

Abstract
Brain injury due to neonatal hypoxia-ischemia (HI) is more homogenously severe in male than in female mice. Because, necrostatin-1 (nec-1) prevents injury progression only in male mice, we hypothesized that changes in BDNF signaling after HI and nec-1 are also sex-specific providing differential conditions to promote recovery of those more severely injured. The increased aromatization of testosterone in male mice during early development and the link between 17-β-estradiol (E2) levels and BDNF transcription substantiate this hypothesis. Hence, we aimed to investigate if sexual differences in BDNF signaling existed in forebrain and diencephalon after HI and HI/ nec-1 and their correlation with estrogen receptors (ER). C57B6 mice (p7) received nec-1(0.1 μL[8μM]) or vehicle (veh) intracerebroventricularly after HI. At 24h after HI, BDNF levels increased in both sexes in forebrain without evidence of TrkB activation. At 96h after HI, BDNF levels in forebrain decreased below those seen in control mice of both sexes. Additionally, only in female mice, truncated TrkB (Tc.TrkB) and p75ntr levels increased in forebrain and diencephalon. In both, forebrain and diencephalon, nec-1 treatment increased BDNF levels and TrkB activation in male mice while, prevented Tc.TrkB and p75ntr increases in female mice. While E2 levels were unchanged by HI or HI/ nec-1 in either sex or treatment, ERα: ERβ ratios were increased in diencephalon of nec-1 treated male mice and directly correlated with BDNF levels. Neonatal HI produces sex-specific signaling changes in the BDNF system, that are differentially modulated by nec-1. The regional differences in BDNF levels may be a consequence of injury severity after HI, but sexual differences in response to nec-1 after HI may represent a differential thalamo-cortical preservation or alternatively off-target regional effect of nec-1. The biological significance of ERα predominance and its correlation with BDNF levels is still unclear.
Keywords: cortex, estradiol, p75ntr, plasticity, thalamus, truncated TrkB receptor
1. INTRODUCTION
Sexually dimorphism have been found in preterm and full-term rodent models of hypoxia, ischemia and hypoxia-ischemia (HI) (Hagberg et al., 2004, Zhu et al., 2006, Renolleau et al., 2008, Mayoral et al., 2009, Arteni et al., 2010, Hill et al., 2011). In female mice, the greater proclivity to caspase-dependent cell death after neonatal HI (Hagberg et al., 2004, Zhu et al., 2006, Chavez-Valdez et al., 2012b) matches the lesser degree of cortical injury compared to males (Northington et al., 2011). Similarly, several neuroprotective agents for neonatal HI also show sexual dimorphism in their effects (Hurn et al., 2005). For example, necrostatin-1 [(nec-1), 5-(1H-indol-3-ylmethyl)-3-methyl-2-sulfanylideneimidazolidin-4-1], an inhibitor of regulated necrosis (Degterev et al., 2005, Lim et al., 2007, You et al., 2008), provides cortical protection only to male mice (Northington et al., 2011) without sexual differences in energy preservation (Chavez-Valdez et al., 2012a, Chavez-Valdez et al., 2012b). Mechanisms behind these sexual differences are unclear but they may involve intrinsic differences in pathways signaling for repair (e.g. BDNF, brain derived neurotrophic factor) following brain injury.
BDNF is a therapeutic target in neonatal brain injury because of its putative role in brain plasticity, enhancing neuronal survival, migration and differentiation, supporting neurogenesis and improving outcomes in adult ischemic and neonatal HI models (Marini et al., 2007, Yasuhara et al., 2010, Douglas-Escobar et al., 2012, Han et al., 2012, Rosenkranz et al., 2012). However, early BDNF exposure after oxidative stress and oxygen-glucose deprivation injury may also exacerbate neuronal death (Gwag et al., 1995, Koh et al., 1995, Kim et al., 2003). This duality is produced by changes in BDNF receptor activation and/ or expression. BDNF exerts trophic effects via phosphorylation of full-length tyrosine-related kinase B (FL.TrkB) receptor and promotes cell death via binding the low affinity p75neurotrophic receptor (p75ntr) if combined with downregulation of FL.TrkB receptor (Frank et al., 1996, Knusel et al., 1997) and/ or upregulation of truncated TrkB (Tc.TrkB) receptor isoforms (Klein et al., 1990, Biffo et al., 1995, Alderson et al., 2000).
Temporal and regional interrogation of the BDNF system is essential when trying to understand its potential effects. BDNF is greatly expressed in forebrain and diencephalon throughout normal development (Schmidt-Kastner et al., 1996, Lush et al., 2005, Webster et al., 2006). Cortex-derived BDNF is necessary for thalamic axonal outgrowth and target identification (Lotto et al., 2001), but not for thalamic neurogenesis (Mooney and Miller, 2011). The influence of BDNF in remote areas (e.g. thalamus) produces a positive loop feedback following the arrival of thalamic axons to the target cortical layer (Hanamura et al., 2004) which provides autocrine support to neocortical neurons increasing neurogenesis and neuronal migration (Pencea et al., 2001, Morcuende et al., 2003, Scharfman, 2005, McCarthy et al., 2011).
An additional layer of complexity is added by the sexual differences in the activation of the BDNF system after neonatal HI and/ or neuroprotective therapies (e.g. nec-1) which have not been studied until now. Therefore, we hypothesize that the BDNF system responds to HI and nec-1 in a sexually dimorphic manner providing the conditions to explain the differences in severity of injury in the forebrain and diencephalon. Because an estrogen response element (ERE)-like motif is found in the BDNF gene (Blurton-Jones et al., 2004), sex differences in BDNF transcription may be linked to either higher levels of 17-β-estradiol (E2) in male mice secondary to greater aromatization of testosterone during early post-natal development or greater influence of E2 due to changes in expression of estrogen receptor (ER) subtypes (Amantea et al., 2005, Roselli et al., 2009).
2. EXPERIMENTAL PROCEDURES
2.1. Animals
Experiments were performed with approval by the Institutional Animal Care and Use Committee at Johns Hopkins University- School of Medicine and they were carried out in accordance with the National Institute of Health Guide for the Care and Use of Laboratory Animals (NIH Publications No. 80-23) revised in 1996. All efforts were made to minimize the number of animals used and their suffering. Male and female mice were compared for these experiments.
2.2. Neonatal HI brain injury model
We used the Vannucci model adapted for neonatal mice to induce HI in C57B6 mice at postnatal day (p) 7 (Ditelberg et al., 1996). Mice were exposed to a second brief period of anesthesia with isofluorane followed by an intracerebroventricular injection of 0.1 μl of 8 μmol nec-1 (Calbiochem-EMD Chemicals Group, Gibbstown, NJ), or vehicle (veh), methyl-β-cyclodextrin (Sigma, St Louis, MO, USA) 15 min after the end of the hypoxic exposure. Pups were returned to the dam until they were killed at 24h, 96h and 21days after HI (n = 4–12 pups/ time/ treatment/ sex) for biochemical analysis. Naive controls were age-matched littermates not exposed to HI or treatments. Mice were killed with an exposure to 20% (v/v) mixture of isoflurane in propylene glycol via one drop exposure method (Markovic and Murasko, 1993) and then decapitated. Brain was microdissected by separating mid-sagittaly the hemispheres at the level of longitudinal cerebral fissure, separating the cerebellum at the level of transverse fissure, and isolating the inner portion of the cerebral hemisphere posterior to the column of the fornix to include thalamus and hypothalamus (diencenphalon) and above and lateral to the fornix to include hippocampus, striatum and cortex (forebrain). Tissues were rapidly microdissected and frozen (−80°C).
2.3. Brain BDNF protein and estradiol (E2) levels by ELISA
Brain homogenates were prepared as previously described (Northington et al., 1996) and protein concentrations determined using the Bradford assay (Bradford, 1976).
BDNF ELISA
BDNF protein levels were measured in ipsilateral forebrain and diencephalon samples from veh- and nec-1-treated mice (n=4–8 pups/ treatment/ time/ sex) at 24h, 96h and 21d after HI and in naive age-matched controls. Mouse sandwich ELISA method was used according to manufacturer’s instructions (Boster Biological Technology LTD. Fremont, CA). This method has a detection range of 2 to 2000 pg/ml and no reported cross-reactivity with other neurotrophins. The ELISA plate, pre-coated with anti-mouse BDNF antibody, was soaked and washed per 3 times with wash buffer T (9 mM Tris, 0.14M NaCl, 1.6M acetic acid in ddH2O, pH 7.4). Samples were clarified by centrifugation at 10000 × g at 4°C for 2 min and then diluted in a 1:3 ratio using sample diluent buffer provided by manufacturer. Blank (diluent buffer alone), standards and clarified diluted samples were plated and incubated for 90 min at 37°C followed by exposure to biotinylated anti-mouse BDNF antibody for 60 min at 37°C. The plate was washed with buffer T and avidin-biotin- peroxidase complex working solution was applied. Following incubation for 30 min at 37°C, plates were washed 5 times with buffer T. 3,3′,5,5′ Tetramethyl- benzidine buffer developing agent was added to each well and after 10 min of incubation at 37°C, stop solution was applied. Plates were read at 450 nm in a microplate reader (Biorad, Hercules, CA) within 30 min after stop solution was applied and analyzed using a linear regression model. Data are reported as pg corrected for mg of protein.
Estradiol ELISA
E2 levels were measured in ipsilateral forebrain and diencephalon samples from veh- and nec-1-treated mice (n=4 pups/ treatment/ sex) at 96h after HI and in naive age-matched controls. A mouse estradiol colorimetric sandwich ELISA method was used according to manufacturer’s instructions (Shanghai Sunred Biological Technology Co., Ltd. Shanghai, China). This assay has a detection range of 1 to 180 pg/ml and no reported cross-reactivity. Following clarification of 100 μl of homogenized sample by centrifugation at 2000 × g for 20 min, 40ul were loaded into the mouse E2 monoclonal antibody pre-coated wells in duplicate followed by addition of biotinylated antibody against E2 and streptavidin-HRP to form an immune complex. Following 60 min of incubation at 37°C, and 3 washes with wash buffer provided by manufacturer, room light was turned off. Chromogen A and B were loaded into the pre-washed wells, mixed into the orbital shaker at RT for 1min, then incubated at 37°C for 10 min before applying stop solution. Optical density was measured at 450 nm wave length within 15 min using a Cook’s 4PL curve in a plate reader.
2.4. Determination of gene expression by real time qRT-PCR
Total RNA was extracted from forebrain and diencephalon harvested from mice exposed to neonatal HI and treatments as described above. PureLink™ RNA mini kit purification system (Invitrogen, Carlsbad, CA) was used according to manufacturer specifications. Approximately 1μg of total RNA was used for generation of complementary DNA (cDNA) using iScript cDNA synthesis kit (BioRad, Hercules, CA). Reverse transcription protocol included 5 min at 25°C; 30 min at 42°C and 5 min at 85°C. cDNA was then used to amplify target gene by real time qRT-PCR using 300 nM concentration of specific primers for: i) BDNF (forward: 5′-TGGCCTAACAATGTTTGCAGAT-3′; reverse: 5′-CCACTCAGAAATTCCTCCTGCT-3′; GenBank: BE101548; 155-bp PCR product); and ii) Parvalbumin (forward 5′-CCTCTTCCTTTCCTTGCAAG-3; reverse: 5′-GGTGTCTGTATCACATTCACCC-3′; GenBank: AC107136.5; 187-bp PCR product). The amplification protocol included 40 cycles of 30 sec at 95.0 °C, 1 min at 61.0 °C and 30 sec at 72.0 °C. GADPH (forward: 5′-TTGTCAAGCTCATTTCCTGGTATG-3′; reverse: 5′-GCCATGTAGGCCATGAGGTC-3′; 76-bp PCR product) and β-actin (forward: 5′-CCCAACTTGATGTATGAAGG-3′; reverse: 5′-TTGTGTAAGGTAAGGTGTGC-3′; 119-bp PCR product) were evaluated as housekeeping genes using the BestKeeper approach to determine stability of gene expression under experimental conditions (Pfaffl et al., 2004) as previously reported (Chavez-Valdez et al., 2012a). Based on their gene stability, β-actin was selected as the housekeeping gene for calculations (stability coefficients were 1.2 for β-actin vs. 2.0 for GAPDH). Fold difference in gene expression was then corrected to β-actin using the Pfaffl method (Pfaffl, 2001). Melting curves confirmed amplification of single PCR products. Results were reported as fold change in gene expression relative to expression in age-matched control mice.
2.5. FL.TrkB, Tc.TrkB, pTrkB, p75ntr, BDNF/ pro-BDNF, parvalbumin, ERα and ERβ protein expression by western blot
Protein homogenates were prepared in homogenization buffer with phosphatase and protease inhibitors and 20% (w/v) glycerol. Concentrations were determined using the Bradford assay (Bradford, 1976). Twenty-five μg-aliquots of homogenized protein were diluted 3:1 (v:v) in 4X loading buffer under reducing conditions and loaded into NuPAGE® Novex® 4–12% Bis-tris Gel (Life Technologies Corporation, Grand Island, NY). Protein was transferred to nitrocellulose membrane using the iBlot® transferring device (Life Technologies Corporation), stained with Ponceau S, blocked with 2.5% nonfat dry milk with 0.1% Tween-20 in 50 mM Tris buffered saline (TBST, 50mM Tris/HCl and 150 mM NaCl, pH 7.4) except for phosphorylated Y817 TrkB (pTrkB; Ab75173, Abcam Inc., Cambridge, MA), BDNF (Ab108319), parvalbumin (Ab114207) and ERα (Ab32063) which were blocked in 2.5% BSA. Nitrocellulose blots were incubated overnight at 4°C with primary antibodies at 1:2000 (parvalbumin), 1:1000 (ERα and ERβ), 1:500 (BDNF and pTrkB) or 1:200 (TrkB and p75ntr). Positive controls were used to ascertain identification of BDNF, TrkB and p75ntr (see details below). After exposure to each primary antibody, membranes were washed with TBST, exposed to secondary antibodies for 1h and then developed with enhanced chemiluminescence using SuperSignal kit (Thermo Scientific, Rockford, IL). To quantify protein immunoreactivity, films were scanned using Adobe Photoshop (Adobe Systems Inc., San Jose, CA), and optical density (OD) was determined with NIH Image J Software (NIH, Bethesda, MD) adjusted for background. The reliability of sample loading and protein transfer was verified by staining nitrocellulose membranes with Ponceau S and by β-actin.
2.6. Positive controls and antibodies
Positive controls
Mature BDNF (ab9794; Abcam Inc, Cambridge, MA): 0.1 μg of human recombinant showing a band at 14 kDa. TrkB (sc-113925; Santa Cruz Biotechnology, Inc., Santa Cruz, CA): 10 μg of human TrkB transfected 293T cell lysate showing a band at 145 kDa (FL.TrkB) and at 95 kDa (Tc.TrkB). p75ntr (sc-2237, Santa Cruz Biotechnology, Inc): 20 μg of human whole neuroepithelioma cell lysate (SK-N-MC cell lysate) showing a band at ~64kDa.
Antibodies
BDNF (ab108319, Abcam Inc.): rabbit monoclonal antibody raised against human BDNF synthetic peptide with no cross reactivity with other cytokines detecting mature BDNF at 14 kDa (under reducing conditions) and the N-glycosylated and sulfated pro-BDNF at 34kDa (1 μg/ mL). TrkB (sc-136990, Santa Cruz Biotechnology Inc.): mouse monoclonal antibody raised against amino acids 209–298 of TrkB of human origin detects the FL.TrkB at ~145 kDa and Tc.TrkB isoform at ~95 kDa (1 μg/ mL) with no cross reactivity with other Trk receptors. pTrkB: two rabbit polyclonal antibodies were used both of which were raised against a short amino acid sequence containing phosphorylated Tyr706 (sc-135645, Santa Cruz Biotechnology Inc.) or phosphorylated Tyr816 (ab75173, Abcam Inc.) of TrkB human origin detecting one band between 95–145 kDa (1 μg/ mL and 0.5 μg/ mL, respectively) with no cross reactivity with other Trk receptors. p75ntr (sc-8317, Santa Cruz Biotechnology Inc): rabbit polyclonal antibody raised against an epitope corresponding to amino acids 29 to 165 mapping within the extracellular domain of p75ntr detects a single band at ~64kDa (1 μg/ mL). Parvalbumin (ab11427, Abcam Inc.): Rabbit polyclonal antibody raised against purified full length parvalbumin of rat origin detects a band at ~12 kDa (0.5 μg/ mL). ERα (Ab32063, Abcam Inc.): Rabbit monoclonal antibody raised against synthetic peptide corresponding to residues surrounding Ser104 and Ser106 of human origin detects a band at 66 kDa (1 μg/ mL). ERβ (Ab16813, Abcam Inc.): Mouse monoclonal antibody raised against recombinant full length protein of human origin detecting a band at ~53–60 kDa (1 μg/ mL) with no cross reactivity with ERα.
2.7. Statistics
For analysis of multiple groups normally distributed, ANOVA was applied with post-hoc pair analysis using Tukey’s test. Those results were reported as mean ± SEM and represented as bar graphs. Gene expression data were not normally distributed, hence non-parametric analysis was applied (Mann-Whitney U test). Fold-change in BDNF and parvalbumin gene expression relative to age-matched naive control were represented as box and whisker plot, where the box was limited by the 25th and 75th percentiles and the solid line represented the median. Pearson product-moment correlation and calculation of a coefficient of determination (R2) were used to determine the relationship between BDNF and estrogen receptors. Significance was assigned by p ≤ 0.05 in all cases. Analysis was performed using IBM SPSS Statistics 18.0 software (IBM Corporation, Armonk, NY).
3. RESULTS
3.1. Changes in overall BDNF levels in forebrain and diencephalon after HI and nec-1 treatment
In forebrain, BDNF protein levels increased by 3.6-fold at 24h after HI (p < 0.001, veh-treated vs. age-matched naive controls; Fig. 1A). This BDNF increase was followed by a decline at 96h after HI by 75% (p=0.005 vs. 24h after HI) and by 20% (p=0.05 vs. age-matched naive controls). Nec-1 treatment did not alter the increase in BDNF levels seen at 24h after HI (Fig. 1A). However, at 96h after HI, mice treated with nec-1 had 45% (p<0.001, vs. age-matched naive controls) and 83% (p<0.001, vs. veh-treated HI mice) higher levels of BDNF (ANOVA, p<0.001). Twenty one days after HI, veh-treated mice had 11% (p=0.05) higher BDNF levels than naive age-matched controls, while nec-1-treated HI mice were not different from either naive controls or veh-treated HI mice (ANOVA, p= 0.03; Fig. 1A).
Figure 1.

Early increase in BDNF protein levels in forebrain after neonatal HI is followed by a delayed decrease and late recovery. Nec-1 prevents the decrease in BDNF levels in forebrain while increases levels in diencephalon at 96h after HI. BDNF gene expression demonstrates a sex-specific-response. BDNF protein levels are presented in bar graphs (pg/mg of protein) in forebrain (A) and diencephalon (B) at 24h, 96h and 21 days after HI. Bars represent the mean ± SEM measured in naive control (white), vehicle (light grey) and nec-1 (dark grey) treated mice. ‡, p ≤ 0.05, one-way ANOVA. p < 0.05 (Tukey’s post-hoc); *, vs. naive control; †, vs. vehicle; n= 8–14 mice/ treatment/ time. Fold-change in gene expression (vs. naive age-matched controls) is shown as box-and-whisker plot, with boxes representing interquartile range (IQR) and median (solid line in boxes) for male (solid color, M) and female (hashed color, F) mice forebrain (C, E) and diencephalon (D,F) at 96h and 21 days after neonatal HI, respectively. Reference (discontinued) line sitting at 1 represents BDNF gene expression in naive age-matched control mice. *, p< 0.05 (Mann Whitney U test vs. naive, n=7–8/ mice/ treatment/ time/ sex).
Basal expression of BDNF was higher in the diencephalon than in the forebrain, e.g. 24h after HI, forebrain: [Mean ± SD] 160.73 ± 39.5 pg/ mg of protein; diencephalon: 337.23 ±108.5 pg/mg (p=0.01). However, in response to HI, BDNF protein level in diencephalon did not increased by 24h after HI in mice treated with either veh or nec-1 (Fig. 1B). Conversely, at 96h after HI, only nec-1-treated mice had 49% and 45% higher BDNF levels than naive controls (p=0.04) and veh-treated HI mice (p=0.04), respectively (ANOVA, p=0.03). No differences in BDNF expression in the diencephalon were found between groups by 21 days after HI.
3.2. Sex differences in BDNF gene expression following HI and nec-1 treatment
Sex differences were first evaluated at the gene level. At 24h after HI, BDNF gene expression was not altered in either forebrain or diencephalon of male or female mice suggesting that acute post-transcriptional events caused the increased in BDNF protein levels in the forebrain after HI injury (Data not shown). At 96h after HI, sex-specific changes in BDNF gene expression were observed in the forebrain. BDNF gene expression in forebrain was downregulated at 96h after HI in veh-treated male (by 35%, p=0.04) and female mice (by 33%, p=0.001) compared to age-matched naive controls (Fig. 1C). However, only in male mice, nec-1 treatment after HI prevented BDNF gene downregulation in the forebrain (Fig 1C) and resulted in BDNF gene upregulation (12.5-fold, p=0.001 vs. age-matched naive controls) in diencephalon (Fig. 1D). By 21 days after HI, BDNF gene expression in veh-treated mice recovered to control levels in the forebrain (Fig. 1E) and diencephalon (Fig. 1F). Conversely, BDNF gene expression in nec-1-treated mice at 21 days after HI was upregulated in both, forebrain (2.1-fold in males, p=0.04; and 3.5-fold in females, p=0.004; Fig. 1E) and diencephalon (4.2-fold in males, p=0.001; and 4.6-fold in female, p=0.006; Fig. 1F) compared to age-matched naive controls.
No sex-differences were identified in the increase of BDNF protein levels in the forebrain at 24h after HI in either veh or nec-1 treated mice (data not shown). Similarly, no sex differences were identified in the decline of BDNF levels in veh-treated mice (by 25% [male], p= 0.05 vs. naive; by 22% [female], p=0.003 vs. naive), and the preservation of BDNF with nec-1-treatment (p<0.001 vs. veh-treated mice; ANOVA p<0.001 in both sexes, n= 4–8 / sex / group, Fig. 2A) in the forebrain at 96h after HI. Conversely, sexual dimorphism characterized the BDNF response at 24h and 96h after HI in diencephalon of nec-1 treated mice. In diencephalon, male mice treated with nec-1 had a 41% higher BDNF level than female mice at 24h after HI (p=0.01; data not shown), a difference that persisted at 96h after HI (34%, p=0.03, Fig 2B). However, it was only at 96h after HI, that nec-1 treated male mice had significantly higher BDNF levels in the diencephalon than age-matched naive controls (42%, p=0.02) and veh-treated male mice (60%, p=0.009, Fig 2B) matching the gene expression data shown above (Fig. 1D). Thus, BDNF levels were preserved locally and remotely in nec-1 treated male mice after HI, while in female mice there was only local preservation (forebrain, Fig. 2A vs. diencephalon, Fig. 2B) in response to nec-1 after HI. We further confirmed, using western blot, that these differences in BDNF levels corresponded to changes in mature BDNF (14 kDa) rather than pro-BDNF (34 kDa). The levels of pro-BDNF were similar in all treatment groups and in forebrain and diencephalon of male and female mice at 96h after HI (n=4/ treatment/ sex/ region, Fig 2C).
Figure 2.

Nec-1 prevents BDNF delayed decrease in forebrain while increasing levels in diencephalon of male mice after HI along with greater TrkB receptor activation. Bar graphs showing BDNF levels (pg/mg of protein) stratified by sex in forebrain (A) and diencephalon (B) at 96h after HI. Representative blots showing a single band for pro-BDNF (34 kDa) and mature BDNF (14kDa) in forebrain and diencephalon are shown (C). BDNF receptors for forebrain and diencephalon are also shown: phospho-TrkB (pTrkB): TrkB ratio (145 kDa) (D, E); truncated TrkB (Tc.TrkB, 95kDa) (F, G); and p75ntr (64 kDa) (H, I). Bars represent the mean ± SEM measured in naive control (white), vehicle (light grey) and nec-1 (dark grey) treated male (solid color, M) and female (hashed color, F) mice. ‡, p ≤ 0.05, one-way ANOVA (M, male; F, female). p < 0.05 (Tukey’s post-hoc); *, vs. naive control; †, vs. vehicle; n= 4–8 mice/ treatment/ time/ sex.
3.3. Sex differences in expression/ activation of BDNF receptors following HI and nec-1 treatment
Phosphorylated TrkB: TrkB receptor ratios, Tc.TrkB isoforms, and p75ntr levels were calculated/ measured to determine if changes in mature BDNF levels (Fig 2A, 2B and 2C) caused alterations in receptor activation or expression. At 24h after HI, pTrkB: TrkB ratios were similar between treatment groups and sexes in both forebrain and diencephalon (data not shown). This suggested that the early increase in BDNF levels in forebrain at 24h after HI was not linked to enhanced trophic support in either veh or nec-1 treated mice of either sex. Conversely, at 96h after HI, forebrain of nec-1 treated mice had 4.3-fold (p=0.004 [male]) and 2.2-fold (p=0.04 [female]) higher levels of TrkB phosphorylation than naive controls; which resulted in higher pTrkB: TrkB ratios (3.4-fold [male], p=0.004; 2.4-fold [female], p=0.03 vs. naive controls; Fig. 2D). In diencephalon, veh-treated HI mice had 48% (male, p=0.005) and 37% (female, p=0.03) lower pTrkB: TrkB ratios than naive controls (Fig. 2E), which matched the decreased BDNF levels in the forebrain (Fig.2A). Only male mice treated with nec-1 had increased pTrkB protein levels (34%, p< 0.001 vs. age-matched naive controls; 50%, p<0.001 vs. veh-treated HI mice) in diencephalon, which resulted in a pTrkB: TrkB ratio similar to naive mice and 52% higher than veh-treated HI mice (p=0.009, Fig 2E). The local (forebrain) increase in BDNF levels (Fig. 2A) and TrkB activation (Fig. 2D) at 96h after HI in nec-1 treated mice was matched by remote (diencephalon) TrkB activation only in male mice (Fig. 2E).
At 24h after HI, Tc.TrkB isoforms and p75ntr levels were not changed in either male or female mice in forebrain or diencephalon regardless of treatment (data not shown). At 96h after HI, only female veh-treated HI mice had higher Tc.TrkB protein levels than age-matched naive controls in forebrain (2.4-fold, p=0.04, Fig. 2F) and diencephalon (2.5-fold, p=0.04, Fig.2G). These increases in Tc.TrkB were prevented by nec-1 treatment. Similarly, at 96h after HI, veh-treated female HI mice had a modest increase in p75ntr levels in forebrain (27%, p=0.05 vs. age-matched naive control, Fig. 2H) and a more significant increase in diencephalon (~2.8-fold, p=0.015 vs. age-matched naive control, Fig 2I). Nec-1 treatment did not prevent the increase in p75ntr in forebrain at 96h after HI but it did in diencephalon. By 21 days after HI, there were no changes in pTrkB: TrkB receptor ratios, Tc.TrkB isoforms, or levels of p75ntr.
3.4. Changes in E2 and ERs following neonatal HI and nec-1 treatment
17-β-estrogen (estradiol, E2) binds to ERs which then translocate to the nucleus and upregulate BDNF expression via binding to the ERE-like sequence in the BDNF gene promoter (Blurton-Jones et al., 2004). Thus, E2 levels and ERα and ERβ protein levels were measured and then correlated with BDNF levels. At 96h after HI, E2 levels were similar in male and female mice forebrain (Fig. 3A) and diencephalon (Fig. 3B) regardless of the treatment (veh- or nec-1) or HI. Similarly, ERα and ERβ protein levels were also similar in diencephalon in all groups (Fig. 3C and 3D) with the exception of male mice treated with nec-1 which had a 29% and 19% higher ERα protein level than age-matched naive control (p=0.003) and veh-treated HI mice (p=0.02), respectively (ANOVA, p=0.003; n=5/ treatment/ sex). However, ERα changes were not directly correlated with mature BDNF levels in either male or female mice (Fig. 3F). Because the relative expression of receptor subtypes influences the final effect of hormones and only ERα is known to co-localize with BDNF in the rodent brain (Blurton-Jones et al., 2004), we evaluated the expression of ERα relative to ERβ. Unlike female mice, male mice treated with nec-1 after HI had 89% and 75% higher ERα: ERβ ratio than age-matched naive controls (p=0.02) and veh-treated male HI mice (p=0.03), respectively (ANOVA [male] p=0.01; Fig. 3E). Furthermore, ERα: ERβ ratio in the diencephalon directly correlated with their respective BDNF levels in male mice (R2 = 0.76, p<0.001; Fig. 3G).
Figure 3.

Estradiol (E2) and estrogen receptor (ER) α and β levels in forebrain and diencephalon are unchanged at 96h after neonatal HI. Only in male mice, nec-1 is linked to higher ERα: ERβ ratio which correlates with BDNF levels. Bar graphs show: E2 levels (ng/ g protein) in forebrain (A) and diencephalon (B) and ERα (C), ERβ (D) and ERα: ERβ ratio (E) adj. for β-actin in diencephalon for male (solid color, M) and female (hashed color, F) at 96h after neonatal HI. ‡, p ≤ 0.05 (one-way ANOVA). p < 0.05 (Tukey’s post-hoc); *, vs. naive age-matched control; †, vs. vehicle; n= 5 mice/ treatment/ sex. Representative blots for ERα and ERβ with bands at 66kDa and 59 kDa, respectively, are shown for male and female mice. Scatterplot of ERα (F) or ERα: ERβ ratio (G) (x-axis) vs. BDNF (y-axis) protein levels in diencephalon of male (closed circles/ continuous line) and female (open circles/ discontinuous line) mice at 96h after HI. Best fitted line by linear regression including constant in equation, p-value by Pearson product-moment correlation and coefficient of determination (R2) calculation are shown (n=5 mice/ treatment/ sex).
3.5. Parvalbumin levels in diencephalon of mice treated with nec-1 after HI
Following neonatal HI, glutamate excitotoxicity can be attenuated by activation of the GABA system which is a suggested mechanism for topiramate-mediated neuroprotection (Schubert et al., 2005). Because BDNF allows maturation of GABAergic circuits (Amateau et al., 2004) promoting differentiation of GABAergic interneurons and increasing vesicular GABA transporter expression (Berghuis et al., 2004), we evaluated parvalbumin as a surrogate for GABAergic inhibitory circuits, in the diencephalon, the region showing sexual dimorphism in BDNF levels in response to nec-1 treatment after HI. At 96h after HI, parvalbumin gene expression did not change in diencephalon of either males or females veh-treated HI mice (vs. age-matched naive control; Fig 4A). However, male mice treated with nec-1 after HI had a 3.7-fold (p=0.001 vs. age-matched naive control) upregulation in parvalbumin gene expression (n=6/ treatment/ sex) and > 40% (p=0.04 vs. age-matched naive control, and p=0.03 vs. veh-treated HI mice) increase in parvalbumin protein levels (ANOVA, p= 0.02; n=5/ treatment/ sex; Fig. 4B). Parvalbumin mRNA and protein levels remained unchanged in female mice after HI regardless of treatment.
Figure 4.
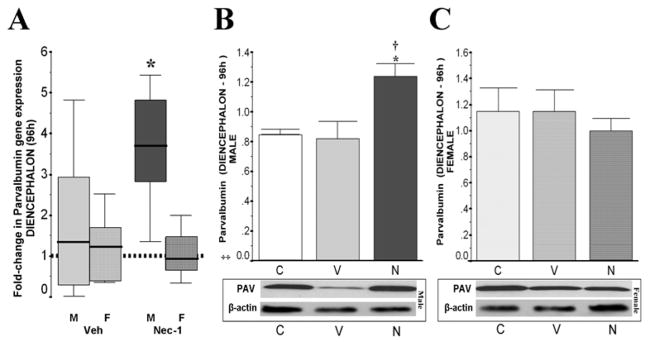
Fold-change in parvalbumin gene expression (vs. naive) is shown as box-and-whisker plot, with boxes representing interquartile range (IQR) and median (solid line in boxes) for male (solid color, M) and female (hashed color, F) mice diencephalon (A) at 96h after neonatal HI. Reference (discontinued) line sitting at 1 represents parvalbumin gene expression in naive age-matched control mice. *, p< 0.05 (Mann Whitney U test vs. naive, n=6 mice/ treatment/ sex). Bar graphs showing parvalbumin protein levels (adjusted for b-actin) in diencephalon in male (B) and female (C) mice at 96h after HI. ‡, p ≤ 0.05, one-way ANOVA. p < 0.05 (Tukey’s post-hoc); *, vs. naive control; †, vs. vehicle; n= 4–6 mice/ treatment/ sex. Representative blots show parvalbumin (PAV, 12 kDa) and loading control.
4. DISCUSSION
The most important findings of the present study are that following neonatal HI, brain BDNF signaling is altered in regional and sex-specific manner (Fig 5). BDNF protein levels in the forebrain, but not in diencephalon, are increased by 24h after HI, followed by a decline by 96h and a recovery to basal levels by 21 days after HI in both male and female mice. However, only in female mice, HI also increases p75ntr and Tc.TrkB in both forebrain and diencephalon. Other differences in BDNF signaling between sexes appear in response to treatment with nec-1 after HI. Male mice treated with nec-1 have preserved BDNF mRNA and protein levels at 96h after HI in forebrain and diencephalon, and increased TrkB activation (pTrkB: TrkB ratio) without changes in Tc.TrkB and p75ntr levels. In contrast in female mice treatment with nec-1 after HI increases BDNF levels and TrkB activation at 96h only in forebrain and prevents the increases in Tc.TrkB and p75ntr levels in forebrain and diencephalon. Although the mechanism is unclear, changes in BDNF expression in diencephalon directly correlate with the relative predominance of ERα over ERβ. Our current findings showing preservation of BDNF levels and TrkB activation in forebrain and diencephalon in male mice, but only in forebrain in female mice after HI, suggest preservation of connectivity between these two regions in male but not in female mice. Alternatively, these findings may represent an off target effect of nec-1 in the different brain regions. This is the first report describing sex-specific changes in BDNF signaling following neonatal HI and a neuroprotectant.
Figure 5.

Changes in BDNF signaling by 96h after HI differ by sex. In both, male (A) and female (B) mice, neonatal HI decreases BDNF levels in forebrain (1) and perhaps by disturbing essential remote trafficking (2), decreases TrkB activation at the diencephalon (3). However, only in female mice, HI also increases p75ntr and truncated TrkB in both forebrain and diencephalon (4), which provides another biochemical mechanism for the known proclivity to caspase-dependent cell death observed in females (vs. males) following neonatal HI (Hagberg et al., 2004, Zhu et al., 2006). Treatment with the neuroprotectant, nec-1, soon after neonatal HI, reverse the BDNF decline in forebrain in both sexes (5); but only in male mice, nec-1 also preserves TrkB activation at the diencephalon (6). We speculate that this male-specific recovery/ preservation of remote receptor activation with nec-1 is an evidence of a preferential protection of connectivity (6). Furthermore, nec-1, indirectly or directly may also facilitate other protective events in the diencephalon of male mice after HI, such as the increase in levels of parvalbumin (PAV, marker of GABAergic neurons) and BDNF (7). The increase in TrkB activation in the forebrain of mice treated with nec- 1 may be explained by both the remote influence by BDNF from the diencephalon (exclusively in male mice) as well as local autocrine influence (in both sexes) (8). Nec-1 prevents pro-cell death changes such as the increases in truncated TrkB and p75ntr in female mice (9). Altogether, these findings provide evidence of a sex differential BDNF response to HI and nec-1 treatment.
4.1. Patterns of BDNF response following neonatal HI and nec-1 treatment
Although studies in whole brain homogenates following neonatal HI show an early increase in BDNF protein levels in rat models (Wang et al., 2013), BDNF responses appear to be regionally specific. An increase in BDNF levels occurs in cortex and striatum but not in hippocampus at 24h after neonatal HI in rats (Jantzie and Todd, 2010). In the present work, BDNF response to neonatal HI is biphasic only in the forebrain, with an early increase (24h after HI) followed by a delayed decrease (96h after HI). The early post-transcriptional BDNF increase (24h after HI) in forebrain is a marker of acute injury which is followed by later transcriptional decline in BDNF expression (96h after HI). The decrease in BDNF levels by 96h after HI only in forebrain may represent the significantly greater extent of injury in cortex and hippocampus compared to thalamus after HI (Northington et al., 2011, and fig. 6). These regional BDNF levels in response to HI are similar in male and female mice and they are followed by a “recovery” to and above age-matched naive control levels by 21 days after HI, which temporally coincides with an increase in neurogenesis and neuronal migration reported in rodent models starting at 2 weeks after neonatal HI (Yang et al., 2007).
Figure 6.

At 96h after HI, nec-1 treated mice have less percent injury than vehicle treated mice in hippocampus (A) of both, male and female mice, and in thalamus (B) of only male mice. Thalamic injury was significantly greater in male than female mice by 96h after HI. Details for methods and results for cortex are reported elsewhere (Northington et al., 2011). Data shown as box-and-whisker plot, with boxes representing interquartile range (IQR, 25th to 75th percentile) and showing median (solid line in boxes) for male (white boxes) and female (grey boxes) mice at 96h after neonatal HI. *, p< 0.05 (Mann Whitney U test vs. naive, n=3–7/ mice/ treatment/ sex).
The effects of neuroprotective treatments on endogenous BDNF responses have not been reported following neonatal HI until now. We found that nec-1 treatment following neonatal HI, like hypothermia following adult ischemia (D’Cruz et al., 2002), preserves BDNF levels by 96h after HI in the forebrain of male and female mice, but in the diencephalon of only male mice. Nec-1 provides histological protection by 96h after HI in cortex (Northington et al., 2011) and thalamus only in male mice, but protects hippocampus in both male and female mice (post-hoc analysis shown in fig. 6). Thus, the sexual differences in BDNF levels in response to nec-1 may be the consequence of a more robust protection of male mice in cortex and thalamus and perhaps it is a marker of preserved connectivity and BDNF trafficking between these two regions (Karpova, 2013) in males. However, the lack of sexual differences in BDNF preservation in forebrain in response to nec-1 after HI, may also represent the equal hippocampal neuroprotection afforded by the treatment in both sexes (Fig. 6A). Therefore, the preservation of BDNF levels may be either a consequence of the degree of neuroprotection as described above or, alternatively, an off target direct effect of nec-1 in the BDNF system in the different brain regions. Regardless of the sequence of events, the delayed preservation of BDNF levels seen with nec-1 treatment at 96h after HI in forebrain and diencephalon of male mice, may have come at an appropriate developmental and post-injury time to support sex preferential repair and later recovery (Northington et al., 2011, Chavez-Valdez et al., 2012a)
The mechanisms behind this male-preferential BDNF response to nec-1 after neonatal HI remain unclear. Because BDNF gene promoter contains a sequence similar to the canonical estrogen response element (Sohrabji et al., 1995), E2 via the estrogen receptor, particularly ERα, may enhance BDNF expression (Blurton-Jones et al., 2004). Here, we show that E2 levels are similar in male and female mice forebrain and diencephalon at 96h after HI regardless of treatment and although ERα protein levels are slightly higher in male mice treated with nec-1 after HI, there is no significant correlation between ERα protein levels and BDNF levels. However, the relative expression of ERα (vs. ERβ) is significantly higher in male mice treated with nec-1 after HI (vs. veh-treated HI mice) and this ratio strongly and directly correlates with BDNF levels in the diencephalon. This correlation suggests that there is a biological significance in the relative predominance of ERα over ERβ, which may explain changes in BDNF despite unchanged E2 levels.
4.2. BDNF receptor response following neonatal HI and nec-1 treatment
BDNF exerts biological effects via its binding to the high-affinity surface receptor, TrkB, and the low-affinity receptor, p75ntr; both of which are highly expressed in the developing rodent brain (Masana et al., 1993, Altar et al., 1994, Lush et al., 2005, Yang et al., 2009). Following binding of BDNF to FL.TrkB, autophosphorylation of the tyrosine kinase domain ensues with downstream signaling that supports neuronal survival and plasticity (Dubus et al., 2000, Luberg et al., 2010). Other TrkB receptor isoforms lacking the tyrosine kinase domain (Tc.TrkB) trigger BDNF sequestration in astrocytes and decrease bio-availability (Altar et al., 1994). The balance between TrkB receptor isoforms may explain the paradoxically enhanced neuronal necrosis caused by exposure to high concentrations of BDNF (Gwag et al., 1995, Koh et al., 1995, Kim et al., 2003) with secondary downregulation of FL.TrkB receptor (Frank et al., 1996, Knusel et al., 1997, Goutan et al., 1998), and upregulation of Tc.TrkB isoforms (Klein et al., 1990, Biffo et al., 1995, Goutan et al., 1998, Alderson et al., 2000). Although nec-1 supports preservation of BDNF expression in the forebrain (Fig. 2A) 96h after HI; it does not produce downregulation of FL.TrkB receptor or upregulation of Tc.TrkB in male mice. Instead, nec-1 treated male mice show increased TrkB phosphorylation in the forebrain (Fig. 2D) and diencephalon (Fig. 2E) 96h after HI. The lack of TrkB phosphorylation in the diencephalon of female mice may suggests sub-optimal preservation of connectivity with forebrain (Lotto et al., 2001) or response to low levels of locally produced BDNF.
P75ntr signaling is linked to: i) cell survival when interacting with highly expressed FL.TrkB receptors to induce autophosphorylation (Hantzopoulos et al., 1994, Bibel et al., 1999, Khursigara et al., 2001), or ii) caspase-dependent cell death when combined with decreasing levels or activation of FL.TrkB (Chao, 1994, Koh et al., 1995, Majdan et al., 1997). NMDA-mediated excitotoxicity, like neonatal HI, increases p75ntr levels and activates caspase-3, while genetic deletion of p75ntr prevents caspase activation (Griesmaier et al., 2010). The present experiments demonstrate that only in female mice p75ntr is increased in forebrain and diencephalon after neonatal HI. The combination of decreased TrkB receptor activation and increased p75ntr expression in forebrain and diencephalon of female mice matches the greater proclivity to caspase-dependent cell death described in females in response to neonatal HI (Hagberg et al., 2004, Zhu et al., 2006, Chavez-Valdez et al., 2012b). Thus, the predominance of caspase-mediated cell death over necrosis after HI in female mice, may explain the milder and more variable cortical (Northington et al., 2011), hippocampal and thalamic (Fig. 6) injuries seen in female mice at 96h after HI. Nec-1 prevents the increase in Tc.TrkB in both forebrain and diencephalon and the increase in p75ntr in diencephalon of female mice, substantially altering the overall balance of receptor subtypes responsive to BDNF. In the histology (Fig.6 and Northington et al., 2011), female mice are not protected by nec-1 after HI in cortex or thalamus; however, the mean percent injury with nec-1 treatment after HI is similar in both sexes. The lack of significant neuroprotection with nec-1 in female mice after HI is explained by their greater injury variability in the different regions at baseline, thus the changes in p75ntr and Tc.TrkB with nec-1 treatment may still provide protection after HI, even if not linked to increased activation of TrkB receptor in the diencephalon of female mice.
4.3. BDNF and parvalbumin expression
BDNF influences the development of both, excitatory (i.e. glutamate) and inhibitory (i.e. GABA) circuits promoting synaptic plasticity (Elmariah et al., 2005, Matsumoto et al., 2006, Laudes et al., 2012). In cultured fast-spiking GABAergic interneurons, BDNF promotes differentiation and increases vesicular GABA transporter expression (Berghuis et al., 2004). In our experiments the levels of parvalbumin, a calcium binding protein that is highly expressed in GABAergic inhibitory interneurons (Destexhe et al., 1998), mirror the increase in BDNF protein level in nec-1 treated male mice after HI suggesting that the increase in parvalbumin expression may be a consequence of the preservation of BDNF. Additionally, because functional potentiation of GABA receptors using propofol increases BDNF in cortex and hippocampus (Ponten et al., 2011), it is possible that the increase in parvalbumin in the diencephalon found in male mice treated with nec-1 provides additional influence to preserve BDNF expression and signaling.
4.4. Conclusions
Conflicting results following exogenous BDNF treatment in models of neonatal HI are likely due to limited understanding of the sex differences in BDNF signaling responses after brain injury. The present results provide a framework to better understand the dynamic changes occurring in BDNF signaling pathway on a regional and sex basis and how these changes might be modulated to improve neuroprotection (Fig 5). The increases in BDNF immediately after HI, may or may not have acute biological effect and it may simply serve as a marker of injury. The decline in BDNF levels in forebrain and TrkB activation in diencephalon in male and female mice by 96h after HI suggest decrease distal BDNF trafficking. The increase in p75ntr and Tc.TrkB levels only in female mice after HI suggest differences in the mechanism of cell death between sexes which explain the lesser degree of injury seen in female mice after HI. The first event modifiable by nec-1 treatment is the preservation of BDNF expression 96h after HI which occurs only in male mice in both forebrain and diencephalon and matches the significant neuroprotection in cortex and thalamus. Using nec-1 treatment as a tool to modulate BDNF signaling after HI, we speculate that delayed treatment with BDNF or BDNF supportive therapies may work to initiate repair and recovery differently in males and females. Other effects of nec-1 treatment after HI appear to prevent the increase levels of receptors known to signal for cell death (Tc.TrkB and p75ntr) in female mice, suggesting that nec-1 may also provide a certain degree of protection to females. The mechanisms behind the effects of nec-1 in BDNF signaling are unknown. We may only speculate that alterations in the relative expression of ER with predominance of ERα over ERβ may be involved. Our results add to the body of literature showing the importance of the evaluation of sex differences when testing potential treatments for neonatal brain injury
HIGHLIGHTS.
In both sexes, neonatal HI acutely increases BDNF levels in forebrain but not diencephalon.
In both sexes, HI decreases BDNF in forebrain and TrkB activation in diencephalon.
Only in female mice, neonatal HI increases p75ntr and truncated TrkB levels.
Only in male mice, nec-1 preserves BDNF levels and TrkB activation.
BDNF changes directly correlate with estrogen receptor α: β ratio.
Acknowledgments
The authors thank Debra Flock and Devin Mack for their expert assistance with ELISA and western blot and Julia Hinojos, Darla Mata, Melissa Valdes-Luna and Veronica Bostic for their administrative assistance.
7. SOURCES OF SUPPORT.
Experiments have been funded in part by a grant from the National Institute of Health (R01HD070996 - FJN).
ABBREVIATIONS
- BDNF
Brain derived neurotrophic factor
- E2
Estradiol
- ER
Estrogen receptor
- FL.TrkB
full-length TrkB
- HI
hypoxia-ischemia
- IQR
interquartile range
- nec-1
necrostatin-1
- p
postnatal day
- p75ntr
p75 neurotrophic receptor
- pTrkB
phosphorylated TrkB
- qRT-PCR
quantitative reverse transcriptase PCR
- Tc.TrkB
truncated TrkB
- TrkB
tyrosine-related kinase B
- veh
vehicle
Footnotes
ETHICS STATEMENT: I have read and have abided by the statement of ethical standards for manuscripts submitted to Neuroscience.
FINAL APPROVAL: All authors have approved the final article
The authors declare no conflict of interest related to the data presented in the manuscript.
6. AUTHOR CONTRIBUTIONS.
RCV participated in experimental design and execution, carried out biochemical studies, analyzed the data, review results and drafted manuscript. LJM participated in experimental design, carried out treatment injections, critically reviewed results and drafted manuscript. SR participated in execution of experiments, review results and drafted the manuscript. EBG participated in experimental design, critically reviewed results and drafted manuscript. FJN participated in experimental design and execution, reviewed results and drafted manuscript. RCV and FJN are responsible for the overall research direction of this and related projects. All authors read and approved the final manuscript.
8. DISCLOSURE/CONFLICT OF INTEREST:
None
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alderson RF, Curtis R, Alterman AL, Lindsay RM, DiStefano PS. Truncated TrkB mediates the endocytosis and release of BDNF and neurotrophin-4/5 by rat astrocytes and schwann cells in vitro. Brain Res. 2000;871:210–222. doi: 10.1016/s0006-8993(00)02428-8. [DOI] [PubMed] [Google Scholar]
- Altar CA, Siuciak JA, Wright P, Ip NY, Lindsay RM, Wiegand SJ. In situ hybridization of trkB and trkC receptor mRNA in rat forebrain and association with high-affinity binding of [125I]BDNF, [125I]NT-4/5 and [125I]NT-3. Eur J Neurosci. 1994;6:1389–1405. doi: 10.1111/j.1460-9568.1994.tb01001.x. [DOI] [PubMed] [Google Scholar]
- Amantea D, Russo R, Bagetta G, Corasaniti MT. From clinical evidence to molecular mechanisms underlying neuroprotection afforded by estrogens. Pharmacol Res. 2005;52:119–132. doi: 10.1016/j.phrs.2005.03.002. [DOI] [PubMed] [Google Scholar]
- Amateau SK, Alt JJ, Stamps CL, McCarthy MM. Brain estradiol content in newborn rats: sex differences, regional heterogeneity, and possible de novo synthesis by the female telencephalon. Endocrinology. 2004;145:2906–2917. doi: 10.1210/en.2003-1363. [DOI] [PubMed] [Google Scholar]
- Arteni NS, Pereira LO, Rodrigues AL, Lavinsky D, Achaval ME, Netto CA. Lateralized and sex-dependent behavioral and morphological effects of unilateral neonatal cerebral hypoxia-ischemia in the rat. Behav Brain Res. 2010;210:92–98. doi: 10.1016/j.bbr.2010.02.015. [DOI] [PubMed] [Google Scholar]
- Berghuis P, Dobszay MB, Sousa KM, Schulte G, Mager PP, Hartig W, Gorcs TJ, Zilberter Y, Ernfors P, Harkany T. Brain-derived neurotrophic factor controls functional differentiation and microcircuit formation of selectively isolated fast-spiking GABAergic interneurons. Eur J Neurosci. 2004;20:1290–1306. doi: 10.1111/j.1460-9568.2004.03561.x. [DOI] [PubMed] [Google Scholar]
- Bibel M, Hoppe E, Barde YA. Biochemical and functional interactions between the neurotrophin receptors trk and p75NTR. EMBO J. 1999;18:616–622. doi: 10.1093/emboj/18.3.616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biffo S, Offenhauser N, Carter BD, Barde YA. Selective binding and internalisation by truncated receptors restrict the availability of BDNF during development. Development. 1995;121:2461–2470. doi: 10.1242/dev.121.8.2461. [DOI] [PubMed] [Google Scholar]
- Blurton-Jones M, Kuan PN, Tuszynski MH. Anatomical evidence for transsynaptic influences of estrogen on brain-derived neurotrophic factor expression. J Comp Neurol. 2004;468:347–360. doi: 10.1002/cne.10989. [DOI] [PubMed] [Google Scholar]
- Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- Chao MV. The p75 neurotrophin receptor. J Neurobiol. 1994;25:1373–1385. doi: 10.1002/neu.480251106. [DOI] [PubMed] [Google Scholar]
- Chavez-Valdez R, Martin LJ, Flock DL, Northington FJ. Necrostatin-1 attenuates mitochondrial dysfunction in neurons and astrocytes following neonatal hypoxia-ischemia. Neuroscience. 2012a;219:192–203. doi: 10.1016/j.neuroscience.2012.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chavez-Valdez R, Martin LJ, Northington FJ. Programmed Necrosis: A Prominent Mechanism of Cell Death following Neonatal Brain Injury. Neurol Res Int. 2012b;2012:257563. doi: 10.1155/2012/257563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Cruz BJ, Fertig KC, Filiano AJ, Hicks SD, DeFranco DB, Callaway CW. Hypothermic reperfusion after cardiac arrest augments brain-derived neurotrophic factor activation. J Cereb Blood Flow Metab. 2002;22:843–851. doi: 10.1097/00004647-200207000-00009. [DOI] [PubMed] [Google Scholar]
- Degterev A, Huang Z, Boyce M, Li Y, Jagtap P, Mizushima N, Cuny GD, Mitchison TJ, Moskowitz MA, Yuan J. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat Chem Biol. 2005;1:112–119. doi: 10.1038/nchembio711. [DOI] [PubMed] [Google Scholar]
- Destexhe A, Contreras D, Steriade M. Mechanisms underlying the synchronizing action of corticothalamic feedback through inhibition of thalamic relay cells. J Neurophysiol. 1998;79:999–1016. doi: 10.1152/jn.1998.79.2.999. [DOI] [PubMed] [Google Scholar]
- Ditelberg JS, Sheldon RA, Epstein CJ, Ferriero DM. Brain injury after perinatal hypoxia-ischemia is exacerbated in copper/zinc superoxide dismutase transgenic mice. Pediatric research. 1996;39:204–208. doi: 10.1203/00006450-199602000-00003. [DOI] [PubMed] [Google Scholar]
- Douglas-Escobar M, Rossignol C, Steindler D, Zheng T, Weiss MD. Neurotrophin-induced migration and neuronal differentiation of multipotent astrocytic stem cells in vitro. PLoS One. 2012;7:e51706. doi: 10.1371/journal.pone.0051706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubus P, Faucheux B, Boissiere F, Groppi A, Vital C, Vital A, Agid Y, Hirsch EC, Merlio JP. Expression of Trk isoforms in brain regions and in the striatum of patients with Alzheimer’s disease. Exp Neurol. 2000;165:285–294. doi: 10.1006/exnr.2000.7447. [DOI] [PubMed] [Google Scholar]
- Elmariah SB, Oh EJ, Hughes EG, Balice-Gordon RJ. Astrocytes regulate inhibitory synapse formation via Trk-mediated modulation of postsynaptic GABAA receptors. J Neurosci. 2005;25:3638–3650. doi: 10.1523/JNEUROSCI.3980-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank L, Ventimiglia R, Anderson K, Lindsay RM, Rudge JS. BDNF down-regulates neurotrophin responsiveness, TrkB protein and TrkB mRNA levels in cultured rat hippocampal neurons. Eur J Neurosci. 1996;8:1220–1230. doi: 10.1111/j.1460-9568.1996.tb01290.x. [DOI] [PubMed] [Google Scholar]
- Goutan E, Marti E, Ferrer I. BDNF, and full length and truncated TrkB expression in the hippocampus of the rat following kainic acid excitotoxic damage. Evidence of complex time-dependent and cell-specific responses. Brain Res Mol Brain Res. 1998;59:154–164. doi: 10.1016/s0169-328x(98)00156-9. [DOI] [PubMed] [Google Scholar]
- Griesmaier E, Schlager G, Wegleiter K, Hermann M, Urbanek M, Simbruner G, Keller M. Role of p75NTR in NMDAR-mediated excitotoxic brain injury in neonatal mice. Brain Res. 2010;1355:31–40. doi: 10.1016/j.brainres.2010.07.095. [DOI] [PubMed] [Google Scholar]
- Gwag BJ, Koh JY, Chen MM, Dugan LL, Behrens MM, Lobner D, Choi DW. BDNF or IGF-I potentiates free radical-mediated injury in cortical cell cultures. Neuroreport. 1995;7:93–96. [PubMed] [Google Scholar]
- Hagberg H, Wilson MA, Matsushita H, Zhu C, Lange M, Gustavsson M, Poitras MF, Dawson TM, Dawson VL, Northington F, Johnston MV. PARP-1 gene disruption in mice preferentially protects males from perinatal brain injury. J Neurochem. 2004;90:1068–1075. doi: 10.1111/j.1471-4159.2004.02547.x. [DOI] [PubMed] [Google Scholar]
- Han J, Pollak J, Yang T, Siddiqui MR, Doyle KP, Taravosh-Lahn K, Cekanaviciute E, Han A, Goodman JZ, Jones B, Jing D, Massa SM, Longo FM, Buckwalter MS. Delayed administration of a small molecule tropomyosin-related kinase B ligand promotes recovery after hypoxic-ischemic stroke. Stroke; a journal of cerebral circulation. 2012;43:1918–1924. doi: 10.1161/STROKEAHA.111.641878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanamura K, Harada A, Katoh-Semba R, Murakami F, Yamamoto N. BDNF and NT-3 promote thalamocortical axon growth with distinct substrate and temporal dependency. Eur J Neurosci. 2004;19:1485–1493. doi: 10.1111/j.1460-9568.2004.03228.x. [DOI] [PubMed] [Google Scholar]
- Hantzopoulos PA, Suri C, Glass DJ, Goldfarb MP, Yancopoulos GD. The low affinity NGF receptor, p75, can collaborate with each of the Trks to potentiate functional responses to the neurotrophins. Neuron. 1994;13:187–201. doi: 10.1016/0896-6273(94)90469-3. [DOI] [PubMed] [Google Scholar]
- Hill CA, Threlkeld SW, Fitch RH. Early testosterone modulated sex differences in behavioral outcome following neonatal hypoxia ischemia in rats. Int J Dev Neurosci. 2011;29:381–388. doi: 10.1016/j.ijdevneu.2011.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurn PD, Vannucci SJ, Hagberg H. Adult or perinatal brain injury: does sex matter? Stroke; a journal of cerebral circulation. 2005;36:193–195. doi: 10.1161/01.STR.0000153064.41332.f6. [DOI] [PubMed] [Google Scholar]
- Jantzie LL, Todd KG. Doxycycline inhibits proinflammatory cytokines but not acute cerebral cytogenesis after hypoxia-ischemia in neonatal rats. J Psychiatry Neurosci. 2010;35:20–32. doi: 10.1503/jpn.090061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karpova NN. Role of BDNF epigenetics in activity-dependent neuronal plasticity. Neuropharmacology. 2013 doi: 10.1016/j.neuropharm.2013.04.002. [DOI] [PubMed] [Google Scholar]
- Khursigara G, Bertin J, Yano H, Moffett H, DiStefano PS, Chao MV. A prosurvival function for the p75 receptor death domain mediated via the caspase recruitment domain receptor-interacting protein 2. J Neurosci. 2001;21:5854–5863. doi: 10.1523/JNEUROSCI.21-16-05854.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HJ, Hwang JJ, Behrens MM, Snider BJ, Choi DW, Koh JY. TrkB mediates BDNF-induced potentiation of neuronal necrosis in cortical culture. Neurobiology of disease. 2003;14:110–119. doi: 10.1016/s0969-9961(03)00103-7. [DOI] [PubMed] [Google Scholar]
- Klein R, Conway D, Parada LF, Barbacid M. The trkB tyrosine protein kinase gene codes for a second neurogenic receptor that lacks the catalytic kinase domain. Cell. 1990;61:647–656. doi: 10.1016/0092-8674(90)90476-u. [DOI] [PubMed] [Google Scholar]
- Knusel B, Gao H, Okazaki T, Yoshida T, Mori N, Hefti F, Kaplan DR. Ligand-induced down-regulation of Trk messenger RNA, protein and tyrosine phosphorylation in rat cortical neurons. Neuroscience. 1997;78:851–862. doi: 10.1016/s0306-4522(96)00616-1. [DOI] [PubMed] [Google Scholar]
- Koh JY, Gwag BJ, Lobner D, Choi DW. Potentiated necrosis of cultured cortical neurons by neurotrophins. Science. 1995;268:573–575. doi: 10.1126/science.7725105. [DOI] [PubMed] [Google Scholar]
- Laudes T, Meis S, Munsch T, Lessmann V. Impaired transmission at corticothalamic excitatory inputs and intrathalamic GABAergic synapses in the ventrobasal thalamus of heterozygous BDNF knockout mice. Neuroscience. 2012;222:215–227. doi: 10.1016/j.neuroscience.2012.07.005. [DOI] [PubMed] [Google Scholar]
- Lim SY, Davidson SM, Mocanu MM, Yellon DM, Smith CC. The cardioprotective effect of necrostatin requires the cyclophilin-D component of the mitochondrial permeability transition pore. Cardiovasc Drugs Ther. 2007;21:467–469. doi: 10.1007/s10557-007-6067-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lotto RB, Asavaritikrai P, Vali L, Price DJ. Target-derived neurotrophic factors regulate the death of developing forebrain neurons after a change in their trophic requirements. J Neurosci. 2001;21:3904–3910. doi: 10.1523/JNEUROSCI.21-11-03904.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luberg K, Wong J, Weickert CS, Timmusk T. Human TrkB gene: novel alternative transcripts, protein isoforms and expression pattern in the prefrontal cerebral cortex during postnatal development. J Neurochem. 2010;113:952–964. doi: 10.1111/j.1471-4159.2010.06662.x. [DOI] [PubMed] [Google Scholar]
- Lush ME, Ma L, Parada LF. TrkB signaling regulates the developmental maturation of the somatosensory cortex. Int J Dev Neurosci. 2005;23:523–536. doi: 10.1016/j.ijdevneu.2005.04.003. [DOI] [PubMed] [Google Scholar]
- Majdan M, Lachance C, Gloster A, Aloyz R, Zeindler C, Bamji S, Bhakar A, Belliveau D, Fawcett J, Miller FD, Barker PA. Transgenic mice expressing the intracellular domain of the p75 neurotrophin receptor undergo neuronal apoptosis. J Neurosci. 1997;17:6988–6998. doi: 10.1523/JNEUROSCI.17-18-06988.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marini AM, Jiang X, Wu X, Pan H, Guo Z, Mattson MP, Blondeau N, Novelli A, Lipsky RH. Preconditioning and neurotrophins: a model for brain adaptation to seizures, ischemia and other stressful stimuli. Amino Acids. 2007;32:299–304. doi: 10.1007/s00726-006-0414-y. [DOI] [PubMed] [Google Scholar]
- Markovic SN, Murasko DM. Anesthesia inhibits interferon-induced natural killer cell cytotoxicity via induction of CD8+ suppressor cells. Cell Immunol. 1993;151:474–480. doi: 10.1006/cimm.1993.1256. [DOI] [PubMed] [Google Scholar]
- Masana Y, Wanaka A, Kato H, Asai T, Tohyama M. Localization of trkB mRNA in postnatal brain development. J Neurosci Res. 1993;35:468–479. doi: 10.1002/jnr.490350503. [DOI] [PubMed] [Google Scholar]
- Matsumoto T, Numakawa T, Yokomaku D, Adachi N, Yamagishi S, Numakawa Y, Kunugi H, Taguchi T. Brain-derived neurotrophic factor-induced potentiation of glutamate and GABA release: different dependency on signaling pathways and neuronal activity. Mol Cell Neurosci. 2006;31:70–84. doi: 10.1016/j.mcn.2005.09.002. [DOI] [PubMed] [Google Scholar]
- Mayoral SR, Omar G, Penn AA. Sex differences in a hypoxia model of preterm brain damage. Pediatric research. 2009;66:248–253. doi: 10.1203/PDR.0b013e3181b1bc34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy DM, Zhang X, Darnell SB, Sangrey GR, Yanagawa Y, Sadri-Vakili G, Bhide PG. Cocaine alters BDNF expression and neuronal migration in the embryonic mouse forebrain. J Neurosci. 2011;31:13400–13411. doi: 10.1523/JNEUROSCI.2944-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mooney SM, Miller MW. Role of neurotrophins on postnatal neurogenesis in the thalamus: prenatal exposure to ethanol. Neuroscience. 2011;179:256–266. doi: 10.1016/j.neuroscience.2011.01.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morcuende S, Gadd CA, Peters M, Moss A, Harris EA, Sheasby A, Fisher AS, De Felipe C, Mantyh PW, Rupniak NM, Giese KP, Hunt SP. Increased neurogenesis and brain-derived neurotrophic factor in neurokinin-1 receptor gene knockout mice. Eur J Neurosci. 2003;18:1828–1836. doi: 10.1046/j.1460-9568.2003.02911.x. [DOI] [PubMed] [Google Scholar]
- Northington FJ, Chavez-Valdez R, Graham EM, Razdan S, Gauda EB, Martin LJ. Necrostatin decreases oxidative damage, inflammation, and injury after neonatal HI. J Cereb Blood Flow Metab. 2011;31:178–189. doi: 10.1038/jcbfm.2010.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Northington FJ, Koehler RC, Traystman RJ, Martin LJ. Nitric oxide synthase 1 and nitric oxide synthase 3 protein expression is regionally and temporally regulated in fetal brain. Brain Res Dev Brain Res. 1996;95:1–14. doi: 10.1016/0165-3806(96)00051-x. [DOI] [PubMed] [Google Scholar]
- Pencea V, Bingaman KD, Wiegand SJ, Luskin MB. Infusion of brain-derived neurotrophic factor into the lateral ventricle of the adult rat leads to new neurons in the parenchyma of the striatum, septum, thalamus, and hypothalamus. J Neurosci. 2001;21:6706–6717. doi: 10.1523/JNEUROSCI.21-17-06706.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001;29:e45. doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfaffl MW, Tichopad A, Prgomet C, Neuvians TP. Determination of stable housekeeping genes, differentially regulated target genes and sample integrity: BestKeeper--Excel-based tool using pair-wise correlations. Biotechnol Lett. 2004;26:509–515. doi: 10.1023/b:bile.0000019559.84305.47. [DOI] [PubMed] [Google Scholar]
- Ponten E, Fredriksson A, Gordh T, Eriksson P, Viberg H. Neonatal exposure to propofol affects BDNF but not CaMKII, GAP-43, synaptophysin and tau in the neonatal brain and causes an altered behavioural response to diazepam in the adult mouse brain. Behav Brain Res. 2011;223:75–80. doi: 10.1016/j.bbr.2011.04.019. [DOI] [PubMed] [Google Scholar]
- Renolleau S, Fau S, Charriaut-Marlangue C. Gender-related differences in apoptotic pathways after neonatal cerebral ischemia. Neuroscientist. 2008;14:46–52. doi: 10.1177/1073858407308889. [DOI] [PubMed] [Google Scholar]
- Roselli CE, Liu M, Hurn PD. Brain aromatization: classic roles and new perspectives. Seminars in reproductive medicine. 2009;27:207–217. doi: 10.1055/s-0029-1216274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenkranz K, Kumbruch S, Tenbusch M, Marcus K, Marschner K, Dermietzel R, Meier C. Transplantation of human umbilical cord blood cells mediated beneficial effects on apoptosis, angiogenesis and neuronal survival after hypoxic-ischemic brain injury in rats. Cell Tissue Res. 2012;348:429–438. doi: 10.1007/s00441-012-1401-0. [DOI] [PubMed] [Google Scholar]
- Scharfman HE. Brain-derived neurotrophic factor and epilepsy--a missing link? Epilepsy Curr. 2005;5:83–88. doi: 10.1111/j.1535-7511.2005.05312.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt-Kastner R, Humpel C, Wetmore C, Olson L. Cellular hybridization for BDNF, trkB, and NGF mRNAs and BDNF-immunoreactivity in rat forebrain after pilocarpine-induced status epilepticus. Exp Brain Res. 1996;107:331–347. doi: 10.1007/BF00230416. [DOI] [PubMed] [Google Scholar]
- Schubert S, Brandl U, Brodhun M, Ulrich C, Spaltmann J, Fiedler N, Bauer R. Neuroprotective effects of topiramate after hypoxia-ischemia in newborn piglets. Brain Res. 2005;1058:129–136. doi: 10.1016/j.brainres.2005.07.061. [DOI] [PubMed] [Google Scholar]
- Sohrabji F, Miranda RC, Toran-Allerand CD. Identification of a putative estrogen response element in the gene encoding brain-derived neurotrophic factor. Proc Natl Acad Sci U S A. 1995;92:11110–11114. doi: 10.1073/pnas.92.24.11110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Cao M, Liu A, Di W, Zhao F, Tian Y, Jia J. Changes of inflammatory cytokines and neurotrophins emphasized their roles in hypoxic-ischemic brain damage. Int J Neurosci. 2013;123:191–195. doi: 10.3109/00207454.2012.744755. [DOI] [PubMed] [Google Scholar]
- Webster MJ, Herman MM, Kleinman JE, Shannon Weickert C. BDNF and trkB mRNA expression in the hippocampus and temporal cortex during the human lifespan. Gene Expr Patterns. 2006;6:941–951. doi: 10.1016/j.modgep.2006.03.009. [DOI] [PubMed] [Google Scholar]
- Yang J, Siao CJ, Nagappan G, Marinic T, Jing D, McGrath K, Chen ZY, Mark W, Tessarollo L, Lee FS, Lu B, Hempstead BL. Neuronal release of proBDNF. Nat Neurosci. 2009;12:113–115. doi: 10.1038/nn.2244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z, Covey MV, Bitel CL, Ni L, Jonakait GM, Levison SW. Sustained neocortical neurogenesis after neonatal hypoxic/ischemic injury. Ann Neurol. 2007;61:199–208. doi: 10.1002/ana.21068. [DOI] [PubMed] [Google Scholar]
- Yasuhara T, Hara K, Maki M, Xu L, Yu G, Ali MM, Masuda T, Yu SJ, Bae EK, Hayashi T, Matsukawa N, Kaneko Y, Kuzmin-Nichols N, Ellovitch S, Cruz EL, Klasko SK, Sanberg CD, Sanberg PR, Borlongan CV. Mannitol facilitates neurotrophic factor up-regulation and behavioural recovery in neonatal hypoxic-ischaemic rats with human umbilical cord blood grafts. J Cell Mol Med. 2010;14:914–921. doi: 10.1111/j.1582-4934.2008.00671.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- You Z, Savitz SI, Yang J, Degterev A, Yuan J, Cuny GD, Moskowitz MA, Whalen MJ. Necrostatin-1 reduces histopathology and improves functional outcome after controlled cortical impact in mice. J Cereb Blood Flow Metab. 2008;28:1564–1573. doi: 10.1038/jcbfm.2008.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu C, Xu F, Wang X, Shibata M, Uchiyama Y, Blomgren K, Hagberg H. Different apoptotic mechanisms are activated in male and female brains after neonatal hypoxia-ischaemia. J Neurochem. 2006;96:1016–1027. doi: 10.1111/j.1471-4159.2005.03639.x. [DOI] [PubMed] [Google Scholar]