mice may reflect compensatory Mre11 complex-dependent ATR activation.
Germ cell dysfunction in Rad50+/46 mice
Rad50+/46 mice exhibited severe fertility defects. All males and 27% of the females were infertile, and the remaining Rad50+/46 females produced fewer and smaller litters than Rad50+/+ controls during the same breeding period (Supplemental Fig. S5A,B). Rad50+/46 exhibited ovarian and testicular atrophy (Fig. 4A,B), and the seminiferous tubules displayed severely reduced cellularity (Fig. 4C). As with the hematopoietic phenotype, testicular defects were ATM-dependent. Testes from Rad50+/46 Atm+/− mice were twofold larger than those from Rad50+/46 (Fig. 4A), and the cellularity of the seminiferous tubules was increased (Fig. 4C).
Figure 4.

Germ cell dysfunction in Rad50+/46 mice. (A) Representative images of testis from mice of the indicated genotypes and average testes weight ± SEM (Rad50+/+, n = 7; Rad50+/46, n = 8; Rad50+/46, Atm+/− n = 5). Bar, 2.5 mm. (B) Representative images of anti-VASA-stained mid-ovary sections from 3-wk-old mice and quantification of the number follicles. Bar, 0.4 mm. Ovaries analyzed: n = 10. P-value was determined by unpaired t-test. (C) Hematoxylin and eosin (H&E), TUNEL, SYCP3, and PLZF immunohistochemical staining of testis sections from adult mice of the indicated genotypes. Bar, 50 μm. (D) Quantification of immunohistochemical staining from C. The TUNEL-positive area is expressed as a ratio to the total testis area. Testis analyzed: Rad50+/+, n = 8; Rad50+/46, n = 10; Rad50+/46 Atm+/−, n = 5. P-values were determined by unpaired t-test.
Immunohistochemical staining for the synaptonemal complex protein SYCP3 revealed that Rad50+/46 mice showed an eightfold reduction in meiotic prophase cells per tubule (Fig. 4C,D). Similar analyses revealed a 23-fold reduction in SYCP3-positive cells from 14-dpp Rad50+/46 mice (Supplemental Fig. S5C,D). TUNEL staining of Rad50+/46 tubules from adult and juvenile mice was unremarkable (Fig. 4C,D; Supplemental Fig. S5C,D), suggesting that the reduction in Rad50+/46 meiotic cells was not due to apoptotic attrition. The reduction in cellularity did not appear to reflect a stage-specific block in meiotic progression, as chromosome spreads from the rare meiotic cells present revealed spermatocytes in all stages of meiosis (Supplemental Fig. S5E).
Spo11 protein initiates meiotic recombination by forming DSBs through a covalent protein–DNA intermediate. Spo11 is cleaved from DSB ends to liberate Spo11–oligonucleotide complexes (Neale et al. 2005) by a process that in yeast requires activity of the Mre11 complex (Neale et al. 2005; Milman et al. 2009). In S. cerevisiae rad50-46 mutants, DSBs are formed, but Spo11 remains covalently bound to genomic DNA (M Hohl, C Tous, T Kochańczyk, A Krężel, A Aguilera, and JHJ Petrini, in prep.). We tested whether a defect in removal of Spo11 from DSB ends accounted for the infertility of Rad50+/46 mice. Spo11–oligonucleotide complexes were nearly absent in adult and juvenile Rad50+/46 mice (Supplemental Fig. S5F). The reduction in Spo11–oligonucleotide complexes in Rad50+/46 testes was not simply a consequence of testicular atrophy because they were evident in Dmc1−/− testes, which, like Rad50+/46, are severely atrophic (Supplemental Fig. S5F). These data suggested that in Rad50+/46 mice, meiotic DSBs are not created, or Spo11 is not released following DSB formation. We could not exclude the possibility that this effect is due to the paucity of meiotic cells in Rad50+/46 testes. To directly address whether defects in the formation or repair of meiotic DSBs were the cause of infertility of Rad50+/46 mice, we generated Rad50+/46 Spo11−/− mice. Seminiferous tubules from Rad50+/46 mice showed reduced cellularity compared with Spo11−/− (Supplemental Fig. S5G). Analysis of sections from Rad50+/46 Spo11−/− mice revealed that Rad50+/46 was epistatic to Spo11−/−. This result indicated that the Rad50+/46 phenotype was independent of Spo11 and thus excluded the possibility of impaired Spo11 removal as a cause of Rad50+/46 infertility.
These data suggest that the Rad50+/46 defect was manifest prior to initiation of the meiotic program. Supporting that interpretation, Rad50+/46 tubules displayed 1.6-fold more cells positive for PLZF (a marker of undifferentiated spermatogonia) (Buaas et al. 2004; Costoya et al. 2004) compared with Rad50+/+ mice (Fig. 4C,D). This expansion of primitive precursors together with the reduction of meiotic cells and the lack of increased cell death was consistent with a block in spermatogonial differentiation, analogizing the block observed in primitive hematopoietic precursors.
Mre11 complex and ATM activation in Rad5046/− cells
Rad5046/ind and Rad50Δ/ind MEFs and ear fibroblast (EF) cultures were established. Lentiviral-mediated delivery of Cre resulted in excision within the Rad50ind allele in 3 d (Supplemental Fig. S6A) to produce Rad50Δ/− and Rad5046/− cells that could be maintained in culture for 7–8 d after Cre infection. This provided a window of time to assess the outcome of limiting cells to Rad5046 as the sole source of Rad50 protein. As shown previously in Rad50Δ/− cell extracts (Adelman et al. 2009), Rad5046/− cells displayed decreased levels of Rad50, Mre11, and Nbs1 (Fig. 5A), suggesting that Mre11 complex proteins were less stable in Rad5046/− cells.
Figure 5.
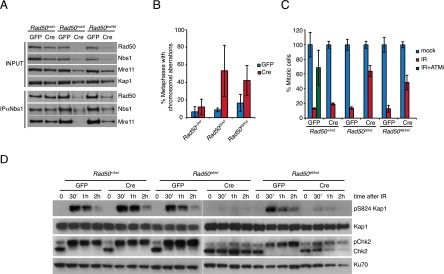
Mre11 complex and ATM activation in Rad5046/− cells. (A) Analysis of Mre11 complex levels in Rad50+/ind, Rad50Δ/ind, and Rad5046/ind SV40-immortalized MEFs 8 d after the lentiviral delivery of Cre or GFP and Nbs1 immunoprecipitation to assess the Mre11 complex integrity in the same cells. Kap1 was used as loading control. (B) Analysis of chromosomal aberrations in Rad50+/ind, Rad50Δ/ind, and Rad5046/ind SV40-immortalized MEFs 8 d after the lentiviral delivery of Cre or GFP. Shown is average ± SD of three independent experiments. Twenty-five or more metaphases were scored per genotype and experiment. (C) Analysis of the G2/M checkpoint in Rad50+/ind, Rad50Δ/ind, and Rad5046/ind SV40-immortalized MEFs 8 d after the lentiviral delivery of Cre or GFP. Mitotic cells were determined in mock-treated cells or 1 h after 5 Gy IR by measuring the phospho-Ser10 Histone H3-positive cells. The ATM inhibitor KU-55933 was added 2 h before IR treatment and kept throughout the experiment. Shown is average mitotic ratio relative to mock-treated ± SD of two independent experiments done in triplicate. (D) Analysis of pS824-Kap1 and Chk2 phosphorylation in Rad50+/ind, Rad50Δ/ind, and Rad5046/ind SV40-immortalized MEFs 8 d after the lentiviral delivery of Cre or GFP after 2 Gy of γ radiation. Time 0 corresponds to untreated cells. Note that Chk2 migrates as a higher-molecular-weight species when phosphorylated (pChk2). Ku70 levels were used as loading control.
Acute disruption of the Mre11 complex results in a dramatic increase in chromosomal aberrations (Demuth et al. 2004; Frappart et al. 2005; Reina-San-Martin et al. 2005; Yang et al. 2006; Adelman et al. 2009). Excision in the Rad50ind allele in Rad5046/ind cells produced a similar outcome, with 53% of Rad50Δ/− and 42% of Rad5046/− cells showing chromosomal aberrations compared with 12% of Rad50+/− (Fig. 5B). Cells with multiple aberrations (three or more) were not seen in Rad50+/− controls but represented up to 30% and 11.8% in Rad50Δ/− and Rad5046/−, respectively (Supplemental Fig. S6B).
Rad50Δ/− MEFs were unable to activate ATM in response to IR, as evidenced by reduced phosphorylation of Kap1 and Chk2 following IR treatment (Fig. 5D). Rad5046/− MEFs were also impaired in ATM activation but to a lesser extent. Whereas phosphorylation of Kap1 and Chk2 was undetectable in Rad50Δ/− MEFs, we could consistently detect it in Rad5046/− MEFs (Fig. 5D). This suggests that Rad5046/− cells retain a degree of Mre11 complex function; however, this observation may reflect the presence of residual wild-type Rad50 protein.
Reduced ATM activation in Rad5046/− cultures was associated with impairment of the DNA damage-dependent G2/M checkpoint. One hour after 5 Gy of IR, cells with at least one copy of wild-type Rad50 showed an 80%–90% reduction in the mitotic cells, indicative of a proficient G2/M checkpoint (Fig. 5C). Excision in the Rad50ind allele to produce Rad50Δ/− or Rad5046/− cells caused defects in the G2/M checkpoint, with cells exhibiting only a 36% and 52% reduction in the proportion of mitotic cells, respectively, after IR (Fig. 5C).
Rad5046 promotes tumorigenesis
The Mre11 complex has been implicated in the suppression of malignancy in several contexts (Stracker and Petrini 2011; Foster et al. 2012; Gupta et al. 2013). Accordingly, cohorts of 30 Rad50+/46, 31 Rad50+/Neo46, and 34 Rad50+/+ mice were followed for 18 mo. Although no signs of morbidity were evident, multiple liver neoplasms were noted upon necropsy. The incidence was 26% in Rad50+/Neo46 mice, 37% in Rad50+/46 mice, and 3% in Rad50+/+ mice (Fig. 6A). Most of the affected mice presented hyperplastic nodules and/or adenomas, but 10% of Rad50+/46 mice developed malignant hepatoblastoma (Fig. 6A,B). Unlike adenomas, which arise from mature hepatocytes, hepatoblastomas arise from hepatic progenitor cells of the portal areas (Marquardt and Thorgeirsson 2010). This indicates that both differentiated hepatocytes and immature progenitors are susceptible to Rad5046-induced malignancy.
Figure 6.

Rad50+/46 and Rad50+/Neo46 mice develop liver tumors. (A) Frequency of liver tumors developed by Rad50+/+, Rad50+/46, and Rad50+/Neo46 mice by 18 mo of age. P-values were calculated with Fisher's exact test comparing with Rad50+/+. (*) P ≤ 0.05; (**) P ≤ 0.01; (***)P ≤ 0.001. (B) Representative images and H&E staining of the hepatic tumors developed by Rad50+/46 and Rad50+/Neo46 mice. Bar, 200 μm for all images.
Discussion
We showed previously that alteration of either invariant cysteine of the S. cerevisiae Rad50 hook domain results in complete inactivation of Rad50, limiting the ability to distinguish hook-specific from general Mre11 complex functions (Hopfner et al. 2002). The residues between the cysteines are also nearly invariant. The first is either Pro (85%) or Tyr (10%), and the second position contains Leu or Val in 80% of the cases (Stracker and Petrini 2011). In this study, the functional significance of the Rad50 hook domain in the mammalian Mre11 complex was examined through the generation of mouse mutants affecting those residues. Neither of the hook mutants encoded gene products that support viability at the organismal or cellular level, indicating that, as in S. cerevisiae, the hook domain is required for Mre11 complex function in mammals. In the heterozygous configuration, we found that Rad5046 and Rad5047 behaved as dominant gain-of-function alleles that promoted chronic DDR signaling. These outcomes at the molecular level were associated with severe phenotypes in vivo, including embryonic lethality, hydrocephalus, defects in the development and maintenance of primitive cells of the bone marrow and germline, and increased incidence of liver neoplasms.
Rad5046 is not a loss-of-function allele
Despite the phenotypic severity observed, the data are inconsistent with Rad5046 being a null allele. First, the phenotypes in Rad5046/− cells are milder than Rad50Δ/−. Although perdurance of wild-type Rad50 protein following Cre-mediated deletion in Rad5046/ind cultures cannot be excluded, Rad5046 may retain limited functionality. Second, Rad5046 exerts dose-dependent dominance; the phenotype of Rad50+/Neo46, in which expression of the mutant gene is lower than in Rad50+/46, was less severe. In contrast, Rad50+/Δ mice and cells are indistinguishable from wild-type (CF Bender, CA Adelman, and JHJ Petrini, unpubl.). Hence, the observed phenotypes do not simply reflect reduced dosage of wild-type Rad50. Finally, genetic ablation of Rad50 in the liver is not associated with detectable pathology (Adelman et al. 2009), whereas liver neoplasia and liver tumors were common in Rad50+/46 mice.
Given that mutations in Rad5046 and Rad5047 affect one of the Mre11 complex's dimerization domains, it is likely that the dominant behavior of these alleles results from interaction of the mutant gene product with the wild-type Rad50 protein. Supporting this hypothesis, we found that Rad50WT and Rad5046 hook domains interact in vitro (Supplemental Fig. S7). In principle, Rad50+/46 cells could contain wild-type homodimers, mutant/wild-type heterodimers, and mutant homodimers. The Rad50+/46 phenotypes observed are likely attributable to heterotypic complexes. Several lines of evidence indicate that alterations of the hook domain exert global effects on Mre11 complex function (Hopfner et al. 2002; Wiltzius et al. 2005; Hohl et al. 2011). The data presented here further suggest that these effects can be exerted in trans within the dimeric assembly of the complex.
Chronic DDR signaling
Rad50+/46 and Rad50+/47 cells exhibit constitutive γH2AX formation. However, Rad50+/46 cells exhibited only a modest reduction in HDR efficiency and a slight sensitivity to camptothecin and did not show increased spontaneous chromosomal aberrations. These facts are inconsistent with spontaneous DNA damage as the cause of γH2AX formation and suggest the possibility that Rad50+/46 behaves as a hypermorph with respect to DNA damage signaling, as previously observed for the Rad50S allele (Usui et al. 2006). Although the underlying mechanisms of Mre11 complex-dependent DDR regulation remain unclear, the increased latency of lymphomas in Rad50+/46 Atm−/− mice supports this interpretation.
The Rad50S and Rad5046 mutations affect domains widely separated yet engender similar DDR signaling phenotypes (Rad50S is the K20M substitution). AFM studies have shown that DNA binding by the globular domain promotes spatial reorganization of the coiled-coil and hook domain (Moreno-Herrero et al. 2005). Conversely, Rad50+/46 and Rad50+/47 mutations as well as mutations that truncate the Rad50 coiled-coil domain affect functions that are likely specified within the globular domain (Hohl et al. 2011). These data illustrate the functional interdependence of the hook and globular domains within the Mre11 complex.
The Rad50+/46 phenotype and progenitor cells
Consistent with the interpretation that the Rad5046 allele chronically activates ATM, the phenotypic outcomes in Rad50+/46 mice are dependent on Atm. Again, this situation is comparable with Rad50S/S mice, and the tissues primarily affected are the same: primitive cells in the hematopoietic and germline lineages. These observations indicate that the Mre11–ATM-dependent signaling pathway is important for the maintenance and function of primitive cells in the bone marrow and germline (Morales et al. 2008; Takubo et al. 2008; Maryanovich et al. 2012). The Mre11 complex–ATM signaling axis likely affects progenitor cells in other organs systems (Shiloh and Ziv 2013). For example, hyperpigmentation, common to both Rad50S/S and Rad50+/46, has also been observed in mice with chronic DDR signaling in the skin due to telomere dysfunction (Hockemeyer et al. 2008; He et al. 2009; Martinez et al. 2010) or DNA repair defects (Zhang et al. 2011). In those instances, melanocyte expansion appears to occur in response to epidermal stem cell depletion (Stout and Blasco 2009).
Tumorigenesis in Rad50+/46 mice
Roughly a quarter (26.7%) of Rad50+/46 mice developed hepatocellular adenomas that were frequently coincident with additional preneoplastic lesions, primarily hyperplastic nodules and areas of disorganized architecture. The Mre11 complex–ATM pathway has been previously implicated in liver cancer, as Atm is required for diethylnitrosamine-induced liver carcinogenesis (Teoh et al. 2010). Notably, almost one-third of the tumor-bearing mice developed hepatoblastoma, a rare tumor that in humans primarily affects infants and children. The blastic and undifferentiated character of this tumor suggests that it arises from hepatic progenitor cells of the portal areas (Marquardt and Thorgeirsson 2010). This outcome represents an additional instance in which Mre11 complex–ATM signaling affects homeostasis by affecting progenitor cells.
Mre11 complex mutations have been identified in ∼4% of human tumors (data from cBioPortal; Cerami et al. 2012). The observation here of dominant Rad50 mutations promoting tumorigenesis supports the possibility that heterozygous Mre11 complex mutations may underlie tumor development, a possibility that should be considered in the genomic analysis of tumors.
Materials and methods
Rad50-HK expression, purification, and analysis
6xHis-SMT3-tagged Rad50-HK proteins (amino acids 591–773) were expressed in Escherichia coli. Soluble extracts were prepared in HK buffer (200 mM NaCl, 50 mM Tris at pH 7.5, 0.2% NP-40, 10% glycerol, 0.5 mM DTT) plus protease and phosphatase inhibitors and was purified by nickel-chelating affinity chromatography followed by Ulp1 protease treatment and a second nickel-chelating affinity purification step to separate the 6xHis-SMT3 tag. Rad50-HK proteins were analyzed by SEC using a 24-mL Superdex200 column equilibrated with 20 mM Tris (pH 7.6), 500 mM NaCl, 0.1 mM EDTA, and 2 mM DTT.
SEC-MALS was performed as described (Rambo and Tainer 2010) with a size exclusion column equilibrated with 20 mM Tris (pH 7.6), 500 mM NaCl, 0.1 mM EDTA, and 1 mM tris(2-carboxyethyl)phosphine. MALS experiments were performed using an 18-angle DAWN HELEOS light-scattering detector connected in tandem to an Optilab refractive index concentration detector (Wyatt Technology). Metal concentrations were measured using a quadrupole-based ICP-MS (7500ce, Agilent Technologies) equipped with a MicroMist nebulizer (Agilent Technologies).
Gene targeting and mice
The targeting vectors were constructed using the recombineering method previously described (Liu et al. 2003). Targeting was carried out via electroporation into the embryonic stem cell AC1 that had a 129/SvEv-background (gift of G. Oltz and E. Rhuley). Selection, clonal expansion, and analysis were performed using previously published procedures (Luo et al. 1999). Chimeras were generated at the SKI Transgenic Facility using standard methods, and male chimeras were crossed with C57BL6 mice to detect germline transmission. Rad50+/Neo46 mice were mated with CAG-Cre transgenic mice (Sakai and Miyazaki 1997) to generate the Rad5046 allele. All mice were maintained on mixed 129/SvEv and C57BL6 background.
Mice were housed in ventilated rack caging in a pathogen-free facility. The Institutional Animal Care and Use Committee of Memorial Sloan-Kettering Cancer Center approved animal use protocols. Atm−/− and Spo11−/− mice were previously described (Barlow et al. 1996; Baudat et al. 2000).
Cellular assays
MEFs and EFs were generated, cultured, and immortalized as described (Bender et al. 2002). Rad50+/47 EFs were generated from Rad50+/Neo47 chimeric mice by growing the cells in medium containing 1 mg/mL G418 followed by lentiviral delivery of Cre and clonal selection.
Western blots were carried out on 40 μg of protein extracted with NTEN (20 mM Tris at pH 8, 150 mM NaCl, 1 mM EDTA, 0.5% NP-40) plus protease and phosphatase inhibitors. For H2AX blots, we used 3 μg of the histone fraction extracted with 0.1 M HCl after a pre-extraction with NTEN. All of the antibodies were incubated overnight at 4°C in 5% milk. The antibodies used were rabbit anti-Rad50 polyclonal (custom Petrini laboratory antibody m84-7), rabbit anti-Nbs1 polyclonal (custom Petrini laboratory antibody 92-5), rabbit anti-Mre11 polyclonal (custom Petrini laboratory antibody 42-5), ATM (Cell Signaling), ATM pS1981 (Cell Signaling), γH2AX (Millipore), H2AX (Bethyl Laboratories), Kap1 pS824 (Abcam), Kap1 (Novus Biologicals), Chk2 (Clone 7, Millipore), and Ku70 (Santa Cruz Biotechnology).
For Nbs1 immunoprecipitation, cell extracts of SV40-immortalized MEFs were prepared with lysis buffer A (300 mM NaCl, 20 mM Tris at pH 8.0, 0.5% sodium deoxicholate, 1% Triton X-100, 5 mM MgCl2 supplemented with protease and phosphatase inhibitors). Total protein (0.5 mg) was incubated with 2 μL of anti-Nbs1 antisera and protein A-agarose beads (Amersham) for 3 h at 4°C, washed extensively, and eluted with Laemmli's buffer.
For colony-forming assays, cells were treated with the indicated clastogens and plated in triplicate, and colonies were assessed 10 d later by staining with crystal violet.
Metaphases were prepared from cultures treated with 2 × 10−7 M colcemid for 1 h. Cells were harvested, hypotonically swelled with 0.075 M KCl for 15 min at 37°C, fixed, washed in ice-cold 3:1 methanol:acetic acid, and dropped on slides. Slides were stained with 5% Giemsa (Sigma) for 10 min and rinsed with distilled water, and coverslips were mounted with Permount (Fisher). Images were captured using an Olympus IX60 microscope and imaged with a Hammamatsu CCD camera. More than 25 spreads were scored for each sample.
G2/M checkpoint was done as previously described (Theunissen and Petrini 2006).
Lentiviral production, concentration, and titering were carried out using previously described methods (Dull et al. 1998; Lois et al. 2002). For lentivirus-Cre and lentivirus-GFP infections, 5 × 104 cells were resuspended in 3 mL of DMEM supplemented with 10% CCS containing 5 μg/mL polybrene and lentivirus at a multiplicity of infection (MOI) 50. Twenty-four hours after infection, fresh medium was added, and cells were trypsinized and replated every 3 d to avoid confluency.
DR-GFP assay
Pim1DR-GFP/DR-GFP mice (Kass et al. 2013) were crossed with Rad50+/46, and heterozygous Pim1+/DR-GFP Rad50+/46 and Pim1+/DR-GFP Rad50+/+ primary skin fibroblast cultures were established. To deliver I-SceI and monitor only the I-SceI-expressing cells, we inserted and IRES-hCD4 cassette into the retroviral vector pMX-ISceI (gift from Fred Alt). I-SceI-hCD4-expressing retrovirus produced in Phoenix cells were used to infect primary cultures in the presence of 5 μg/mL polybrene for 24 h. Seventy-two hours after the infection, cells were harvested, stained with anti-CD4 PE-Cy5 (BD-Pharmingen), and analyzed by flow cytometry. The number of GFP-positive cells was expressed as a ratio to the CD4-positive (infected) cells.
Hematopoietic lineage analysis
Hematopoietic lineages were determined as previously described (Brenet et al. 2013). Briefly, bone marrow single-cell suspensions prepared from both back legs were depleted of lineage-positive cells using a lineage cell depletion kit (Miltenyi Biotec) and simultaneously stained with different antibodies. Hematopoietic populations were identified as follows: long-term HSCs, LKS+ CD34−Flk2−; short-term HSCs, LKS+ CD34+ Flk2−; MPPs, LKS+ CD34+ Flk2+; CMPs, LKS− CD34+ FcRlow; granulocyte macrophage progenitors, LKS− CD34+ FcR+; and megakaryocyte erythroid progenitors, LKS− CD34− FcR−. The CLP population was defined as LKS+ Flk2+, IL7Rα+. DAPI was used to exclude dead cells during flow cytometric analysis.
MPPs were enumerated using the day 13 CFU-S assay. In brief, recipient mice were lethally irradiated and injected with bone marrow cells. Spleens were isolated 13 d after transplantation and fixed in Bouin's solution, and macroscopic colonies were counted.
Histological sample preparation, staining, and analysis
Tissue samples for histological analyses were fixed overnight at 4°C with 4% paraformaldehyde, rinsed, stored at 4°C in 70% ethanol, and then processed for paraffin embedding. Eight-micrometer sections were prepared, and slides were processed and stained at the Memorial Sloan-Kettering molecular cytogenetics core facility for hematoxylin and eosin (H&E), TUNEL, anti-VASA, anti-SYCP3, and anti-PLZF.
H&E-stained and immunohistochemically stained slides were digitally scanned using a Mirax scanner. TUNEL staining was quantified relative to the total area using standardized color filter settings in the Metamorph image analysis software. Anti-PLZF- and anti-SYCP3-positive cells per tubule were manually scored from at least 25 tubules per testis.
Acknowledgments
We thank Maria Jasin for providing Pim1DR-GFP mice; Fred Alt for the pMX-ISceI plasmid; Linda Johnson and Julie White from the Center for Comparative Medicine and Pathology for assistance with pathological analysis; David Klimstra for help with liver pathology; members of the J.H.J.P. laboratory for critical reading of the manuscript, discussions, and helpful insight; Katelynd Vanness for technical assistance; and Thomas J. Kelly for critical reading of the manuscript. This work was supported by the following grants: RO1-GM56888 (to J.H.J.P.); National Cancer Institute CA159175 (to J.M.S.); National Heart, Lung, and Blood Institute (NHLBI) HL119872 (to J.M.S.); NHLBI HL055748 (to J.M.S.); P01 CA092584 (to J.A.T.); R01 CA117638 (to J.A.T); and R01 GM105421 (to Maria Jasin and S.K.). R.R. was supported in part by Fundación Alfonso Martín Escudero. J.L.V. is recipient of a fellowship from Le Fonds de la Recherche en Santé du Québec. J.M.S. is a Leukemia and Lymphoma Society Scholar. J.L. was supported in part by an American Cancer Society post-doctoral fellowship.
Footnotes
Supplemental material is available for this article.
Article published online ahead of print. Article and publication date are online at http://www.genesdev.org/cgi/doi/10.1101/gad.236745.113.
References
- Adelman CA, De S, Petrini JH 2009. Rad50 is dispensable for the maintenance and viability of postmitotic tissues. Mol Cell Biol 29: 483–492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barlow C, Hirotsune S, Paylor R, Liyanage M, Eckhaus M, Collins F, Shiloh Y, Crawley JN, Ried T, Tagle D, et al. 1996. Atm-deficient mice: A paradigm of ataxia telangiectasia. Cell 86: 159–171 [DOI] [PubMed] [Google Scholar]
- Baudat F, Manova K, Yuen JP, Jasin M, Keeney S 2000. Chromosome synapsis defects and sexually dimorphic meiotic progression in mice lacking Spo11. Mol Cell 6: 989–998 [DOI] [PubMed] [Google Scholar]
- Bender CF, Sikes ML, Sullivan R, Huye LE, Le Beau MM, Roth DB, Mirzoeva OK, Oltz EM, Petrini JH 2002. Cancer predisposition and hematopoietic failure in Rad50(S/S) mice. Genes Dev 16: 2237–2251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenet F, Kermani P, Spektor R, Rafii S, Scandura JM 2013. TGFβ restores hematopoietic homeostasis after myelosuppressive chemotherapy. J Exp Med 210: 623–639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bressan DA, Baxter BK, Petrini JH 1999. The Mre11-Rad50-Xrs2 protein complex facilitates homologous recombination-based double-strand break repair in Saccharomyces cerevisiae. Mol Cell Biol 19: 7681–7687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buaas FW, Kirsh AL, Sharma M, McLean DJ, Morris JL, Griswold MD, de Rooij DG, Braun RE 2004. Plzf is required in adult male germ cells for stem cell self-renewal. Nat Genet 36: 647–652 [DOI] [PubMed] [Google Scholar]
- Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E et al. 2012. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer discovery 2: 401–404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costoya JA, Hobbs RM, Barna M, Cattoretti G, Manova K, Sukhwani M, Orwig KE, Wolgemuth DJ, Pandolfi PP 2004. Essential role of Plzf in maintenance of spermatogonial stem cells. Nat Genet 36: 653–659 [DOI] [PubMed] [Google Scholar]
- de Jager M, van Noort J, van Gent DC, Dekker C, Kanaar R, Wyman C 2001. Human Rad50/Mre11 is a flexible complex that can tether DNA ends. Mol Cell 8: 1129–1135 [DOI] [PubMed] [Google Scholar]
- de Jager M, Trujillo KM, Sung P, Hopfner KP, Carney JP, Tainer JA, Connelly JC, Leach DR, Kanaar R, Wyman C 2004. Differential arrangements of conserved building blocks among homologs of the Rad50/Mre11 DNA repair protein complex. J Mol Biol 339: 937–949 [DOI] [PubMed] [Google Scholar]
- Demuth I, Frappart PO, Hildebrand G, Melchers A, Lobitz S, Stockl L, Varon R, Herceg Z, Sperling K, Wang ZQ, et al. 2004. An inducible null mutant murine model of Nijmegen breakage syndrome proves the essential function of NBS1 in chromosomal stability and cell viability. Hum Mol Genet 13: 2385–2397 [DOI] [PubMed] [Google Scholar]
- Dull T, Zufferey R, Kelly M, Mandel RJ, Nguyen M, Trono D, Naldini L 1998. A third-generation lentivirus vector with a conditional packaging system. J Virol 72: 8463–8471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duursma AM, Driscoll R, Elias JE, Cimprich KA 2013. A role for the MRN complex in ATR activation via TOPBP1 recruitment. Mol Cell 50: 116–122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster SS, De S, Johnson LK, Petrini JH, Stracker TH 2012. Cell cycle- and DNA repair pathway-specific effects of apoptosis on tumor suppression. Proc Natl Acad Sci 109: 9953–9958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frappart PO, Tong WM, Demuth I, Radovanovic I, Herceg Z, Aguzzi A, Digweed M, Wang ZQ 2005. An essential function for NBS1 in the prevention of ataxia and cerebellar defects. Nat Med 11: 538–544 [DOI] [PubMed] [Google Scholar]
- Gonzalez-Barrera S, Cortes-Ledesma F, Wellinger RE, Aguilera A 2003. Equal sister chromatid exchange is a major mechanism of double-strand break repair in yeast. Mol Cell 11: 1661–1671 [DOI] [PubMed] [Google Scholar]
- Gupta GP, Vanness K, Barlas A, Manova-Todorova KO, Wen YH, Petrini JH 2013. The Mre11 complex suppresses oncogene-driven breast tumorigenesis and metastasis. Mol Cell 52: 353–365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartsuiker E, Vaessen E, Carr AM, Kohli J 2001. Fission yeast Rad50 stimulates sister chromatid recombination and links cohesion with repair. EMBO J 20: 6660–6671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He H, Wang Y, Guo X, Ramchandani S, Ma J, Shen MF, Garcia DA, Deng Y, Multani AS, You MJ, et al. 2009. Pot1b deletion and telomerase haploinsufficiency in mice initiate an ATR-dependent DNA damage response and elicit phenotypes resembling dyskeratosis congenita. Mol Cell Biol 29: 229–240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He J, Shi LZ, Truong LN, Lu CS, Razavian N, Li Y, Negrete A, Shiloach J, Berns MW, Wu X 2012. Rad50 zinc hook is important for the Mre11 complex to bind chromosomal DNA double-stranded breaks and initiate various DNA damage responses. J Biol Chem 287: 31747–31756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hockemeyer D, Palm W, Wang RC, Couto SS, de Lange T 2008. Engineered telomere degradation models dyskeratosis congenita. Genes Dev 22: 1773–1785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hohl M, Kwon Y, Galvan SM, Xue X, Tous C, Aguilera A, Sung P, Petrini JH 2011. The Rad50 coiled-coil domain is indispensable for Mre11 complex functions. Nat Struct Mol Biol 18: 1124–1131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopfner KP, Craig L, Moncalian G, Zinkel RA, Usui T, Owen BA, Karcher A, Henderson B, Bodmer JL, McMurray CT, et al. 2002. The Rad50 zinc-hook is a structure joining Mre11 complexes in DNA recombination and repair. Nature 418: 562–566 [DOI] [PubMed] [Google Scholar]
- Kass EM, Helgadottir HR, Chen CC, Barbera M, Wang R, Westermark UK, Ludwig T, Moynahan ME, Jasin M 2013. Double-strand break repair by homologous recombination in primary mouse somatic cells requires BRCA1 but not the ATM kinase. Proc Natl Acad Sci 110: 5564–5569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lammens K, Bemeleit DJ, Mockel C, Clausing E, Schele A, Hartung S, Schiller CB, Lucas M, Angermuller C, Soding J, et al. 2011. The Mre11:Rad50 structure shows an ATP-dependent molecular clamp in DNA double-strand break repair. Cell 145: 54–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim HS, Kim JS, Park YB, Gwon GH, Cho Y 2011. Crystal structure of the Mre11–Rad50–ATPγS complex: Understanding the interplay between Mre11 and Rad50. Genes Dev 25: 1091–1104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu P, Jenkins NA, Copeland NG 2003. A highly efficient recombineering-based method for generating conditional knockout mutations. Genome Res 13: 476–484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lois C, Hong EJ, Pease S, Brown EJ, Baltimore D 2002. Germline transmission and tissue-specific expression of transgenes delivered by lentiviral vectors. Science 295: 868–872 [DOI] [PubMed] [Google Scholar]
- Luo G, Yao MS, Bender CF, Mills M, Bladl AR, Bradley A, Petrini JH 1999. Disruption of mRad50 causes embryonic stem cell lethality, abnormal embryonic development, and sensitivity to ionizing radiation. Proc Natl Acad Sci 96: 7376–7381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marquardt JU, Thorgeirsson SS 2010. Stem cells in hepatocarcinogenesis: Evidence from genomic data. Semin Liver Dis 30: 26–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez P, Thanasoula M, Carlos AR, Gomez-Lopez G, Tejera AM, Schoeftner S, Dominguez O, Pisano DG, Tarsounas M, Blasco MA 2010. Mammalian Rap1 controls telomere function and gene expression through binding to telomeric and extratelomeric sites. Nat Cell Biol 12: 768–780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maryanovich M, Oberkovitz G, Niv H, Vorobiyov L, Zaltsman Y, Brenner O, Lapidot T, Jung S, Gross A 2012. The ATM-BID pathway regulates quiescence and survival of haematopoietic stem cells. Nat Cell Biol 14: 535–541 [DOI] [PubMed] [Google Scholar]
- Milman N, Higuchi E, Smith GR 2009. Meiotic DNA double-strand break repair requires two nucleases, MRN and Ctp1, to produce a single size class of Rec12 (Spo11)-oligonucleotide complexes. Mol Cell Biol 29: 5998–6005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morales M, Theunissen JW, Kim CF, Kitagawa R, Kastan MB, Petrini JH 2005. The Rad50S allele promotes ATM-dependent DNA damage responses and suppresses ATM deficiency: Implications for the Mre11 complex as a DNA damage sensor. Genes Dev 19: 3043–3054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morales M, Liu Y, Laiakis EC, Morgan WF, Nimer SD, Petrini JH 2008. DNA damage signaling in hematopoietic cells: A role for Mre11 complex repair of topoisomerase lesions. Cancer Res 68: 2186–2193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno-Herrero F, de Jager M, Dekker NH, Kanaar R, Wyman C, Dekker C 2005. Mesoscale conformational changes in the DNA-repair complex Rad50/Mre11/Nbs1 upon binding DNA. Nature 437: 440–443 [DOI] [PubMed] [Google Scholar]
- Neale MJ, Pan J, Keeney S 2005. Endonucleolytic processing of covalent protein-linked DNA double-strand breaks. Nature 436: 1053–1057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rambo RP, Tainer JA 2010. Improving small-angle X-ray scattering data for structural analyses of the RNA world. RNA 16: 638–646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reina-San-Martin B, Nussenzweig MC, Nussenzweig A, Difilippantonio S 2005. Genomic instability, endoreduplication, and diminished Ig class-switch recombination in B cells lacking Nbs1. Proc Natl Acad Sci 102: 1590–1595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakai K, Miyazaki J 1997. A transgenic mouse line that retains Cre recombinase activity in mature oocytes irrespective of the cre transgene transmission. Biochem Biophys Res Commun 237: 318–324 [DOI] [PubMed] [Google Scholar]
- Shiloh Y, Ziv Y 2013. The ATM protein kinase: Regulating the cellular response to genotoxic stress, and more. Nat Rev Mol Cell Biol 14: 197–210 [PubMed] [Google Scholar]
- Shiotani B, Nguyen HD, Hakansson P, Marechal A, Tse A, Tahara H, Zou L 2013. Two distinct modes of ATR activation orchestrated by Rad17 and Nbs1. Cell reports 3: 1651–1662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spangrude GJ, Heimfeld S, Weissman IL 1988. Purification and characterization of mouse hematopoietic stem cells. Science 241: 58–62 [DOI] [PubMed] [Google Scholar]
- Stiff T, Reis C, Alderton GK, Woodbine L, O'Driscoll M, Jeggo PA 2005. Nbs1 is required for ATR-dependent phosphorylation events. EMBO J 24: 199–208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stout GJ, Blasco MA 2009. Genetic dissection of the mechanisms underlying telomere-associated diseases: Impact of the TRF2 telomeric protein on mouse epidermal stem cells. Dis Model Mech 2: 139–156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stracker TH, Petrini JH 2011. The MRE11 complex: Starting from the ends. Nat Rev Mol Cell Biol 12: 90–103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takubo K, Ohmura M, Azuma M, Nagamatsu G, Yamada W, Arai F, Hirao A, Suda T 2008. Stem cell defects in ATM-deficient undifferentiated spermatogonia through DNA damage-induced cell-cycle arrest. Cell Stem Cell 2: 170–182 [DOI] [PubMed] [Google Scholar]
- Teoh N, Pyakurel P, Dan YY, Swisshelm K, Hou J, Mitchell C, Fausto N, Gu Y, Farrell G 2010. Induction of p53 renders ATM-deficient mice refractory to hepatocarcinogenesis. Gastroenterology 138: 1155–1165.e2 [DOI] [PubMed] [Google Scholar]
- Theunissen JW, Petrini JH 2006. Methods for studying the cellular response to DNA damage: Influence of the Mre11 complex on chromosome metabolism. Methods Enzymol 409: 251–284 [DOI] [PubMed] [Google Scholar]
- Theunissen JW, Kaplan MI, Hunt PA, Williams BR, Ferguson DO, Alt FW, Petrini JH 2003. Checkpoint failure and chromosomal instability without lymphomagenesis in Mre11(ATLD1/ATLD1) mice. Mol Cell 12: 1511–1523 [DOI] [PubMed] [Google Scholar]
- Usui T, Petrini JH, Morales M 2006. Rad50S alleles of the Mre11 complex: Questions answered and questions raised. Exp Cell Res 312: 2694–2699 [DOI] [PubMed] [Google Scholar]
- van Noort J, van Der Heijden T, de Jager M, Wyman C, Kanaar R, Dekker C 2003. The coiled-coil of the human Rad50 DNA repair protein contains specific segments of increased flexibility. Proc Natl Acad Sci 100: 7581–7586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ventura A, Kirsch DG, McLaughlin ME, Tuveson DA, Grimm J, Lintault L, Newman J, Reczek EE, Weissleder R, Jacks T 2007. Restoration of p53 function leads to tumour regression in vivo. Nature 445: 661–665 [DOI] [PubMed] [Google Scholar]
- Williams RS, Moncalian G, Williams JS, Yamada Y, Limbo O, Shin DS, Groocock LM, Cahill D, Hitomi C, Guenther G, et al. 2008. Mre11 dimers coordinate DNA end bridging and nuclease processing in double-strand-break repair. Cell 135: 97–109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams RS, Dodson GE, Limbo O, Yamada Y, Williams JS, Guenther G, Classen S, Glover JN, Iwasaki H, Russell P, et al. 2009. Nbs1 flexibly tethers Ctp1 and Mre11-Rad50 to coordinate DNA double-strand break processing and repair. Cell 139: 87–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiltzius JJ, Hohl M, Fleming JC, Petrini JH 2005. The Rad50 hook domain is a critical determinant of Mre11 complex functions. Nat Struct Mol Biol 12: 403–407 [DOI] [PubMed] [Google Scholar]
- Yang YG, Saidi A, Frappart PO, Min W, Barrucand C, Dumon-Jones V, Michelon J, Herceg Z, Wang ZQ 2006. Conditional deletion of Nbs1 in murine cells reveals its role in branching repair pathways of DNA double-strand breaks. EMBO J 25: 5527–5538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, Yajima H, Huynh H, Zheng J, Callen E, Chen HT, Wong N, Bunting S, Lin YF, Li M, et al. 2011. Congenital bone marrow failure in DNA-PKcs mutant mice associated with deficiencies in DNA repair. J Cell Biol 193: 295–305 [DOI] [PMC free article] [PubMed] [Google Scholar]