

Many loci maintain parent-of-origin DNA methylation only briefly after fertilization during mammalian development. Whether this form of transient genomic imprinting could functionally impact mammalian genome regulation is unknown. Here, Duffié et al. describe a mechanism by which a transient maternal imprint at the mouse Gpr1/Zdbf2 locus regulates the paternal-specific expression of long isoforms of Zdbf2 (Liz) in the early embryo. This study demonstrates the potential for short-term and long-term effects of genomic imprinting on mammalian genome regulation.
Keywords: genomic imprinting, DNA methylation, epigenetics, mouse development
Abstract
Many loci maintain parent-of-origin DNA methylation only briefly after fertilization during mammalian development: Whether this form of transient genomic imprinting can impact the early embryonic transcriptome or even have life-long consequences on genome regulation and possibly phenotypes is currently unknown. Here, we report a maternal germline differentially methylated region (DMR) at the mouse Gpr1/Zdbf2 (DBF-type zinc finger-containing protein 2) locus, which controls the paternal-specific expression of long isoforms of Zdbf2 (Liz) in the early embryo. This DMR loses parental specificity by gain of DNA methylation at implantation in the embryo but is maintained in extraembryonic tissues. As a consequence of this transient, tissue-specific maternal imprinting, Liz expression is restricted to the pluripotent embryo, extraembryonic tissues, and pluripotent male germ cells. We found that Liz potentially functions as both Zdbf2-coding RNA and cis-regulatory RNA. Importantly, Liz-mediated events allow a switch from maternal to paternal imprinted DNA methylation and from Liz to canonical Zdbf2 promoter use during embryonic differentiation, which are stably maintained through somatic life and conserved in humans. The Gpr1/Zdbf2 locus lacks classical imprinting histone modifications, but analysis of mutant embryonic stem cells reveals fine-tuned regulation of Zdbf2 dosage through DNA and H3K27 methylation interplay. Together, our work underlines the developmental and evolutionary need to ensure proper Liz/Zdbf2 dosage as a driving force for dynamic genomic imprinting at the Gpr1/Zdbf2 locus.
The process of germline differentiation produces mature gametes with sexually dimorphic morphologies, functions, and epigenomes. In mammals, the sperm and oocyte genomes display notably different DNA methylation density and distribution (Smallwood et al. 2011; Kobayashi et al. 2012a; Smith et al. 2012) despite reliance on the same enzymatic machinery formed by the de novo DNA methyltransferase Dnmt3A and its cofactor, Dnmt3L (Bourc'his et al. 2001; Bourc'his and Bestor 2004; Kaneda et al. 2004). Intergenic regions and transposable repeats are densely methylated in sperm, while DNA methylation preferentially occurs inside genes in the oocyte, with deeper density at CpG islands (CGIs) coinciding with intragenic promoters. Genetic and genome-wide studies have suggested a role for active transcription in oocyte DNA methylation whereby transcription promotes DNA methylation deposition in gene bodies (Chotalia et al. 2009; Kobayashi et al. 2012a). In sperm, small RNAs are instead responsible for targeting methylation to transposons (Aravin and Bourc'his 2008).
Given the heritable nature of DNA methylation, gametic sex-specific patterns are transmitted upon fertilization; this creates tremendous opportunity for parental asymmetry in the progeny (Duffié and Bourc'his 2013). However, as part of the early embryonic program, sequential events of DNA demethylation and remethylation remodel most gametically inherited methylation. Only a handful of regions endowed with favorable nucleotide sequence and chromatin environments are capable of maintaining parent-of-origin DNA methylation after fertilization. These include regions subject to genomic imprinting. Around 20 imprinting control regions (ICRs) have been identified to date, and these dictate the parent-specific monoallelic expression of ∼130 genes in cis (Schulz et al. 2008).
All known ICRs harbor binding motifs for ZFP57, a zinc finger protein that recruits the KRAB-associated protein 1 (KAP1)-dependent heterochromatin complex (Schultz et al. 2002; Quenneville et al. 2011). This property confers the ability of ICRs to maintain methylation on one parental allele during the genome-wide demethylation that accompanies preimplantation development (Li et al. 2008; Quenneville et al. 2011; Messerschmidt et al. 2012; Zuo et al. 2012). What protects the unmethylated allele of ICRs from global de novo methylation after implantation has not been determined precisely; occupancy by trans-acting factors and/or features linked to active transcription such as permissive histone modifications and R-loop structures are likely to be involved (Ooi et al. 2007; Lienert et al. 2011; Ginno et al. 2012). Once these critical periods of intense DNA methylation remodeling have reached completion, canonical ICRs perpetuate as parental differentially methylated regions (DMRs) in a life-long manner.
We recently uncovered new forms of genomic imprinting, which exist only transiently during preimplantation development or specifically in certain tissues during adulthood (Proudhon et al. 2012). The underlying regions all have ZFP57-binding motifs and are protected against DNA demethylation after fertilization. However, they are susceptible to processes of de novo methylation that occur in the post-implantation embryo or during tissue differentiation and consequently lose imprinting by gain of DNA methylation. Transient genomic imprinting is intriguing: Whether short-term differential methylation is only a byproduct of the divergent sperm and oocyte methylomes or could functionally impact mammalian genome regulation is currently unknown.
Here we report the case of a region of transient germline DMRs (gDMRs) inherited from the oocyte at the mouse Gpr1/Zdbf2 (DBF-type zinc finger-containing protein 2) locus (1qC2) (Kobayashi et al. 2009). We found that this maternal gDMR coincides with a promoter, which initiates transcription of long isoforms of Zdbf2 (Liz) in a paternal-specific manner but is shut down upon DNA methylation gain after implantation. Importantly, Liz transient expression is associated with acquisition of a paternal somatic DMR (sDMR) and paternal-specific transcription of Zdbf2 from its canonical promoter, both of which are stably maintained for the rest of life. We further reveal the ubiquitous availability of Liz/Zdbf2 transcripts throughout development, the fine-tuned regulation of Zdbf2 promoter activity by interplay between DNA methylation and histone modifications, and the conservation of GPR1/ZDBF2 gene regulation in humans. In conclusion, we demonstrate for the first time the potential for short-term and long-term effects of transient genomic imprinting on mammalian genome regulation.
Results
Two gDMRs at the Gpr1/Zdbf2 locus exhibit dynamic allele-specific methylation during development
The Gpr1/Zdbf2 locus was first described to be paternally imprinted, with three intergenic paternally methylated DMRs (DMR1, DMR2, and DMR3) that are located between 8.5 and 16 kb upstream of the Zdbf2 transcription start site (TSS) and can be considered as a single entity (Fig. 1A; Hiura et al. 2010). However, recent work, including our own, suggested that this locus may be primarily under maternal imprinting control (Kobayashi et al. 2012b; Proudhon et al. 2012). Accordingly, we found that the originally described paternal DMR lacks important hallmarks of all ICRs identified so far: TGCCGC motifs for ZFP57 binding and local ZFP57/KAP1 enrichment, as seen in available embryonic stem (ES) cell chromatin immunoprecipitation (ChIP) sequencing (ChIP-seq) data (Quenneville et al. 2011). Instead, ZFP57/KAP1 enrichment was found at a CGI in the second intron of the closest neighboring gene, Gpr1 (G protein-coupled receptor 1), and localized ∼65 kb upstream of the paternal DMR (Supplemental Fig. S1A). An in silico search allowed us to uncover three ZFP57 recognition motifs at this intragenic CGI.
Figure 1.

DNA methylation profiling at two parental DMRs of the Gpr1/Zdbf2 locus during mouse development. (A) Mouse Gpr1/Zdbf2 locus indicating CGIs (green bars) and positions of maternal DMRs (red bar) and tripartite paternal DMRs (blue bars) (DMR1, DMR2, DMR3 from left to right). DNA methylation was analyzed by bisulfite cloning/sequencing in gametes and embryos of wild-type (WT) and maternal methylation-free Dnmt3L−/+ background (A), adult somatic tissues (B), and extraembryonic tissues (C). The maternal DMR is a transient gDMR: It acquires methylation in oocytes, maintains maternal-specific DNA methylation during preimplantation development, and is lost by methylation gain after implantation, except in extraembryonic tissues. Of note, CpGs in positions 2, 5, and 7 of the amplified maternal gDMR amplicon are embedded into hexanucleotidic ZFP57-binding motifs. The paternal DMR is methylated in sperm and lost in the blastocyst but is re-established at implantation and provides paternal-specific information in embryonic and adult somatic tissues, qualifying as both gDMR and sDMR (g/sDMR). Results for paternal DMR3 are depicted here; similar patterns were obtained with DMR2 (data not shown). (Red lines) Maternal alleles; (blue lines) paternal alleles; (white circles) unmethylated CpG; (black circles) methylated CpG; (dash) absent CpG due to SNP or, rarely, sequencing error. Mouse strains used were as follows: C57Bl6/J (designated B) and CAST/Ei (designated C).
To resolve the parental origin of imprinting control at the Gpr1/Zdbf2 locus, we analyzed DNA methylation by bisulfite cloning/sequencing through mouse development. Crosses between the C57Bl6/J (B) and CAST/Ei (C) mouse strains permitted parental distinction based on the presence of single nucleotide polymorphisms (SNPs). Investigation of Dnmt3L−/+ embryos derived from DNA methylation-free Dnmt3L−/− oocytes further allowed us to infer the role of maternally inherited DNA methylation (Bourc'his et al. 2001). Reciprocal gDMRs were identified, with the paternal DMR harboring sperm-specific methylation (paternal gDMR) and the ZFP57/KAP1 site harboring oocyte-specific methylation (maternal gDMR) (Fig. 1A; Supplemental Fig. S1B). Of note, the maternal gDMR bisulfite amplicon includes the three ZFP57 sequence motifs, which are conserved among our different mouse strains. In embryonic day 3.5 (E3.5) blastocysts, DNA methylation remained at maternal alleles of the maternal gDMR, likely due to protection by the ZFP57/KAP1 complex. In contrast, the paternal gDMR underwent demethylation and loss of parent specificity.
After implantation, the two parental gDMRs were subjected to de novo DNA methylation. At the maternal gDMR, gain of methylation on the paternal allele abolished maternal specificity by E9.5 (Fig. 1A; Supplemental Fig. S1B). Full biallelic methylation was consistently maintained in all somatic tissues examined (fibroblasts, kidney, muscle, cortex, pituitary gland, and liver) (Fig. 1B; data not shown). This classifies this locus as a transient maternal gDMR, only present during preimplantation development (Proudhon et al. 2012). As previously observed for transient gDMRs, de novo methylation occurred indiscriminately on both parental alleles in Dnmt3L−/+ embryos, which lacked methylation at the maternal gDMR before implantation (Fig. 1A; Supplemental Fig. S1B; Proudhon et al. 2012).
Remarkably, from a totally methylation-free status, the paternal DMR returned to paternal-specific methylation after implantation (Fig. 1A; Supplemental Fig. S1B). This classifies this paternal DMR as both a gDMR established in sperm and a sDMR established post-fertilization. Paternal-specific methylation at the sDMR was extremely stable, as it was maintained in all adult tissues examined (Fig. 1B; data not shown). Interestingly, in the Dnmt3L−/+ context, both parental alleles were targeted for de novo methylation, and the locus was fully methylated by E9.5. This demonstrates that paternal sDMR specificity depends on maternally inherited DNA methylation. Finally, the two CGIs flanking the paternal DMR—one encompassing the Zdbf2 promoter and the other at an intergenic location ∼25 kb upstream—were consistently devoid of DNA methylation throughout development and adulthood in both the wild-type and Dnmt3L−/+ contexts (Supplemental Fig. S1C).
De novo methylation after implantation globally spares the extraembryonic lineage (Monk et al. 1987). Accordingly, we found that the maternal gDMR was maintained in a preimplantation, blastocyst-like pattern in extraembryonic tissues: Preferential maternal methylation and paternal hypomethylation were observed in trophoblast giant cells (TGCs) and visceral yolk sacs (VYSs) at E9.5 and later, during gestation, in E17.5 placenta (Fig. 1C). Some de novo methylation occurred at the paternal sDMR, but while the paternal allele reached full methylation in the embryo, the density was lower in extraembryonic tissues (30%–60%), and maternal alleles were also targeted, suggesting relaxed imprinted methylation at the paternal sDMR in these tissues.
In summary, two DMRs at the Gpr1/Zdbf2 locus are targeted by de novo DNA methylation in a parent-of-origin manner during gametogenesis. During preimplantation, only the maternal gDMR is maintained, placing the locus under maternal imprinting. In post-implantation embryos, a switch in parent-of-origin information occurs in the wake of embryonic de novo DNA methylation: The paternal DMR is somatically reestablished at E6.5, just before the maternal gDMR disappears by methylation gain (Supplemental Fig. S1B). Once acquired, the paternal sDMR provides lifelong, paternal specificity to the locus. Extraembryonic tissues are an exception, where maternal specificity is maintained at the maternal gDMR.
Characterization of Liz, a transcript linking the two DMRs in ES cells
We next explored ES cells as a model system to investigate the molecular events that underlie the DNA methylation dynamics at the Gpr1/Zdbf2 locus in embryos. Early passages of ES cells derived in “ground-state” pluripotency 2i conditions (Ying et al. 2008) showed faithful maintenance of blastocyst methylation patterns; i.e., maternal-specific methylation of the maternal gDMR and lack of methylation at the paternal sDMR (Fig. 2A). However, the maternal gDMR rapidly lost DNA methylation upon passage (above passage 5 [P5]) or differentiation, a feature we systematically observed in several independent 2i lines. Furthermore, biallelic methylation was acquired at the paternal sDMR upon differentiation, as observed by day 4 (D4) of retinoic acid (RA) treatment (Fig. 2A). These data underscore, as in in vivo embryos, the dependence of the paternal sDMR methylation upon the maternal gDMR methylation status. ES cells grown in classic serum conditions showed even more aberrant methylation patterns, already demonstrating full paternal sDMR methylation at undifferentiated states (D0) (Supplemental Fig. S2A). In conclusion, no ES cell model is likely to fully recapitulate the epigenetic changes that occur in vivo at the Gpr1/Zdbf2 locus. However, despite loss of imprinting, we were able to use 2i ES cells as a model system to understand what may signal de novo methylation at the sDMR during embryonic differentiation.
Figure 2.
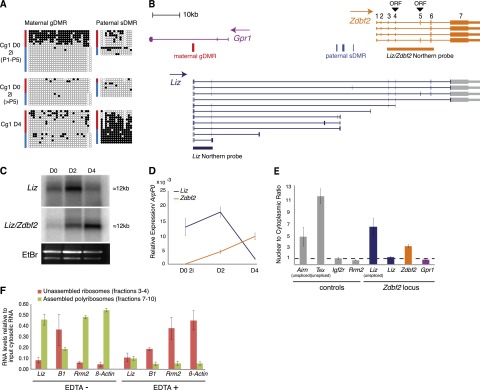
Identification of Liz transcripts in ES cells. (A) Bisulfite analysis of maternal gDMR and paternal sDMR methylation in 2i-derived ES cells (Cg1, Cx129 hybrid background) in undifferentiated states (D0) before and after five passages (P5) and after differentiation (D4). (B) Transcription map of the mouse Gpr1/Zdbf2 locus, with positions of putative ORFs for Zdbf2 translation. Transcripts originating from the maternal gDMR were characterized by sequencing of RT–PCR and RACE products from 2i ES cell RNA. Many transcripts incorporate Zdbf2 exons, called long isoforms of Zdbf2 (Liz) (blue). (C) Northern blot analysis over ES cell differentiation using a probe specific to Liz and a probe that recognizes Liz and Zdbf2 (probe positions in B). (D) RT-qPCR quantification of Liz (Liz exons 1 and 2) or Zdbf2 (Zdbf2 exons 1– 3) transcripts confirms reverse abundance of these transcripts over differentiation. Error bars indicate SEM of two individual ES cell lines: Cg1 and gC6. (E) RT-qPCR quantification of nuclear-to-cytoplasmic RNA enrichment. Unless otherwise specified, primers detecting spliced RNA forms (spanning introns) were used. Error bars are as above. (F) RT-qPCR quantification of spliced Liz levels after sucrose gradient fractionation of cytosolic RNA without or with EDTA treatment (EDTA− and EDTA+). Unassembled ribosomal fractions (3 and 4) and assembled polyribosomal fractions (7–10) were determined by 28S/18S ratio measurement. Error bars indicate SEM of two biological 2i ES cell grown replicates.
Many sDMRs have been proposed to gain methylation as a result of their position along the path of transcripts controlled by promoter-associated gDMRs (John and Lefebvre 2011). Transcriptome reconstruction from RNA sequencing (RNA-seq) data shows evidence of a plethora of transcriptional activity arising from the maternal gDMRs (extending antisense to Gpr1 and sense to Zdbf2) and present in ES cells but not in differentiated cell types (Guttman et al. 2010). Using RT–PCR and 3′ and 5′ RACE in 2i ES cells, we confirmed the presence of a TSS within the maternal gDMR, identified additional exons and splicing events, and demonstrated that these transcripts can be polyadenylated (Fig. 2B). While the shortest variants stopped before or within the paternal sDMR domain, the longest extended over and linked the Gpr1 and Zdbf2 genes, covering ∼100 kb in genomic distance. These long transcripts shared multiple exons with Zdbf2, which are of relatively small sizes (110 base pairs [bp] on average), except the large 12-kb exon 7 and 3′ untranslated region (UTR). This led us to postulate that the maternal gDMR coincides with an alternative promoter for Zdbf2 transcription, located ∼73 kb upstream of the annotated RefSeq Zdbf2 TSS. We termed these extended transcripts Liz, for long isoforms of Zdbf2. Liz spliced variants contain at least two 5′ exons that do not overlap with Zdbf2, and Zdbf2 exons 1 and 2 were never incorporated in Liz transcripts (Fig. 2B).
Northern blotting with a probe specific for Liz detected a unique band, slightly larger than 12 kb (Fig. 2C), indicative of a long spliced variant extending over the last exon of Zdbf2, not significantly different in size from canonical Zdbf2 mRNA. RT-qPCR confirmed that spliced forms of Liz were fivefold to 10-fold more abundant than unspliced forms, with ratios similar to protein-coding transcripts (data not shown). Comparison with a probe recognizing both Zdbf2 and Liz revealed dynamic expression changes over RA-induced ES cell differentiation. In undifferentiated D0 and early D2 differentiating ES cells, Liz was the dominant transcript. As Liz levels declined in abundance by differentiation D4, the canonical form of Zdbf2 became up-regulated, suggesting a switch from Liz to Zdbf2 promoter usage. Using primers specific to Liz or Zdbf2, we confirmed the reverse abundance of Liz and Zdbf2 RNAs before and after differentiation in multiple differentiation conditions (Fig. 2D; data not shown).
In ES cell ChIP-seq data sets, the Liz TSS is occupied by RNA polymerase II (Pol II) and the pluripotency transcription factor Sox2, which could explain pluripotency-associated Liz expression (Supplemental Fig. S2C; Mikkelsen et al. 2008). Importantly, using an RT–PCR pyrosequencing assay exploiting strain-specific SNPs, we found Liz to be dominantly paternally expressed in early passage 2i ES cells (Supplemental Fig. S2B), which harbor maternal-specific methylation at the maternal gDMR/Liz promoter (Fig. 2A). Upon passages, as methylation was lost, Liz expression became biallelic, and this coincided with biallelic methylation at the paternal sDMR later upon differentiation (Figs. 1A, 2A; Supplemental Fig. S2B). As the paternal sDMR lies within the path of Liz transcription, we hypothesize that Liz mediates the effect of the maternal gDMR on the downstream paternal DMR methylation via either the act of transcription or the RNA product itself.
Liz and Zdbf2 transcripts have similar coding potential
Liz was previously reported to be a long intergenic RNA with no coding potential (GenBank accession no. AB777270) (Guttman et al. 2010, 2011; Kobayashi et al. 2012b). While this may be true for the short isoforms that do not extend into the Zdbf2 gene, we found the long isoforms containing multiple Zdbf2-derived exons to be the most abundant isoforms in ES cells. ZDBF2 is a protein of unknown function. The C2H2-type zinc finger motif at its N terminus is the only annotated domain (pfam07535) that relates to the budding yeast DBF4 (dumbbell-forming protein 4) protein. Two putative ORFs exist within the RefSeq mouse Zdbf2 sequence, initiated in exon 4 or exon 5 (Fig. 2B), and have similarly high coding potential calculator (CPC) scores (23.1 vs. 23.2) (Kong et al. 2007). Although the main initiating codon is reported to be in exon 5 (University of California at Santa Cruz [UCSC] and Ensembl genome browsers), the corresponding ORF lacks the complete zinc finger motif (UniProt Q5S200). Importantly, a large fraction of canonical Zdbf2 and nearly all Liz spliced transcripts skip exon 5; this suggests that the exon 4 initiating codon is frequently accessible, which would potentiate translation of a 2498-amino-acid-long protein containing the conserved DBF4 zinc finger (Supplemental Fig. S2D). Importantly, Liz and canonical Zdbf2 exon 4-derived ORF scores are nearly identical (22.9 vs. 23.1), making it likely that Liz is an alternative source of zinc finger-containing Zdbf2 protein during development, with a different 5′ UTR region.
To further characterize the translational properties of Liz, we performed subcellular fractionation of RNA from D2 differentiating ES cells. The known noncoding Tsix and Airn RNAs showed high nuclear to cytoplasmic ratios in RT-qPCR assays, in contrast to the protein-coding Igf2r and Rrm2 RNA controls, which were dominantly cytoplasmic, thus potentiating their translation (Fig. 2E; Seidl et al. 2006; Nora et al. 2012). Using Liz-specific primers, we found unspliced forms of Liz to be retained in the nucleus, while spliced forms were preferentially exported to the cytoplasm, suggesting potential ribosome association (Fig. 2E). Importantly, Liz cytoplasmic enrichment was higher compared with canonical spliced Zdbf2 at this stage of differentiation. Using sucrose gradient fractionation of cytosolic RNAs, we demonstrated that spliced Liz RNAs are indeed bound to translating ribosomes (Fig. 2F). The Liz polyribosome-association profile was similar to that of the protein-coding Rrm2 and β-actin RNAs, while the SINEs B1 retrotransposon noncoding transcripts were preferentially found in unassembled ribosome fractions. Moreover, Liz RNA showed a clear redistribution toward unassembled ribosomes upon EDTA treatment, which disrupts polysomes, further confirming the specificity of the polyribosome association and the possibility that Liz is efficiently translated in ES cells. In summary, while Liz RNA may have cis-acting function on DNA methylation, its conserved coding potential and polyribosome association strongly suggest that it may dually function as a message for the Zdbf2 protein.
Liz and Zdbf2 promoter usage is dynamically regulated during embryonic development
We hypothesized that the switch from Liz to canonical Zdbf2 transcription observed upon ES cell differentiation would also occur in vivo during early embryonic development. RT-qPCR analysis revealed that the Zdbf2 mRNA is abundantly produced in the oocyte and transmitted upon fertilization (Fig. 3A). While the maternal Zdbf2 transcript was rapidly degraded, Liz was activated and accumulated in the pluripotent preimplantation embryo from the eight-cell stage to the blastocyst stage. As in ES cells, the long exon 4-containing form was produced during this period. In the E6.5 epiblast, Liz levels sharply dropped, and canonical Zdbf2 transcripts became the prevailing form in post-implantation embryos, consistent with what was observed in vitro in differentiating ES cells. Therefore, Zdbf2-related transcripts are continuously present in the early embryo but originate from distinct sources: first from the canonical Zdbf2 promoter, then from Liz, and finally, from Zdbf2 again.
Figure 3.

Liz and Zdbf2 expression is dynamically regulated during mouse embryonic development. (A) Liz- and Zdbf2-specific detection by RT-qPCR during mouse embryonic development. Error bars indicate SEM of technical replicates in pooled oocytes or embryos. (B) RNA FISH detection of nascent Liz transcripts in wild-type (WT) and Dnmt3L−/+ E3.5 blastocysts. (Left panel) Representative microscopy images. Yellow arrows point to Liz FISH signals (red). Dashed white lines indicate nucleus borders delineated by DAPI staining (blue). Bar, 5μm. (Right panel) Cell counting of biallelic and monoallelic Liz signals. Wild type, n = 25 blastocysts/1488 cells; Dnmt3L−/+, n = 13 blastocysts/659 cells. (C, left panel) RT–PCR pyrosequencing detects Liz paternal-specific expression in E3.5 BxC wild-type blastocysts and biallelic expression in Dnmt3L−/+ blastocysts. Genomic DNA (gDNA) was used to exclude assay-specific biases. (Right panel) RT-qPCR shows increased Liz levels in Dnmt3L−/+ blastocysts. Error bars indicate SEM technical replicates of pooled blastocysts. (*) P < 0.03, Student's t-test. (D) Same analyses as in C, for Zdbf2 in E8.5 wild-type and Dnmt3L−/+ embryos. (**) P < 0.002, Student's t-test. (E) RT-qPCR analysis of Liz expression in various mouse tissues. Error bars indicate SEM of technical replicates. (F) RT-qPCR analysis of Liz expression over spermatogenesis. Levels were measured in testes at nine different ages, from embryonic (E) to postnatal stages (days post-partum [dpp]), to follow representative male germ cell types appearing during the course of the first spermatogenesis wave, as indicated below the graph. Dnmt3L and Scp3 expression peaks denote periods of de novo DNA methylation and meiotic recombination, respectively. Highest values for each transcript are set to 1. Error bars are as above. (G) Stability of Zdbf2 paternal-specific expression measured by RT–PCR pyrosequencing in tissues collected in embryonic E8.5 and E17.5 stages and after birth at 3 and 60 dpp. Error bars indicate SEM in reverse cross BxC and CxB tissues.
To visualize Liz allelic expression on a single-cell level, we performed RNA fluorescent in situ hybridization (FISH) of nascent Liz transcripts with an intronic BAC probe, spanning the intergenic region 3′ of Gpr1 and 5′ of the Zdbf2 promoter. Single pinpoints indicative of monoallelic expression were visible in blastomeres of E3.5 blastocysts from a pure C57Bl6/J background (Fig. 3B). Liz expression was not lineage-restricted, as we observed predominant monoallelic signals in both the inner cell mass (ICM) and the trophectoderm (TE). Determination of allelic specificity by RT–PCR pyrosequencing further revealed that Liz is paternally expressed in blastocysts (Fig. 3C), concordant with maternal methylation of the Liz promoter at this stage (Fig. 1A). The repressive effect of DNA methylation was further demonstrated by biallelic expression of Liz in maternal methylation-free Dnmt3L−/+ E3.5 blastocysts (Fig. 1A), as seen by RNA FISH and RT–pyrosequencing. Biallelic expression resulted in a doubling of Liz mRNA levels (Fig. 3B,C).
After implantation, in E8.5 embryos, as paternal sDMR methylation is acquired (Figs. 1A, 3A), we also found Zdbf2 to be paternally expressed (Fig. 3D). We validated the positive correlation between intergenic paternal sDMR methylation and the canonical Zdbf2 promoter activity with the observation of biallelic and twofold increase of Zdbf2 transcripts in E8.5 Dnmt3L−/+ embryos (Fig. 3D), where paternal sDMR methylation is biallelic (Fig. 1A; Supplemental Fig. S1B). Moreover, reactivation of the normally silent maternal allele of Zdbf2 in the Dnmt3L−/+ background is strong evidence that the hypomethylated state of the Liz promoter and/or Liz expression before implantation is associated with Zdbf2 promoter activation after implantation.
Liz expression occurs in extraembryonic tissues and during spermatogenesis
We reasoned that Liz should also exist in male germ cells and extraembryonic tissues, two contexts outside of the preimplantation window where the Liz promoter is totally or partially DNA methylation-free (Fig. 1A). We successfully amplified Liz mRNA by RT-qPCR in testes and placenta in levels similar to 2i ES cells but not in any other tested somatic or postnatal tissue (Fig. 3E). Because we were able to amplify long Liz transcripts that extend over the Zdbf2 RefSeq gene and include exon 4, we concluded that, like in early embryonic cells, Liz mRNA can serve as a source of Zdbf2 protein in the placenta and testis.
As a maternally imprinted gDMR, the Liz promoter does not gain methylation in the male germline. Accordingly, Liz expression was biallelic in fetal and adult testes (Supplemental Fig. S3A). Interestingly, Liz expression exhibited developmentally regulated dynamics throughout spermatogenesis (Fig. 3F). In fetal testes, Liz transcripts accumulated from E14.5 to E18.5, concomitant with the wave of male germline de novo DNA methylation (Kato et al. 2007) and in particular with germline methylation of the paternal DMR (Hiura et al. 2010). After birth, Liz transcription increased at the onset of meiosis. Considering the consistently fully methylated status of the Liz promoter in all somatic cell types, Liz expression in the testis is likely to emanate from germ cells. This was confirmed in sorted primordial germ cells from fetal testes (Supplemental Fig. S3B). Additionally, in Dnmt3L mutant adult testes, which exhibit a severe germ cell depletion phenotype (Bourc'his and Bestor 2004), Liz expression was dramatically reduced, behaving as typical germ cell markers such as Vasa (Supplemental Fig. S3C). In extraembryonic tissues, maternal-specific methylation is globally maintained at the Liz promoter. Accordingly, a strong paternal bias in Liz expression was found in hybrid E9.5 VYSs and TGCs and E17.5 placenta (Fig. 1A; Supplemental Fig. S3D). Liz expression therefore occurs in preimplantation embryos, extraembryonic tissues throughout gestation, and male germ cells, all of which are types in which the Liz promoter is not completely obstructed by DNA methylation. Besides these specific contexts, we only observed use of the Zdbf2 promoter.
Stability and extent of imprinted control by the paternal sDMR
As Zdbf2 monoallelic expression occurs without allele-specific DNA methylation at its TSS (Supplemental Fig. S1C), we questioned the stability of Zdbf2 paternal-specific expression throughout life. To this end, we conducted quantitative RT–PCR pyrosequencing using a bank of 50 hybrid tissues from reciprocal BxC and CxB crosses collected at fetal, neonatal, and adult stages. According to microarray profiling data (BioGPS), Zdbf2 is most highly expressed in brain tissues. We found paternal Zdbf2 expression bias to be extremely stable throughout life and in a tissue-wide manner, independent of Zdbf2 expression level (Fig. 3G).
As many imprinted DMRs exert their control over large gene clusters (Barlow 2011), we further assessed the imprinted status of the three most proximal genes relative to Liz/Zdbf2. We first focused on the Gpr1 gene (45 kb 5′ of the paternal sDMR), which houses the transient maternal gDMR and was formerly reported to have kidney-specific paternal expression (Hiura et al. 2010). Despite an extensive search, we could not find evidence of parent-specific expression in kidney or other somatic tissues, in extraembryonic tissues where Gpr1 expression levels are the highest (BioGPS), or in late blastocysts, where we detected a slight peak of Gpr1 expression (Supplemental Fig. S3E–H). Biallelic expression was also consistently observed tissue- and stage-wide for Eef1b2 (87 kb 5′ of the sDMR) and Adam23 (182 kb 3′ of the sDMR) despite a report of moderately biased paternal expression in fetal brains for the latter (Supplemental Fig. S3E; DeVeale et al. 2012). Contrary to previous conclusions, our findings suggest that the two regions of differential DNA methylation of the Gpr1/Zdbf2 locus, the maternal gDMR and the paternal sDMR, are exclusively linked to the local regulation of Zdbf2 imprinted expression in both its long (Liz) and canonical forms.
LIZ expression and coding potential are evolutionarily conserved in humans
As an indication of functional relevance of Liz, we checked its conservation from mice to humans. Paternal-specific methylation at the paternal g/sDMR and paternal ZDBF2 expression were previously described in humans, showing imprinting at the syntenic GPR1/ZDBF2 locus (2q3.3) (Kobayashi et al. 2009; Hiura et al. 2010). We designed RT-qPCR primers that mapped to a predicted transcript (Aceview nehura.cAug10), which arises from a relatively CpG-rich region within GPR1 intron 2 and is antisense to GPR1 (similar to mouse Liz) but is interrupted before the ZDBF2 RefSeq sequence (Fig. 4A). As in mice, we detected this transcript in the placenta and testis but not in somatic adult tissues (Fig. 4B). Unlike in mice, however, we failed to measure significant expression in different human ES lines.
Figure 4.

DNA methylation and expression at the human GPR1/ZDBF2 locus. (A) Map of the GPR1/ZDBF2 locus indicating RT–PCR primers designed for a predicted transcript (green) that detect multiple LIZ-like transcripts (blue) in the placenta, including isoforms sharing exons with ZDBF2 and incorporating ZDBF2 putative ORFs. (B) RT-qPCR analysis of LIZ in human tissues and ES cells. Error bars indicate SEM of technical replicates for tissues and HU1 and H9 biological replicates for ES cells. (C) RT–PCR of short and long forms of ZDBF2 in placenta and fetal testes. (D) Bisulfite-based DNA methylation analysis in the sperm, liver, and placenta of the Liz promoter and four intergenic regions (regions 1–4) upstream of the ZDBF2 promoter (primer positions in A). (E) DNA methylation in HUES1 and H9 ES cells.
We confirmed the production of short truncated versions of LIZ in both the placenta and fetal testis by RT–PCR (Fig. 4A,C). Importantly, primers anchored in the first putative exon of LIZ and the last exon of ZDBF2 amplified long multiexonic LIZ variants in placenta but not in testes. These extended over the full ZDBF2 RefSeq sequence, a feature that we confirmed by the detection of a >12-kb band by Northern blotting, which indicates the frequent inclusion of the large last exon/3′ UTR of ZDBF2 (Supplemental Fig. S4A). As in mice, ATG codons exist in exons 4 and 5 of ZDBF2, but only exon 4 can potentiate a zinc finger-containing ZDBF2 ORF (Supplemental Fig. S2D). Exon 4 is present in the majority of LIZ forms (Fig. 4A); we concluded that LIZ also has the coding potential for a functional ZDBF2 protein in humans, at least in placenta.
Bisulfite-based methylation analysis around the putative LIZ promoter revealed absence of methylation in sperm, full allelic methylation in somatic tissues (liver), and differentially methylated alleles in placenta, indicative of a DMR, although we could not infer parental origin due to the lack of informative SNPs in our samples (Fig. 4D; Supplemental Fig. S4B). Therefore, DNA methylation at the putative human LIZ promoter shows developmental dynamics similar to the mouse Liz promoter, reminiscent of a tissue-specific maternal gDMR maintained in extraembryonic lineages only. However, unlike in mice, the LIZ promoter was fully methylated in several human ES cell lines (Fig. 4E; Supplemental Fig. S4B), in agreement with the more advanced, post-implantation-like state of human versus mouse ES cells (McEwen et al. 2013). These results explain our ability to detect LIZ transcription in the testis and placenta but not in human ES cells.
Finally, we analyzed the developmental dynamics of DNA methylation at the paternal DMR in humans. The 25-kb intergenic region upstream of the ZDBF2 promoter exhibits low conservation across 32 eutherian mammals (PhyloP) (Pollard et al. 2010) compared with the entire GPR1/ZDBF2 locus (Supplemental Fig. S4C). This indicates poor evolutionary constraint over the paternal sDMR sequence, except for a 1-kb region, which maps 3′ to the short BC028329 EST and shares 74% homology between mice and humans. We investigated four regions within the putative paternal DMR locus, including the conserved BC0288329 sequence (region 4) and a region of relative homology with the mouse paternal DMR2 (58%, region 3) (Fig. 4A). In the liver and placenta, we consistently found two distinct populations of alleles, methylated and unmethylated, indicative of a single extended DMR or several discrete DMRs in the region (Fig. 4D). In human ES cells, while allelic specificity was conserved in HUES1 and H1, H9 showed complete DNA methylation (Fig. 4E; Supplemental Fig. S4B). This may reflect either loss of imprinting or genetic loss of maternal chromosome 2. In sperm from two fertile men, only one subregion (region 2) was 100% methylated, as expected for a paternal gDMR (Fig. 4D; Supplemental Fig. S4B); the paternal DMR of the GPR1/ZDBF2 locus is therefore more constrained in the germline than in somatic tissues, a feature also manifested in publically available whole-genome bisulfite sequencing data (Molaro et al. 2011). The other regions ranged from 0% (region 1) to variable methylation (30%–60%, regions 3 and 4) with extensive allelic variability.
Although pluripotent embryonic states were not accessible due to the advanced epigenetic features of human ES cells, the human GPR1/ZDBF2 locus seems to conserve the same DNA methylation dynamics that we found in mice. Importantly, LIZ transcripts also have the potential to function as regulatory and protein-coding RNA species in humans.
Dynamic and unusual chromatin features at the imprinted Gpr1/Zdbf2 locus
To gain insight into the chromatin regulation of promoter usage and imprinting of the Gpr1/Zdbf2 locus, we analyzed histone modification patterns in mouse undifferentiated 2i ES cells and in a differentiated tissue—the whole brain from neonates—by ChIP-qPCR. We found enrichment of histone H3 Lys 4 di- and trimethylation (H3K4me2 and H3K4me3) at the Liz promoter in ES cells, while the canonical Zdbf2 promoter was preferentially enriched in the brain, reflecting differential promoter activity in these two cell types (Fig. 5A; Supplemental Fig. S5A,B). The intergenic CGI was occupied by these active marks in both ES cells and the brain, in agreement with its ubiquitous DNA methylation-free status.
Figure 5.
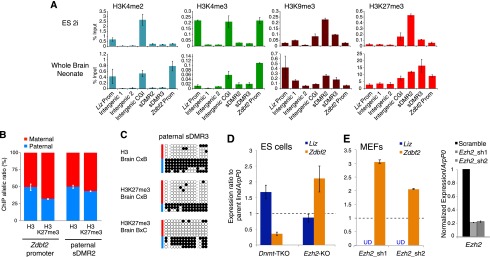
Interplay between histone modifications and DNA methylation at the mouse Gpr1/Zdbf2 locus. (A) Histone modification profiling by ChIP-qPCR in 2i Cg1 ES cells and neonate whole brains (WBs) over the Gpr1/Zdbf2 locus (primer positions in Supplemental Fig. S5A). Error bars indicate SEM of two biological replicates for ES cells and SEM from reverse cross BxC and CxB neonates for whole brains. (B) H3K27me3 allelic enrichment detected by ChIP pyrosequencing at the Zdbf2 promoter and at the paternal sDMR in neonate brains. Error bars indicate SEM from reverse crosses BxC and CxB. (C) Bisulfite-based DNA methylation analysis of H3- and H3K27me3-bound chromatin at the paternal sDMR (DMR3) in neonate brains from CxB and BxC crosses. (D) RT-qPCR analysis of Liz and Zdbf2 RNA levels in Dnmt-TKO and Ezh2-KO ES cells, normalized to wild-type (WT) parental cell lines. Error bars indicate SEM from two (Dnmt-TKO) and four (Ezh2-KO) biological replicates. (E, left panel) RT-qPCR analysis of Liz and Zdbf2 levels after Ezh2 shRNA-mediated knockdown in MEFs. Liz levels were undetectable (UD), as expected from Liz TSS full DNA methylation in somatic cells. (Right panel) RT-qPCR analysis shows 80% depletion of Ezh2 levels by two independent hairpins. Error bars indicate SEM from two biological replicates.
Co-occurrence of H3K4 and H3K9 methylation is considered to be a hallmark of imprinted DMRs, in correlation with the differential allelic activity of these loci (McEwen and Ferguson-Smith 2010). In concordance with the rapid loss of imprinting in culture, we did not find this typical chromatin signature anywhere throughout the Gpr1/Zdbf2 locus in ES cells. In the brain, the maternal gDMR was enriched in H3K9me3 only, reflecting the extinction and full DNA methylation of the Liz promoter upon differentiation. Surprisingly, the intergenic paternal sDMR also failed to display patterns of dual H3K4/H3K9 methylation, despite imprinted DNA methylation in the brain (sDMR2 and sDMR3) (Fig. 5A; Supplemental Fig. S5A,B). Instead, there was significant enrichment of H3K9me3 compared with the rest of the locus, but the level of H3K4 methylation was low and similar to random intergenic sites located 5′ of the intergenic CGI border. Finally, co-occurrence of H3K4/H3K9 methylation was not found at the Zdbf2 promoter despite consistent monoallelic paternal activity in all somatic tissues. In summary, while histone modification patterns are consistent with the developmental promoter switch in Zdbf2 transcription, the imprinted status of the Gpr1/Zdbf2 locus is not apparent at the chromatin level.
Interestingly, at a region overlapping the paternal sDMR, we found a compact block of repressive H3K27me3 marks that was delineated by the intergenic CGI in 5′ and the Zdbf2 promoter CGI in 3′ (Fig. 5A). This broad local H3K27me3 enrichment was constitutively observed in all cell types available in public ChIP-seq data sets (Supplemental Fig. S5A; Mikkelsen et al. 2007; Marks et al. 2012). However, we noted some developmental differences: The paternal sDMR was 50-fold more enriched in brain cells compared with 2i ES cells, and H3K27me3 marks extended into the Zdbf2 gene in brain cells but not in ES cells (Fig. 5A).
Dual roles of DNA and H3K27 methylation on Zdbf2 expression
We then tested the allelism of H3K27me3 enrichment by ChIP followed by quantitative pyrosequencing of whole neonate brains from hybrid mouse crosses. At the Zdbf2 promoter, which is constitutively devoid of DNA methylation, the maternal allele was substantially more enriched in H3K27me3 than the paternal allele (70/30 ratio) (Fig. 5B), in agreement with maternal-specific Zdbf2 silencing in differentiated tissues. However, at the paternal sDMR, there was no parental distinction in H3K27me3 enrichment. Using sequential bisulfite sequencing after ChIP, we confirmed the presence of both paternally DNA methylated and maternally unmethylated alleles in H3- and H3K27me3-captured chromatin (Fig. 5C; Supplemental Fig. S5C); this indicates that, at least in brains, DNA and H3K27 methylation can co-occur on the paternal allele of the paternal sDMR. While DNA and H3K27 methylation are usually antagonistic at CGIs, recent studies have shown their compatibility in CG-poor regions (Tanay et al. 2007; Brinkman et al. 2012; Statham et al. 2012), which is the case for the paternal sDMR.
We functionally tested the respective role of DNA and H3K27 methylation on Zdbf2 expression by comparing ES cell lines that are genetically deficient for the three active DNA methyltransferases (Dnmt1, Dnmt3A, and Dnmt3B; Dnmt triple knockout [TKO]) or the H3K27 methyltransferase Ezh2 (Ezh2 knockout [KO]) (Fig. 5D). The J1 ES cells, from which the Dnmt-TKO cells were derived (Tsumura et al. 2006), showed full DNA methylation at the paternal sDMR, as consistently observed for serum-grown ES cells (Supplemental Figs. S2A, S5D). In DNA methylation-free Dnmt-TKO J1 cells, RT-qPCR assays measured increased Liz transcript levels, confirming the repressive role of DNA methylation on the Liz promoter. In contrast, levels of canonical Zdbf2 were reduced, arguing toward a positive role of the paternal sDMR methylation on Zdbf2 promoter activity. Alternatively, higher Liz transcripts may also be responsible for reduced Zdbf2 expression via cross-talk between the two promoters.
The effect of H3K27me3 marks was inferred after tamoxifen-induced deletion of the Ezh2 gene in 2i-derived ES cells. Ten days after Ezh2 deletion, EZH2 protein and H3K27me3 marks were barely detectable, while the paternal sDMR maintained low DNA methylation levels, comparable with the nondeleted parental 2i cell line (Supplemental Fig. S5D,E). Upon Ezh2 knockout, we found a highly reproducible up-regulation of Zdbf2 (Fig. 5D), which was not related to altered pluripotency state of these cells (normal Oct4 and Nanog mRNA levels) (data not shown). This suggests that H3K27me3 acts by repressing the Zdbf2 promoter. To determine whether H3K27me3 also tunes down Zdbf2 expression in differentiated cells where the paternal sDMR is DNA methylated, we performed an Ezh2 shRNA knockdown in immortalized hybrid mouse embryonic fibroblasts (MEFs) (Fig. 5E; Supplemental Fig. S5D,E). With two independent hairpins inducing 80% reduction of Ezh2 mRNA, we observed a more than twofold increase in Zdbf2 levels, confirming that H3K27me3 consistently down-regulates Zdbf2 output in different cell types. Interestingly, we did not find a relaxation of imprinted Zdbf2 expression in Ezh2 knockdown MEFs by RT–PCR pyrosequencing (data not shown). Together, these results imply that DNA and H3K27me3 methylation oppositely regulate the activity of the Zdbf2 promoter, with DNA methylation likely acting on expression allelism, and H3K27me3 modulating expression levels in a direct or indirect manner. These two marks do not seem to be spatially competing: Loss of DNA methylation does not increase H3K27me3 levels at the paternal sDMR in Dnmt-TKO ES cells, as manifested in available ChIP-seq data sets (Brinkman et al. 2012), and H3K27me3 loss does not promote ectopic gain of DNA methylation at this locus in ES cells or MEFs (Supplemental Fig. S5C).
Discussion
Our study of the Gpr1/Zdbf2 locus provides new important paradigms in mammalian genomic imprinting. It illustrates the functional relevance of transient imprinting for short- and long-term genome regulation. It also reveals the existence of dynamic genomic imprinting occurring via a parent-of-origin switch in DNA methylation. Together, our work demonstrates how intricate mechanisms can evolve to ensure proper gene dosage regulation through life despite the dramatic epigenetic reprogramming undergone by the embryonic genome.
Genomic imprinting is currently defined by epigenetic characteristics universally shared by all lifelong imprinted loci: (1) acquisition of a gDMR in one of the parental germline only, (2) an uninterrupted continuum of paternal- or maternal-specific methylation from gametes to adult progeny, and (3) the combination of both repressive and permissive histone marks. The Gpr1/Zdbf2 locus breaks these rules: It contains two gDMRs of opposite parental origins, neither of which is permanent after fertilization, and they lack dual patterns of H3K4/H3K9 methylation. Importantly, the Gpr1/Zdbf2 locus demonstrates a lifelong imprinted status (Fig. 6): The maternal gDMR provides maternal-specific methylation in the preimplantation embryo and in the placenta after implantation, while the paternal DMR is somatically re-established in the embryo after implantation and maintained in somatic tissues. Discovering whether the maternal DMR alone or both parental DMRs together act as ICRs (i.e., sequences regulating imprinted gene expression) will require genetic testing. Incidentally, our study raises the possibility that transient gDMRs may be tissue-specific gDMRs of the most extreme sort: those that diverge at the time of the first lineage differentiation and are maintained in extraembryonic but not embryonic tissues. This implies that the number of imprinted gDMRs may be greater in the placenta than in the embryo.
Figure 6.

Model for developmental dynamics of promoter usage and parental DNA methylation at the mouse Gpr1/Zdbf2 locus. Zdbf2-related transcripts are produced from alternative promoters: Liz (dark blue) is available in the male germline, preimplantation embryos, and extraembryonic tissues; canonical Zdbf2 (orange) is expressed in the female germline, post-implantation embryos, and somatic tissues. Parent-of-origin methylation occurs in a lifelong manner: Maternal-specific (red) information is provided by the maternal DMR in preimplantation embryos and extraembryonic tissues, and paternal-specific information (light blue) is carried by the paternal intergenic DMR in post-implantation embryos and somatic tissues. Sex-specific methylation occurs in the male and female germlines.
Transcriptional events likely drive the cross-talk between the two parental DMRs, which allows a parent-of-origin switch in DNA methylation at the Gpr1/Zdbf2 locus: The maternal gDMR hosts a starting site for transcripts that are paternally expressed in early embryos and 2i ES cells. Among them, the Liz spliced variants, which extend across the paternal DMR and incorporate many Zdbf2 exons, are the dominantly produced isoforms. Liz expression positively correlates with paternal DMR methylation in cis: Paternal-specific Liz expression is associated with paternal-specific sDMR methylation. When Liz transcription is biallelic, as in Dnmt3L−/+ embryos, cultured 2i ES cells, or fetal male germ cells, biallelic methylation is acquired. However, Liz expression is not sufficient: The height of Liz expression coincides with the lowest DNA methylation of the paternal sDMR in blastocysts. More likely, Liz expression primes paternal sDMR accessibility to de novo DNA methyltransferases for when they become available: after implantation or during ES cell differentiation. Interestingly, ChIP-seq data sets show evidence of H3K36me3 marks lining the path of Liz transcription in ES cells (Supplemental Fig. S5A; Mikkelsen et al. 2007). Given the ability of the PWWP domain of DNMT3 enzymes to interact with H3K36me3-modified tails (Dhayalan et al. 2010), changes in histone modifications likely provide intermediates between Liz expression and paternal DMR de novo methylation. Whether the act of Liz transcription or the Liz RNA per se is involved in this process will be an important question to address.
Chromatin remodeling properties have been traditionally attributed to noncoding RNAs, including at imprinted loci such as Airn or Kcnq1ot1 (Pandey et al. 2008; Khalil et al. 2009; Latos et al. 2012). Accordingly, Liz was originally described as a long intergenic noncoding RNA (lincRNA) (Guttman et al. 2010, 2011) despite the lack of experimental proof (Kobayashi et al. 2012b, 2013). In contrast, we show here that Liz RNA can be polyadenylated, is efficiently spliced and exported to the cytoplasm, associates with translating polyribosomes, includes multiple exons of Zdbf2, and, most importantly, has the same coding potential as Zdbf2 for an ORF, which is evolutionarily conserved in humans. Because Liz and canonical Zdbf2 transcripts share the same putative ORF, it is impossible to distinguish their respective association efficiency and occupancy timing with the translation machinery by ribosome footprinting (Ingolia et al. 2011; Guttman et al. 2013). However, as Liz and Zdbf2 vary by their 5′ UTR regions, differential secondary structure or stability could entail additional levels of post-transcriptional regulation. In conclusion, we postulate that Liz likely functions as an alternative mRNA source for Zdbf2 protein translation during development, depending on promoter availability (Fig. 6).
Nothing is known about Zdbf2 protein function, except for the inclusion of a DBF4-like zinc finger when translation is initiated from exon 4. The eponymous DBF4 protein is an activator of the CDC7 serine/threonine kinase, which has a conserved role in DNA replication initiation from yeast to humans (Hughes et al. 2012). In yeast, the dbf4/cdc7 complex was moreover linked to chromosome segregation during meiosis (Kovacikova et al. 2013; Le et al. 2013). Coincidently, we found that Liz expression peaks at meiosis during mouse spermatogenesis, and canonical Zdbf2 is highly transcribed in the oocyte, leading to the exciting possibility that Zdbf2 may fulfill meiotic roles in mammals.
Alternative promoter usage is a common process of tissue- and stage-specific gene regulation, which may affect 40% of protein-coding genes in mouse and human genomes (Sun et al. 2011). Very little is known about the nature of the mutual regulation between alternative promoters. We found that the downstream Zdbf2 promoter is turned on when the upstream Liz promoter is turned off upon differentiation: Liz transcription may prevent activity from the Zdbf2 canonical promoter in a competitive model where the promoter located the most upstream is dominant over the one located the most downstream (Maunakea et al. 2010). However, our results further suggest that Liz activity prior to differentiation promotes Zdbf2 promoter activation in cis after differentiation: Liz expression precedes Zdbf2 expression from the same, paternal allele, and in cases of biallelic Liz expression, biallelic Zdbf2 expression occurs. As discussed above, in addition to functioning as a protein-coding RNA, Liz may prepare Zdbf2 promoter activity via chromatin changes, therefore classifying as a bifunctional RNA.
Finally, we identified an unusual interplay between DNA and H3K27 methylation that highlights the developmental complexity of Liz and Zdbf2 regulation. Using Dnmt3L−/+ embryos, Dnmt-TKO ES cells, and 2i versus serum ES cells, we demonstrated the functional link between DNA methylation and transcriptional activity but with opposite effects on Liz and canonical Zdbf2. DNA methylation at the Liz promoter is negatively correlated with Liz transcription levels. In contrast, Zdbf2 paternal-specific expression is extremely stable throughout somatic life yet does not rely on differential DNA methylation or H3K9 methylation at its promoter, as is usually the case for imprinted genes. The only form of parent-specific DNA methylation found in the vicinity is at the intergenic paternal DMR located a few tens of kilobases upstream. Here, DNA methylation positively correlates with activity of the canonical Zdbf2 promoter: When DNA methylation is biallelically gained, biallelic Zdbf2 expression is observed. When DNA methylation is absent at the sDMR, Zdbf2 expression is down-regulated. The other important chromatin component of Zdbf2 regulation is linked to H3K27me3 marks, which occur on the maternal allele at the Zdbf2 promoter specifically, while it densely accumulates on both maternal and paternal alleles over the paternal sDMR located upstream. Importantly, we found that upon deletion or knockdown of the H3K27 methyltransferase of the Polycomb complex (Ezh2), Zdbf2 expression levels are increased. These results suggest that DNA methylation and H3K27 methylation coregulate Zdbf2 expression, with intergenic DNA methylation linked to the active Zdbf2 allele, while H3K27me3 down-regulates Zdbf2 output.
Here we provide evidence for multiple conserved mechanisms acting at the Gpr1/Zdbf2 locus for the sole regulation of Zdbf2 dosage, which is the only gene to show imprinted expression over 400 kb. We show here that besides serving as a new paradigm of dynamic genomic imprinting, the Gpr1/Zdbf2 locus can provide an important model system for dissecting the signals shaping DNA methylation patterns, the interplay between DNA and histone modifications, and alternative promoter usage.
Materials and methods
Additional materials and methods can be found in the Supplemental Material.
Isolation of mouse gametes, embryos, and tissues
The care and use of animals used here strictly adhered to European and National Regulation for the Protection of Vertebrate Animals used for Experimental and other Scientific Purposes (directive 86/609 and 2010/63). Mice were bred in the pathogen-free Animal Care Facility of the Institut Curie (agreement no. C 75-05-18). MII stage oocytes from superovulated females, epididymal sperm, preimplantation embryos, post-implantation embryos, extraembryonic tissues, and postnatal tissues were prepared as described previously (Proudhon et al. 2012). Purity of gamete samples was assessed by bisulfite-based DNA methylation analysis of maternally imprinted KvDMR and paternally imprinted H19/Igf2 DMD. The Mus musculus C57Bl/6J strain was used in a wild-type or Dnmt3L mutant background (Bourc'his et al. 2001). Allelic information was obtained from crosses between C57Bl6/J and Mus castaneus CAST/Ei or Mus molossinus JF1 strains.
Bisulfite-based DNA methylation analysis
Genomic DNA was extracted according to Smith et al. (2009), except for sperm DNA (Jeffreys et al. 1994). The Epitect kit (Qiagen) was used for bisulfite conversion of DNA, except for limited quantities (oocytes and embryos from E3.5 to E6.5) where DNA was embedded in agarose beads before processing for bisulfite conversion according to Proudhon et al. (2012). Nested or seminested PCR was performed using the primers listed in Supplemental Table S1. PCR amplicons were gel-purified (Qiagen) and subsequently cloned in pCR2.1 Topo-TA vector (Invitrogen) before Sanger sequencing of at least 30 clones. BiQ Analyzer software was used for sequence alignments (Bock et al. 2005).
Polysome fractionation
Sucrose gradient polysome fractionation was performed as described in Gu et al. (2012), with some modifications. ES cells grown in 15-mm plates in 2i medium (5 × 107 cells per plate) were incubated with 100 μg/mL cycloheximide (Sigma) for 15 min to arrest ribosome movement on mRNAs. Cells were then incubated for 5 min on ice in lysis buffer (20 mM Tris-HCl at pH 7.5, 50 mM KCl, 10 mM MgCl2, 1% Triton, 1 mM DTT, 40 U/mL RNase inhibitors [Promega], 100 μg/mL cycloheximide), harvested by scraping off the dish, transferred to Eppendorf tubes, homogenized by three passages through a 26-gauge needle, and further incubated for 5 min on ice. Nuclei and insolubilized material were pelleted by two rounds of centrifugation at 13,000 rpm for 6 min. Clarified lysates were then kept as total cytosolic RNA fraction (input) or processed for fractionation by loading 1 mL onto an 11-mL linear 10%–50% sucrose gradient in 20 mM Tris-HCl (pH 7.5), 50 mM KCl, 10 mM MgCl2, 1 mM DTT, and 100 μg/mL cycloheximide prepared using the gradient master (Biocomp). A control experiment of polyribosome disruption was performed by adding 15 mM EDTA before sucrose fractionation. Twelve fractions were collected from top to bottom after ultracentrifugation at 36,000 rpm for 2 h at 4°C using an SW41 swinging-bucket rotor (Beckman Coulter). To determine the distribution of nonassembled ribosomal fractions and translation-engaged polyribosomes (without EDTA), 28S/18S ratios were measured after RNA extraction using the 2000 Bioanalyzer (Agilent). Nonassembled ribosomes have a 28S/18S ratio below or above 2, while assembled 80S ribosomes show 28S/18S ratios close to 2. Based on these measurements and RNA integrity, layers three and four were pooled as the nonassembled ribosome fraction, and layers seven to 10 were pooled as the polyribosome-loaded fraction.
RNA FISH
RNA FISH on preimplantation embryos was performed as previously described (Okamoto et al. 2004) using the intron-spanning BAC probe WI1-2819H7 (BacPac Consortium at Children's Hospital Oakland Research Institute). The probe maps to chr1: 63266216–63306435 (UCSC mm9 alignment). Probe labeling was done with SpectrumRed-dUTP by nick translation (Vysis). Images were acquired using a spinning-disk confocal microscope (Zeiss) at 63× magnification with 0.3 μm between each image obtained in Z dimension. Images were analyzed using ImageJ software (NIH).
Acknowledgments
We thank Patricia Diabangouaya, Michel Wassef, Ronan Chaligné, Manuela Portoso, Elphège Nora, Veronica Rodilla, and Marius Walter for experimental help; Raphaël Margueron for the anti-EZH2 antibody and Ezh2-shRNAs; Claire Rougeulle for human ES material; Stéphane Viville for Oct4-GFP-sorted PGCs; the imaging facility Bioimaging Cell and Tissue Core Facility of the Institut Curie (PICT-IBiSA) at BDD; and the Institut Curie animal facility for excellent mouse husbandry. D.B.'s laboratory is part of the Laboratoire d'Excellence (LABEX) entitled DEEP (11-LBX-0044). This research was supported by a European Young Investigator Award (EURYI) and the Fondation Schlumberger. R.D. and S.A. are the recipients of PhD fellowships from Institut Curie and Canceropôle Ile-de-France, respectively.
Footnotes
Supplemental material is available for this article.
Article is online at http://www.genesdev.org/cgi/doi/10.1101/gad.232058.113.
References
- Aravin AA, Bourc'his D 2008. Small RNA guides for de novo DNA methylation in mammalian germ cells. Genes Dev 22: 970–975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barlow DP 2011. Genomic imprinting: A mammalian epigenetic discovery model. Annu Rev Genet 45: 379–403 [DOI] [PubMed] [Google Scholar]
- Bock C, Reither S, Mikeska T, Paulsen M, Walter J, Lengauer T 2005. BiQ Analyzer: Visualization and quality control for DNA methylation data from bisulfite sequencing. Bioinformatics 21: 4067–4068 [DOI] [PubMed] [Google Scholar]
- Bourc'his D, Bestor TH 2004. Meiotic catastrophe and retrotransposon reactivation in male germ cells lacking Dnmt3L. Nature 431: 96–99 [DOI] [PubMed] [Google Scholar]
- Bourc'his D, Xu GL, Lin CS, Bollman B, Bestor TH 2001. Dnmt3L and the establishment of maternal genomic imprints. Science 294: 2536–2539 [DOI] [PubMed] [Google Scholar]
- Brinkman AB, Gu H, Bartels SJ, Zhang Y, Matarese F, Simmer F, Marks H, Bock C, Gnirke A, Meissner A, et al. 2012. Sequential ChIP-bisulfite sequencing enables direct genome-scale investigation of chromatin and DNA methylation cross-talk. Genome Res 22: 1128–1138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chotalia M, Smallwood SA, Ruf N, Dawson C, Lucifero D, Frontera M, James K, Dean W, Kelsey G 2009. Transcription is required for establishment of germline methylation marks at imprinted genes. Genes Dev 23: 105–117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeVeale B, van der Kooy D, Babak T 2012. Critical evaluation of imprinted gene expression by RNA-Seq: A new perspective. PLoS Genet 8: e1002600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhayalan A, Rajavelu A, Rathert P, Tamas R, Jurkowska RZ, Ragozin S, Jeltsch A 2010. THE DNMT3A PWWP domain reads histone 3 lysine 36 trimethylation and guides DNA methylation. J Biol Chem 285: 26114–26120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duffié R, Bourc'his D 2013. Parental epigenetic asymmetry in mammals. Curr Top Dev Biol 104: 293–328 [DOI] [PubMed] [Google Scholar]
- Ginno PA, Lott PL, Christensen HC, Korf I, Chedin F 2012. R-loop formation is a distinctive characteristic of unmethylated human CpG island promoters. Mol Cell 45: 814–825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu L, Zhang H, He J, Li J, Huang M, Zhou M 2012. MDM2 regulates MYCN mRNA stabilization and translation in human neuroblastoma cells. Oncogene 31: 1342–1353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guttman M, Garber M, Levin JZ, Donaghey J, Robinson J, Adiconis X, Fan L, Koziol MJ, Gnirke A, Nusbaum C, et al. 2010. Ab initio reconstruction of cell type-specific transcriptomes in mouse reveals the conserved multi-exonic structure of lincRNAs. Nat Biotechnol 28: 503–510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guttman M, Donaghey J, Carey BW, Garber M, Grenier JK, Munson G, Young G, Lucas AB, Ach R, Bruhn L, et al. 2011. lincRNAs act in the circuitry controlling pluripotency and differentiation. Nature 477: 295–300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guttman M, Russell P, Ingolia NT, Weissman JS, Lander ES 2013. Ribosome profiling provides evidence that large noncoding RNAs do not encode proteins. Cell 154: 240–251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiura H, Sugawara A, Ogawa H, John RM, Miyauchi N, Miyanari Y, Horiike T, Li Y, Yaegashi N, Sasaki H, et al. 2010. A tripartite paternally methylated region within the Gpr1-Zdbf2 imprinted domain on mouse chromosome 1 identified by meDIP-on-chip. Nucleic Acids Res 38: 4929–4945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes S, Elustondo F, Di Fonzo A, Leroux FG, Wong AC, Snijders AP, Matthews SJ, Cherepanov P 2012. Crystal structure of human CDC7 kinase in complex with its activator DBF4. Nat Struct Mol Biol 19: 1101–1107 [DOI] [PubMed] [Google Scholar]
- Ingolia NT, Lareau LF, Weissman JS 2011. Ribosome profiling of mouse embryonic stem cells reveals the complexity and dynamics of mammalian proteomes. Cell 147: 789–802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeffreys AJ, Tamaki K, MacLeod A, Monckton DG, Neil DL, Armour JA 1994. Complex gene conversion events in germline mutation at human minisatellites. Nat Genet 6: 136–145 [DOI] [PubMed] [Google Scholar]
- John RM, Lefebvre L 2011. Developmental regulation of somatic imprints. Differentiation 81: 270–280 [DOI] [PubMed] [Google Scholar]
- Kaneda M, Okano M, Hata K, Sado T, Tsujimoto N, Li E, Sasaki H 2004. Essential role for de novo DNA methyltransferase Dnmt3a in paternal and maternal imprinting. Nature 429: 900–903 [DOI] [PubMed] [Google Scholar]
- Kato Y, Kaneda M, Hata K, Kumaki K, Hisano M, Kohara Y, Okano M, Li E, Nozaki M, Sasaki H 2007. Role of the Dnmt3 family in de novo methylation of imprinted and repetitive sequences during male germ cell development in the mouse. Hum Mol Genet 16: 2272–2280 [DOI] [PubMed] [Google Scholar]
- Khalil AM, Guttman M, Huarte M, Garber M, Raj A, Rivea Morales D, Thomas K, Presser A, Bernstein BE, van Oudenaarden A, et al. 2009. Many human large intergenic noncoding RNAs associate with chromatin-modifying complexes and affect gene expression. Proc Natl Acad Sci 106: 11667–11672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi H, Yamada K, Morita S, Hiura H, Fukuda A, Kagami M, Ogata T, Hata K, Sotomaru Y, Kono T 2009. Identification of the mouse paternally expressed imprinted gene Zdbf2 on chromosome 1 and its imprinted human homolog ZDBF2 on chromosome 2. Genomics 93: 461–472 [DOI] [PubMed] [Google Scholar]
- Kobayashi H, Sakurai T, Imai M, Takahashi N, Fukuda A, Yayoi O, Sato S, Nakabayashi K, Hata K, Sotomaru Y, et al. 2012a. Contribution of intragenic DNA methylation in mouse gametic DNA methylomes to establish oocyte-specific heritable marks. PLoS Genet 8: e1002440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi H, Sakurai T, Sato S, Nakabayashi K, Hata K, Kono T 2012b. Imprinted DNA methylation reprogramming during early mouse embryogenesis at the Gpr1-Zdbf2 locus is linked to long cis-intergenic transcription. FEBS Lett 586: 827–833 [DOI] [PubMed] [Google Scholar]
- Kobayashi H, Yanagisawa E, Sakashita A, Sugawara N, Kumakura S, Ogawa H, Akutsu H, Hata K, Nakabayashi K, Kono T 2013. Epigenetic and transcriptional features of the novel human imprinted lncRNA GPR1AS suggest it is a functional ortholog to mouse Zdbf2linc. Epigenetics 8: 635–645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong L, Zhang Y, Ye ZQ, Liu XQ, Zhao SQ, Wei L, Gao G 2007. CPC: Assess the protein-coding potential of transcripts using sequence features and support vector machine. Nucleic Acids Res 35: W345–W349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovacikova I, Polakova S, Benko Z, Cipak L, Zhang L, Rumpf C, Miadokova E, Gregan J 2013. A knockout screen for protein kinases required for the proper meiotic segregation of chromosomes in the fission yeast Schizosaccharomyces pombe. Cell Cycle 12: 618–624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latos PA, Pauler FM, Koerner MV, Senergin HB, Hudson QJ, Stocsits RR, Allhoff W, Stricker SH, Klement RM, Warczok KE, et al. 2012. Airn transcriptional overlap, but not its lncRNA products, induces imprinted Igf2r silencing. Science 338: 1469–1472 [DOI] [PubMed] [Google Scholar]
- Le AH, Mastro TL, Forsburg SL 2013. The C terminus of S. pombe DDK subunit Dfp1 is required for meiosis-specific transcription and cohesin cleavage. Biol Open 2: 728–738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Ito M, Zhou F, Youngson N, Zuo X, Leder P, Ferguson-Smith AC 2008. A maternal-zygotic effect gene, Zfp57, maintains both maternal and paternal imprints. Dev Cell 15: 547–557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lienert F, Wirbelauer C, Som I, Dean A, Mohn F, Schubeler D 2011. Identification of genetic elements that autonomously determine DNA methylation states. Nat Genet 43: 1091–1097 [DOI] [PubMed] [Google Scholar]
- Marks H, Kalkan T, Menafra R, Denissov S, Jones K, Hofemeister H, Nichols J, Kranz A, Stewart AF, Smith A, et al. 2012. The transcriptional and epigenomic foundations of ground state pluripotency. Cell 149: 590–604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maunakea AK, Nagarajan RP, Bilenky M, Ballinger TJ, D'Souza C, Fouse SD, Johnson BE, Hong C, Nielsen C, Zhao Y, et al. 2010. Conserved role of intragenic DNA methylation in regulating alternative promoters. Nature 466: 253–257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McEwen KR, Ferguson-Smith AC 2010. Distinguishing epigenetic marks of developmental and imprinting regulation. Epigenetics Chromatin 3: 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McEwen KR, Leitch HG, Amouroux R, Hajkova P 2013. The impact of culture on epigenetic properties of pluripotent stem cells and pre-implantation embryos. Biochem Soc Trans 41: 711–719 [DOI] [PubMed] [Google Scholar]
- Messerschmidt DM, de Vries W, Ito M, Solter D, Ferguson-Smith A, Knowles BB 2012. Trim28 is required for epigenetic stability during mouse oocyte to embryo transition. Science 335: 1499–1502 [DOI] [PubMed] [Google Scholar]
- Mikkelsen TS, Ku M, Jaffe DB, Issac B, Lieberman E, Giannoukos G, Alvarez P, Brockman W, Kim TK, Koche RP, et al. 2007. Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature 448: 553–560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikkelsen TS, Hanna J, Zhang X, Ku M, Wernig M, Schorderet P, Bernstein BE, Jaenisch R, Lander ES, Meissner A 2008. Dissecting direct reprogramming through integrative genomic analysis. Nature 454: 49–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molaro A, Hodges E, Fang F, Song Q, McCombie WR, Hannon GJ, Smith AD 2011. Sperm methylation profiles reveal features of epigenetic inheritance and evolution in primates. Cell 146: 1029–1041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monk M, Boubelik M, Lehnert S 1987. Temporal and regional changes in DNA methylation in the embryonic, extraembryonic and germ cell lineages during mouse embryo development. Development 99: 371–382 [DOI] [PubMed] [Google Scholar]
- Nora EP, Lajoie BR, Schulz EG, Giorgetti L, Okamoto I, Servant N, Piolot T, van Berkum NL, Meisig J, Sedat J, et al. 2012. Spatial partitioning of the regulatory landscape of the X-inactivation centre. Nature 485: 381–385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamoto I, Otte AP, Allis CD, Reinberg D, Heard E 2004. Epigenetic dynamics of imprinted X inactivation during early mouse development. Science 303: 644–649 [DOI] [PubMed] [Google Scholar]
- Ooi SK, Qiu C, Bernstein E, Li K, Jia D, Yang Z, Erdjument-Bromage H, Tempst P, Lin SP, Allis CD, et al. 2007. DNMT3L connects unmethylated lysine 4 of histone H3 to de novo methylation of DNA. Nature 448: 714–717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandey RR, Mondal T, Mohammad F, Enroth S, Redrup L, Komorowski J, Nagano T, Mancini-Dinardo D, Kanduri C 2008. Kcnq1ot1 antisense noncoding RNA mediates lineage-specific transcriptional silencing through chromatin-level regulation. Mol Cell 32: 232–246 [DOI] [PubMed] [Google Scholar]
- Pollard KS, Hubisz MJ, Rosenbloom KR, Siepel A 2010. Detection of nonneutral substitution rates on mammalian phylogenies. Genome Res 20: 110–121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proudhon C, Duffie R, Ajjan S, Cowley M, Iranzo J, Carbajosa G, Saadeh H, Holland ML, Oakey RJ, Rakyan VK, et al. 2012. Protection against de novo methylation is instrumental in maintaining parent-of-origin methylation inherited from the gametes. Mol Cell 47: 909–920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quenneville S, Verde G, Corsinotti A, Kapopoulou A, Jakobsson J, Offner S, Baglivo I, Pedone PV, Grimaldi G, Riccio A, et al. 2011. In embryonic stem cells, ZFP57/KAP1 recognize a methylated hexanucleotide to affect chromatin and DNA methylation of imprinting control regions. Mol Cell 44: 361–372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz DC, Ayyanathan K, Negorev D, Maul GG, Rauscher FJ 3rd 2002. SETDB1: A novel KAP-1-associated histone H3, lysine 9-specific methyltransferase that contributes to HP1-mediated silencing of euchromatic genes by KRAB zinc-finger proteins. Genes Dev 16: 919–932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz R, Woodfine K, Menheniott TR, Bourc'his D, Bestor T, Oakey RJ 2008. WAMIDEX: A Web atlas of murine genomic imprinting and differential expression. Epigenetics 3: 89–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seidl CI, Stricker SH, Barlow DP 2006. The imprinted Air ncRNA is an atypical RNAPII transcript that evades splicing and escapes nuclear export. EMBO J 25: 3565–3575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smallwood SA, Tomizawa S, Krueger F, Ruf N, Carli N, Segonds-Pichon A, Sato S, Hata K, Andrews SR, Kelsey G 2011. Dynamic CpG island methylation landscape in oocytes and preimplantation embryos. Nat Genet 43: 811–814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith ZD, Gu H, Bock C, Gnirke A, Meissner A 2009. High-throughput bisulfite sequencing in mammalian genomes. Methods 48: 226–232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith ZD, Chan MM, Mikkelsen TS, Gu H, Gnirke A, Regev A, Meissner A 2012. A unique regulatory phase of DNA methylation in the early mammalian embryo. Nature 484: 339–344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Statham AL, Robinson MD, Song JZ, Coolen MW, Stirzaker C, Clark SJ 2012. Bisulfite sequencing of chromatin immunoprecipitated DNA (BisChIP-seq) directly informs methylation status of histone-modified DNA. Genome Res 22: 1120–1127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun H, Wu J, Wickramasinghe P, Pal S, Gupta R, Bhattacharyya A, Agosto-Perez FJ, Showe LC, Huang TH, Davuluri RV 2011. Genome-wide mapping of RNA Pol-II promoter usage in mouse tissues by ChIP-seq. Nucleic Acids Res 39: 190–201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanay A, O'Donnell AH, Damelin M, Bestor TH 2007. Hyperconserved CpG domains underlie Polycomb-binding sites. Proc Natl Acad Sci 104: 5521–5526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsumura A, Hayakawa T, Kumaki Y, Takebayashi S, Sakaue M, Matsuoka C, Shimotohno K, Ishikawa F, Li E, Ueda HR, et al. 2006. Maintenance of self-renewal ability of mouse embryonic stem cells in the absence of DNA methyltransferases Dnmt1, Dnmt3a and Dnmt3b. Genes Cells 11: 805–814 [DOI] [PubMed] [Google Scholar]
- Ying QL, Wray J, Nichols J, Batlle-Morera L, Doble B, Woodgett J, Cohen P, Smith A 2008. The ground state of embryonic stem cell self-renewal. Nature 453: 519–523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuo X, Sheng J, Lau HT, McDonald CM, Andrade M, Cullen DE, Bell FT, Iacovino M, Kyba M, Xu G, et al. 2012. Zinc finger protein ZFP57 requires its co-factor to recruit DNA methyltransferases and maintains DNA methylation imprint in embryonic stem cells via its transcriptional repression domain. J Biol Chem 287: 2107–2118 [DOI] [PMC free article] [PubMed] [Google Scholar]