

ABSTRACT
The cyclic AMP receptor protein (Crp) is a transcriptional regulator that controls the expression of numerous bacterial genes, usually in response to environmental conditions and particularly by sensing the availability of carbon. In the plague pathogen Yersinia pestis, Crp regulates the expression of multiple virulence factors, including components of the type III secretion system and the plasminogen activator protease Pla. The regulation of Crp itself, however, is distinctly different from that found in the well-studied Escherichia coli system. Here, we show that at physiological temperatures, the synthesis of Crp in Y. pestis is positively regulated at the posttranscriptional level. The loss of the small RNA chaperone Hfq results in decreased Crp protein levels but not in steady-state Crp transcript levels, and this regulatory effect occurs within the 5′ untranslated region (UTR) of the Crp mRNA. The posttranscriptional activation of Crp synthesis is required for the expression of pla, and decoupling crp from Hfq through the use of an exogenously controlled promoter and 5′ UTR increases Pla protein levels as well as partially rescues the growth defect associated with the loss of Hfq. Finally, we show that both Hfq and the posttranscriptional regulation of Crp contribute to the virulence of Y. pestis during pneumonic plague. The Hfq-dependent, posttranscriptional regulation of Crp may be specific to Yersinia species, and thus our data help explain the dramatic growth and virulence defects associated with the loss of Hfq in Y. pestis.
IMPORTANCE
The Crp protein is a major transcriptional regulator in bacteria, and its synthesis is tightly controlled to avoid inappropriate induction of the Crp regulon. In this report, we provide the first evidence of Crp regulation in an Hfq-dependent manner at the posttranscriptional level. Our discovery that the synthesis of Crp in Yersinia pestis is Hfq dependent adds an additional layer of regulation to catabolite repression in this bacterium. Our work provides a mechanism by which the plague pathogen links not just the sensing of glucose or other carbon sources but also other signals that influence Crp abundance via the expression of small RNAs to the induction of the Crp regulon. In turn, this allows Y. pestis to fine-tune Crp levels to optimize virulence gene expression during plague infection and may allow the bacterium to adapt to its unique environmental niches.
INTRODUCTION
Over the past two millennia, Yersinia pestis, the bacterium responsible for the disease plague, has been associated with significant morbidity and mortality (1, 2). When infected with Y. pestis via the respiratory route, individuals can develop primary pneumonic plague, a severe, rapidly progressing purulent exudative bronchopneumonia that, if untreated, is almost always fatal. In an intranasal mouse model of infection, the progression of primary pneumonic plague in the lungs is biphasic: the first half of the infection is characterized by the lack of a significant immune response (the anti-inflammatory phase) in the lungs that transitions rapidly to a highly inflammatory state (the proinflammatory phase) (3–5). This transition occurs approximately midway through the infection.
We have previously shown that the plasminogen activator Pla, an atypical aspartate protease located in the outer membrane of Y. pestis bacteria, is required to cause primary pneumonic plague (6). In the absence of Pla, the bacterial load in the lungs does not increase significantly over time, and yet Y. pestis is still able to escape the respiratory system to cause a systemic infection. In addition, Pla is needed for the pulmonary disease to transition to the proinflammatory stage but is not necessary to maintain the early stage of the infection (6). The major activity of Pla during infection is hypothesized to be the direct conversion of the mammalian zymogen protein plasminogen to the active plasmin form, thereby accelerating fibrinolysis (7, 8), although other recently identified targets of Pla proteolysis may also contribute to pathogenesis (9–11).
While pla is thought to be constitutively expressed, several reports have demonstrated multiple factors that affect Pla synthesis and activity. For instance, Pla protein levels are moderately influenced by temperature, and higher temperatures result in increased Pla protein compared to the lower temperatures associated with the flea (12). In addition, the catalytic activity of Pla requires bound lipopolysaccharide (LPS), specifically of the “rough” form that has minimal or no O-antigen side chains (such as the LPS of Y. pestis) (13, 14). Furthermore, the transcription of pla is activated by the cyclic AMP (cAMP) receptor protein (Crp), a global transcription factor that is found in many pathogenic and nonpathogenic bacteria (15–17).
Crp regulates a large number of genes (over 100 genes in Escherichia coli and 292 genes in the 201 strain of Y. pestis) via a process known as catabolite repression (17, 18). Crp-regulated genes can often be identified by the presence of a conserved Crp box sequence within their promoter regions, and Crp, in conjunction with its coactivator molecule cAMP, binds to this sequence to activate or repress gene transcription (18, 19). Thus, Crp and cAMP together allow the bacterium to modulate the expression of numerous genes in order to appropriately meet the needs of the cell (20). This occurs through the coordinated regulation of the expression of factors involved in general housekeeping and metabolism, cell division, and pathogenesis, including virulence determinants such as the type I fimbriae of uropathogenic E. coli and the HapR regulon of Vibrio cholerae (18, 21–23). The regulation of virulence factors via catabolite repression is particularly important in Yersinia species, as Crp has been shown to influence not only pla gene expression but also the sycO-ypkA-yopJ operon of the Yop-Ysc type III secretion system (T3SS) in Y. pestis, the Ysa T3SS and flagellum of Yersinia enterocolitica, and the virulence-associated Csr small RNAs (sRNAs) in Yersinia pseudotuberculosis (15, 24–26). Indeed, mutants of all three pathogenic Yersinia species lacking crp are highly attenuated in animal models of infection, although this may be due in part to the severe growth defect associated with the loss of Crp in addition to the dysregulation of virulence factor expression (17, 24, 26, 27).
Both cAMP and Crp levels fluctuate due to changing environmental states or nutritional status, often via the sensing of carbon levels and through the effects of other regulatory proteins, such as Fis (28–30). For instance, in E. coli, Crp represses its own gene expression and that of the adenylate cyclase gene cyaA in response to glucose (31, 32). In Y. pestis, Crp is also a repressor of the cyaA gene, but the crp gene itself is not autoregulated; instead, crp is directly regulated via the PhoPQ two-component system (33, 34). This suggests that, while there are similarities between the regulation of catabolite repression in Y. pestis and E. coli, differences have emerged between the plague bacillus and other species in the mechanisms by which the Crp regulon is controlled, although as yet a posttranscriptional component has not been reported.
In a screen to discover additional factors that regulate Pla activity, we identified the gene encoding Hfq. Hfq is a chaperone of bacterial small, noncoding regulatory RNAs that enhances the interaction between sRNAs and their target mRNAs. This results in the up- or downregulation of target gene expression, usually through posttranscriptional mechanisms (35). Hfq often binds to the 5′ region of regulated mRNAs, and in conjunction with sRNAs, these interactions can have dramatic effects on translation, transcript stability, and protein activity (36). By acting as a homohexamer, Hfq contains at least 2 distinct RNA-binding surfaces which may explain why the protein is able to facilitate RNA-RNA interactions (35).
Hfq has been implicated in the virulence of multiple bacteria (37), and as a pleiotropic mediator of sRNA-mRNA interactions, is known to affect the regulation of multiple virulence-associated genes (38–40). Both Hfq and sRNAs are significant contributors to the posttranscriptional regulation of gene expression in Yersinia species and are required for the virulence of Y. pestis and Y. pseudotuberculosis in multiple animal models of infection (41–44). This study describes the contribution of Hfq to the regulation of pla expression via the direct, posttranscriptional control of Crp synthesis and addresses the contribution of Hfq-mediated regulation of Crp to the virulence of Y. pestis during primary pneumonic plague.
RESULTS
Dynamic expression of pla during primary pneumonic plague.
Previous studies have shown a modest increase in Pla protein levels when Y. pestis is cultured in vitro at 37°C compared to that at lower temperatures (12). As primary pneumonic plague is a biphasic syndrome and dependent on the activity of Pla during the proinflammatory phase of the disease (6), we hypothesized that Y. pestis also controls the expression of pla to coincide with the temporal requirement of the protease during the infection. Therefore, we examined the relative levels of the pla transcript in the lungs of mice at various times postinfection compared with transcript levels in bacteria when cultured in vitro in brain heart infusion (BHI) broth at 37°C. In the early phase of the infection, pla transcript levels are decreased by up to 16-fold compared to those in bacteria grown in broth culture, but by 48 h, pla transcript levels recover to approximately in vitro levels, and this expression pattern continues into the terminal phase of the infection (Fig. 1). Notably, transcript levels of the control gene gyrB remain relatively constant over the course of the infection compared with those of either proS or the 16S rRNA (see Table S1 in the supplemental material), thus demonstrating that pla expression is specifically and dynamically altered in a manner that is consistent with the Pla-dependent biphasic inflammatory response during primary pneumonic plague.
FIG 1 .
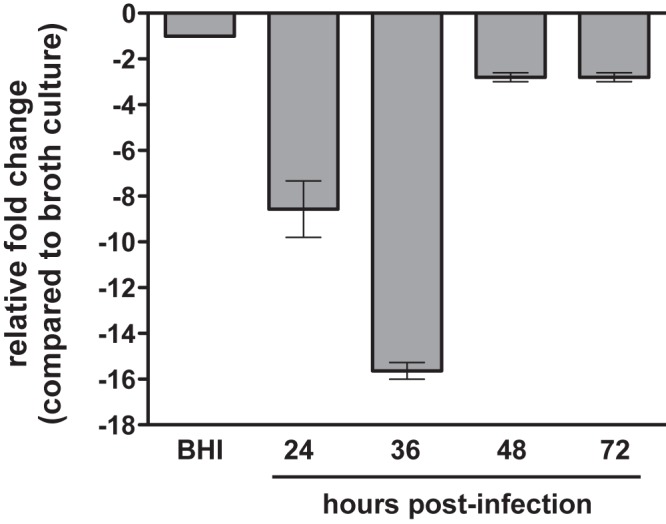
Changes in relative pla mRNA levels during the progression of primary pneumonic plague. C57BL/6 mice were infected via the intranasal (i.n.) route with Y. pestis, and the relative levels of the pla transcript in the lungs at the times indicated were compared to BHI broth after 12 h at 37°C (set at −1) by qRT-PCR. Data are representative of two independent experiments.
Decreased plasminogen activation by Y. pestis in the absence of Hfq.
The changes in pla transcript levels in vivo led us to conduct a genetic screen for regulators of Pla activity. In this screen, Y. pestis was mutagenized with the Tn5 transposon TnMod-RKm′ (45), plated onto BHI agar, and cultured at 37°C for 2 days. Then, colonies were overlaid with agar containing milk and human Glu-plasminogen (Plg) and incubated for an additional day. Since milk (casein) is a substrate for plasmin, changes in the ability of Y. pestis to activate plg can be assessed by examining altered zones of clearance surrounding the bacteria (Fig. 2A, left). This activity is dependent upon Pla (Fig. 2A, middle).
FIG 2 .

Hfq contributes to the regulation of Pla activity. (A) Y. pestis was grown on BHI agar and then overlaid with a top agar containing fat-free milk and human Glu-Plg. Activation of plg by Pla results in a zone of clearance surrounding the bacteria (left), while Y. pestis lacking Pla does not produce equivalent zones (middle). Reduced zones of clearance produced by one of the Y. pestis::Tn5 mutants recovered from the screen (right). (B) Y. pestis was mutagenized with the transposon TnMod-RKm′, and mutants were assessed for altered zones of clearance. Two mutants with reduced zones of clearance had independent insertions (indicated by black triangles) in the gene encoding the small RNA chaperone Hfq. (C) The plg-activating ability of Y. pestis, an isogenic Y. pestis Δhfq mutant, a ∆hfq strain complemented with a wild-type copy of hfq integrated onto the chromosome (Δhfq + hfq), and a ∆pla mutant cultured at 37°C are shown. Data are representative of at least 3 independent experiments.
We screened approximately 20,000 mutants from 13 independent matings for altered zones of clearance. Of those, 169 mutants with reduced zones compared to those of the wild type (Fig. 2A, right) were rescreened for plg-activating ability in liquid culture. Among the mutants with confirmed reductions in Pla activity compared to that of wild-type bacteria were those with transposon insertions in crp and cyaA, confirming earlier reports (15). In addition, we identified two independent mutants with reduced ability to activate plg containing insertions in the gene encoding the small RNA chaperone Hfq (Fig. 2B). To examine this further, we used a strain of Y. pestis with a previously generated isogenic, unmarked deletion of hfq (46) and performed a plg activation assay with bacteria cultured at 37°C. The hfq mutant was unable to activate plg to the same extent as the wild-type strain, confirming the transposon insertion phenotype (Fig. 2C).
Hfq-dependent, transcriptional regulation of pla.
To determine if the absence of Hfq affects Pla protein levels, we cultured wild-type Y. pestis, ∆hfq Y. pestis, a ∆hfq strain of Y. pestis complemented with a wild-type copy of hfq integrated onto the chromosome (∆hfq + hfq Y. pestis), and ∆pla Y. pestis for 6 h at 37°C and performed immunoblot analyses on cell lysates with an anti-Pla antibody. We observed that the loss of Hfq reduces Pla protein abundance compared to that in wild-type bacteria (Fig. 3A). To establish if Hfq has an effect on the steady-state levels of pla mRNA, we then compared the relative levels of the pla transcript between the wild-type, ∆hfq, and ∆hfq + hfq strains cultured under the same conditions. The level of pla mRNA in the ∆hfq strain was reduced by 7.5-fold compared to that of wild-type Y. pestis, demonstrating that Hfq affects the expression and/or stability of pla at the transcript level (Fig. 3B) and providing a potential explanation for the reduced abundance of the Pla protein under our conditions.
FIG 3 .

Indirect, transcriptional control of pla via Hfq. (A) Wild-type, ∆hfq, Δhfq + hfq, and ∆pla Y. pestis were grown at 37°C, and equal concentrations of cell lysates were separated by SDS-PAGE followed by immunoblot analysis with an anti-Pla or anti-RpoA antibody. Pla is indicated with an arrow; the lower band is nonspecific. Immunodetection of RpoA is shown as a loading control. The relative density of the Pla band compared with the wild type is shown below the RpoA panel. NA, not applicable. Data are representative of 3 independent experiments. (B) Steady-state levels of the pla transcript after 6 h at 37°C. Strains of Y. pestis were grown in triplicate, and the relative fold change of the pla transcript in the ∆hfq and Δhfq + hfq strains compared to that of the wild-type bacteria (set at 1) was determined by qRT-PCR using the ∆∆CT method. Data are representative of 3 independent experiments. (C) Half-life of pla mRNA. Wild-type and ∆hfq Y. pestis were cultured as described above, and at time 0 rifampin was added to prevent de novo RNA synthesis. Percent remaining mRNA at each time point was measured by qRT-PCR and compared with time 0. pla half-life: 1.66 ± 0.27 min for the wild type, 1.70 ± 0.24 min for the ∆hfq mutant, difference not significant. Data are representative of 2 independent experiments. (D) Wild-type or ∆hfq Y. pestis strains with the chromosomal-integrated Ppla-gfp or PtetO-pla 5′ UTR-gfp reporter constructs were cultured at 37°C for 6 h, and fold change in fluorescence compared with that of the wild type (set at 1), normalized to the optical density of the culture, was determined. For the PtetO-pla 5′ UTR-gfp reporters, ATc was added at time 0. **, P < 0.005; NS, not significant. Data represent the combination of 3 independent experiments. For all panels, error bars indicate standard deviation of the mean.
To test if the reduced steady-state transcript levels of pla in the absence of Hfq are due to changes in pla mRNA stability or half-life (t1/2), we cultured wild-type and ∆hfq Y. pestis for 6 h at 37°C, after which rifampin was added to the cultures to inhibit the further initiation of de novo transcription. We found no significant difference in pla transcript levels between the wild-type and ∆hfq strains (Fig. 3C), indicating that the loss of Hfq has no impact on the stability of the pla mRNA.
As direct effects on protein production mediated by Hfq and sRNAs typically occur within the 5′ untranslated region (UTR) of regulated transcripts (38), we generated two green fluorescence protein (GFP)-based reporter constructs to measure Hfq-dependent, posttranscriptional or transcriptional and posttranscriptional activity from the pla promoter and/or 5′ UTR. To measure transcriptional and posttranscriptional effects, we cloned 500 nucleotides (nt) upstream of the pla translational start site, including 27 nt of the pla coding sequence (CDS) immediately upstream of the gfp CDS (Ppla-gfp). To measure solely posttranscriptional effects at the pla 5′ UTR, this region (15) and 27 nt of the pla CDS were cloned downstream of a modified PtetO promoter that lacks the PtetO 5′ UTR, followed by the CDS for gfp (PtetO-pla 5′ UTR-gfp). The transcription of this construct is controlled by the addition of anhydrotetracycline (ATc), and thus any differences in GFP fluorescence can be attributed to effects at the pla 5′ UTR. Both reporters were integrated onto the chromosomes of wild-type and ∆hfq Y. pestis, bacteria were cultured for 6 h at 37°C (and in the case of the PtetO-pla 5′ UTR-gfp reporter strains, ATc was added at time 0), and fluorescence was measured and normalized to the optical densities of the cultures. Although GFP fluorescence from the Ppla-gfp reporter was significantly reduced in the ∆hfq strain compared to that in the wild type, we found that there was no difference in fluorescence between the strains carrying the PtetO-pla 5′ UTR-gfp reporter (Fig. 3D). This indicates that Hfq does not contribute to the posttranscriptional regulation of pla at the 5′ UTR. In total, the data presented in Fig. 3 indicate that the Hfq-dependent effects on pla are likely at the transcriptional rather than the posttranscriptional level and therefore indirect.
Hfq-dependent, posttranscriptional regulation of crp.
Based on these findings, we hypothesized that Hfq may contribute to the regulation of a factor upstream of Pla. Previous studies have demonstrated that the transcription of pla is directly activated by Crp (15, 17). Therefore, to test if Hfq contributes to the regulation of Crp, we replaced the native crp gene with a C-terminal hemagglutinin (HA)-tagged version by allelic exchange in the wild-type, ∆hfq, and hfq-complemented strains of Y. pestis. These bacteria were cultured for 6 h at 37°C, and whole-cell lysates were examined by immunoblot analysis with an anti-HA antibody. We found that the loss of Hfq results in decreased Crp protein levels compared to that of the wild type (Fig. 4A). To determine the impact of Hfq on the steady-state levels of the crp transcript, we performed quantitative reverse transcription-PCR (qRT-PCR) on RNA extracted from the same cultures. Unlike that observed for pla, here the loss of Hfq has no effect on the relative level of the crp mRNA compared to that of the wild type (Fig. 4B), indicating that Hfq does not affect the turnover of this transcript in Y. pestis. Thus, the data from Fig. 4A and B together suggest that Hfq contributes to the posttranscriptional regulation of Crp.
FIG 4 .

Hfq-dependent, posttranscriptional control of Crp. (A) Wild-type, ∆hfq, and Δhfq + hfq Y. pestis strains carrying an HA-tagged version of the crp gene were cultured for 6 h in BHI broth at 37°C, and whole-cell lysates were analyzed by immunoblotting with an anti-HA antibody. RpoA (bottom) is shown as a loading control. The relative density of the Crp-HA band compared with that of the wild type is shown below the RpoA panel. Data are representative of at least 3 independent experiments. (B) Steady-state levels of the crp transcript after 6 h at 37°C. Strains of Y. pestis were grown in triplicate, and the relative fold change of the crp transcript in the ∆hfq and Δhfq + hfq strains compared to that of the wild-type bacteria (set at 1) was determined by qRT-PCR using the ∆∆CT method. Data represent the combination of 3 independent experiments. (C) Wild-type or ∆hfq Y. pestis strains with the chromosomal-integrated Pcrp-gfp or PtetO-crp 5′ UTR-gfp reporter constructs were cultured at 37°C for 6 h, and fold change in fluorescence compared with that of the wild type (set at 1), normalized to the optical density of the culture, was determined. For the PtetO-crp 5′ UTR-gfp reporters, ATc was added at time 0. **, P < 0.005. Data represent the combination of 3 independent experiments. For all panels, error bars indicate standard deviation of the mean.
To assess this more directly, we generated transcriptional and posttranscriptional (Pcrp-gfp) and posttranscriptional (PtetO-crp 5′ UTR-gfp) GFP reporter constructs for crp in the same manner as described for pla. To create the posttranscriptional reporter, we first mapped the transcriptional start site of the Y. pestis CO92 crp mRNA to 79 nt upstream of the translation start site (ATG) by the technique of 5′/3′ rapid amplification of cDNA ends (RACE) (43). The reporter constructs were integrated in single copy onto the chromosomes of the wild-type and ∆hfq strains of Y. pestis, and fluorescence was measured after 6 h at 37°C (ATc was added to the PtetO-crp 5′ UTR-gfp reporter strains at time 0). For both the Pcrp-gfp and PtetO-crp 5′ UTR-gfp reporters, fluorescence intensity was significantly lower in the absence of Hfq than in the wild type (Fig. 4C). This indicates that Hfq promotes Crp synthesis through posttranscriptional regulatory effects at the 5′ UTR and/or proximal region of the crp CDS. A kinetic analysis of this effect over the growth curve of Y. pestis shows that fluorescence produced from the PtetO-crp 5′ UTR-gfp reporter in the wild-type strain rapidly increases until approximately 6 h postinduction, while fluorescence in the ∆hfq bacteria increases between 2 and 4 h to approximately 40 to 50% of the normalized signal intensity of the wild type (see Fig. S1 in the supplemental material).
Decoupling crp expression from Hfq increases Pla levels and activity.
If the Hfq-dependent posttranscriptional regulation of Crp subsequently influences the expression of pla, we hypothesized that unlinking Crp from the regulatory effects mediated by Hfq would result in increased Pla protein levels and activity. To test this, we generated a construct in which crp expression is decoupled from Hfq by linking the crp CDS to the PtetO promoter and tetO 5′ UTR, a promoter and UTR that are unaffected by the absence of Hfq (46). This construct was integrated in a single copy onto the chromosomes of wild-type, ∆hfq, and ∆crp strains of Y. pestis. In the absence of ATc, Pla protein levels are reduced in both mutant strains compared to those of the wild type as measured by immunoblotting, confirming that both Hfq and Crp positively influence the production of Pla (Fig. 5A). In the presence of ATc, however, Pla protein levels in the ∆hfq and ∆crp strains are increased over −ATc conditions, demonstrating that the defect in Pla levels in the absence of Hfq can be compensated for by the increased production of Crp, although we cannot rule out that the residual levels of endogenous Crp in the Hfq mutant may contribute to Pla synthesis indirectly through the regulation of another factor via Hfq or Crp. Consistent with this, we confirmed by immunoblotting that Crp-HA is produced by these same bacteria only in the presence of ATc. As expected, the addition of ATc to the same strains lacking the inducible PtetO-crp-HA construct has no impact on plg activation (see Fig. S2).
FIG 5 .

Hfq-decoupled expression of crp increases Pla synthesis and activity. (A) Wild-type, ∆crp, and ∆hfq Y. pestis strains, all containing a chromosomal-integrated PtetO-crp-HA construct, were cultured in the presence or absence of ATc for 6 h at 37°C, and cell lysates were analyzed by immunoblotting for Pla (top), the HA tag (middle), or RpoA (as a loading control; bottom). The ∆pla strain is shown as a negative control for the presence of Pla. The relative density of the Pla band compared with that of the wild type is shown below the Pla panel. Data are representative of 3 independent experiments. (B) A plg activation assay was performed as described in Fig. 2C on the same strains as described above in the presence or absence of ATc. Data are representative of 3 independent experiments.
We then tested if the expression of crp-HA in the Hfq mutant could increase the Pla-dependent plg-activating ability of Y. pestis. Here, we cultured the same Y. pestis strains as described above in the presence or absence of ATc for 6 h at 37°C and performed a plg activation assay. We found that the addition of ATc to the wild-type strain has no influence on plg activation compared to that of the equivalent strain in the absence of ATc (Fig. 5B), confirming our previous observations and demonstrating that excess Crp does not adversely affect Pla activity. The addition of equivalent amounts of ATc to the ∆crp PtetO-crp-HA strain fully restores plg activation to wild-type levels, indicating that Pla activity can be controlled by manipulating Crp levels in this strain. The addition of ATc to the ∆hfq PtetO-crp-HA strain is able to partially restore Pla activity, although not to the same extent as wild-type bacteria or the ∆crp PtetO-crp-HA strain in the presence of ATc (Fig. 5B). In total, the data presented in Fig. 5 indicate that, although the Hfq-dependent regulation of Crp contributes to Pla protein production, there may be additional Hfq-regulated factors beyond Crp that are involved in the ability of Pla to maximally activate plg.
Hfq-dependent regulation of Crp promotes the growth of Y. pestis in rich medium.
The loss of Hfq from Y. pestis results in a significant growth defect compared to growth of wild-type bacteria when cultured in vitro in rich medium, such as BHI broth (41, 46, 47). Upon generating a ∆crp mutant of Y. pestis, we also noted a large growth defect under the same conditions (Fig. 6), although the growth defect is greater than that of the Hfq mutant. Considering the impact of Hfq on the regulation of Crp, we hypothesized that these phenomena may be linked, in that the growth defect of the ∆hfq mutant may be due in part to the dysregulation of Crp synthesis. To test this, we used the same PtetO-crp-HA-inducible strains described for experiments in Fig. 5 to assess whether the induction of Crp in the Hfq mutant could restore the growth of this strain. Bacteria were cultured at 37°C with shaking in BHI broth, and the optical density at 620 nm (OD620) was measured every 2 h. We found that, as expected, the addition of ATc to the ∆crp PtetO-crp-HA strain restores growth to that of the wild-type strain (Fig. 6). The addition of ATc to the ∆hfq PtetO-crp-HA strain increases the growth rate of this strain over vehicle alone but does not fully restore growth to that of the wild type. Thus, these data indicate that the growth defect associated with the loss of Hfq from Y. pestis is in part due to the reduced synthesis of Crp but that other Hfq-regulated factors also participate in the full growth of the plague bacillus in vitro.
FIG 6 .

Induction of Crp synthesis partially restores the growth of Δhfq Y. pestis in rich broth. The same bacterial strains containing the PtetO-crp-HA construct as described in Fig. 5 were cultured in BHI broth with shaking at 37°C for 12 h in the presence or absence of ATc, and at the indicated times the OD620 was measured. The growth of the strains containing the PtetO-crp-HA construct in the absence of ATc was equivalent to the parent strains without the construct, and the addition of ATc had no impact on the growth of wild-type bacteria (not shown). Data are representative of 3 independent experiments.
Contribution of Hfq and the posttranscriptional regulation of Crp to primary pneumonic plague.
Hfq has been implicated in the virulence of numerous pathogenic bacteria, and as a pleiotropic participant in gene regulation, Hfq may have effects on multiple factors involved in virulence. Therefore, in order to test the contribution of Hfq to the virulence of Y. pestis during primary pneumonic plague, we intranasally infected C57BL/6 mice with pCD1+ wild-type or ∆hfq Y. pestis. While all mice infected with the wild-type strain succumbed to the infection by day 4, none of the Y. pestis ∆hfq-infected mice died, even after 7 days (Fig. 7A). In addition, we examined the kinetics of infection by enumerating CFUs in the lungs at various times postinfection and found that the Y. pestis ∆hfq mutant is unable to persist in the pulmonary compartment (see Fig. S3).
FIG 7 .
Contribution of Hfq and the posttranscriptional control of Crp to pneumonic plague. (A) Survival of mice infected via the i.n. route with wild-type or ∆hfq Y. pestis. (B) Impact of decoupling Crp synthesis from Hfq on Y. pestis CFUs in the lungs. Mice were infected via the i.n. route with wild-type Y. pestis or the ∆hfq or ∆crp strains of Y. pestis carrying either the constitutively expressed PtetO-crp-HA construct or the Tn7 vector only. After 48 h, CFUs in the lungs were determined. Each point represents the numbers of bacteria recovered from a single mouse. The limit of detection is indicated by a dashed line. Symbols below the limit of detection represent mice that did not have detectable numbers of bacteria. A solid line indicates the median of CFU recovered. **, P < 0.005. Data are representative of 2 independent experiments.
As previous work has demonstrated the contribution of Crp to the virulence of Y. pestis (17, 27), we hypothesized that the attenuation of the Hfq mutant may be in part due to the dysregulation of Crp. To test this, we generated strains of pCD1+ Y. pestis carrying the PtetO-crp-HA construct as described above, but in this case, the gene encoding the tet repressor TetR was not included. In the absence of TetR, the addition of ATc to induce gene expression is not required, and thus transcription from the PtetO promoter is constitutive. Production of Crp-HA from both the ∆hfq and ∆crp strains of Y. pestis carrying the constitutive PtetO-crp-HA construct was confirmed by immunoblotting (see Fig. S4). Mice were intranasally infected with wild-type Y. pestis, the ∆hfq and ∆crp strains of Y. pestis carrying the constitutive PtetO-crp-HA, or the equivalent mutants carrying the Tn7 vector only. After 48 h, mice were sacrificed and CFUs in the lungs were enumerated. Three of 4 mice infected with the ∆hfq strain containing the vector only had no bacteria in the lungs (below the limit of detection), while the ∆crp strain had a median of 55 CFUs (Fig. 7B). On the other hand, mice infected with wild-type Y. pestis or the ∆crp PtetO-crp-HA strain had a median of 5.5 × 107 or 2.5 × 107 CFUs in the lungs, respectively. Although full virulence was not restored to the Y. pestis ∆hfq strain carrying PtetO-crp-HA, we found that the constitutive production of Crp in the ∆hfq mutant significantly increased the number of CFUs in the lungs compared to that of the same ∆hfq strain carrying the vector insertion only. Thus, our results indicate that a portion of the attenuation associated with the loss of Hfq is directly attributable to the dysregulation of Crp synthesis.
DISCUSSION
The influence of catabolite repression on gene expression via Crp and cAMP is well known, and many decades of research, particularly in E. coli, have established the paradigm by which this occurs. The expression, synthesis, and activity of both crp and cyaA must be appropriately controlled for the optimal physiology of the cell, and multiple studies have identified a variety of mechanisms by which these events occur. For instance, in E. coli, both crp expression and Crp activity (via cAMP binding) are regulated by the levels of extracellular glucose; indeed, Crp can positively or negatively affect its own expression in an autoregulatory feedback circuit (28, 48). Extracellular osmolarity also influences crp expression and Crp activity, linking not just nutrient levels but also salt concentration with Crp-dependent gene expression (49). In addition, Crp itself is acetylated in E. coli, suggesting that posttranslational modifications may also influence Crp activity (50, 51).
While Crp is a repressor of the cyaA gene in Y. pestis, crp expression is not autoregulated, suggesting that Crp levels themselves may be less sensitive to glucose levels in the plague pathogen than in E. coli (34, 52). In addition, Zhang et al. recently showed that the two-component response regulator PhoP directly activates the transcription of both crp and cyaA (33). Thus, considering the distinct environments that Y. pestis inhabits compared to E. coli or even other bacterial pathogens, these data suggest the possibility that divergent mechanisms of crp regulation have evolved that are specific or unique to Yersinia species. Indeed, the dependence of crp expression on the PhoPQ regulatory system indicates that Y. pestis has adapted multiple mechanisms to link catabolite repression with other environmental cues, such as low Mg2+ or the presence of antimicrobial peptides (53). This report adds to our understanding of Crp biology in the plague pathogen by demonstrating that at physiological temperatures, Y. pestis positively regulates the synthesis of Crp at the posttranscriptional level via the activity of the sRNA chaperone Hfq. This additional level of regulation to control Crp synthesis may be confined to a limited number of bacterial species, as Hfq-dependent, posttranscriptional mechanisms of Crp regulation have not been previously described, despite the many years of study of both Hfq and catabolite repression. Furthermore, these data explain why inducible promoters such as PBAD and PlacO are not fully functional in the hfq mutant of Y. pestis (not shown), since these promoters rely on Crp as a coactivator for induction (54).
An alignment of the E. coli strain MG1655 and Y. pestis CO92 crp 5′ UTR sequences reveals that, except for 15 of the 17 most proximal nucleotides to the translational start codon, no significant homology exists between the two, even though the crp CDS between the species are 84% identical. In addition, the E. coli crp 5′ UTR is 167 nt in length, while the equivalent Y. pestis CO92 sequence is only 79 nt long. An mFOLD analysis (55) of the crp 5′ UTR and proximal coding region transcript used to generate the PtetO-crp 5′ UTR-gfp reporter predicts a secondary structure with extensive base pairing, including the region of the putative ribosome-binding site (see Fig. S5). Considering that the regulatory effect of Hfq occurs within the Y. pestis crp 5′ UTR, these differences may explain the adaptation of Crp synthesis in Y. pestis to include a posttranscriptional layer. As Hfq usually acts in conjunction with sRNAs to regulate protein synthesis, the Yersinia-specific crp 5′ UTR may enable sRNAs to bind to this region of the transcript and alleviate possible inhibitory secondary structures, such as that observed for the E. coli rpoS transcript (56, 57), whereas other bacteria may lack this mechanism of regulation due to the differences in the UTR.
In many species, Hfq contributes to the regulation of a large number of genes; however, the regulons controlled through Hfq are distinct between various genera and species, as are the subsets of sRNAs encoded by different bacteria. For example, we found that approximately 80% of the putative sRNAs expressed by Y. pseudotuberculosis, the most recent ancestor of modern Y. pestis strains (58), are unique to Yersinia species and not found in other bacteria, such as E. coli or Salmonella (43). This suggests that, although there is overlap between some sRNAs and regulated genes between closely related species, many bacteria have adapted individual posttranscriptional mechanisms of gene regulation to meet the specific needs of the cell, be it due to differences in environmental niche, nutrient availability, or stress conditions.
In particular, Y. pestis has adapted to a lifestyle in which it is thought to exist predominantly as a parasite—the plague bacillus cycles between infection of an arthropod vector and a mammal and is not believed to spend significant time outside a host (59). During infection of the flea, Y. pestis expresses genes required for survival, colonization, and transmission by the flea, while other genes that are required for mammalian infection are downregulated (60). Conversely, upon entry into a mammalian host, flea-specific genes are downregulated and a different subset of genes is expressed. These include many mammalian virulence factors, including the Yop-Ysc T3SS, the F1 antigen pilus, the antiphagocytic pH 6 antigen, and Pla (3, 61). Not only are the levels of Pla protein reduced at lower temperatures compared to those under physiological conditions (12), here we show that pla is also differentially regulated during pneumonic plague in a manner that is consistent with its requirement during the latter stage of the respiratory infection, when Pla is needed to induce the proinflammatory phase of disease (6). As Crp acts as a direct positive transcriptional regulator of pla, and both Hfq and Crp are required for the full virulence of Y. pestis, the data presented here suggest the possibility that Y. pestis may optimize metabolic and virulence gene expression via the posttranscriptional regulation of Crp synthesis to link catabolite repression with Hfq/sRNA-dependent environmental sensing. These regulatory connections could work in both directions, as Heroven et al. recently showed that in Y. pseudotuberculosis, Crp influences the expression of the Hfq-independent sRNAs CsrB and CsrC (26).
Multiple regulatory pathways may synergize to induce the Crp regulon at the appropriate time and/or location. This might occur as follows: first, transcription of crp (and cyaA) is induced in a PhoPQ-dependent manner in response to external signals such as ion concentrations or the presence of antimicrobial peptides, to result in “priming” of the crp mRNA for translation. Full Crp synthesis, however, would not proceed until a second signal is detected that results in the expression of Hfq-dependent sRNA(s) that interacts with the crp 5′ UTR. Finally, once Crp is synthesized, sensing of the appropriate nutrient conditions would allow for the production of cAMP by CyaA, thus linking the levels of carbon/glucose with optimal Crp-dependent gene expression, but only after both the PhoPQ system and the required sRNAs have been activated and expressed. While the overproduction of Crp does not seem to adversely affect wild-type Y. pestis under our conditions, it is possible that excessive Crp induction during mammalian or flea infection could be detrimental to the bacterium and that these or other regulatory pathways may prevent this from occurring in vivo. The kinetics of the posttranscriptional regulation of Crp presented in Fig. S1 suggest that the sRNA(s) that activates Crp synthesis may become limiting as the bacteria transition into stationary phase, for instance.
During infection, these regulatory mechanisms could allow Y. pestis not only to modulate highly energetic processes such as type III secretion but also to induce virulence gene expression only when specific conditions are met. While it is not yet known what specific signals stimulate these changes in gene expression during pneumonic plague, it is possible that a low-level induction of the inflammatory response (6) may be sufficient to initiate the process. For example, Salmonella specifically stimulates gastrointestinal inflammation during infection in part to acquire nutrients (62), which consequently could alter the availability of carbon and thus the expression of the Crp regulon. Alternatively, variations in oxygenation, temperature, or even bacterial load during infection could serve as signals for expression of the sRNA(s) that influence the induction of Crp in Y. pestis. Our data suggest that, whatever these signal(s) may be, they are likely to posttranscriptionally stimulate Crp synthesis, thereby allowing for increased pla expression and presumably other components of the Crp regulon during infection.
Indeed, the posttranscriptional regulation of Crp in Y. pestis contributes significantly to virulence during pneumonic plague, as decoupling Crp synthesis from Hfq partially restores bacterial levels in a ∆hfq mutant. Not surprisingly, other Hfq-dependent, Crp-independent genes must also be required for disease, as the loss of Hfq results in the clearance of bacteria from the lungs over time. Indeed, our data are among the first to define the specific factors directly regulated through Hfq that are needed during plague infection. However, additional studies are needed to further delineate the genes within the Hfq regulon that are required for virulence during pneumonic plague, the sRNA(s) that contributes to the posttranscriptional regulation of Crp, and the host signals that Y. pestis senses to influence the proper transcriptional and posttranscriptional control of both Crp and other factors while in the mammalian host.
MATERIALS AND METHODS
Reagents, bacterial strains, and growth conditions.
All chemicals were obtained from the Sigma Chemical Company (St. Louis, MO) unless otherwise indicated. Bacterial strains are listed in Table S2 in the supplemental material, plasmids are described in Table S3, and oligonucleotide sequences are given in Table S4. The fully virulent, wild-type Y. pestis strain CO92 and the avirulent, pCD1-negative derivative were obtained previously from the U.S. Army, Fort Detrick, MD. The presence or absence of pCD1, pMT1, pPCP1, and the pgm locus was confirmed by PCR. Unless otherwise indicated, all experiments were conducted with the pCD1− derivative of Y. pestis CO92. Experiments with select agent strains of Y. pestis were performed in CDC-approved biosafety level 3 (BSL3)/animal BSL3 (ABSL3) laboratories at Northwestern University. Y. pestis was routinely cultivated on BHI agar (Difco) at 26°C for 2 to 3 days. For liquid cultures, Y. pestis was grown in BHI broth at 26°C in a roller drum overnight before being diluted to an OD620 of 0.1 to 0.2 in 10 to 15 ml BHI broth in a 125-ml Erlenmeyer flask or in a roller drum and cultured with shaking at 250 rpm at 37°C, unless otherwise indicated. Ampicillin (100 µg/ml) or kanamycin (50 µg/ml) was added to the medium as needed. For animal infections, Y. pestis was cultured as described above at 37°C with the addition of 2.5 mM CaCl2.
Animal experiments.
All animal experiments were approved by the Northwestern University ACUC. Pathogen-free, 6- to 8-week-old female C57BL/6 mice were obtained from the Jackson Laboratory (Bar Harbor, ME) and were housed in HEPA-filtered barrier units kept inside a ventilated animal cage rack for the duration of the experiments. Mice were given food and water ad libitum and were housed at ambient temperature with alternating 12-hour periods of light and dark. pCD1+ Y. pestis and derivatives were prepared as described above, washed once in sterile phosphate-buffered saline (PBS), and maintained at 37°C. Mice were anesthetized with ketamine and xylazine and inoculated by the intranasal route with approximately 1 × 104 CFU of the indicated bacterial strains in a volume of 20 µl as described previously (3, 43). Actual numbers of CFU inoculated were determined by plating serial dilutions on BHI agar. At various times, animals were sacrificed with an overdose of sodium pentobarbital. For CFU determination experiments, lungs were surgically removed and homogenized in PBS, and serial dilutions were plated on BHI agar. For survival experiments, mice were monitored twice daily for 7 days, and any surviving mice at the end of the experiment were euthanized as described above. For the analysis of gene expression in vivo, lungs were removed at the indicated times and immediately submerged in an excess of RNAlater RNA stabilization solution (Ambion) before proceeding.
RNA extraction and quantitative RT-PCR.
For experiments described for Fig. 1, pCD1+ Y. pestis was cultured under the same conditions used to prepare the bacteria for infection, and after 12 h, bacteria were mixed with 2 volumes of RNAprotect-bacteria RNA stabilization solution (Qiagen). For all other experiments, pCD1− strains were cultured at 37°C for 6 h, and RNA was stabilized as described above. Total RNA was then purified from lung tissue or cultured bacteria with the RiboPure RNA extraction kit (Ambion), treated with DNase (Ambion), and cDNA synthesized using the Superscript II reverse transcriptase (Invitrogen) and random primers (Invitrogen). qRT-PCR for the target genes was performed in triplicate with the SYBR green dye (Bio-Rad) by using the primer sets listed in Table S4 in the supplemental material. The calculated threshold cycle was normalized to the CT of the gyrB gene from the same cDNA sample before calculating the fold change using the ∆∆CT method (63); in Table S1, the CT of gyrB was normalized to either proS or the 16S rRNA. Statistical analyses were performed using Student’s t test.
Transposon mutagenesis and screen for altered Pla activity.
Mutagenesis was performed by electroporating the pTnMod-RKm plasmid carrying the Tn5 minitransposon into pCD1− Y. pestis (45); transposon mutants were plated onto BHI agar containing kanamycin and incubated at 37°C for 2 days. Plates were then overlaid with a top agar containing 5% skim milk and 200-µg human Glu-Plg (Haematologic Technologies) and incubated at 37°C for an additional day. Colonies that showed altered zones of clearance were purified and examined in a liquid plg activation assay as described below.
Mutagenesis, HA tagging, and Tn7-based chromosomal integration.
Unmarked, isogenic mutants of hfq and crp were generated by lambda red recombination in the fully virulent or pCD1− strains of Y. pestis CO92 as described previously (43, 46). Regions of homology upstream and downstream of the genes were PCR-amplified using the primer sets listed in Table S4. The kanamycin resistance cassette used for the selection of recombinants was excised as described earlier (46).
The crp gene was replaced by allelic exchange in pCD1− strains of Y. pestis with a variant carrying the sequence for the HA tag on the 3′ end immediately preceding the stop codon, as described previously (46), with the primers listed in Table S4 in the supplemental material. Incorporation of the HA tag at the appropriate locus in kanamycin-sensitive colonies was confirmed by PCR.
Construction of ATc-inducible crp-HA strains was performed similarly as described (46). Here, the CDS for crp was amplified by PCR from the Y. pestis genome using a 3′ primer containing the HA sequence immediately prior to the stop codon, and the PtetO promoter and 5′ UTR was PCR amplified from the plasmid pWL213 (6) before subsequently joining the fragments using the technique of splicing by overlap extension (SOE)-PCR. The resulting product was then subcloned into the Tn7-based integration plasmid pWL212 (6).
All Tn7-based constructs destined for integration onto the chromosome of Y. pestis were introduced to the attTn7 site contained within the glmS-gstS intergenic region via the Tn7 site-specific transposon as described (46, 64). The kanamycin resistance cassette was resolved as described above, and integration was confirmed by PCR.
Plasminogen activation assays.
The plg-activating ability of Y. pestis was assessed as described previously (6). Strains were grown for 6 h at 37°C before being diluted to 1 × 106 CFU in PBS and combined with purified human Glu-Plg (Haematologic Technologies; 4 µg) and the chromogenic substrate D-AFK-ANSNH-iC 4H9-2HBr (SN5; Hematologic; 50 µM) in a total volume of 200 µl PBS. Reaction mixtures were incubated in triplicate for 3 h at 37°C, and the fluorescence at 460 nm was measured every 10 to 11 min in a Molecular Devices SpectraMax M5 microplate reader. For experiments measuring the effect of crp expression on plg activation, ATc (0.25 µg/ml) was added to the cultures at time 0, as appropriate. Data shown are representative of at least 3 independent experiments.
mRNA half-life determination.
The decay rate and half-life of the pla transcript were determined essentially as described (46). Y. pestis strains were cultured in triplicate for 6 h at 37°C, following which rifampin (50 µg/ml) was added to the cultures to prevent de novo transcription. Aliquots of bacteria were removed immediately (time 0) and every minute thereafter for 4 min and immediately mixed with 2 volumes of RNAprotect-bacteria reagent. Relative transcript levels at each time point were measured by qRT-PCR as described above, and the half-life of the mRNA was determined by plotting the relative fold change compared with time 0 for each strain on a semilog plot. The first 4 data points were then fit with a linear curve, and the equation t1/2 = 0.693/k was used, where k = the slope of the line. Data are represented as a percentage of mRNA remaining over time. Statistical analysis was performed using Student’s t test.
Transcriptional start site determination.
The transcriptional start site of the crp mRNA was performed using RACE as previously described (43). The oligonucleotides used for RACE are listed in Table S4. In each case, the generated PCR products of correct size were directly cloned into the plasmid pCR2.1 (Invitrogen). Plasmids were sequenced using M13 forward and reverse primers.
GFP assays.
To construct Ppla-gfp and Pcrp-gfp reporters, the promoter regions of pla and crp (500 bp upstream of the ATG and including the proximal 27 bp of the pla or crp CDS) were amplified by PCR. The gene for GFP was also PCR-amplified, and the resulting products were subsequently joined by SOE-PCR before being subcloned into pUC18R6K-min-Tn7T-kan. To construct the PtetO-pla 5′ UTR-gfp and PtetO-crp 5′ UTR-gfp reporters, the PtetO sequence lacking the 5′ UTR (54) was PCR amplified from pWL213, and the 5′ UTR of pla (15) or crp was amplified from the genome of Y. pestis (77 bp upstream of the ATG of pla and including the proximal 27 bp of the pla CDS, or 79 bp upstream of the ATG of crp and including the proximal 27 bp of the crp CDS). These products were subsequently joined by SOE-PCR and cloned into pWL212. The reporters were then integrated onto the chromosome of Y. pestis as described above.
Strains carrying the GFP reporters were assessed for relative fluorescence at 37°C in triplicate as described previously (46). ATc (0.25 µg/ml) was added to the cultures where appropriate. Background fluorescence subtraction and normalization were performed as described (46). Statistical differences were determined by Student’s t test.
Immunoblot analyses.
For analysis of proteins by immunoblotting, bacteria were cultured as described above (at time 0, ATc was added to a concentration of 0.25 µg/ml unless otherwise indicated, when appropriate). Aliquots of the cultures were removed and centrifuged at 5,000 × g for 10 min at 4°C, and bacterial pellets were washed once with PBS and incubated for 30 min on ice with lysozyme (50 µg/ml). Cells were then sonicated using 3 30-s pulses, and lysates were centrifuged at 10,000 × g for 10 min at 4°C. Protein concentrations were measured by the Bradford assay (Bio-Rad), and equal concentrations of lysates were mixed with reducing sample buffer, separated by SDS-PAGE, transferred to nitrocellulose, and analyzed by immunoblotting with antibodies to the HA tag (Roche), Pla (65), or RpoA.
SUPPLEMENTAL MATERIAL
Kinetics of PtetO-crp 5′ UTR-gfp induction during the growth of Y. pestis. Wild-type or Δhfq Y. pestis strains with the chromosomal-integrated PtetO-crp 5′ UTR-gfp reporter construct were cultured at 37°C in the presence of ATc, and every 2 h, relative fluorescence normalized to the optical density of the culture was determined. Data are representative of at least 3 independent experiments. Download
ATc has no impact on plasminogen activation, a measure of Pla activity. Wild-type, Δhfq, or Δcrp Y. pestis bacteria were cultured in the presence of ATc or vehicle, and plg activation was measured as described for Fig. 2C. The Δpla strain of Y. pestis is shown as a control for Pla activity. Download
Kinetics of Δhfq Y. pestis during lung infection. Mice were infected i.n. with either Y. pestis or Δhfq Y. pestis, and at the indicated times, CFUs in the lungs were enumerated. Each point represents the numbers recovered from a single mouse. The limit of detection is indicated by a dashed line. Symbols below the limit of detection represent mice that did not have detectable numbers of bacteria. A solid line indicates the median of CFU recovered. **, P < 0.005. Data are representative of 2 independent experiments. Download
Crp-HA production in pCD1+ strains of Y. pestis. Wild-type pCD1+ Y. pestis, the Δhfq and Δcrp strains of Y. pestis carrying the constitutively expressed PtetO-crp-HA construct, or the equivalent mutants carrying the chromosomal-integrated Tn7 vector only were cultured for 6 h at 37°C in the presence of 2.5 mM CaCl2, and cell lysates were analyzed by immunoblotting for the HA tag (top) or RpoA (as a loading control; bottom). Download
Predicted Mfold secondary structure of the Y. pestis crp 5′ UTR and proximal coding region. The most probable prediction with the lowest free energy state is shown. The transcriptional start site is indicated with an arrow, the putative ribosome binding site (RBS) is highlighted with a box, and the translational start site is shown with a black line. Download
ΔCT values of the gyrB transcript normalized to proS or 16S rRNA during pneumonic plague.
Bacterial strains used in this study.
Plasmids used in this study.
Primers used in this study.
ACKNOWLEDGMENTS
We thank Chelsea Schiano and Lauren Priniski for helpful discussions, Roger Pechous for technical assistance, and Melanie Marketon for the kind gift of the RpoA antibody.
This work was supported by the Northwestern University Driskill Graduate Program, NIH grant U54 AI057157 to the Southeastern Regional Center of Excellence for Emerging Infections and Biodefense, and NIH grants F32 AI069688 and R01 AI093727 to W.W.L.
Footnotes
Citation Lathem WW, Schroeder JA, Bellows LE, Ritzert JT, Koo JT, Price PA, Caulfield AJ, Goldman WE. 2014. Posttranscriptional regulation of the Yersinia pestis cyclic AMP receptor protein Crp and impact on virulence. mBio 5(1):e01038-13. doi:10.1128/mBio.01038-13.
REFERENCES
- 1. Perry RD, Fetherston JD. 1997. Yersinia pestis—etiologic agent of plague. Clin. Microbiol. Rev. 10:35–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Stenseth NC, Atshabar BB, Begon M, Belmain SR, Bertherat E, Carniel E, Gage KL, Leirs H, Rahalison L. 2008. Plague: past, present, and future. PLoS Med. 5:e3. 10.1371/journal.pmed.1000003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lathem WW, Crosby SD, Miller VL, Goldman WE. 2005. Progression of primary pneumonic plague: a mouse model of infection, pathology, and bacterial transcriptional activity. Proc. Natl. Acad. Sci. U. S. A. 102:17786–17791. 10.1073/pnas.0506840102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bubeck SS, Cantwell AM, Dube PH. 2007. Delayed inflammatory response to primary pneumonic plague occurs in both outbred and inbred mice. Infect. Immun. 75:697–705. 10.1128/IAI.00403-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Price PA, Jin J, Goldman WE. 2012. Pulmonary infection by Yersinia pestis rapidly establishes a permissive environment for microbial proliferation. Proc. Natl. Acad. Sci. U. S. A. 109:3083–3088. 10.1073/pnas.1112729109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lathem WW, Price PA, Miller VL, Goldman WE. 2007. A plasminogen-activating protease specifically controls the development of primary pneumonic plague. Science 315:509–513. 10.1126/science.1137195 [DOI] [PubMed] [Google Scholar]
- 7. Degen JL, Bugge TH, Goguen JD. 2007. Fibrin and fibrinolysis in infection and host defense. J. Thromb. Haemost. 5(Suppl 1):24–31. 10.1111/j.1538-7836.2007.02519.x [DOI] [PubMed] [Google Scholar]
- 8. Korhonen TK, Haiko J, Laakkonen L, Järvinen HM, Westerlund-Wikström B. 2013. Fibrinolytic and coagulative activities of Yersinia pestis. Front. Cell. Infect. Microbiol. 3:35. 10.3389/fcimb.2013.00035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Caulfield AJ, Lathem WW. 2012. Substrates of the plasminogen activator protease of Yersinia pestis. Adv. Exp. Med. Biol. 954:253–260. 10.1007/978-1-4614-3561-7_32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lawrenz MB, Pennington J, Miller VL. 2013. Acquisition of omptin reveals cryptic virulence function of autotransporter YapE in Yersinia pestis. Mol. Microbiol. 89:276–287. 10.1111/mmi.12273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Järvinen HM, Laakkonen L, Haiko J, Johansson T, Juuti K, Suomalainen M, Buchrieser C, Kalkkinen N, Korhonen TK. 2013. Human single-chain urokinase is activated by the omptins PgtE of Salmonella enterica and Pla of Yersinia pestis despite mutations of active site residues. Mol. Microbiol. 89:507–517. 10.1111/mmi.12293 [DOI] [PubMed] [Google Scholar]
- 12. Chromy BA, Choi MW, Murphy GA, Gonzales AD, Corzett CH, Chang BC, Fitch JP, McCutchen-Maloney SL. 2005. Proteomic characterization of Yersinia pestis virulence. J. Bacteriol. 187:8172–8180. 10.1128/JB.187.23.8172-8180.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Eren E, van den Berg B. 2012. Structural basis for activation of an integral membrane protease by lipopolysaccharide. J. Biol. Chem. 287:23971–23976. 10.1074/jbc.M112.376418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kukkonen M, Suomalainen M, Kyllönen P, Lähteenmäki K, Lång H, Virkola R, Helander IM, Holst O, Korhonen TK. 2004. Lack of O-antigen is essential for plasminogen activation by Yersinia pestis and Salmonella enterica. Mol. Microbiol. 51:215–225 [DOI] [PubMed] [Google Scholar]
- 15. Kim TJ, Chauhan S, Motin VL, Goh EB, Igo MM, Young GM. 2007. Direct transcriptional control of the plasminogen activator gene of Yersinia pestis by the cyclic AMP receptor protein. J. Bacteriol. 189:8890–8900. 10.1128/JB.00972-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Botsford JL, Harman JG. 1992. Cyclic AMP in prokaryotes. Microbiol. Rev. 56:100–122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zhan L, Han Y, Yang L, Geng J, Li Y, Gao H, Guo Z, Fan W, Li G, Zhang L, Qin C, Zhou D, Yang R. 2008. The cyclic AMP receptor protein, CRP, is required for both virulence and expression of the minimal CRP regulon in Yersinia pestis biovar Microtus. Infect. Immun. 76:5028–5037. 10.1128/IAI.00370-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Fic E, Bonarek P, Gorecki A, Kedracka-Krok S, Mikolajczak J, Polit A, Tworzydlo M, Dziedzicka-Wasylewska M, Wasylewski Z. 2009. cAMP receptor protein from Escherichia coli as a model of signal transduction in proteins—a review. J. Mol. Microbiol. Biotechnol. 17:1–11. 10.1159/000178014 [DOI] [PubMed] [Google Scholar]
- 19. Busby S, Ebright RH. 1999. Transcription activation by catabolite activator protein (CAP). J. Mol. Biol. 293:199–213. 10.1006/jmbi.1999.3161 [DOI] [PubMed] [Google Scholar]
- 20. Saier MH. 1998. Multiple mechanisms controlling carbon metabolism in bacteria. Biotechnol. Bioeng. 58:170–174. 10.1002/(SICI)1097-0290(19980420)58:2/3 [DOI] [PubMed] [Google Scholar]
- 21. Crasnier M. 1996. Cyclic AMP and catabolite repression. Res. Microbiol. 147:479–482. 10.1016/0923-2508(96)84002-2 [DOI] [PubMed] [Google Scholar]
- 22. Silva AJ, Benitez JA. 2004. Transcriptional regulation of Vibrio cholerae hemagglutinin/protease by the cyclic AMP receptor protein and RpoS. J. Bacteriol. 186:6374–6382. 10.1128/JB.186.19.6374-6382.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Müller CM, Aberg A, Straseviçiene J, Emody L, Uhlin BE, Balsalobre C. 2009. Type 1 fimbriae, a colonization factor of uropathogenic Escherichia coli, are controlled by the metabolic sensor CRP-cAMP. PLoS Pathog. 5:e1000303. 10.1371/journal.ppat.1000303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Petersen S, Young GM. 2002. Essential role for cyclic AMP and its receptor protein in Yersinia enterocolitica virulence. Infect. Immun. 70:3665–3672. 10.1128/IAI.70.7.3665-3672.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zhan L, Yang L, Zhou L, Li Y, Gao H, Guo Z, Zhang L, Qin C, Zhou D, Yang R. 2009. Direct and negative regulation of the sycO-ypkA-yopJ operon by cyclic AMP receptor protein (CRP) in Yersinia pestis. BMC Microbiol. 9:178. 10.1186/1471-2180-9-178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Heroven AK, Sest M, Pisano F, Scheb-Wetzel M, Steinmann R, Böhme K, Klein J, Münch R, Schomburg D, Dersch P. 2012. Crp induces switching of the CsrB and CsrC RNAs in Yersinia pseudotuberculosis and links nutritional status to virulence. Front. Cell Infect. Microbiol. 2:158. 10.3389/fcimb.2012.00158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sun W, Roland KL, Kuang X, Branger CG, Curtiss R. 2010. Yersinia pestis with regulated delayed attenuation as a vaccine candidate to induce protective immunity against plague. Infect. Immun. 78:1304–1313. 10.1128/IAI.01122-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ishizuka H, Hanamura A, Inada T, Aiba H. 1994. Mechanism of the down-regulation of cAMP receptor protein by glucose in Escherichia coli: role of autoregulation of the crp gene. EMBO J. 13:3077–3082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. González-Gil G, Kahmann R, Muskhelishvili G. 1998. Regulation of crp transcription by oscillation between distinct nucleoprotein complexes. EMBO J. 17:2877–2885. 10.1093/emboj/17.10.2877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Berthoumieux S, de Jong H, Baptist G, Pinel C, Ranquet C, Ropers D, Geiselmann J. 2013. Shared control of gene expression in bacteria by transcription factors and global physiology of the cell. 10.1038/msb.2012.70. Mol. Syst. Biol. 9:634. PubMed. [DOI] [PMC free article] [PubMed]
- 31. Aiba H. 1985. Transcription of the Escherichia coli adenylate cyclase gene is negatively regulated by cAMP-cAMP receptor protein. J. Biol. Chem. 260:3063–3070 [PubMed] [Google Scholar]
- 32. Hanamura A, Aiba H. 1991. Molecular mechanism of negative autoregulation of Escherichia coli crp gene. Nucleic Acids Res. 19:4413–4419. 10.1093/nar/19.16.4413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zhang Y, Wang L, Han Y, Yan Y, Tan Y, Zhou L, Cui Y, Du Z, Wang X, Bi Y, Yang H, Song Y, Zhang P, Zhou D, Yang R. 2013. Autoregulation of PhoP/PhoQ and positive regulation of the cyclic AMP receptor protein-cyclic AMP complex by PhoP in Yersinia pestis. J. Bacteriol. 195:1022–1030. 10.1128/JB.01530-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Qu S, Zhang Y, Liu L, Wang L, Han Y, Yang R, Zhou D, Liu M. 2013. Cyclic AMP receptor protein is a repressor of adenylyl cyclase gene cyaA in Yersinia pestis. Can. J. Microbiol. 59:304–310. 10.1139/cjm-2012-0705 [DOI] [PubMed] [Google Scholar]
- 35. Rajkowitsch L, Schroeder R. 2007. Dissecting RNA chaperone activity. RNA 13:2053–2060. 10.1261/rna.671807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Aiba H. 2007. Mechanism of RNA silencing by Hfq-binding small RNAs. Curr. Opin. Microbiol. 10:134–139. 10.1016/j.mib.2007.03.010 [DOI] [PubMed] [Google Scholar]
- 37. Chao Y, Vogel J. 2010. The role of Hfq in bacterial pathogens. Curr. Opin. Microbiol. 13:24–33. 10.1016/j.mib.2010.01.001 [DOI] [PubMed] [Google Scholar]
- 38. Gottesman S, McCullen CA, Guillier M, Vanderpool CK, Majdalani N, Benhammou J, Thompson KM, FitzGerald PC, Sowa NA, FitzGerald DJ. 2006. Small RNA regulators and the bacterial response to stress. Cold Spring Harb. Symp. Quant. Biol. 71:1–11. 10.1101/sqb.2006.71.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lenz DH, Mok KC, Lilley BN, Kulkarni RV, Wingreen NS, Bassler BL. 2004. The small RNA chaperone Hfq and multiple small RNAs control quorum sensing in Vibrio harveyi and Vibrio cholerae. Cell 118:69–82. 10.1016/j.cell.2004.06.009 [DOI] [PubMed] [Google Scholar]
- 40. Sittka A, Pfeiffer V, Tedin K, Vogel J. 2007. The RNA chaperone Hfq is essential for the virulence of Salmonella typhimurium. Mol. Microbiol. 63:193–217. 10.1111/j.1365-2958.2006.05489.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Geng J, Song Y, Yang L, Feng Y, Qiu Y, Li G, Guo J, Bi Y, Qu Y, Wang W, Wang X, Guo Z, Yang R, Han Y. 2009. Involvement of the post-transcriptional regulator Hfq in Yersinia pestis virulence. PLoS One 4:e6213. 10.1371/journal.pone.0006213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Schiano CA, Bellows LE, Lathem WW. 2010. The small RNA chaperone Hfq is required for the virulence of Yersinia pseudotuberculosis. Infect. Immun. 78:2034–2044. 10.1128/IAI.01046-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Koo JT, Alleyne TM, Schiano CA, Jafari N, Lathem WW. 2011. Global discovery of small RNAs in Yersinia pseudotuberculosis identifies Yersinia-specific small, noncoding RNAs required for virulence. Proc. Natl. Acad. Sci. U. S. A. 108:E709–E717. 10.1073/pnas.1101655108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Schiano CA, Lathem WW. 2012. Post-transcriptional regulation of gene expression in Yersinia species. Front Cell. Infect. Microbiol. 2:129. 10.3389/fcimb.2012.00129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Dennis JJ, Zylstra GJ. 1998. Plasposons: modular self-cloning minitransposon derivatives for rapid genetic analysis of Gram-negative bacterial genomes. Appl. Environ. Microbiol. 64:2710–2715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Bellows LE, Koestler BJ, Karaba SM, Waters CM, Lathem WW. 2012. Hfq-dependent, co-ordinate control of cyclic diguanylate synthesis and catabolism in the plague pathogen Yersinia pestis. Mol. Microbiol. 86:661–674. 10.1111/mmi.12011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Bai G, Golubov A, Smith EA, McDonough KA. 2010. The importance of the small RNA chaperone Hfq for growth of epidemic Yersinia pestis, but not Yersinia pseudotuberculosis, with implications for plague biology. J. Bacteriol. 192:4239–4245. 10.1128/JB.00504-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Ishizuka H, Hanamura A, Kunimura T, Aiba H. 1993. A lowered concentration of cAMP receptor protein caused by glucose is an important determinant for catabolite repression in Escherichia coli. Mol. Microbiol. 10:341–350. 10.1111/j.1365-2958.1993.tb01960.x [DOI] [PubMed] [Google Scholar]
- 49. Balsalobre C, Johansson J, Uhlin BE. 2006. Cyclic AMP-dependent osmoregulation of crp gene expression in Escherichia coli. J. Bacteriol. 188:5935–5944. 10.1128/JB.00235-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Zhang J, Sprung R, Pei J, Tan X, Kim S, Zhu H, Liu CF, Grishin NV, Zhao Y. 2009. Lysine acetylation is a highly abundant and evolutionarily conserved modification in Escherichia coli. Mol. Cell. Proteomics 8:215–225. 10.1074/mcp.M800187-MCP200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Yu BJ, Kim JA, Moon JH, Ryu SE, Pan JG. 2008. The diversity of lysine-acetylated proteins in Escherichia coli. J. Microbiol. Biotechnol. 18:1529–1536 [PubMed] [Google Scholar]
- 52. Gao H, Zhang Y, Yang L, Liu X, Guo Z, Tan Y, Han Y, Huang X, Zhou D, Yang R. 2011. Regulatory effects of cAMP receptor protein (CRP) on porin genes and its own gene in Yersinia pestis. BMC Microbiol. 11:40. 10.1186/1471-2180-11-40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Groisman EA, Mouslim C. 2006. Sensing by bacterial regulatory systems in host and non-host environments. Nat. Rev. Microbiol. 4:705–709. 10.1038/nrmicro1478 [DOI] [PubMed] [Google Scholar]
- 54. Lutz R, Bujard H. 1997. Independent and tight regulation of transcriptional units in Escherichia coli via the LacR/O, the TetR/O and AraC/I1-I2 regulatory elements. Nucleic Acids Res. 25:1203–1210. 10.1093/nar/25.6.1203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Zuker M. 1989. Computer prediction of RNA structure. Methods Enzymol. 180:262–288 [DOI] [PubMed] [Google Scholar]
- 56. Sledjeski DD, Gupta A, Gottesman S. 1996. The small RNA, DsrA, is essential for the low temperature expression of RpoS during exponential growth in Escherichia coli. EMBO J. 15:3993–4000 [PMC free article] [PubMed] [Google Scholar]
- 57. Majdalani N, Hernandez D, Gottesman S. 2002. Regulation and mode of action of the second small RNA activator of RpoS translation, RprA. Mol. Microbiol. 46:813–826. 10.1046/j.1365-2958.2002.03203.x [DOI] [PubMed] [Google Scholar]
- 58. Achtman M, Zurth K, Morelli G, Torrea G, Guiyoule A, Carniel E. 1999. Yersinia pestis, the cause of plague, is a recently emerged clone of Yersinia pseudotuberculosis. Proc. Natl. Acad. Sci. U. S. A. 96:14043–14048. 10.1073/pnas.96.24.14043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Ligon BL. 2006. Plague: a review of its history and potential as a biological weapon. Semin. Pediatr. Infect. Dis. 17:161–170. 10.1053/j.spid.2006.07.002 [DOI] [PubMed] [Google Scholar]
- 60. Vadyvaloo V, Jarrett C, Sturdevant D, Sebbane F, Hinnebusch BJ. 2007. Analysis of Yersinia pestis gene expression in the flea vector. Adv. Exp. Med. Biol. 603:192–200. 10.1007/978-0-387-72124-8_16 [DOI] [PubMed] [Google Scholar]
- 61. Sebbane F, Lemaître N, Sturdevant DE, Rebeil R, Virtaneva K, Porcella SF, Hinnebusch BJ. 2006. Adaptive response of Yersinia pestis to extracellular effectors of innate immunity during bubonic plague. Proc. Natl. Acad. Sci. U. S. A. 103:11766–11771. 10.1073/pnas.0601182103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Thiennimitr P, Winter SE, Winter MG, Xavier MN, Tolstikov V, Huseby DL, Sterzenbach T, Tsolis RM, Roth JR, Bäumler AJ. 2011. Intestinal inflammation allows Salmonella to use ethanolamine to compete with the microbiota. Proc. Natl. Acad. Sci. U. S. A. 108:17480–17485. 10.1073/pnas.1107857108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Biosystems Applied. 1997. ABI Prism 7700 sequence detection system user bulletin 2. Applied Biosystems, Grand Island, NY [Google Scholar]
- 64. Choi KH, Gaynor JB, White KG, Lopez C, Bosio CM, Karkhoff-Schweizer RR, Schweizer HP. 2005. A Tn7-based broad-range bacterial cloning and expression system. Nat. Methods 2:443–448. 10.1038/nmeth765 [DOI] [PubMed] [Google Scholar]
- 65. Houppert AS, Bohman L, Merritt PM, Cole CB, Caulfield AJ, Lathem WW, Marketon MM. 2013. RfaL is required for Yersinia pestis type III secretion and virulence. Infect. Immun. 81:1186–1197. 10.1128/IAI.01417-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Kinetics of PtetO-crp 5′ UTR-gfp induction during the growth of Y. pestis. Wild-type or Δhfq Y. pestis strains with the chromosomal-integrated PtetO-crp 5′ UTR-gfp reporter construct were cultured at 37°C in the presence of ATc, and every 2 h, relative fluorescence normalized to the optical density of the culture was determined. Data are representative of at least 3 independent experiments. Download
ATc has no impact on plasminogen activation, a measure of Pla activity. Wild-type, Δhfq, or Δcrp Y. pestis bacteria were cultured in the presence of ATc or vehicle, and plg activation was measured as described for Fig. 2C. The Δpla strain of Y. pestis is shown as a control for Pla activity. Download
Kinetics of Δhfq Y. pestis during lung infection. Mice were infected i.n. with either Y. pestis or Δhfq Y. pestis, and at the indicated times, CFUs in the lungs were enumerated. Each point represents the numbers recovered from a single mouse. The limit of detection is indicated by a dashed line. Symbols below the limit of detection represent mice that did not have detectable numbers of bacteria. A solid line indicates the median of CFU recovered. **, P < 0.005. Data are representative of 2 independent experiments. Download
Crp-HA production in pCD1+ strains of Y. pestis. Wild-type pCD1+ Y. pestis, the Δhfq and Δcrp strains of Y. pestis carrying the constitutively expressed PtetO-crp-HA construct, or the equivalent mutants carrying the chromosomal-integrated Tn7 vector only were cultured for 6 h at 37°C in the presence of 2.5 mM CaCl2, and cell lysates were analyzed by immunoblotting for the HA tag (top) or RpoA (as a loading control; bottom). Download
Predicted Mfold secondary structure of the Y. pestis crp 5′ UTR and proximal coding region. The most probable prediction with the lowest free energy state is shown. The transcriptional start site is indicated with an arrow, the putative ribosome binding site (RBS) is highlighted with a box, and the translational start site is shown with a black line. Download
ΔCT values of the gyrB transcript normalized to proS or 16S rRNA during pneumonic plague.
Bacterial strains used in this study.
Plasmids used in this study.
Primers used in this study.