

Abstract
The human APOBEC3 proteins are a family of DNA-editing enzymes that play an important role in the innate immune response and have broad activity against retroviruses and retrotransposons. APOBEC3G is a member of this family that inhibits HIV-1 replication in the absence of the viral infectivity factor Vif. Inhibition of HIV replication occurs by both deamination of viral single-stranded DNA and a deamination-independent mechanism. Efficient deamination requires rapid binding to and dissociation from ssDNA. However, a relatively slow dissociation rate is required for the proposed deaminase-independent roadblock mechanism in which APOBEC3G binds the viral template strand and blocks reverse transcriptase-catalyzed DNA elongation. Here we show that APOBEC3G initially binds ssDNA with rapid on-off rates and subsequently converts to a slowly dissociating mode. In contrast, an oligomerization-deficient APOBEC3G mutant did not exhibit a slow off rate. We propose that catalytically active monomers or dimers slowly oligomerize on the viral genome and inhibit reverse transcription.
Keywords: APOBEC, DNA binding, HIV-1, restriction factor, roadblock mechanism, optical tweezers, single molecule force spectroscopy
TEXT
APOBEC3 proteins are DNA-editing enzymes that are part of the innate human immune response to viral pathogens, including retroviruses and retrotransposons1–4. Of all the A3 proteins, A3G5 is the most potent inhibitor of HIV-1 replication1,4,6, reducing viral infectivity by several orders of magnitude in the absence of the HIV-1 viral infectivity factor Vif7–9. In fact, the function of Vif is to specifically counteract the antiviral activity of A3G1,9.
Although A3G is the most studied of all the APOBEC proteins, the molecular mechanism for A3G-mediated HIV-1 restriction is still not fully understood. A3G is a deoxcytidine deaminase, which converts deoxycytidine bases in single-stranded DNA (ssDNA) to deoxyuridine5,10–14. A3G deamination of minus-strand viral DNA formed during reverse transcription results in G to A hypermutation in the plus-strand11,12,15, which effectively impairs viral replication. However, there are several lines of evidence that suggest that a deaminase-independent mechanism is also involved1,6,20. First, A3G catalytic mutants retain antiviral activity16–19. Second, A3G inhibits hepatitis B virus replication without G to A hypermutation21. Third, A3A inhibits LINE-1 and Alu retrotransposition22–28 and parvovirus replication24,29 independent of deaminase activity. Further evidence for a non-editing mechanism is based on the reduction of minus-strand viral DNA levels in HIV-1 particles during endogenous reverse transcription30, inhibition of reverse transcriptase (RT)-catalyzed viral DNA elongation in vitro by catalytic A3G mutants31,32, inhibition of strand transfer reactions in vitro and in cell-based assays32–34, and A3G-induced inhibition of reverse transcription in viruses from human CD4+ T cells35. A roadblock model, in which A3G molecules bind the template strand at one or a few locations and physically block viral DNA synthesis, has therefore been proposed as a molecular mechanism for deaminase-independent inhibition32.
Since only 7 (± 4) A3G molecules are incorporated into each vif-deficient virion36, RT inhibition by an A3G roadblock requires a slow A3G off-rate from single-stranded nucleic acids. In contrast, these few A3G molecules must have fast on-off rates to deaminate up to 1000 sites in several minutes14 using a rapid search mechanism on viral ssDNA37,38. To resolve this apparent paradox, we hypothesize that A3G exhibits fast binding kinetics as a monomer or dimer in order to function as an efficient enzyme, and slow kinetics upon oligomerization in order to block RT from elongating viral DNA. To test this idea, we used optical tweezers to monitor A3G binding kinetics on a single DNA molecule.
RESULTS
Single molecule measurements of A3G binding to ssDNA
For these studies, a single double-stranded λ-DNA molecule was tethered to two polystyrene beads, with one bead held in an optical trap and another on a micropipette tip. As the fixed bead is gradually moved away from the optically-trapped bead, the force on the DNA molecule is measured at each extension, yielding a force-extension curve (Fig. 1, solid black line). The solution surrounding the single DNA molecule can be exchanged in order to measure the effects of DNA binding ligands on the properties of DNA.
Figure 1.

The force-dependent difference in length between DNA and a saturated A3G-DNA complex allows us to measure A3G binding. Typical extension (solid black) and return (dashed black) of a single DNA molecule. At 61.0 ± 0.5 pN, the molecule undergoes a force-induced melting transition from dsDNA (green, Eq. S1) to ssDNA (blue). A3G-saturated ssDNA (200 nM A3G, t > 500 s, data points fit to Eq. S4, solid purple) is longer than dsDNA (Δxb below the melting transition) and shorter than ssDNA (Δxa above the melting transition). A3G-saturated ssDNA is significantly shorter than ssDNA only (blue, Eq. S2), which suggests that A3G may wrap ssDNA upon binding.
In the absence of binding ligands, force-induced melting occurs at a constant force of 61.0 ± 0.5 pN, generating ssDNA, either by peeling from the ends or forming melting bubbles39. Although a stretched form of dsDNA called S-DNA may form upon overstretching at high salt, it is well-established that overstretching is force-induced melting in the presence of ssDNA binding proteins and at the ionic strength used in this work39,40. At the end of the overstretching transition, the bead movement reverses direction and DNA tension is gradually released (Fig. 1, dashed black line). The minimal hysteresis, or mismatch between DNA extension and release, indicates that the ssDNA generated by force reanneals immediately into dsDNA during the return. At any given point along the transition, the molecule is a well-characterized combination of dsDNA and ssDNA (Supplementary Eq. S3)40. Pausing at fixed extension during the melting transition allows precise control of the fraction of ssDNA substrate available for protein binding.
The force-extension curve probes the length of the captured DNA molecule at a given force. At forces above 7 pN, ssDNA is longer than dsDNA. We exploit this force-dependent difference in length to measure the ssDNA binding properties of A3G. A3G-saturated ssDNA, obtained at high protein concentration by first overstretching DNA and allowing A3G to fully bind and stabilize the DNA in its single-stranded form, is longer than dsDNA and shorter than ssDNA (Fig. 1). Therefore, A3G bound to ssDNA increases the molecule in length by Δxb below the melting transition, and decreases the length by Δxa above the melting transition.
In the presence of 50 nM A3G, the extension curve follows the DNA-only curve before the melting transition, reflecting no measurable binding to dsDNA (Fig. 2, panel a, solid line). A3G only binds after force-induced melting generates ssDNA. Based on the observed hysteresis (Fig. 2, panel a, dashed line), most of the protein does not dissociate upon DNA release and prevents the two strands from fully reannealing. A3G-bound ssDNA is longer than dsDNA (Fig. 1), so the change in length at a given force (Fig. 2, panel a, Δxt) describes the total fraction of A3G bound to ssDNA (ftotal, see Supplementary Fig. 1, panel a online).
Figure 2.

Single molecule method to measure fast and slow fractions of A3G binding. (A) Without protein (black), a single DNA molecule reanneals immediately upon release, exhibiting minimal hysteresis, or mismatch between stretch (solid) and release (dashed) curves. In the presence of 50 nM A3G, the stretch curve (solid green) follows the dsDNA-only curve, indicating negligible A3G-dsDNA binding. A3G binds the exposed ssDNA, and prohibits the DNA strands from reannealing, resulting in hysteresis (dashed green). For a given force (40 pN shown), there is a corresponding change in DNA length Δxt between A3G-free dsDNA (left arrow, drawing 1) and partially A3G-bound ssDNA (right arrow, drawing 2). This force-dependent length change measures A3G-ssDNA binding (Supplementary Fig. 1). (B) The second stretch (solid blue) lies between the first stretch and release curves, distinguishing the fraction of A3G that remains bound (fslow) from the fraction that dissociated (ffast) before the second stretch. The A3G that dissociates rapidly allows the strands to reanneal immediately into dsDNA (drawing 3), resulting in length decrease Δxf. (C) Pausing at fixed DNA extension after incubating ssDNA with 50 nM A3G results in additional binding (drawing 4), indicated by the corresponding length increase Δxi measured during DNA release. (D) A3G binding increases with total exposure time to ssDNA (dashed lines).
A second stretch of the same molecule does not retrace the release curve, revealing that some fraction of the protein has dissociated during the 30 s incubation at zero force between stretch-release cycles (Fig. 2, panel b). As soon as any A3G dissociates at forces below the force-induced melting transition, the two strands reanneal into dsDNA, which is shorter than A3G-bound ssDNA (Fig. 2, panel b, Δxf). Therefore the second stretch reflects the fraction of A3G that remains ssDNA-bound (fslow), which allows the fraction that dissociates quickly (ffast) to be quantified as ffast = ftotal − fslow. The DNA was held at zero force between the first release and the second stretch for 30 s, but longer wait times up to 120 minutes do not lead to further measurable dissociation (data not shown for clarity).
A3G was exposed to ssDNA for 50 s in this experiment. However, A3G oligomerization observed in bulk experiments occurs on much longer timescales37. To measure slow binding, A3G was incubated for 250 s with ssDNA generated by force-induced melting (Fig. 2, panel c). The DNA release curve obtained after this incubation exhibits a length increase relative to the initial release curve (Fig. 2, panel c, Δxi) because additional A3G binds ssDNA during incubation. This effect increases at longer incubation times (Fig. 2, panel d), approaching the A3G-saturated ssDNA curve (shown in Fig. 1).41 Fits to the DNA release curves at increasing incubation time yield ftotal(t), while the subsequent stretch (data not shown for clarity) quantifies fslow(t), and ffast(t) is the difference between the two. These measurements for 50 nM A3G are presented in Fig. 3, panel a.
Figure 3.

Quantifying A3G binding reveals association and dissociation rates for fast and slow binding modes. (a) Total binding at 50 nM A3G (ftotal, red) separated into a fast fraction (ffast, blue) and slow fraction (fslow, green), as a function of ssDNA-A3G incubation time. Fits to the binding model (solid lines, Supplementary Eqs. S9–S11) yield observed rates kfast and kslow. (b) Slow fraction bound as a function of time for five A3G concentrations. Solid lines are fits to Supplementary Eq. S10. Error bars (panels a, b) are standard error (N≥3) for 50–200 nM A3G and propagated error for 10–20 nM A3G. (c) Fast rates (kfast, blue data points) obtained from fits to the binding model (shown in panel a for 50 nM A3G). The linear fit (solid blue line, Supplementary Eq. S13) yields k1 and k−1. k1c (purple data points) and k−1 (red data points) were also calculated from the binding model. Linear fits (solid lines, Supplementary Eqs. S15 and S17) yield consistent values of k1 and k−1. (d) Slow rates (kslow, green data points) from fits to the binding model (panel b). Fits to Supplementary Eq. S21 (solid green line, see Supplementary Fig. 2, panel b) yield k2 and k−2. Separate calculations of k2 (purple) and k−2 (red) from the binding model (Supplementary Eqs. S24 and S25) are also shown.
Quantitative binding model
The slow binding component increases at the expense of the fast component, suggesting that the A3G-ssDNA reaction may be modeled as a two-step process:
| 1 |
in which an initial bimolecular process leads to a fast complex that converts to a slow, more stable complex in the second unimolecular step. Binding rates were obtained from fits to this model (Supplementary Eqs. S9–S11) at five A3G concentrations (Fig. 3, panel b). (A3G precipitates at high concentrations18, so experiments were calibrated using force-extension curves at known protein concentrations.) As expected, the observed fast rate kfast and the on rate k1c are both linear with A3G concentration (Fig. 3, panel c). The bimolecular rate constant k1 = 1.5 (± 0.1) × 105 M−1s−1 and off rate k−1 = 1.2 (± 0.1) × 10−2 s−1 are consistent with single molecule FRET42 and fluorescence spectroscopy37 measurements, considering differences in solution conditions. The observed slow rate kslow saturates at high A3G concentration (Fig. 3, panel d), and both the on and off rates for the second, unimolecular step are concentration-independent (k2 = 6.7 (± 0.6) × 10−3 s−1 and k−2 = 2.8 (± 0.5) × 10−5 s−1). Elementary reaction rates were obtained from the data in several different ways, and agreement of the resulting values (see Supplementary Table 2 online) supports the binding model.
Oligomerization is responsible for slow binding
To determine whether slow binding is due to A3G oligomerization, we expressed and purified the F126A/W127A A3G mutant (A3G FW), which is severely defective in oligomerization41. When this mutant was incubated with ssDNA for 1050 s, the release curve exhibited minimal hysteresis (Fig. 4, panel a), and all the bound protein dissociated prior to the subsequent stretch (Fig. 4, panel b). A direct comparison of the hysteresis observed for both wild type and mutant A3G is shown in Fig. 4, panel c. The lack of a slow ssDNA bound fraction observed for the mutant, along with the striking difference between the hysteresis observed for the two proteins, shows that the oligomerization-defective mutant does not exhibit slow ssDNA binding kinetics. Thus, we conclude that the slow kinetics observed for wild type A3G is due to oligomerization.
Figure 4.

Oligomerization-defective mutant F126A/W127A (FW) demonstrates that the slow kinetics observed for wild type A3G is due to oligomerization. (a) In the absence of protein (black), a single DNA molecule reanneals immediately upon release, exhibiting minimal hysteresis between extension (solid) and release (dashed). In the presence of 50 nM F126A/W127A A3G (orange), the stretch curve (solid) follows the dsDNA-only curve, indicating no measurable A3G FW binding to dsDNA (drawing 1). Pausing at fixed DNA extension after the melting transition to incubate the ssDNA with the protein results in ssDNA binding (drawing 2), indicated by the corresponding increase in length Δxt measured during DNA release at a given force (shown for 40 pN). (b) The subsequent stretch (dark blue) follows the initial stretch curve (solid orange), indicating that all the mutant A3G bound during incubation dissociates rapidly (drawing 3), resulting in a decrease in length Δxf. (c) Wild type A3G (drawing 4) exhibits a greater change in length Δxo relative to the FW mutant (drawing 2) at 1050 s incubation due to oligomerization on ssDNA.
DISCUSSION
Here we use a single molecule method that allows us to quantify two distinct modes of A3G binding to ssDNA and characterize the conversion of a fast state into a slow state. These results suggest a binding mechanism in which monomers or dimers initially bind ssDNA and rapidly reach equilibrium (1/kfast = 24 ± 1 s at 200 nM), before slowly converting to oligomers (1/kslow = 206 ± 20 s) (Fig. 5, panel a). Previous bulk solution experiments have established that A3G oligomerizes in the presence of single-stranded nucleic acids41,43, which inhibits efficient deaminase activity41. We also demonstrate that an A3G mutant (F126A/W127A) that is severely compromised in oligomerization, but retains deaminase activity, does not exhibit slow binding kinetics. Furthermore, a recent study shows that a similar oligomerization-defective mutant (W127A) inhibits deamination-independent viral restriction44. These observations support the hypothesis that A3G oligomerization is responsible for deaminase-independent inhibition of viral replication.
Figure 5.
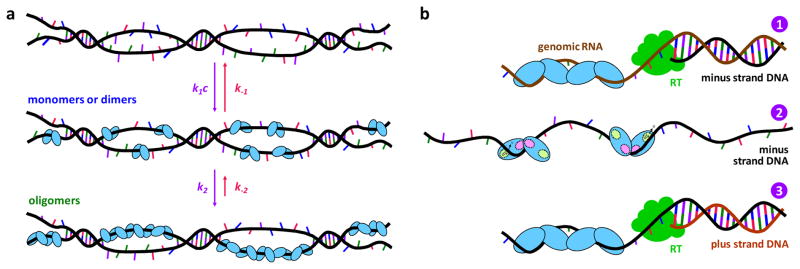
Models for A3G oligomerization (A) in vitro and (B) in virio. (a) Initially monomers or dimers bind ssDNA with on rate k1c and off rate k−1. These forward and backward rates are on similar timescales (1/k1c = 33 ± 1 s at 200 nM A3G, and 1/k−1 = 85 ± 5 s), so fast binding reaches equilibrium before the monomers or dimers convert to oligomers (1/k2 = 149 ± 13 s) on ssDNA. Oligomer dissociation is significantly slower (1/k−2 = 10 ± 2 h) in vitro. (b) Inside the virus, A3G oligomerizes on the RNA genome, blocking minus-strand DNA synthesis by RT (panel 1). Once the oligomer dissociates, the monomers or dimers released bind ssDNA within a second, allowing rapid enzymatic activity (panel 2) until A3G oligomerizes on the ssDNA template in 150 s and blocks plus strand synthesis (panel 3).
In light of these quantitative results in vitro, we propose a model for A3G’s effects on HIV-1 replication. A3G oligomerizes on viral RNA when it is packaged inside the virion43,45, stalling RT during minus-strand synthesis. RT pauses until the oligomer dissociates, or switches to the other RNA template strand, circumventing the A3G roadblock and leading to partial inhibition of reverse transcription35. In contrast to our in vitro studies, the maximum size of the A3G oligomer may be limited by the small number of A3G molecules packaged in virions36. These limitations on oligomer size may lead to higher off rates in virio. Once the A3G oligomer dissociates during minus-strand synthesis, the monomers or dimers that are released have low affinity for the newly formed RNA-DNA duplex14,18. As RNase H activity exposes the minus strand, all A3G molecules bind the ssDNA template within a second, as indicated by 1/k1 = 0.7 ± 0.1 s when fast binding rates are extrapolated to the estimated 13 ± 8 μM A3G concentration in the virion32. A3G remains bound in a rapid sliding mode37,42 for 1/k−1 = 85 ± 5 s, which allows high deamination rates37,38 until oligomerization forms a roadblock to plus-strand synthesis after 1/k2 = 149 ± 13 s (Fig. 5, panel b).
The in virio model that we propose based on our measurements is consistent with the available data on A3G function. Recent cell-based experiments demonstrate that A3G uniformly blocks minus-strand synthesis in the absence of preferred RT termination sites along the viral template35. However, this does not necessarily indicate that A3G binds throughout the length of the genomic RNA. Oligomerization is nucleic acid sequence-independent and therefore we expect the roadblock to form at a small number of random sites along the viral genome, as observed in virio. Although A3G may bind RT in an RNA-independent interaction46, a recent report concludes that there is no direct interaction between A3G and RT31. In any case, this type of binding is equally probable for any of the 100 RT heterodimers present in the virion47 and the probability that one of 7 ± 4 A3G molecules binds and inhibits the catalytically active RT molecule is therefore less than 10%. Thus, a possible A3G-RT interaction appears unlikely to be primarily responsible for A3G-induced inhibition of reverse transcription.
The data we present resolve seemingly contradictory mechanisms for A3G inhibition of viral replication by demonstrating that A3G can function as both a fast deaminase and a slow nucleic acid binding protein. Regulation of enzymatic activity via protein oligomerization may be a general property of other APOBEC family members that inhibit replication of retroviruses and retrotransposons independent of deaminase activity. The single molecule method described here does not require labeling of DNA or protein and is optimal for measuring the biologically important process of slow protein oligomerization on ssDNA.
METHODS
A3G preparation and purification
Recombinant WT A3G and A3G FW were expressed in a baculovirus expression system using an N-terminal glutathione S-transferase (GST) tag and purified. The GST tag was then removed using a Novagen Enterokinase Cleavage Capture Kit. The deaminase activity of purified wild type A3G was verified using a gel-based uracil DNA glycosylase assay after removal of the GST tag. A similar method was used to verify the enzymatic activity of purified GST-tagged A3G FW. Additional protein preparation details are supplied in the Supplementary Methods.
Single molecule experiments
Biotin-labeled bacteriophage λ DNA was captured between two streptavidin-coated polystyrene beads, one on a fixed micropipette tip and the other held in an optical trap, as described in Supplementary Methods. The DNA was extended by moving the micropipette tip at a rate of 100 nm/s, and the resulting force on the bead in the trap was recorded in order to obtain the force-extension curve for DNA alone. The buffer surrounding the DNA molecule was then exchanged for a solution of fixed protein concentration. Force-extension curves in the presence and absence of protein were fit to the Worm-Like Chain model for dsDNA (Supplementary Eq. S1) and the Freely-Jointed Chain model (Supplementary Eq. S2) for ssDNA. The A3G-saturated ssDNA force-extension curve was fit to Supplementary Eq. S4. Measurements of the fraction of ssDNA bound by protein were then determined by fitting force-extension curves to the linear combination of dsDNA extension and A3G-bound ssDNA extension, as shown in Supplementary Eqs S6 and S7.
Two-step binding model
The kinetics of protein-DNA binding was fit to a two-step binding model, in which it was assumed that an initial fast bimolecular binding event was followed by a slow unimolecular binding event, as described by Supplementary Eqs. S9–S11, yielding measured fast and slow binding rates kfast and kslow. By then fitting the concentration-dependent measurements of kfast and kslow to the reaction shown in Eq. 1, we determine the elementary reaction rates for each step k1, k−1, k2, and k−2, as well as the elementary equilibrium constants for each step K1 and K2, as described in Supplementary Methods.
Supplementary Material
Acknowledgments
The authors thank Darja Pollpeter, Michael H. Malim, and David Rueda for valuable discussions, and Myron F. Goodman for his generous gift of the F126A/W127A mutant clone. This work was supported in part by NIH [GM072462] and NSF [MCB-1243883] (MCW), NIH [GM065056] (KMF), and the NIH Intramural Research Program (NICHD) (JGL). KRC was supported by the NSF IGERT Program [DGE-0504331].
Footnotes
AUTHOR CONTRIBUTIONS
MCW, KRC, and IR designed experiments; KRC performed experiments and analyzed data; MM performed experiments with the mutant; HG performed preliminary experiments; SK, WW, DFQ, TW, YI, DC, and AH prepared the proteins; IR developed the binding model; KRC, MCW, IR, JGL, and KMF wrote the manuscript.
References
- 1.Malim MH. APOBEC proteins and intrinsic resistance to HIV-1 infection. Philos Trans R Soc Lond B Biol Sci. 2009;364(1517):675–687. doi: 10.1098/rstb.2008.0185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Harris RS, Liddament MT. Retroviral restriction by APOBEC proteins. Nat Rev Immunol. 2004;4(11):868–877. doi: 10.1038/nri1489. [DOI] [PubMed] [Google Scholar]
- 3.Duggal NK, Emerman M. Evolutionary conflicts between viruses and restriction factors shape immunity. Nat Rev Immunol. 2012;12(10):687–695. doi: 10.1038/nri3295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chiu YL, Greene WC. The APOBEC3 cytidine deaminases: An innate defensive network opposing exogenous retroviruses and endogenous retroelements. Annu Rev Immunol. 2008;26:317–353. doi: 10.1146/annurev.immunol.26.021607.090350. [DOI] [PubMed] [Google Scholar]
- 5.Sheehy AM, Gaddis NC, Choi JD, Malim MH. Isolation of a human gene that inhibits HIV-1 infection and is suppressed by the viral Vif protein. Nature. 2002;418(6898):646–650. doi: 10.1038/nature00939. [DOI] [PubMed] [Google Scholar]
- 6.Holmes RK, Malim MH, Bishop KN. APOBEC-mediated viral restriction: not simply editing? Trends Biochem Sci. 2007;32(3):118–128. doi: 10.1016/j.tibs.2007.01.004. [DOI] [PubMed] [Google Scholar]
- 7.Fisher AG, Ensoli B, Ivanoff L, Chamberlain M, Petteway S, Ratner L, et al. The sor gene of HIV-1 is required for efficient virus transmission in vitro. Science. 1987;237(4817):888–893. doi: 10.1126/science.3497453. [DOI] [PubMed] [Google Scholar]
- 8.Strebel K, Daugherty D, Clouse K, Cohen D, Folks T, Martin MA. The HIV ‘A’ (sor) gene product is essential for virus infectivity. Nature. 1987;328(6132):728–730. doi: 10.1038/328728a0. [DOI] [PubMed] [Google Scholar]
- 9.Goila-Gaur R, Strebel K. HIV-1 Vif, APOBEC, and intrinsic immunity. Retrovirology. 2008;5(1):1–16. doi: 10.1186/1742-4690-5-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lecossier D, Bouchonnet F, Clavel F, Hance AJ. Hypermutation of HIV-1 DNA in the absence of the Vif protein. Science. 2003;300(5622):1112. doi: 10.1126/science.1083338. [DOI] [PubMed] [Google Scholar]
- 11.Mangeat B, Turelli P, Caron G, Friedli M, Perrin L, Trono D. Broad antiretroviral defence by human APOBEC3G through lethal editing of nascent reverse transcripts. Nature. 2003;424(6944):99–103. doi: 10.1038/nature01709. [DOI] [PubMed] [Google Scholar]
- 12.Zhang H, Yang B, Pomerantz RJ, Zhang CM, Arunachalam SC, Gao L. The cytidine deaminase CEM15 induces hypermutation in newly synthesized HIV-1 DNA. Nature. 2003;424(6944):94–98. doi: 10.1038/nature01707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Suspène R, Sommer P, Henry M, Ferris S, Guetard D, Pochet S, et al. APOBEC3G is a single-stranded DNA cytidine deaminase and functions independently of HIV reverse transcriptase. Nucleic Acids Res. 2004;32(8):2421–2429. doi: 10.1093/nar/gkh554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yu Q, Konig R, Pillai S, Chiles K, Kearney M, Palmer S, et al. Single-strand specificity of APOBEC3G accounts for minus-strand deamination of the HIV genome. Nat Struct Mol Biol. 2004;11(5):435–442. doi: 10.1038/nsmb758. [DOI] [PubMed] [Google Scholar]
- 15.Harris RS, Bishop KN, Sheehy AM, Craig HM, Petersen-Mahrt SK, Watt IN, et al. DNA deamination mediates innate immunity to retroviral infection. Cell. 2004;116(4) doi: 10.1016/s0092-8674(03)00423-9. [DOI] [PubMed] [Google Scholar]
- 16.Newman ENC, Holmes RK, Craig HM, Klein KC, Lingappa JR, Malim MH, et al. Antiviral function of APOBEC3G can be dissociated from cytidine deaminase activity. Curr Biol. 2005;15(2):166–170. doi: 10.1016/j.cub.2004.12.068. [DOI] [PubMed] [Google Scholar]
- 17.Holmes RK, Koning FA, Bishop KN, Malim MH. APOBEC3F can inhibit the accumulation of HIV-1 reverse transcription products in the absence of hypermutation - Comparisons with APOBEC3G. J Biol Chem. 2007;282(4):2587–2595. doi: 10.1074/jbc.M607298200. [DOI] [PubMed] [Google Scholar]
- 18.Iwatani Y, Takeuchi H, Strebel K, Levin JG. Biochemical activities of highly purified, catalytically active human APOBEC3G: Correlation with antiviral effect. J Virol. 2006;80(12):5992–6002. doi: 10.1128/JVI.02680-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Luo K, Wang T, Liu BD, Tian CJ, Xiao ZX, Kappes J, et al. Cytidine deaminases APOBEC3G and APOBEC3F interact with human immunodeficiency virus type 1 integrase and inhibit proviral DNA formation. J Virol. 2007;81(13):7238–7248. doi: 10.1128/JVI.02584-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Levin JG, Mitra M, Mascarenhas A, Musier-Forsyth K. Role of HIV-1 nucleocapsid protein in HIV-1 reverse transcription. RNA Biol. 2010;7(6):754–774. doi: 10.4161/rna.7.6.14115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Turelli P, Mangeat B, Jost S, Vianin S, Trono D. Inhibition of hepatitis B virus replication by APOBEC3G. Science. 2004;303(5665):1829–1829. doi: 10.1126/science.1092066. [DOI] [PubMed] [Google Scholar]
- 22.Bogerd HP, Wiegand HL, Doehle BP, Lueders KK, Cullen BR. APOBEC3A and APOBEC3B are potent inhibitors of LTR-retrotransposon function in human cells. Nucleic Acids Res. 2006;34(1):89–95. doi: 10.1093/nar/gkj416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bogerd HP, Wiegand HL, Hulme AE, Garcia-Perez JL, O’Shea KS, Moran JV, et al. Cellular inhibitors of long interspersed element 1 and Alu retrotransposition. P Natl Acad Sci USA. 2006;103(23):8780–8785. doi: 10.1073/pnas.0603313103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen H, Lilley CE, Yu Q, Lee DV, Chou J, Narvaiza I, et al. APOBEC3A is a potent inhibitor of adeno-associated virus and retrotransposons. Curr Biol. 2006;16(5):480–485. doi: 10.1016/j.cub.2006.01.031. [DOI] [PubMed] [Google Scholar]
- 25.Muckenfuss H, Hamdorf M, Held U, Perkovic M, Lower J, Cichutek K, et al. APOBEC3 proteins inhibit human LINE-1 retrotransposition. J Biol Chem. 2006;281(31):22161–22172. doi: 10.1074/jbc.M601716200. [DOI] [PubMed] [Google Scholar]
- 26.Kinomoto M, Kanno T, Shimura M, Ishizaka Y, Kojima A, Kurata T, et al. All APOBEC3 family proteins differentially inhibit LINE-1 retrotransposition. Nucleic Acids Res. 2007;35(9):2955–2964. doi: 10.1093/nar/gkm181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Niewiadomska AM, Tian C, Tan L, Wang T, Sarkis PTN, Yu X-F. Differential inhibition of Long Interspersed Element 1 by APOBEC3 does not correlate with high-molecular-mass-complex formation or P-body association. J Virol. 2007;81(17):9577–9583. doi: 10.1128/JVI.02800-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bulliard Y, Narvaiza I, Bertero A, Peddi S, Röhrig UF, Ortiz M, et al. Structure-function analyses point to a polynucleotide-accommodating groove essential for APOBEC3A restriction activities. J Virol. 2011;85(4):1765–1776. doi: 10.1128/JVI.01651-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Narvaiza I, Linfesty DC, Greener BN, Hakata Y, Pintel DJ, Logue E, et al. Deaminase-independent inhibition of parvoviruses by the APOBEC3A cytidine deaminase. PLoS Pathog. 2009 May;(5) doi: 10.1371/journal.ppat.1000439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bishop KN, Verma M, Kim EY, Wolinsky SM, Malim MH. APOBEC3G inhibits elongation of HIV-1 reverse transcripts. PLoS Pathog. 2008;4(12) doi: 10.1371/journal.ppat.1000231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Adolph MB, Webb J, Chelico L. Retroviral restriction factor APOBEC3G delays the initiation of DNA synthesis by HIV-1 reverse transcriptase. Plos One. 2013;8(5):e64196. doi: 10.1371/journal.pone.0064196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Iwatani Y, Chan DSB, Wang F, Maynard KS, Sugiura W, Gronenborn AM, et al. Deaminase-independent inhibition of HIV-1 reverse transcription by APOBEC3G. Nucleic Acids Res. 2007;35(21):7096–7108. doi: 10.1093/nar/gkm750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li XY, Guo F, Zhang L, Kleiman L, Cen S. APOBEC3G inhibits DNA strand transfer during HIV-1 reverse transcription. J Biol Chem. 2007;282(44):32065–32074. doi: 10.1074/jbc.M703423200. [DOI] [PubMed] [Google Scholar]
- 34.Mbisa JL, Barr R, Thomas JA, Vandegraaff N, Dorweiler IJ, Svarovskaia ES, et al. Human immunodeficiency virus type 1 cDNAs produced in the presence of APOBEC3G exhibit defects in plus-strand DNA transfer and integration. J Virol. 2007;81(13):7099–7110. doi: 10.1128/JVI.00272-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gillick K, Pollpeter D, Phalora P, Kim EY, Wolinsky SM, Malim MH. Suppression of HIV-1 infection by APOBEC3 proteins in primary human CD4+ T cells Is associated with inhibition of processive reverse transcription as well as excessive cytidine deamination. J Virol. 2013;87(3):1508–1517. doi: 10.1128/JVI.02587-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xu HZ, Chertova E, Chen JB, Ott DE, Roser JD, Hu WS, et al. Stoichiometry of the antiviral protein APOBEC3G in HIV-1 virions. Virology. 2007;360(2):247–256. doi: 10.1016/j.virol.2006.10.036. [DOI] [PubMed] [Google Scholar]
- 37.Chelico L, Sacho EJ, Erie DA, Goodman MF. A model for oligomeric regulation of APOBEC3G cytosine deaminase-dependent restriction of HIV. J Biol Chem. 2008;283(20):13780–13791. doi: 10.1074/jbc.M801004200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nowarski R, Britan-Rosich E, Shiloach T, Kotler M. Hypermutation by intersegmental transfer of APOBEC3G cytidine deaminase. Nat Struct Mol Biol. 2008;15(10):1059–1066. doi: 10.1038/nsmb.1495. [DOI] [PubMed] [Google Scholar]
- 39.King GA, Gross P, Bockelmann U, Modesti M, Wuite GJL, Peterman EJG. Revealing the competition between peeled ssDNA, melting bubbles, and S-DNA during DNA overstretching using fluorescence microscopy. P Natl Acad Sci USA. 2013 doi: 10.1073/pnas.1213676110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chaurasiya KR, Paramanathan T, McCauley MJ, Williams MC. Biophysical characterization of DNA binding from single molecule force measurements. Phys Life Rev. 2010;7(3):299–341. doi: 10.1016/j.plrev.2010.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chelico L, Prochnow C, Erie DA, Chen XS, Goodman MF. Structural model for deoxycytidine deamination mechanisms of the HIV-1 inactivation enzyme APOBEC3G. J Biol Chem. 2010;285(21):16195–16205. doi: 10.1074/jbc.M110.107987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Senavirathne G, Jaszczur M, Auerbach PA, Upton TG, Chelico L, Goodman MF, et al. Single-stranded DNA scanning and deamination by APOBEC3G cytidine deaminase at single molecule resolution. J Biol Chem. 2012;287(19):15826–15835. doi: 10.1074/jbc.M112.342790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Huthoff H, Autore F, Gallois-Montbrun S, Fraternali F, Malim MH. RNA-dependent oligomerization of APOBEC3G is required for restriction of HIV-1. PLoS Pathog. 2009;5(3):e1000330. doi: 10.1371/journal.ppat.1000330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bélanger K, Savoie M, Rosales Gerpe MC, Couture J-F, Langlois M-A. Binding of RNA by APOBEC3G controls deamination-independent restriction of retroviruses. Nucleic Acids Res. doi: 10.1093/nar/gkt527. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Soros V, Greene W. APOBEC3G and HIV-1: Strike and counterstrike. Current HIV/AIDS Reports. 2007;4(1):3–9. doi: 10.1007/s11904-007-0001-1. [DOI] [PubMed] [Google Scholar]
- 46.Wang XX, Ao ZJ, Chen LY, Kobinger G, Peng JY, Yao XJ. The cellular antiviral protein APOBEC3G interacts with HIV-1 reverse transcriptase and inhibits its function during viral replication. J Virol. 2012;86(7):3777–3786. doi: 10.1128/JVI.06594-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Coffin JM, Hughes SH, Varmus HE. Retroviruses. Cold Spring Harbor Laboratory Press; Cold Spring Harbor NY: 1997. [PubMed] [Google Scholar]
- 48.Vo MN, Barany G, Rouzina I, Musier-Forsyth K. Mechanistic studies of mini-TAR RNA/DNA annealing in the absence and presence of HIV-1 nucleocapsid protein. J Mol Biol. 2006;363(1):244–261. doi: 10.1016/j.jmb.2006.08.039. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.