

Eukaryotic initiator tRNA (tRNAi) contains several highly conserved, unique sequence features, yet their importance in accurate start codon selection is unknown. Using genetic and biochemical analyses, Dong et al. show that conserved bases throughout tRNAi, from the anticodon stem to the acceptor stem, play key roles in ensuring the fidelity of start codon recognition. This work delineates specific molecular functions for signature initiator tRNA residues and establishes their importance for initiation accuracy in living eukaryotic cells.
Keywords: accuracy, initiation, initiator, scanning, tRNA, translation, yeast
Abstract
Eukaryotic initiator tRNA (tRNAi) contains several highly conserved unique sequence features, but their importance in accurate start codon selection was unknown. Here we show that conserved bases throughout tRNAi, from the anticodon stem to acceptor stem, play key roles in ensuring the fidelity of start codon recognition in yeast cells. Substituting the conserved G31:C39 base pair in the anticodon stem with different pairs reduces accuracy (the Sui− [suppressor of initiation codon] phenotype), whereas eliminating base pairing increases accuracy (the Ssu− [suppressor of Sui−] phenotype). The latter defect is fully suppressed by a Sui− substitution of T-loop residue A54. These genetic data are paralleled by opposing effects of Sui− and Ssu− substitutions on the stability of methionylated tRNAi (Met-tRNAi) binding (in the ternary complex [TC] with eIF2-GTP) to reconstituted preinitiation complexes (PICs). Disrupting the C3:G70 base pair in the acceptor stem produces a Sui− phenotype and also reduces the rate of TC binding to 40S subunits in vitro and in vivo. Both defects are suppressed by an Ssu− substitution in eIF1A that stabilizes the open/POUT conformation of the PIC that exists prior to start codon recognition. Our data indicate that these signature sequences of tRNAi regulate accuracy by distinct mechanisms, promoting the open/POUT conformation of the PIC (for C3:G70) or destabilizing the closed/PIN state (for G31:C39 and A54) that is critical for start codon recognition.
Identification of the translation initiation codon in eukaryotic mRNA typically occurs by a scanning mechanism where the 40S ribosomal subunit recruits methionylated initiator tRNA (Met-tRNAi) in a ternary complex (TC) with eIF2-GTP, the resulting 43S preinitiation complex (PIC) attaches to the mRNA 5′ end, and the leader sequence is inspected for complementarity with the anticodon of Met-tRNAi to identify the AUG start codon (Supplemental Fig. S1; Hinnebusch 2011). The GTP in TC is hydrolyzed in the scanning complex, dependent on eIF5, but Pi release is blocked by eIF1, which also impedes stable binding of Met-tRNAi in the P site. AUG recognition triggers dissociation of eIF1 from the 40S subunit (Maag et al. 2005), which allows interaction between eIF5 and the C-terminal tail (CTT) of eIF1A (Nanda et al. 2013), Pi release (Algire et al. 2005), and stable binding of TC to the P site (Passmore et al. 2007). Subsequent dissociation of eIF2-GDP and other eIFs enables eIF5B-catalyzed subunit joining and formation of an 80S initiation complex with Met-tRNAi base-paired to AUG in the P site (Supplemental Fig. S1; Pestova et al. 2007).
Both eIF1 and scanning enhancer (SE) elements in the eIF1A CTT promote an open, scanning-conducive conformation of the PIC and metastable mode of TC binding (the POUT state) that allows inspection of P-site triplets during scanning. A scanning inhibitor (SI) element in the eIF1A N-terminal tail (NTT) antagonizes SE function and promotes rearrangement to the closed state (Fekete et al. 2007), with dissociation of eIF1 (Cheung et al. 2007) and more stable binding of TC in the PIN conformation (Supplemental Fig. S2A; Saini et al. 2010). Biochemical mapping experiments for the eIF1A CTT (Yu et al. 2009) and X-ray crystal structures of PICs containing eIF1, eIF1A, or tRNAi (Rabl et al. 2011; Lomakin and Steitz 2013; Weisser et al. 2013) suggest that the eIF1A CTT and eIF1 physically obstruct Met-tRNAi binding in the PIN state, thus favoring POUT, whereas the eIF1A NTT likely stabilizes TC binding in the PIN state (Supplemental Fig. S2A).
Genetic experiments have implicated eIF1, eIF1A, eIF5, and eIF2 in accurate AUG selection in living cells. Sui− (suppressor of initiation codon) mutations in these factors enable initiation at the third, UUG codon in his4-301 mRNA, lacking the wild-type AUG codon, to restore growth on medium lacking histidine (His+/Sui− phenotype) (Yoon and Donahue 1992; Donahue 2000; Saini et al. 2010). Most Sui− mutations in eIF1 weaken its 40S binding and likely enable eIF1 release at near-cognate triplets (Valasek et al. 2004; Cheung et al. 2007; Martin-Marcos et al. 2013). Sui− mutations in the eIF1A SEs destabilize the open/POUT conformation, allowing transition from the open/POUT to closed/PIN state at near-cognates, and also reduce the rate of TC loading (Saini et al. 2010), as TC binds most rapidly to the open conformation (Supplemental Fig. S2B; Passmore et al. 2007). Substitution of residues 17–21 in the eIF1A SI element stabilizes the open/POUT state, which reduces UUG initiation in Sui− mutants—the Ssu− (suppressor of Sui−) phenotype (Fekete et al. 2007)—and also increases the rate of TC binding (Saini et al. 2010) while decreasing the rate of eIF1 dissociation (Supplemental Fig. S2C; Cheung et al. 2007).
tRNAi contains highly conserved sequences not present in elongator tRNAs (Fig. 1A; RajBhandary and Chow 1995; Marck and Grosjean 2002), with important functions in initiation. The A1:U72 base pair of the acceptor stem enhances eIF2-GTP binding to Met-tRNAi (Farruggio et al. 1996; Kapp and Lorsch 2004) and TC binding to 40S PICs (Kapp et al. 2006) and is required for wild-type tRNAi function in yeast cells (von Pawel-Rammingen et al. 1992; Astrom et al. 1993). The three consecutive G:C pairs in the anticodon stem–loop (ASL) promote P-site binding of tRNAi in eubacteria (Varshney et al. 1993; Mandal et al. 1996). They also confer efficient initiation in mammalian extracts (Drabkin et al. 1993) and enhance the stability of mammalian PICs reconstituted in vitro (Lomakin et al. 2006). In the reconstituted yeast system, the first (G29:C41) and third (G31:C39) of these G:C pairs were found to be required for the stabilizing effect of AUG on the affinity of TC for 43S·mRNA PICs. The deleterious effect on TC binding of substituting G31:C39 with the corresponding U:U pair in elongator Met-tRNA (tRNAeMet) (Fig. 1A) was mitigated by replacing conserved T-loop residues A54 and A60, suggesting interplay between the T-loop and ASL in AUG recognition by Met-tRNAi in the P site (Kapp et al. 2006). Surprisingly, however, G31:C39, G29:C41, A54, and A60 were altered to their tRNAeMet identities without affecting yeast growth (von Pawel-Rammingen et al. 1992), making it unclear whether the function of these residues identified in vitro is important in living cells for the efficiency or accuracy of initiation. To address this last question, we investigated whether substitutions in these and other conserved residues created by site-directed mutagenesis confer Sui− or Ssu− phenotypes in yeast cells. We also screened a library of substitutions produced by random mutagenesis for the Sui− phenotype. Our findings demonstrate that the identities of the third G:C pair of the ASL, T-loop residue A54, and the invariant C3:G70 pair in the acceptor stem are crucial for accurate AUG selection and that these signature residues use distinct molecular mechanisms to discriminate against near-cognate start codons.
Figure 1.

Loss of W:C pairing at 31:39 increases the accuracy of start codon recognition. (A) Secondary structures of yeast Met-tRNAi (left) and tRNAeMet (right). Arrows indicate substitutions analyzed below. (B) his4-301 strains with the indicated IMT2 alleles and harboring HIS4-lacZ fusions (shown schematically) with AUG or UUG start codons (on plasmids p367 and p391, respectively) were cultured in SD+His medium, and β-galactosidase activities were measured in whole-cell extracts. Ratios of mean activities from three transformants are plotted with error bars indicating SEMs. Asterisks indicate significant differences between mutant and wild type (WT) as judged by a Student’s t-test (P < 0.005). (C) his4-301 strains with the indicated IMT2 alleles and harboring a single-copy (sc) SUI5 plasmid or empty vector (Vec) were spotted on SD+His and incubated for 3 d (+His) or 6 d (−His) at 30°C. (D) UUG:AUG initiation ratios were determined as in B for strains harboring the indicated IMT2 alleles and sc SUI5, except using HIS4-lacZ reporters on p367 (AUG) and p4957 (UUG). Asterisks indicate significant differences between mutant and wild type or between two mutants (connected by a bracket) (P < 0.005). (E) In vivo analysis of aminoacylation. Total RNA was extracted and resolved by electrophoresis under acidic conditions and subjected to Northern analysis using [32P]-labeled oligonucleotides complementary to tRNAi (top) or tRNAeMet (bottom), and signal intensities were quantified by PhosphorImaging. For in vitro deacylation, an aliquot of each RNA was deacylated at pH 9.0. Normalized percentages of tRNAi aminoacylation were determined by calculating the ratio of signals Met-tRNAi/(Met-tRNAi + tRNAi) for each nondeacylated sample, normalizing to the same ratio determined for tRNAeMet, and expressing the results as a fraction of the value determined for wild type. (F) ASL structures and phenotypes for substitutions (in bold) at the 31:39 base pair. (G) Phenotypic analysis of strains with the indicated IMT2 alleles and sc SUI5 or empty vector conducted as in C. (H) UUG:AUG initiation ratios determined as in D for strains harboring the indicated IMT2 alleles and sc SUI5.
Results
Disrupting Watson-Crick pairing at G31:C39 in the ASL increases initiation accuracy
We examined substitutions of tRNAi for Sui− or Ssu− phenotypes using a his4-301 strain lacking all four genes (IMT1–IMT4) encoding the same wild-type tRNAi and harboring wild-type IMT4 on a URA3 plasmid. The latter was replaced with high-copy (hc) LEU2 plasmids containing the mutant IMT2 alleles of interest by counterselection with 5-fluoroorotic acid (5-FOA) (Boeke et al. 1987). Sui− phenotypes were recognized by the ability to grow on medium lacking histidine (−His), whereas Ssu− phenotypes were identified by the ability to suppress the dominant His+/Sui− phenotype conferred by the SUI5 allele of eIF5 introduced on a plasmid. Adverse effects of the IMT2 mutations on cell viability were quantified by measuring the efficiency of plating (EOP) on 5-FOA medium (Table 1; von Pawel-Rammingen et al. 1992). Viable mutants displaying significant reductions in EOP were characterized for slow-growth phenotypes (Slg−) by spotting serial dilutions on +His medium (Supplemental Fig. S3).
Table 1.
EOP measurements of IMT2 alleles
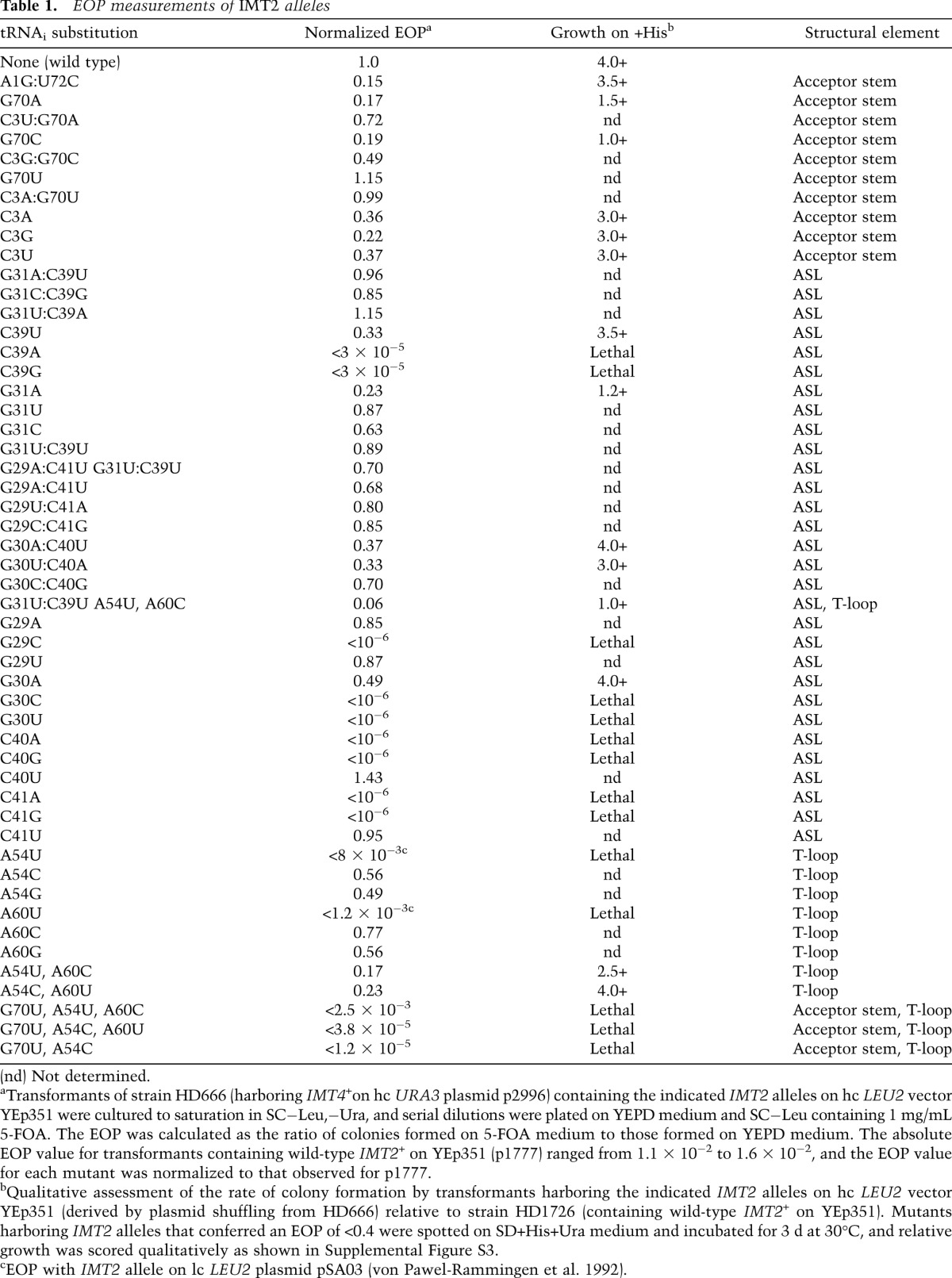
We began by analyzing replacements of the first and third of the three consecutive G:C pairs in the ASL, unique to tRNAi, with the A:U and U:U pairs found at these positions in tRNAeMet, generated by site-directed mutagenesis of IMT2 (Fig. 1A). These replacements, G29A:C41U and G31U:C39U, have little effect on the EOP (Table 1) or cell growth rate (von Pawel-Rammingen et al. 1992) and do not increase growth on −His medium in the manner expected for Sui− substitutions (data not shown). However, comparing expression of matched HIS4-lacZ reporters containing AUG or UUG start codons revealed ∼50% reduced UUG:AUG initiation ratios for both G31U:C39U and the G29A:C41U,G31U:C39U double substitution (Fig. 1B), suggesting that G31U:C39U increases initiation accuracy. This possibility is supported by the fact that G31U:C39U and the double substitution, but not G29A:C41U alone, are Ssu−, suppressing the His+ phenotype of SUI5 (Fig. 1C, rows 1,4,5) and reducing by ∼50% the elevated UUG:AUG ratio conferred by SUI5 (Fig. 1D). (These last measurements involved a HIS4-lacZ UUG reporter that mimics his4-301 in containing an ACG at codon-1 and UUG at codon-3.) Thus, converting G31:C39 to U31:U39 increases the requirement for an AUG start codon. Consistent with previous findings (von Pawel-Rammingen et al. 1992), Northern analysis of total RNA under acidic conditions shows that G31U:C39U does not diminish tRNAi abundance or the proportion aminoacylated in vivo (Fig. 1E, seventh and eight lanes vs. first and second lanes). (Unless otherwise stated, none of the tRNAi variants that we analyzed significantly reduce tRNAi abundance or aminoacylation [Fig. 5A {below}; Supplemental Fig. S4A–G]).
Figure 5.

Evidence that the Sui− and Gcd− phenotypes of the G70A substitution have a common molecular basis. (A) Substitutions of C3:G70 do not affect aminoacylation in vivo. Analysis conducted as in Figure 1E. (B–D) Substitutions of C3:G70 do not impair TC assembly in vivo. (B) Whole-cell extracts from strains harboring a SUI3-FL plasmid or empty vector were immunoprecipitated with Flag antibodies, and 5% of each immune complex was subjected to Western analysis with Flag or Gcd11/eIF2γ antibodies. (C) RNA extracted from the remainder was subjected to Northern analysis of tRNAi. The second lane in C derives from the IMT2+ strain with untagged SUI3; all others derive from SUI3-FL strains. Northern signals in C quantified by PhosphorImaging were normalized for FL-Sui3/eIF2β Western signals in B and quantified with the Odyssey Infrared imaging system, and the resulting ratios were normalized to those determined for the IMT2+SUI3-FL strain. Mean ratios and SEMs from three independent immunoprecipitations were plotted. (E) his4-301 strains harboring chromosomal PGAL1-TIF11 and plasmid-borne TIF11+ or tif11-17-21 and the indicated IMT2 allele analyzed as in Figure 2B. (F) GCN4-lacZ expression was assayed in cells cultured in SD+His (“0”) or with 1 mg/L sulfometuron methyl (SM) for 3 h or 6 h. (G) UUG:AUG initiation ratios were determined as in Figure 1B. (H) GCN4-lacZ expression was measured as in Figure 1B.
Interestingly, any of the three possible substitutions of G31, which disrupt Watson-Crick pairing (W:C) at 31:39 (Fig. 1F), resembled G31U:C39U in conferring Ssu− phenotypes, diminishing the His+ phenotype (Fig. 1G), and, at least for G31U and G31C, substantially lowering the elevated UUG:AUG ratio (Fig. 1H) in SUI5 cells. The weaker Ssu− phenotype of G31A might be attributable to the fact that it introduces an A:C wobble pair at 31:39. Although G31A evokes a 20%–30% reduction in the proportion of tRNAi aminoacylated in vivo (Fig. 1E), this is unlikely to contribute to its Ssu− phenotype because a strain lacking two of four IMT genes (IMT3 and IMT4), with substantially reduced levels of tRNAi and TC (Dever et al. 1995), displays essentially wild-type UUG:AUG initiation (Supplemental Fig. S5). C39A and C39G, which also disrupt W:C pairing at 31:39, are lethal (Table 1; Fig. 1F). These findings are consistent with the possibility that W:C pairing at 31:39 is important for efficient start codon recognition such that viable Ssu− substitutions disrupting 31:39 increase the requirement for AUG and thereby diminish UUG initiation. In this view, the lethal substitutions would substantially reduce recognition of AUG as well as near-cognates.
We also examined the effects of disrupting W:C pairing at G29:C41 and G30:C40. As summarized in Figure 2F, seven of eight single-base substitutions that introduce purine:purine or pyrimidine:pyrimidine pairs at positions 29:41 or 30:40 are lethal. In contrast, substitutions that generate A:C or G:U wobble replacements are viable and either have no effect on initiation accuracy (G29A, G30A, and C40U) or moderately increase accuracy and confer a weak Ssu− phenotype (C41U) (Table 1; Supplemental Fig. S6A–E).
Figure 2.

W:C replacements at 31:39 and A54 substitutions in the T-loop reduce the accuracy of start codon recognition. (A) ASL structures and phenotypes (in order of severity: Sui− > Sui−/+ > Sui+/−) for W:C substitutions of G:C pairs. (B,D) his4-301 strains with the indicated IMT2 alleles were replica-plated to +His medium (0.3 mM His) or 0.5% His (1.5 µM His) and incubated for 3 d (+His) or 7–10 d (0.5% His). (C,E,G) UUG:AUG initiation ratios were determined as in Figure 1B for strains with the indicated IMT2 alleles. (F) Phenotypes of substitutions that disrupt W:C pairing at positions 29:41 and 30:40. (H) Structures of the T-loop with substitutions at A54 or A60 and the associated phenotypes.
Thus, the integrity of all three G:C base pairs in the ASL stem is critical in vivo, as substitutions that generate purine:purine pairs at any of these positions are lethal, and pyrimidine:pyrimidine pairs are either lethal (first and second base pairs) or confer marked hyperaccuracy (Ssu−) phenotypes (third base pair). In contrast, most wobble replacements appear to have little or no effect on initiation accuracy.
W:C substitutions of 31:39 in the ASL strongly decrease initiation accuracy
We next analyzed the effects of double substitutions that replace the conserved G:C base pairs with other W:C pairs (Fig. 2A). Remarkably, all three W:C replacements of G31:C39 as well as C39U, which produces a wobble G:U at this position, substantially reduce initiation accuracy, conferring His+/Sui− phenotypes (Fig. 2B) and increasing the UUG:AUG ratio sixfold to sevenfold for the W:C substitutions and ∼2.5-fold for the G:U replacement (Fig. 2C). In contrast, W:C substitutions of the first G:C pair did not produce His+ phenotypes (Fig. 2D; data not shown) and evoked <30% increases in the UUG:AUG ratio (Fig. 2E). W:C replacements of the second G:C pair conferred somewhat greater increases in the UUG:AUG ratio (Fig. 2E) and a His+ phenotype only for G30C:C40G (Fig. 2D). As noted above, C40U and C41U substitutions that introduce G:U wobble pairs at 29:41 and 30:40 do not confer Sui− phenotypes (Fig. 2F). Thus, W:C substitutions at each of the ASL G:C pairs reduce initiation accuracy, with the strongest defects for the 31:39 substitutions adjacent to the anticodon loop and the weakest defects for the 29:41 substitutions furthest from the anticodon loop (Fig. 2A). One way to explain these findings is to propose that the ASL G:C pairs promote initiation accuracy by affecting the conformation of the anticodon loop, with the 31:39 pair closest to the loop having the greatest effect on accuracy.
Ssu− disruptions of 31:39 destabilize the closed/PIN conformation of the PIC
The fact that G31C and G31U evoke Ssu− phenotypes suggests that these mutations destabilize the closed/PIN state of the PIC normally triggered by start codon recognition (Supplemental Fig. S2C). To test this interpretation, we examined the effects of these substitutions on the equilibrium and rate constants governing TC binding to the 40S subunit. We first determined that [35S]-Met-tRNAi variants harboring these substitutions all efficiently form TC with eIF2 and GDPNP (Supplemental Table S1). Subsequently, we measured the affinities of the TCs for the 40S subunit in the presence of saturating eIF1, eIF1A, and a model mRNA containing an AUG or UUG start codon using native gel electrophoresis to separate bound and unbound fractions of TC. Interestingly, G31C and G31U increased the affinity of TCs for 40S complexes lacking mRNA while greatly reducing affinity in the presence of mRNA(AUG) (Fig. 3A; Table 2). Strikingly, the affinity of these mutant TCs for the 43S·mRNA(UUG) complex is so low that no complex formation could be detected at the highest concentrations of 40S subunits employed (≥250 nM) (Table 2; data not shown). These findings support the prediction that G31C and G31U destabilize the closed/PIN state, and the fact that TC affinity is much lower for the UUG versus AUG complex is consistent with the Ssu− phenotype of these mutations (Fig. 1H). Moreover, considering that the wild-type TC has a much lower affinity for 43S versus 43S·mRNA(AUG) complexes (Table 2; Kapp et al. 2006), the fact that G31C/G31U essentially eliminate this differential in stability indicates that they abolish thermodynamic coupling between Met-tRNAi and the start codon.
Figure 3.

Disrupting ASL base pair G31:C39 and replacing it with other W:C base pairs have opposite effects on the stability of 43S·mRNA complexes. (A,B) Determination of Kd values for TC (with wild-type [WT] or the indicated variant of [35S]-Met-tRNAi) binding to 40S•eIF1•eIF1A complexes assembled with mRNA(AUG) (A) or mRNA(UUG) (B). (C,D) Determination of kon values for TC binding to 40S•eIF1•eIF1A complexes from plots of observed rate constants (kobs) versus 40S concentration with mRNA(AUG) (C) or mRNA(UUG) (D). (E,F) Analysis of TC dissociation from 43S•mRNA complexes for mRNA(AUG) (E) or mRNA(UUG) complexes (F). Representative curves from at least two independent experiments are shown. koff values and end-points for dissociable complexes are given in Supplemental Table S2.
Table 2.
Affinity of TC for 40S·eIF1·eIF1A·mRNA complexes
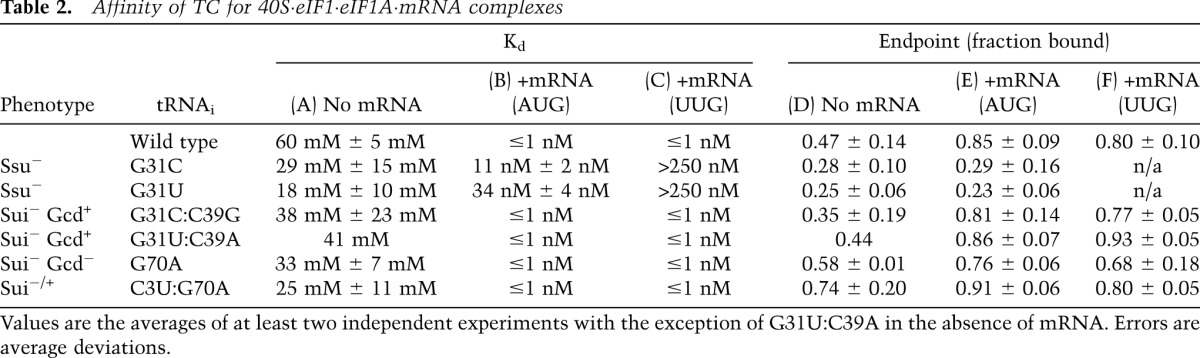
We previously proposed that the endpoints of curves for TC binding at high concentrations of 40S subunits reflect the distribution of PICs in the open versus closed states; the open state was proposed to be unstable during electrophoresis and therefore could not be visualized, leading to endpoints of less than one (measured as fractions of TC bound to 40S complexes) in cases where open complex persists (Kapp et al. 2006; Kolitz et al. 2009). Consistent with this idea, G31C and G31U, which we propose bias the system toward the open/POUT state, decrease the endpoints of TC binding in both the absence and presence of mRNA(AUG) (Fig. 3A; Table 2).
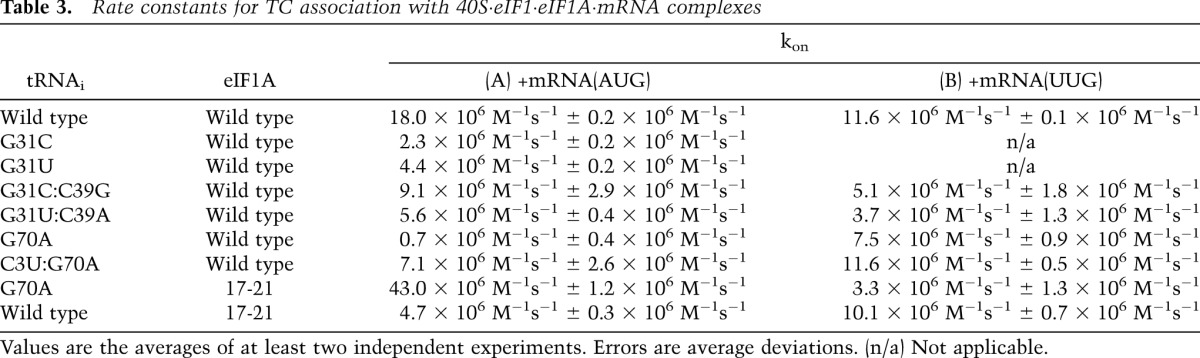
To gain more insight into the effect of these mutations on formation and stability of the PIC, we measured the rate constants for TC forming and dissociating from PICs. The kinetics of TC binding was measured by mixing TC containing [35S]-Met-tRNAi with varying concentrations of 40S subunits and saturating eIF1, eIF1A, and mRNA(AUG) or mRNA(UUG). Time points were removed, reactions were terminated with excess unlabeled TC, and the amount of labeled TC in PICs was measured by native gel electrophoresis. The slope of the plot of the pseudo-first-order rate constants (kobs) for PIC formation versus 40S concentration yields the second-order rate constant (kon) (Kolitz et al. 2009).
The G31C and G31U mutations decrease kon for TC in the presence of mRNA(AUG) by eightfold and fourfold, respectively (Fig. 3C; Table 3). The corresponding values with mRNA(UUG) could not be determined because TC binding with these mutants is too weak to measure. One possible interpretation of these data is that the mutations slow conversion of the open/POUT state of the PIC to the closed/PIN state, which is dramatically accelerated by start codon recognition in wild-type PICs and has a strong influence on the observed rate of PIC formation (Kolitz et al. 2009). Slowing of this step would be consistent with the mutants’ Ssu− phenotypes.
Table 3.
Rate constants for TC association with 40S·eIF1·eIF1A·mRNA complexes
Next, we measured the rate at which TC made with the mutant tRNAs dissociates from PICs. After assembling 43S·mRNA complexes as above, we quantified the amount of [35S]-Met-tRNAi remaining in the slowly migrating PIC band as a function of time after adding a chase of excess unlabeled TC made with wild-type Met-tRNAi. With wild-type PICs formed with mRNA(AUG), TC dissociates from ∼40% of the PICs with a rate constant of 0.4 h−1, whereas the remaining ∼60% of the complexes are completely stable over this time period (Fig. 3E). We presume that the former fraction of PICs contain Met-tRNAi bound in the PIN state, whereas the latter arise by isomerization of Met-tRNAi from PIN to a new state where it is fully locked in to the P site. This putative highly stable state might be closer to the classical P/P state than the P/I state observed recently in reconstituted mammalian PICs (Hashem et al. 2013; Lomakin and Steitz 2013), which can be regarded as the PIN conformation. (As the AUG codon-dependent conversion to the initial closed/PIN state is rapid [Kolitz et al. 2009], it is unlikely that the two states represent open/POUT and closed/PIN because if the rate of reversion of the closed/PIN state back to the open/POUT state was slow, all of the complexes should be in the closed/PIN state, whereas if the rate of reversion was fast, all of the complexes should dissociate on chasing with unlabeled TC.) Importantly, dissociation of wild-type TC from PICs assembled on mRNA(UUG) goes to completion and occurs with a rate constant of 1.2 h−1 (Fig. 3F), suggesting that PICs do not achieve the highly stable state with a UUG codon in the P site.
Interestingly, both G31C and G31U increase the fraction of AUG complexes from which TC can dissociate, from ∼40% with wild type to 100% with the mutants (Fig. 3E), resembling the behavior of wild-type complexes at UUG (Fig. 3F). Thus, the Ssu− mutations decrease the ability of the PIC to enter the highly stable state accessible to the wild-type complex.
Sui− W:C substitutions of 31:39 stabilize the closed/PIN state
As described above, the Ssu− phenotypes of the G31C and G31U mutations in the ASL are suppressed by the compensatory C39G and C39A mutations that restore W:C pairing at this position; and these double mutants produce Sui− phenotypes instead, suggesting that they shift the balance in favor of the closed/PIN state. Consistent with this proposal, G31C:C39G and G31U:C39A dramatically reduce the Kds for TC binding to the 40S complex with both mRNA(AUG) and mRNA(UUG) relative to the Kds with the G31C and G31U single substitutions and also restore the endpoints of TC binding (Fig. 3A,B; Table 2). These data suggest that the double mutations stabilize the closed/PIN state.
Analysis of dissociation kinetics revealed that G31C:C39G and G31U:C39A produce complexes from which >80% of the TC does not dissociate with mRNA(AUG) or mRNA(UUG) (Fig. 3E,F), indicating that they not only favor the closed/PIN state but lead to more complexes entering the highly stable state. The magnitude of these changes appear to be bigger with mRNA(UUG) (Fig. 3E,F, cf. curves for wild type vs. G31C:C39G and G31U:C39A), consistent with the elevated UUG:AUG initiation ratios observed in vivo for these variants.
Analysis of association kinetics showed that the kon value in the presence of mRNA(AUG) with the G31C:C39G mutant was increased fourfold relative to that with G31C (Fig. 3C; Table 3), suggesting that restoring this base pair speeds up conversion of the open/POUT state to the closed/PIN state on start codon recognition. The G31U:C39A mutation does not enhance the rate of TC loading relative to that seen with the Ssu− G31U mutant, however (Fig. 3C; Table 3), suggesting that the key effect of the Sui− mutations is on stability of the closed/PIN state, which is reflected in the dissociation rates. Stable complexes could not be formed on mRNA(UUG) with the G31C and G31U mutants, but restoring base-pairing at position 31:39 restores stable complex formation, as noted above. The kon values for mRNA(UUG) complexes with the G31C:C39G and G31U:C39A mutants were twofold to threefold lower than with wild type (Fig. 3D; Table 3), but because kon values could not be measured with the single mutants, we cannot determine the extent to which restoring the base pair increases kon.
T-loop residue A54 contributes to stringent AUG selection
The results described above indicate that Ssu− substitutions G31C and G31U destabilize TC binding to 43S·mRNA complexes in vitro (Table 2). We observed the same outcome previously on replacing G31:C39 with the U:U pair found in tRNAeMet (Kapp et al. 2006), and, importantly, we concluded above that G31U:C39U likewise confers an Ssu− phenotype in vivo (Fig. 1B–D). We also reported that substitutions A54U and A60C of the two signature T-loop residues of tRNAi reduced the deleterious effect of G31U:C39U on the affinity of TC for 43S·mRNA(AUG) complexes (Kapp et al. 2006). We reasoned that if the Ssu− phenotype of G31U:C39U results from less stable binding of Met-tRNAi to the closed/PIN state of the PIC, as proposed above for G31C and G31U, then the A54U,A60C substitutions should suppress the Ssu− phenotype of G31U:C39U.
Remarkably, combining A54U,A60C with G31U:C39U restores the His+ phenotype (Fig. 1C, cf. rows 4,7 vs. row 1) and reinstates the elevated UUG:AUG ratio conferred by SUI5 in IMT2+ cells (Fig. 1D, cf. columns 3,6 vs. column 1). A54U,A60C also produces a modest Sui− phenotype in otherwise wild-type cells, increasing the UUG:AUG ratio (Fig. 1B). Consistent with this, A54U,A60C exacerbates the Sui− phenotype of SUI5, decreasing growth on +His but not −His medium (Fig. 1C, rows 1,6) and elevating the UUG:AUG ratio above that seen in SUI5 IMT2+cells (Fig. 1D, columns 1,5). Note also that G31U:C39U reverses the Slg− phenotype of the A54U,A60C substitution in SUI5 cells on +His medium (Fig. 1C, +His, rows 1,6,7), which likely reflects the ability of G31U:C39U to mitigate the elevated UUG initiation conferred by A54U,A60C in SUI5 cells (Fig. 1D, columns 5,6). Thus, replacing both highly conserved T-loop residues with the corresponding bases in tRNAeMet decreases the accuracy of AUG selection (Sui−) and suppresses the hyperaccurate (Ssu−) phenotype of the ASL substitution G31U:C39U, and these substitutions mutually suppress their opposing effects on initiation accuracy. The fact that A54U,A60C suppresses the destabilizing effect on TC binding to 43S·mRNA PICs in vitro (Kapp et al. 2006) as well as the Ssu− phenotype of G31U:C39U in cells (Fig. 1C,D) provides strong evidence that the stability of the closed/PIN state of the PIC is a critical determinant of initiation accuracy in vivo.
We went on to explore which T-loop substitution confers the moderate Sui− phenotype of A54U,A60C. Neither A60C nor A60G single substitutions affected the UUG:AUG ratio (summarized in Fig. 2H; data not shown). These findings, together with the fact that A54C alone confers a Sui− phenotype (Fig. 2G), suggest that A54U is responsible for the Sui− phenotype of A54U,A60C. However, we cannot eliminate the possibility that A60C contributes to the Sui− phenotype of the A54U,A60C double mutant. Interestingly, both A54U and A60U are lethal (Table 1), which might derive from their ability to extend the T-stem as depicted in Figure 2H. However, A54C alone or in combination with A60U increases the UUG:AUG ratio by a factor of 2.0–2.5 (Fig. 2G), whereas A54G does not significantly affect the UUG:AUG ratio (data not shown). We conclude that a purine residue is required at position 54 in the T-loop for wild-type discrimination against UUG start codons.
Acceptor stem residue G70 is crucial for stringent AUG selection
To identify additional determinants of initiation accuracy, we screened a library of mutant IMT2 plasmids produced by random mutagenesis for a His+ phenotype in the his4-301 strain and identified the G70U substitution in the acceptor stem as a novel Sui− mutation (Fig. 4C [−His, rows 1,2], D [columns 1,6]). C3:G70 is a highly conserved feature of tRNAi in all kingdoms of life (Marck and Grosjean 2002), but its possible function in initiation was unknown. Interestingly, site-directed mutagenesis showed that G70A, G70C, and C3G confer even stronger His+/Sui− phenotypes (Fig. 4C) and larger (ninefold or greater) increases in UUG:AUG initiation compared with G70U (Fig. 4D). However, C3A and C3U confer smaller increases in the UUG:AUG ratio (∼2.5-fold) (Fig. 4D) and little growth on −His medium (Fig. 4C). The strength of the His+/Sui− and elevated UUG:AUG initiation phenotypes of these substitutions (Fig. 4C [−His], D) correlate well with their effects on cell growth (Fig. 4C, +His). Thus, disrupting W:C pairing at 3:70 confers a Sui− phenotype whose severity varies with the substitution (summarized in Fig. 4A).
Figure 4.

C3:G70 in the acceptor stem is crucial for accurate AUG selection and rapid TC binding to PICs in vivo. (A) Acceptor stem structures and phenotypes (in order of severity: Sui3− > Sui2− > Sui− > Sui−/+) for 3:70 substitutions. (B) Slg− phenotypes on +His medium analyzed as in Figure 1C. (C) His+/Sui− phenotypes analyzed as in Figure 2B. (Results in B and C were obtained in parallel from the same plates and rearranged only for ease of interpretation.) (D) UUG:AUG initiation ratios were determined as in Figure 1B. (E) β-Galactosidase expressed from the GCN4-lacZ reporter on p180 measured as in Figure 1B.
C3U, which introduces a U:G wobble pair (Fig. 4A), produces one of the weakest Sui− phenotypes among the single-base changes in the 3:70 base pair (Fig. 4C,D). Consistent with this, combining C3U with G70A to introduce a W:C replacement at the 3:70 base pair (Fig. 4A) effectively diminishes the Slg−, His+, and elevated UUG:AUG initiation phenotypes conferred by G70A (Fig. 4B–D). Similar findings were obtained when G70C and G70U were combined with the appropriate C3 substitutions to reinstate W:C pairing, although suppression of the strong Sui− phenotypes of G70C and G70U was less pronounced, as the double substitutions retained weak His+ phenotypes and significantly elevated UUG:AUG ratios (Fig. 4A–D). Nevertheless, it is noteworthy that combining these mutations mitigated rather than exacerbated their respective Sui−/Slg− phenotypes. We conclude that base-pairing per se and the identity of the W:C pair at position 3:70 both contribute to discrimination against UUG start codons.
C3:G70 substitutions confer Gcd− phenotypes without decreasing TC abundance in vivo
Interestingly, we found that the Sui− substitutions at C3:G70 confer constitutive derepression of a GCN4-lacZ reporter, the Gcd− phenotype, indicating a reduced rate of TC binding to 40S subunits in vivo. A decrease in the rate of TC binding to 40S subunits derepresses GCN4-lacZ expression because scanning 40S subunits that have already translated uORF1 can bypass the start codons of the inhibitory uORF2–4 before rebinding TC and then reinitiate further downstream at the GCN4 AUG codon instead (Hinnebusch 2005). Whereas G70A, G70C, G70U, C3A, and C3G, all disrupting C3:G70, confer twofold to threefold increases in GCN4-lacZ reporter expression in nonstarvation conditions, the C3U:G70 wobble substitution and C3U:G70A, C3G:G70C, and C3A:G70U double substitutions (producing W:C replacements) have smaller (<1.7-fold) effects on GCN4-lacZ expression (Fig. 4E). In particular, C3U:G70A suppresses the marked derepression of GCN4-lacZ conferred by G70A (Fig. 4E, columns 2,3). None of the G70 or C3 single substitutions significantly affected expression of a GCN4-lacZ reporter lacking all four uORFs (Supplemental Fig. S7A), indicating that they alter translational control of GCN4-lacZ expression. The severity of the Gcd− phenotypes provoked by disrupting or altering C3:G70 is generally correlated with that of the Sui− phenotypes produced by these mutations (Fig. 4, cf. D and E), suggesting that these defects are mechanistically linked. Moreover, none of the strong Sui− substitutions involving W:C replacements of G31:C39 confers a Gcd− phenotype (Supplemental Fig. S7B), suggesting distinct mechanisms underlying the Sui− phenotypes of 31:39 versus 3:70 substitutions.
Importantly, none of the Gcd− G70 substitutions (G70A, G70C, and G70U) reduces tRNAi abundance (Supplemental Fig. S4E) or tRNAi aminoacylation in vivo (Fig. 5A; data not shown). Given the location of C3:G70 in the acceptor stem, which contacts eIF2 (Shin et al. 2011), G70 substitutions might reduce TC formation as the means of derepressing GCN4 translation. In fact, our in vitro measurements of Met-tRNAi binding to eIF2 revealed approximately fourfold increases in Kd for C3U and G70A that were mitigated in the C3U:G70A double mutant (Supplemental Table S1). Accordingly, we measured native TC levels in cell extracts by immunoprecipitating Flag-tagged eIF2β (FL-Sui3) expressed in the IMT2 mutants of interest, probed immune complexes by Northern analysis for tRNAi levels, and normalized the tRNAi signal for amounts of immunoprecipitated FL-Sui3 (Fig. 5B–D). We verified that only a low background level of tRNAi was immunoprecipitated from the parental IMT2+ strain containing untagged eIF2β (Fig. 5C,D, [−]tag) and that the eIF2γ-N135D substitution (Alone et al. 2008) reduces the amount of tRNAi coimmunoprecipitating with eIF2β-FL (Supplemental Fig. S8A–C). Using this assay, we observed no difference between wild type and the G70U, G70A, and G70C mutants (Fig. 5B–D), suggesting that G70 substitutions do not significantly reduce TC abundance in vivo. Accordingly, their Gcd− phenotypes likely result instead from reducing the rate of TC binding to 40S subunits scanning downstream from uORF1, which induces reinitiation at GCN4 (Hinnebusch 2005).
eIF1A mutation 17-21 cosuppresses Sui− and Gcd− phenotypes of C3:G70 substitutions
We showed previously that mutations in the SEs of the eIF1A CTT confer Sui− and Gcd− phenotypes that are cosuppressed by the Ssu− mutation 17-21 in the SI element of the eIF1A NTT. This and other findings led us to conclude that SE mutations destabilize the open conformation of the 40S subunit and POUT mode of TC binding. Destabilization of the open/POUT state reduces the rate of TC binding and confers the Gcd− phenotype, as TC binds most rapidly to the open conformation and also shifts the balance from the open/POUT to closed/PIN state to permit more frequent initiation at UUG codons for the Sui− phenotype (Supplemental Fig. S2B). The 17-21 substitution suppresses both defects by stabilizing the open/POUT state, restoring rapid TC loading and maintaining the scanning-conducive conformation at UUG codons (Supplemental Fig. S2C; Saini et al. 2010).
Thus, it was of interest to determine whether the 17-21 mutation can also cosuppress the Sui− and Gcd− phenotypes of the G70A substitution. To this end, we constructed strains in which expression of wild-type eIF1A from a chromosomal PGAL1-TIF11 allele is repressed on glucose medium, and either the wild-type or 17-21 forms of eIF1A are expressed constitutively from plasmid-borne alleles under the native promoter. Remarkably, the His+/Sui− phenotype (Fig. 5E) and elevated UUG:AUG initiation ratio (Fig. 5G) as well as the Gcd− phenotype (Fig. 5H) conferred by G70A were essentially eliminated in the tif11-17-21 strain. This cosuppression of G70A phenotypes suggests that, like eIF1A SEs, the C3:G70 base pair preferentially stabilizes the open/POUT conformation of the PIC.
We also demonstrated that tif11-17-21 has little effect on induction of GCN4-lacZ expression in cells expressing wild-type tRNAi in response to starvation for isoleucine and valine, which lowers TC abundance via phosphorylation of eIF2α (Fig. 5F). The ability of tif11-17-21 to block derepression of GCN4-lacZ in response to the G70A substitution (Fig. 5H), but not in response to amino acid starvation (Fig. 5F), supports our proposal that G70A reduces the rate of TC binding to the PIC rather than reducing TC abundance.
Evidence that substituting G70 destabilizes the open/POUT conformation of the PIC
We sought to test our interpretation of the genetic data that G70A reduces the rate of TC binding to the PIC in a manner mitigated by the eIF1A 17-21 mutation. Measuring the rate constant for TC binding to PICs containing model mRNA(AUG) as described above revealed a dramatic ∼25-fold reduction in kon for TC assembled with the G70A variant of Met-tRNAi. This defect was strongly diminished in the C3U:G70A double mutant and, remarkably, was fully suppressed by the 17-21 variant of eIF1A (Fig. 6A; Table 3). The fact that the C3U substitution and eIF1A-17-21 both overcome the defect in TC binding in vitro and the Gcd− phenotype in vivo conferred by G70A strongly suggests that a W:C pair at position 3:70 is required for rapid binding of TC to the open/PIN conformation of the PIC (Saini et al. 2010).
Figure 6.

Disrupting acceptor stem base pair C3:G70 shifts the equilibrium from POUT to PIN. (A,B) Determination of kon values for TC association with 40S•eIF1•eIF1A complexes and mRNA(AUG) (A) or mRNA(UUG) (B). Each value is the average of at least two independent experiments. Errors are average deviations. (C,D) Analysis of TC dissociation from 43S·mRNA complexes for mRNA(AUG) (C) or mRNA(UUG) (D) complexes. Representative curves from at least two independent experiments are shown. koff values and endpoints for dissociable complexes are given in Supplemental Table S2. (E) PyMol rendering of the crystal structure of yeast tRNAi (Protein Data Bank [PDB]: 1YFG) using color-coding to designate the acceptor stem (red), T-stem–loop (green), D-stem–loop (blue), and ASL (gold) and depicting by spheres the bases or base pairs implicated here in start codon recognition. (F) Model summarizing the deduced roles of conserved tRNAi residues in start codon recognition. See Supplemental Figure S2 for description of the open/POUT and closed/PIN states of the PIC and roles of eIF1 and the SEs/SI elements of eIF1A in regulating conformational rearrangements and reactions accompanying AUG recognition. Results in this study indicate that base pair C3:G70 functions together with eIF1 and eIF1A SEs to stabilize the POUT conformation of TC binding, whereas residues A54/A60 impede rearrangement to the PIN state in a manner overcome efficiently only with the perfect codon:anticodon duplex formed at AUG. G31:C39 and most likely the other two ASL G:C pairs are required for thermodynamic coupling between AUG and tRNAi in the PIN state. Not summarized here is the fact that replacing G31:C39 with other Watson-Crick pairs further stabilizes PIN and thereby increases initiation at NUG near-cognates (see Supplemental Figs. S11–S13 for further details.)
Despite the ∼20-fold decrease in kon, the G70A substitution does not show a detectable increase in the Kd for TC in 43S·mRNA(AUG) complexes (Table 2), which implies that it also substantially reduces the koff value for these PICs. Indeed, G70A eliminates detectable dissociation of TC from 43S·mRNA(AUG) complexes, leading to nearly 100% of the complexes being in the highly stable state (Fig. 6C). G70A also significantly increases the fraction of 43S·mRNA(UUG) complexes in the highly stable state from almost none with wild type to ∼70% for the mutant (Fig. 6D). These findings support the idea that, by destabilizing TC binding to the open/POUT conformation, G70A shifts the equilibrium toward the closed/PIN state, which increases the probability of UUG initiation. Supporting this interpretation, combining C3U with G70A in the double mutant, which diminishes the Sui− phenotype of G70A, also diminishes the increased formation of the highly stable state by 43S·mRNA(UUG) complexes conferred by G70A alone from ∼70% with G70A to ∼35% with C3U:G70A (Fig. 6D).
It is intriguing that the G70A substitution has little effect on the kon for 43S·mRNA(UUG) complexes despite the fact that G70A reduces kon for 43S·mRNA(AUG) complexes by ∼20-fold (Fig. 6A,B; Table 3). If we adhere to our conclusion above that G70A decreases kon for 43S·mRNA(AUG) complexes by reducing occupancy of the open/POUT state, we would expect a similar decrease for 43S·mRNA(UUG) complexes, as codons are not recognized in the open/POUT conformation. One explanation might be that the predicted reduction in kon for 43S·mRNA(UUG) complexes conferred by slower TC loading to the open/POUT state is offset by an increase of nearly equal magnitude in the rate of POUT-to-PIN isomerization at UUG but not AUG codons. To explain why G70A would selectively accelerate the POUT-to-PIN transition at UUG, it could be proposed that G70A perturbs interaction of Met-tRNAi with eIF2 in a way that alters the orientation of TC in the P site to favor base-pairing with UUG, which entails a U:U mismatch at the first position of the codon:anticodon duplex but not for the perfect codon:anticodon duplex at AUG. We came to a similar conclusion recently regarding the SUI5 mutation in eIF5 that stabilizes PIN at UUG while destabilizing it at AUG (Martin-Marcos et al. 2014).
G31:C39 discriminates preferentially against near-cognates with first position mismatches
Results above indicate that Sui− substitutions in the ASL and acceptor stem decrease initiation accuracy by distinct mechanisms. We asked whether they also differ in their effects on utilization of different near-cognates by comparing expression of firefly luciferase reporters harboring different start codons normalized for expression of a renilla luciferase reporter bearing an AUG codon (Takacs et al. 2011). Interestingly, the G31A:C39U ASL substitution elevates utilization of UUG, CUG, or GUG triplets, all first-base mismatches, but not AUA or ACG near-cognates with second- or third-base mismatches; whereas this bias does not exist for the G70A substitution in the acceptor stem (Supplemental Fig. S9). Thus, G31A:C39U differs from G70A by increasing utilization of near-cognates with first position mismatches.
ASL Sui− substitution G31A:C39U does not alter N6-threonylcarbamoyl modification of A37
N6-threonylcarbamoyl modification of A37 (t6A37) immediately adjacent to the anticodon triplet in tRNA is thought to stabilize the first base pair of the codon:anticodon duplex for the subset of tRNAs that decode ANN triplets (Agris 2008), which includes decoding of AUG by tRNAi. Consistent with this, mutations that reduce t6A37 formation in yeast impair recognition of AUG codons (Lin et al. 2009; Daugeron et al. 2011; Srinivasan et al. 2011) and can increase the ratio of GUG to AUG initiation (El Yacoubi et al. 2011). To eliminate the possibility that G31:C39 substitutions confer Sui− phenotypes by impairing t6A37 formation, we purified wild-type and G31A:C39U mutant tRNAi, digested them with nuclease P1, and resolved the nucleoside products by high-performance liquid chromatography (HPLC). Quantification of the HPLC tracings revealed that both mutant and wild-type tRNAi contain ∼1 mol of t6A per mole of tRNA (Supplemental Fig. S10).
Discussion
In this study, we probed the roles of highly conserved, signature residues of tRNAi in the ASL, T-loop, and acceptor stem (Fig. 6E) in determining the stability of TC binding to the PIC in vitro and the accuracy of translation initiation in vivo. Our findings implicate the ASL base pair G31:C39, T-loop residues A54,A60, and acceptor stem base pair C3:G70 in stringent AUG selection in yeast cells and indicate that these residues function by distinct biochemical mechanisms. All substitutions introducing purine:purine mismatches at any of the three G:C pairs in the ASL are lethal, as are most substitutions creating pyrimidine:pyrimidine mismatches at the first or second G:C pair. In contrast, pyrimidine:pyrimidine mismatches are tolerated at the third G:C pair, as are A:C or G:U wobble pairs at all three positions (Figs. 1F, 2F). We interpret these findings to indicate that disruption of the ASL helix is lethal and that substitutions eliminating both W:C and wobble pairing have a greater effect on helix stability for the first or second G:C pairs, owing to their internal locations, versus the third G:C pair at the end of the helix. With the exception of the G:U replacement of the third G:C pair, nonlethal substitutions introducing wobble pairs at any of these positions either have no effect on initiation accuracy or, for C41U and G31A, confer moderate hyperaccuracy (Ssu−) phenotypes. Much stronger Ssu− phenotypes were observed for the viable replacements of the third G:C pair with the pyrimidine:pyrimidine mismatches C:C, U:C, or U:U. One way to explain these findings is to propose that any substitution that prevents both W:C and wobble pairing at any of these three positions impairs start codon recognition. For the lethal substitutions, AUG as well as near-cognate recognition would be substantially reduced, whereas nonlethal Ssu− substitutions would impair AUG recognition less dramatically while still conferring a marked reduction in near-cognate (UUG) initiation.
Our biochemical analysis of the Ssu− substitutions G31C and G31U, which introduce C:C or U:C mismatches at the third G:C pair, supports this view by revealing order-of-magnitude increases in Kd for TC in PICs with mRNA(AUG) and an inability to form stable 43S·mRNA(UUG) complexes. In contrast, these substitutions do not reduce the affinity of TC for 43S PICs lacking mRNA, indicating that they disrupt thermodynamic coupling between Met-tRNAi and the start codon in the closed/PIN state. The fact that G31C and G31U evoke a more extensive reduction in the stability of the closed/PIN state at UUG versus AUG codons is consistent with the reduced UUG:AUG initiation ratio conferred by these Ssu− mutations in vivo (Fig. 6F; Supplemental Fig. S11).
Further support for this interpretation comes from the fact that the U:U substitution of the third G:C pair, found here to confer an Ssu− phenotype, was shown previously to increase the Kd for TC in mRNA(AUG) complexes but not in 43S complexes lacking mRNA (Kapp et al. 2006)—the same finding made here for G31C and G31U Ssu− substitutions. This defect in TC binding was fully reversed by T-loop substitutions A54U,A60C (Kapp et al. 2006), and we found here that the Ssu− phenotype of the U31:U39 substitution is likewise reversed by A54U,A60C. The strong concordance between these biochemical and genetic data provides compelling evidence that the hyperaccuracy phenotypes of disrupting the third G:C pair result from a diminished contribution of base-pairing between the start codon and Met-tRNAi to the stability of the PIN state, which is exacerbated by the less stable codon:anticodon duplex formed at UUG triplets (Fig. 6F; Supplemental Fig. S11). It is possible that these mutations produce this effect by increasing the energetic barrier to a conformational change in the ASL that is required to attain the PIN state.
How might T-loop substitutions compensate for the reduced ability to access the PIN state at UUG codons conferred by the U31:U39 replacement? Perhaps altering the T-loop removes a structural impediment to the PIN state that is normally overcome by the perfect AUG:anticodon duplex (Fig. 6F). Transition to PIN might require deforming Met-tRNAi structure, and A54C/U substitutions would increase the flexibility of Met-tRNAi to reduce the energetic cost of this transition and increase its frequency at UUG codons. Interestingly, A54, A60, and m1A58 in the T-loop and A20 in the D-loop participate in hydrogen bonds that rigidify the T-loop and its connection to the D-loop (Fig. 6E; Basavappa and Sigler 1991). Thus, weakening T-loop/D-loop interaction by A54 substitutions might facilitate the proposed distortion of Met-tRNAi required to achieve PIN in the absence of a perfect codon:anticodon match at near-cognates.
In crystal structures of bacterial 70S·mRNA·tRNA complexes, G1338 and A1339 of 16S rRNA are poised to make A-minor interactions with the minor grooves of the first and second G:C pairs in the ASL (Korostelev et al. 2006; Selmer et al. 2006), and there is evidence that these interactions stabilize Met-tRNAi binding to the 70S P site (Lancaster and Noller 2005; Qin et al. 2007). Our finding that purine:purine and most pyrimidine:pyrimidine mismatches are not tolerated at the first and second G:C pairs is consistent with this mechanism operating in yeast. This model can also explain our previous results indicating that nearly all substitutions of the corresponding yeast 18S rRNA residues (G1575 and A1576) are lethal and impair AUG recognition in cells coexpressing wild-type rRNA (Dong et al. 2008). The lethal or Ssu− phenotypes of substitutions disrupting W:C pairing at the third G:C pair might also be attributed to an indirect disruption of A-minor interactions made by the adjacent G:C pairs. Our finding that W:C substitutions at 29:41 or 30:40 have little effect on cell growth is not inconsistent with the model because, with few exceptions, the stabilities of A-minor interactions vary little with different W:C pairs as receptors (Doherty et al. 2001; Battle and Doudna 2002). Harder to explain, however, is the absence of strong phenotypes associated with various substitutions that introduce wobble pairs at 29:41 or 30:40, which should strongly destabilize A-minor interactions (Battle and Doudna 2002). Therefore, more work is required to determine whether A-minor interactions of G1575 and A1576 with the ASL G:C pairs play a critical role in stabilizing the PIN conformation and account for the lethal or hyperaccuracy phenotypes of disrupting the ASL G:C base pairs.
In contrast to the Ssu− phenotypes produced by mutations that eliminate base-pairing in the ASL, it is striking that replacing the third G:C pair with any other W:C pair reduces accuracy and confers a marked Sui− phenotype. Our biochemical analysis of Sui− substitutions G31C:C39G and G31U:C39A showed that they increase the stability of TC binding with AUG and UUG codons in the P site to the point where dissociation of TC from the PICs was undetectable. Because TC formed with wild-type Met-tRNAi dissociates more rapidly from UUG than from AUG complexes, the stabilization of PICs conferred by these substitutions appears to be relatively greater at UUG codons, consistent with their Sui− phenotypes. Interestingly, the G31A:C39U replacement specifically increased utilization of near-cognates with first position mismatches (CUG and GUG in addition to UUG) but not second or third position mismatches (AUA and ACG). Thus, while a base pair per se is required at position 31:39 for thermodynamic coupling between the start codon and anticodon of tRNAi, a G:C pair is needed specifically to enforce a requirement for an A:U pair at the first position of the codon:anticodon helix.
One way to explain our finding that W:C replacements of G31:C39 stabilize PIN at AUG or UUG codons is to propose that, compared with other Watson-Crick base pairs, the wild-type G31:C39 base pair imposes an impediment to PIN that can be overcome efficiently only with the perfect codon:anticodon duplex formed at AUG codons. In this view, W:C replacements at 31:39 reduce this impediment, allowing “NUG” near-cognates to overcome the impediment more effectively. We envision that G31:C39, being the last base pair of the ASL, promotes a rigid conformation of the anticodon loop that clashes with a P-site element, and this clash is eliminated by a conformational change triggered by the first base pair of the codon:anticodon duplex. G31:C39 would be optimized to impart this inhibitory conformation of the anticodon loop in a way that could not be replaced by other W:C pairs at this position. The fact that W:C replacements at the first or second G:C pairs evoke considerably weaker Sui− phenotypes could be explained by proposing that they act indirectly to diminish the critical function of G31:C39 in blocking the transition to PIN at NUG near-cognates (Supplemental Fig. S12). Interestingly, the ASL in the crystal structure of Escherichia coli tRNAi displays a noncanonical conformation in which A37 interacts with G29:C41 instead of stacking on residue 36 of the anticodon loop, the ASL helix is extended by a C32:A38 base pair, and its major groove is obscured (Barraud et al. 2008). If this noncanonical conformation occurs in yeast tRNAi, it might be stabilized by G31:C39, and an A:U base pair at the first position of the codon:anticodon duplex could be required for isomerization to the conformation needed for a stable PIN state.
There is evidence that the G1338A substitution of 16S rRNA, which appears to enhance its A-minor interaction with the ASL of tRNAi, decreases initiation fidelity by compensating for mismatches in the start codon:anticodon helix (Qin et al. 2007). It is unlikely that this phenomenon is involved in the moderate Sui− phenotypes of W:C replacements at G29:C41 and G30:C40 because these W:C replacements should, if anything, decrease the stability of A-minor interactions and increase accuracy by discriminating against mismatched codon:anticodon duplexes. Hence, we favor the alternative explanation that the W:C replacements at G29:C41 and G30:C40 decrease accuracy indirectly by impairing the critical function of G31:C39 in discriminating against near-cognates.
Although the C3:G70 base pair in the acceptor stem is a highly conserved feature of tRNAi (Marck and Grosjean 2002), no function had been ascribed to it. We found that mutations disrupting W:C pairing at C3:G70 decrease accuracy but, unlike Sui− substitutions of G31:C39, also confer the Gcd− phenotype signifying slower TC loading to the open conformation of the PIC. Importantly, the Sui− and Gcd− phenotypes of G70 substitutions were cosuppressed by the 17-21 substitution in the eIF1A NTT. The 17-21 mutation was shown previously to stabilize the open/POUT conformation of the PIC (Fekete et al. 2007; Saini et al. 2010) and cosuppress Gcd− and Sui− phenotypes conferred by mutations inactivating the SEs in the eIF1A CTT (Supplemental Fig. S2; Saini et al. 2010) and by other mutations eliminating 40S contact sites in eIF1 (Martin-Marcos et al. 2013). Hence, an attractive model is that C3:G70 in tRNAi acts together with eIF1 and the eIF1A SEs to promote TC binding in the POUT conformation. Disrupting C3:G70 would destabilize POUT, reducing the rate of TC loading (for the Gcd− phenotype), and allow more frequent rearrangement to the PIN conformation at UUG codons (for the Sui− phenotype) (Fig. 6F; Supplemental Fig. S13).
Strong support for this model comes from our in vitro findings that G70A greatly reduces the kon for TC binding to 43S·mRNA(AUG) complexes in a manner mitigated by the C3U substitution (which restores W–C pairing) and fully suppressed by eIF1A-17-21. Because 17-21 stabilizes the open/POUT conformation (Cheung et al. 2007; Fekete et al. 2007; Saini et al. 2010), its ability to suppress the kon defect of G70A supports the idea that G70A decreases the rate of TC binding specifically to the POUT conformation. Consistent with this, G70A stabilizes the PIN state, as it reduces the rate of TC dissociation from PICs reconstituted with mRNA(AUG) or mRNA(UUG), and this defect for mRNA(UUG) complexes was diminished by C3U. This last finding supports the idea that by disrupting C3:G70, G70A elevates UUG initiation by stabilizing PIN at UUG codons. Because G70A has a much smaller effect on the kon of TC in PICs reconstituted with mRNA(UUG) versus mRNA(AUG), we inferred that it also increases the rate of POUT-to-PIN isomerization specifically at UUG codons, which should contribute to its Sui− phenotype (Supplemental Fig. S13).
One way to explain the ability of G70A to disfavor the POUT mode of TC binding is to propose that eliminating the C3:G70 pair destabilizes the acceptor stem and renders the connection between Met-tRNAi and eIF2 more flexible in a manner that is particularly detrimental for binding in the POUT state. The recent crystal structure of a Tetrahymena 40S·eIF1·eIF1A complex reveals that the SI element of the eIF1A NTT bridges a connection between the head and body of the 40S subunit (Weisser et al. 2013), which might indicate that the 17-21 substitution in the NTT shifts the equilibrium from PIN to POUT and thereby restores rapid TC loading by the G70A variant by weakening this connection within the 40S subunit. However, why this enhancement occurs at AUG but not UUG codons is not yet clear.
In summary, our results provide compelling evidence that the distinctive C3:G70 base pair in the acceptor stem is important for rapid TC binding in the POUT conformation of the PIC and functions with eIF1 and the eIF1A SEs to discriminate against UUG start codons by impeding the POUT-to-PIN transition at near-cognate triplets. In contrast, G31:C39 in the ASL and A54 in the T-loop appear to function differently to block UUG initiation by imposing an impediment to PIN that can be overcome only with a perfect AUG:anticodon duplex, with G31:C39 specifically enforcing the requirement for an A:U pair at the first position of the AUG:anticodon duplex. While G31:C39 discriminates against NUG near-cognates, a Watson-Crick base pair is required at this position in the ASL for PIN stability and efficient start codon recognition, and disrupting base pairing at this position discriminates against near-cognates. Thus, different regions of tRNAi perform distinct functions in the PIC to promote AUG recognition in vivo. Considering that the signature residues of tRNAi are conserved in all kingdoms of life, the functions ascribed here to these residues of yeast tRNAi likely apply to tRNAi in mammals and other eukaryotes.
Materials and methods
Plasmids and yeast strains
Plasmids and yeast strains are listed in Supplemental Tables S3 and S5, respectively, and details of their construction are provided in the Supplemental Material.
Biochemical analyses of yeast cells
Northern analyses of tRNAi expression were conducted as described previously (Anderson et al. 1998). tRNA aminoacylation in vivo was analyzed using RNA isolated (Zaborske et al. 2009) and subjected to Northern analysis (Varshney et al. 1991) as previously described using oligonucleotide probes listed in the Supplemental Material. Assays of β-galactosidase activity in whole-cell extracts were performed as described previously (Moehle and Hinnebusch 1991), as were measurements of luminescence in whole-cell extracts (Dyer et al. 2000). Coimmunoprecipitation analysis of the TC was performed as described previously (Dev et al. 2010) using antibodies against Flag (Sigma) and Gcd11 (provided by E. Hannig).
Biochemical analysis in the reconstituted yeast translation system
Purification of 40S ribosomal subunits and eIF1, eIF1A, and eIF2 as well as preparation of mRNA and tRNAi variants charged with [35S]-labeled or unlabeled methionine were performed as described (Acker et al. 2007). A double filter-binding assay was used to measure Kd values of [35S]-Met-tRNAi variants and eIF2-GDPNP essentially as described previously (Acker et al. 2007). The Kd values of TC (assembled with [35S]-Met-tRNAi variants) and 40S·eIF1·eIF1A·mRNA PICs and the rate constants of TC association/dissociation for the same PICs were determined by gel shift assays as described previously (Kolitz et al. 2009) with slight modifications described in the Supplemental Material.
Acknowledgments
We are indebted to Sonia D’Silva and Eric Phizicky for analysis of t6A37 in tRNAi, Anders Bystrom and Katsura Asano for strains and plasmids, Ernest Hannig for Gcd11 antibodies, and Tom Dever for critical comments. This work was supported by the Intramural Research Program of the National Institutes of Health (NIH) and NIH grant GM62128 to J.R.L.
Footnotes
Supplemental material is available for this article.
Article is online at http://www.genesdev.org/cgi/doi/10.1101/gad.236547.113.
References
- Acker MG, Kolitz SE, Mitchell SF, Nanda JS, Lorsch JR 2007. Reconstitution of yeast translation initiation. Methods Enzymol 430: 111–145 [DOI] [PubMed] [Google Scholar]
- Agris PF 2008. Bringing order to translation: The contributions of transfer RNA anticodon-domain modifications. EMBO Rep 9: 629–635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Algire MA, Maag D, Lorsch JR 2005. Pi release from eIF2, not GTP hydrolysis, is the step controlled by start-site selection during eukaryotic translation initiation. Mol Cell 20: 251–262 [DOI] [PubMed] [Google Scholar]
- Alone PV, Cao C, Dever TE 2008. Translation initiation factor 2γ mutant alters start codon selection independent of Met-tRNA binding. Mol Cell Biol 28: 6877–6888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson J, Phan L, Cuesta R, Carlson BA, Pak M, Asano K, Bjork GR, Tamame M, Hinnebusch AG 1998. The essential Gcd10p–Gcd14p nuclear complex is required for 1-methyladenosine modification and maturation of initiator methionyl-tRNA. Genes Dev 12: 3650–3662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Astrom SU, von Pawel-Rammingen U, Bystrom AS 1993. The yeast initiator tRNAMet can act as an elongator tRNAMet in vivo. J Mol Biol 233: 43–58 [DOI] [PubMed] [Google Scholar]
- Barraud P, Schmitt E, Mechulam Y, Dardel F, Tisne C 2008. A unique conformation of the anticodon stem–loop is associated with the capacity of tRNAfMet to initiate protein synthesis. Nucleic Acids Res 36: 4894–4901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basavappa R, Sigler PB 1991. The 3 Å crystal structure of yeast initiator tRNA: Functional implications in initiator/elongator discrimination. EMBO J 10: 3105–3111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Battle DJ, Doudna JA 2002. Specificity of RNA-RNA helix recognition. Proc Natl Acad Sci 99: 11676–11681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boeke JD, Trueheart J, Natsoulis G, Fink GR 1987. 5-fluoroorotic acid as a selective agent in yeast molecular genetics. Methods Enzymol 154: 164–175 [DOI] [PubMed] [Google Scholar]
- Cheung YN, Maag D, Mitchell SF, Fekete CA, Algire MA, Takacs JE, Shirokikh N, Pestova T, Lorsch JR, Hinnebusch AG 2007. Dissociation of eIF1 from the 40S ribosomal subunit is a key step in start codon selection in vivo. Genes Dev 21: 1217–1230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daugeron MC, Lenstra TL, Frizzarin M, El Yacoubi B, Liu X, Baudin-Baillieu A, Lijnzaad P, Decourty L, Saveanu C, Jacquier A, et al. 2011. Gcn4 misregulation reveals a direct role for the evolutionary conserved EKC/KEOPS in the t6A modification of tRNAs. Nucleic Acids Res 39: 6148–6160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dev K, Qiu H, Dong J, Zhang F, Barthlme D, Hinnebusch AG 2010. The β/Gcd7 subunit of eukaryotic translation initiation factor 2B (eIF2B), a guanine nucleotide exchange factor, is crucial for binding eIF2 in vivo. Mol Cell Biol 30: 5218–5233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dever TE, Yang W, Åström S, Byström AS, Hinnebusch AG 1995. Modulation of tRNAiMet, eIF-2 and eIF-2B expression shows that GCN4 translation is inversely coupled to the level of eIF-2·GTP·Met-tRNAiMet ternary complexes. Mol Cell Biol 15: 6351–6363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doherty EA, Batey RT, Masquida B, Doudna JA 2001. A universal mode of helix packing in RNA. Nat Struct Biol 8: 339–343 [DOI] [PubMed] [Google Scholar]
- Donahue T. 2000. Genetic approaches to translation initiation in Saccharomyces cerevisiae. In Translational control of gene expression (ed. N Sonenberg, et al.), pp. 487–502. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- Dong J, Nanda JS, Rahman H, Pruitt MR, Shin BS, Wong CM, Lorsch JR, Hinnebusch AG 2008. Genetic identification of yeast 18S rRNA residues required for efficient recruitment of initiator tRNA(Met) and AUG selection. Genes Dev 22: 2242–2255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drabkin HJ, Helk B, RajBhandary UL 1993. The role of nucleotides conserved in eukaryotic initiator methionine tRNAs in initiation of protein synthesis. J Biol Chem 268: 25221–25228 [PubMed] [Google Scholar]
- Dyer BW, Ferrer FA, Klinedinst DK, Rodriguez R 2000. A noncommercial dual luciferase enzyme assay system for reporter gene analysis. Anal Biochem 282: 158–161 [DOI] [PubMed] [Google Scholar]
- El Yacoubi B, Hatin I, Deutsch C, Kahveci T, Rousset JP, Iwata-Reuyl D, Murzin AG, de Crecy-Lagard V 2011. A role for the universal Kae1/Qri7/YgjD (COG0533) family in tRNA modification. EMBO J 30: 882–893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farruggio D, Chaudhuri J, Maitra U, RajBhandary UL 1996. The A1 U72 base pair conserved in eukaryotic initiator tRNAs is important specifically for binding to the eukaryotic translation initiation factor eIF2. Mol Cell Biol 16: 4248–4256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fekete CA, Mitchell SF, Cherkasova VA, Applefield D, Algire MA, Maag D, Saini A, Lorsch JR, Hinnebusch AG 2007. N-and C-terminal residues of eIF1A have opposing effects on the fidelity of start codon selection. EMBO J 26: 1602–1614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashem Y, des Georges A, Dhote V, Langlois R, Liao HY, Grassucci RA, Hellen CU, Pestova TV, Frank J 2013. Structure of the mammalian ribosomal 43S preinitiation complex bound to the scanning factor DHX29. Cell 153: 1108–1119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinnebusch AG 2005. Translational regulation of GCN4 and the general amino acid control of yeast. Annu Rev Microbiol 59: 407–450 [DOI] [PubMed] [Google Scholar]
- Hinnebusch AG 2011. Molecular mechanism of scanning and start codon selection in eukaryotes. Microbiol Mol Biol Rev 75: 434–467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapp LD, Lorsch JR 2004. GTP-dependent recognition of the methionine moiety on initiator tRNA by translation factor eIF2. J Mol Biol 335: 923–936 [DOI] [PubMed] [Google Scholar]
- Kapp LD, Kolitz SE, Lorsch JR 2006. Yeast initiator tRNA identity elements cooperate to influence multiple steps of translation initiation. RNA 12: 751–764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolitz SE, Takacs JE, Lorsch JR 2009. Kinetic and thermodynamic analysis of the role of start codon/anticodon base pairing during eukaryotic translation initiation. RNA 15: 138–152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korostelev A, Trakhanov S, Laurberg M, Noller HF 2006. Crystal structure of a 70S ribosome-tRNA complex reveals functional interactions and rearrangements. Cell 126: 1065–1077 [DOI] [PubMed] [Google Scholar]
- Lancaster L, Noller HF 2005. Involvement of 16S rRNA nucleotides G1338 and A1339 in discrimination of initiator tRNA. Mol Cell 20: 623–632 [DOI] [PubMed] [Google Scholar]
- Lin CA, Ellis SR, True HL 2009. The Sua5 protein is essential for normal translational regulation in yeast. Mol Cell Biol 30: 354–363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lomakin IB, Steitz TA 2013. The initiation of mammalian protein synthesis and mRNA scanning mechanism. Nature 500: 307–311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lomakin IB, Shirokikh NE, Yusupov MM, Hellen CU, Pestova TV 2006. The fidelity of translation initiation: Reciprocal activities of eIF1, IF3 and YciH. EMBO J 25: 196–210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maag D, Fekete CA, Gryczynski Z, Lorsch JR 2005. A conformational change in the eukaryotic translation preinitiation complex and release of eIF1 signal recognition of the start codon. Mol Cell 17: 265–275 [DOI] [PubMed] [Google Scholar]
- Mandal N, Mangroo D, Dalluge JJ, McCloskey JA, Rajbhandary UL 1996. Role of the three consecutive G:C base pairs conserved in the anticodon stem of initiator tRNAs in initiation of protein synthesis in Escherichia coli. RNA 2: 473–482 [PMC free article] [PubMed] [Google Scholar]
- Marck C, Grosjean H 2002. tRNomics: Analysis of tRNA genes from 50 genomes of Eukarya, Archaea, and Bacteria reveals anticodon-sparing strategies and domain-specific features. RNA 8: 1189–1232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin-Marcos P, Nanda J, Luna RE, Wagner G, Lorsch JR, Hinnebusch AG 2013. β-Hairpin loop of eIF1 mediates 40S ribosome binding to regulate initiator tRNAMet recruitment and accuracy of AUG selection in vivo. J Biol Chem 38: 27546–27562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin-Marcos P, Nanda JS, Luna RE, Zhang F, Saini AK, Cherkasova VA, Wagner G, Lorsch JR, Hinnebusch AG 2014. Enhanced eIF1 binding to the 40S ribosome impedes conformational rearrangements of the preinitiation complex and elevates initiation accuracy. RNA 20: 150–167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moehle CM, Hinnebusch AG 1991. Association of RAP1 binding sites with stringent control of ribosomal protein gene transcription in Saccharomyces cerevisiae. Mol Cell Biol 11: 2723–2735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nanda JS, Saini AK, Munoz AM, Hinnebusch AG, Lorsch JR 2013. Coordinated movements of eukaryotic translation initiation factors eIF1, eIF1A, and eIF5 trigger phosphate release from eIF2 in response to start codon recognition by the ribosomal preinitiation complex. J Biol Chem 288: 5316–5329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Passmore LA, Schmeing TM, Maag D, Applefield DJ, Acker MG, Algire MA, Lorsch JR, Ramakrishnan V 2007. The eukaryotic translation initiation factors eIF1 and eIF1A induce an open conformation of the 40S ribosome. Mol Cell 26: 41–50 [DOI] [PubMed] [Google Scholar]
- Pestova TV, Lorsch JR, Hellen CUT 2007. The mechanism of translation initiation in eukaryotes. In Translational control in biology and medicine (ed. M Mathews, et al.), pp. 87–128. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- Qin D, Abdi NM, Fredrick K 2007. Characterization of 16S rRNA mutations that decrease the fidelity of translation initiation. RNA 13: 2348–2355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabl J, Leibundgut M, Ataide SF, Haag A, Ban N 2011. Crystal structure of the eukaryotic 40S ribosomal subunit in complex with initiation factor 1. Science 331: 730–736 [DOI] [PubMed] [Google Scholar]
- RajBhandary UL. Chow CM. 1995. Initiator tRNAs and initiation of protein synthesis. In tRNA structure, biosynthesis, and function (ed. D Soll and UL RajBhandary), pp. 511–528. American Society for Microbiology Press, Washington, DC. [Google Scholar]
- Saini AK, Nanda JS, Lorsch JR, Hinnebusch AG 2010. Regulatory elements in eIF1A control the fidelity of start codon selection by modulating tRNA(i)(Met) binding to the ribosome. Genes Dev 24: 97–110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selmer M, Dunham CM, Murphy FV IV, Weixlbaumer A, Petry S, Kelley AC, Weir JR, Ramakrishnan V 2006. Structure of the 70S ribosome complexed with mRNA and tRNA. Science 313: 1935–1942 [DOI] [PubMed] [Google Scholar]
- Shin BS, Kim JR, Walker SE, Dong J, Lorsch JR, Dever TE 2011. Initiation factor eIF2 promotes eIF2-GTP-Met-tRNA(i)(Met) ternary complex binding to the 40S ribosome. Nat Struct Mol Biol 18: 1227–1234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivasan M, Mehta P, Yu Y, Prugar E, Koonin EV, Karzai AW, Sternglanz R 2011. The highly conserved KEOPS/EKC complex is essential for a universal tRNA modification, t6A. EMBO J 30: 873–881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takacs JE, Neary TB, Ingolia NT, Saini AK, Martin-Marcos P, Pelletier J, Hinnebusch AG, Lorsch JR 2011. Identification of compounds that decrease the fidelity of start codon recognition by the eukaryotic translational machinery. RNA 17: 439–452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valasek L, Nielsen KH, Zhang F, Fekete CA, Hinnebusch AG 2004. Interactions of eukaryotic translation initiation factor 3 (eIF3) subunit NIP1/c with eIF1 and eIF5 promote preinitiation complex assembly and regulate start codon selection. Mol Cell Biol 24: 9437–9455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varshney U, Lee CP, RajBhandary UL 1991. Direct analysis of aminoacylation levels of tRNA as in vivo. J Biol Chem 266: 24712–24718 [PubMed] [Google Scholar]
- Varshney U, Lee CP, RajBhandary UL 1993. From elongator tRNA to initiator tRNA. Proc Natl Acad Sci 90: 2305–2309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Pawel-Rammingen U, Astrom S, Bystrom AS 1992a. Mutational analysis of conserved positions potentially important for initiator tRNA function in Saccharomyces cerevisiae. Mol Cell Biol 12: 1432–1442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisser M, Voigts-Hoffmann F, Rabl J, Leibundgut M, Ban N 2013. The crystal structure of the eukaryotic 40S ribosomal subunit in complex with eIF1 and eIF1A. Nat Struct Mol Biol 20: 1015–1017 [DOI] [PubMed] [Google Scholar]
- Yoon HJ, Donahue TF 1992. The sui1 suppressor locus in Saccharomyces cerevisiae encodes a translation factor that functions during tRNAiMet recognition of the start codon. Mol Cell Biol 12: 248–260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Y, Marintchev A, Kolupaeva VG, Unbehaun A, Veryasova T, Lai SC, Hong P, Wagner G, Hellen CU, Pestova TV 2009. Position of eukaryotic translation initiation factor eIF1A on the 40S ribosomal subunit mapped by directed hydroxyl radical probing. Nucleic Acids Res 37: 5167–5182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaborske JM, Narasimhan J, Jiang L, Wek SA, Dittmar KA, Freimoser F, Pan T, Wek RC 2009. Genome-wide analysis of tRNA charging and activation of the eIF2 kinase Gcn2p. J Biol Chem 284: 25254–25267 [DOI] [PMC free article] [PubMed] [Google Scholar]