

ABSTRACT
Cryptococcosis is an infectious disease of global significance for which new therapies are needed. Repurposing previously developed drugs for new indications can expedite the translation of new therapies from bench to beside. Here, we characterized the anti-cryptococcal activity and antifungal mechanism of estrogen receptor antagonists related to the breast cancer drugs tamoxifen and toremifene. Tamoxifen and toremifene are fungicidal and synergize with fluconazole and amphotericin B in vitro. In a mouse model of disseminated cryptococcosis, tamoxifen at concentrations achievable in humans combines with fluconazole to decrease brain burden by ~1 log10. In addition, these drugs inhibit the growth of Cryptococcus neoformans within macrophages, a niche not accessible by current antifungal drugs. Toremifene and tamoxifen directly bind to the essential EF hand protein calmodulin, as determined by thermal shift assays with purified C. neoformans calmodulin (Cam1), prevent Cam1 from binding to its well-characterized substrate calcineurin (Cna1), and block Cna1 activation. In whole cells, toremifene and tamoxifen block the calcineurin-dependent nuclear localization of the transcription factor Crz1. A large-scale chemical genetic screen with a library of C. neoformans deletion mutants identified a second EF hand-containing protein, which we have named calmodulin-like protein 1 (CNAG_05655), as a potential target, and further analysis showed that toremifene directly binds Cml1 and modulates its ability to bind and activate Cna1. Importantly, tamoxifen analogs (idoxifene and methylene-idoxifene) with increased calmodulin antagonism display improved anti-cryptococcal activity, indicating that calmodulin inhibition can be used to guide a systematic optimization of the anti-cryptococcal activity of the triphenylethylene scaffold.
IMPORTANCE
Worldwide, cryptococcosis affects approximately 1 million people annually and kills more HIV/AIDS patients per year than tuberculosis. The gold standard therapy for cryptococcosis is amphotericin B plus 5-flucytosine, but this regimen is not readily available in regions where resources are limited and where the burden of disease is highest. Herein, we show that molecules related to the breast cancer drug tamoxifen are fungicidal for Cryptococcus and display a number of pharmacological properties desirable for an anti-cryptococcal drug, including synergistic fungicidal activity with fluconazole in vitro and in vivo, oral bioavailability, and activity within macrophages. We have also demonstrated that this class of molecules targets calmodulin as part of their mechanism of action and that tamoxifen analogs with increased calmodulin antagonism have improved anti-cryptococcal activity. Taken together, these results indicate that tamoxifen is a pharmacologically attractive scaffold for the development of new anti-cryptococcal drugs and provide a mechanistic basis for its further optimization.
INTRODUCTION
Cryptococcosis is one of the most important human fungal infections. Worldwide, approximately 1,000,000 new infections occur each year, and over 620,000 deaths are attributable to cryptococcosis (1). The largest burden of disease occurs in people living with HIV/AIDS, and cryptococcosis causes more deaths in this patient population than tuberculosis. The two most common clinical manifestations of cryptococcosis in humans are meningoencephalitis and pneumonia (2). Cryptococcosis also occurs in patients with other forms of immune system compromise, including solid organ transplant patients and, less commonly, hematopoietic stem cell transplant recipients (2). Most infections are due to serotype A Cryptococcus neoformans var. grubii, with serotype D Cryptococcus neoformans var. neoformans being isolated in a minority of cases. Recently, Cryptococcus gattii has emerged as the etiologic agent of an ongoing outbreak of cryptococcosis in immune-competent people in the Pacific Northwest region of North America (3).
The gold standard therapy for cryptococcal meningitis (CM) is a combination of amphotericin B (AMB) and flucytosine (FC) during the initial phase of treatment (4). Indeed, a recently reported clinical trial showed that the combination of AMB and FC is superior to AMB alone (5). Unfortunately, AMB and FC are not generally available in regions where resources are limited and where the burden of disease is highest (6). In these regions, fluconazole (FLU) is the mainstay of therapy because it is widely available and inexpensive and, unlike AMB/FC, does not require intravenous administration or laboratory monitoring for toxicity. However, outcomes associated with FLU-based treatment are much poorer (20 to 60%) and are likely to contribute to the overall worse prognosis for patients with cryptococcal meningitis in regions where resources are limited (6).
A key difference between the AMB- and FLU-based regimens for CM is that AMB-based combinations have fungicidal activity and lead to relatively rapid sterilization of the cerebrospinal fluid. This so-called early fungicidal activity has been associated with better prognosis clinically (7). In contrast, FLU is not fungicidal and, even at high doses, has poor early fungicidal activity as well as much poorer clinical efficacy. In order to improve the treatment of CM in regions where resources are limited, an orally bioavailable agent with fungicidal activity for Cryptococcus is needed. However, the pace of development of new antifungal drugs has been extremely slow (8).
Previously approved drugs with activities other than that for which they were designed can be valuable scaffolds for further optimization. This approach to new lead identification has been called selective optimization of side activities (SOSA) or, more recently, drug repurposing (9, 10). In the ideal scenario, a repurposed molecule is sufficiently active in its new application to be directly used in the clinic without new formulation or drastic changes in dosage. However, even if this ideal scenario does not come to fruition, the strategy may ultimately prove successful because the initial scaffold can be used as a starting point for medicinal chemistry-based optimization of the new activity in the context of a pharmacologically attractive structure (10). Recently, our laboratory screened a library of off-patent drugs and biologically active small molecules for agents that directly kill Cryptococcus (11). One of the most active molecules identified by the screen was the estrogen receptor antagonist tamoxifen. The antifungal activity of tamoxifen had been previously described (12, 13), and it was also recently identified in a screen for molecules synergistic with fluconazole (14). In a previous study we showed that tamoxifen had anti-candidal activity in vitro and in vivo (13).
Tamoxifen and other triphenylethylene-based estrogen receptor antagonists have a number of features that are particularly attractive for the treatment of cryptococcosis. First, the molecules are orally bioavailable (15) and, therefore, are amenable to use in regions where resources are limited. Second, they cross the blood-brain barrier and accumulate in the brain, reaching concentrations 10- to 100-fold higher than that found in serum (15). Third, this class of molecules is lysosomotropic and accumulates within the lysosome/phagolysosome (16). Cryptococcus replicates within the phagolysosome of macrophages, a niche not accessible to either FLU or AMB (17, 18). Because the triphenylethylenes have pharmacological features that are well suited to potential therapy for cryptococcosis, we characterized further the anti-cryptococcal activity and mechanism of action of this scaffold.
RESULTS
Estrogen receptor antagonists are fungicidal anti-cryptococcal molecules in vitro.
Three triphenylethylenes are currently in clinical use (tamoxifen, toremifene, and clomiphene; structures of all molecules are shown in Fig. S1A in the supplemental material), and we first determined the MIC of each for C. neoformans var. grubii (strain H99; strain information is tabulated in Table S1 in the supplemental material) using standard CLSI microdilution methods (19). As shown in Fig. 1A, tamoxifen and toremifene have identical activities (8 µg/ml), while clomiphene is much less active (64 µg/ml). Tamoxifen is a prodrug that is metabolized to two derivatives (15), 4-hydroxy-tamoxifen and endoxifen (see Fig. S1A in the supplemental material). Interestingly, both tamoxifen metabolites are slightly more active (4 µg/ml) than the parent molecule (Fig. 1A). Because these molecules represent important species circulating within the host and since molecules within this class are metabolized similarly (15), the fact that the metabolites retain anti-cryptococcal activity supports their potential utility as candidate drugs.
FIG 1 .

Anti-cryptococcal activity of triphenylethylenes. (A) MICs of the indicated molecules against C. neoformans var. grubii strain H99 as determined by CLSI standard methods. CLM, clomiphene; TAM, tamoxifen; TOR, toremifene; 4-HO-TAM, 4-hydroxy-tamoxifen; ENDO, endoxifen; IDX, idoxifene; M-IDX, methylene-idoxifene. The chemical structures for each molecule are shown in Fig. S1A. The y axis of the graph is a modified log2 scale. (B) The combination of subinhibitory concentrations of toremifene (TOR; 2 µg/ml) and fluconazole (FLU; 2 µg/ml) reduces the initial inoculum of C. neoformans (4 log10 CFU/ml) more than 2 log10 CFU/ml at 24 h and is therefore a fungicidal combination. (C) J774 mouse macrophages were infected with opsonized C. neoformans, washed to remove nonadherent/nonphagocytosed cells, and incubated with fresh medium containing either DMSO (1%) or drug for 24 h. After 24 h, macrophages were lysed and the number of C. neoformans CFU per well determined as described in Materials and Methods. Data are means of triplicates, and error bars represent standard deviations. Differences between groups were statistically significant (Student’s t test; P < 0.05). (D) Male A/JCr mice (7 per group) were infected with serotype A C. neoformans H99 by tail vein injection. The control group (CTRL) received saline by intraperitoneal injection and peanut oil by oral gavage. The fluconazole group (FLU) received 5 mg/kg FLU by intraperitoneal injection and peanut oil by oral gavage. The tamoxifen group (TAM) received saline by intraperitoneal injection and 200 mg/kg tamoxifen suspended in peanut oil by oral gavage. The combination treatment group (TAM+FLU) received 5 mg/kg fluconazole by intraperitoneal injection and 200 mg/kg tamoxifen suspended in peanut oil by oral gavage. The bars represent the median for each group, and each dot represents an individual animal. The number of CFU/gram of brain tissue was determined and transformed into log10 units, and differences between groups were analyzed by ANOVA; statistical significance was set at a P value of <0.05 (SigmaPlot software).
Interest in combination therapies for the treatment of fungal infections has increased (20, 21), and therefore, we examined the interaction of toremifene and tamoxifen with the three antifungal drugs used to treat cryptococcal meningoencephalitis (AMB, FLU, and FC) by means of a standard checkerboard interaction assay (20). The combination of toremifene (2 µg/ml; 1/4 MIC) and FLU (2 µg/ml; 1/4 MIC) inhibited all growth, giving a fractional inhibitory concentration index (FICI) of 0.50 (synergy is defined as an FICI of ≤0.5 [20]). Toremifene was additive in combination with AMB (FICI, 1.0; 4 µg/ml toremifene–0.5 µg/ml AMB; AMB MIC, 1 µg/ml). In contrast, tamoxifen was additive with FLU (FICI, 0.75; 4 µg/ml tamoxifen–2 µg/ml FLU) and synergistic with AMB (FICI, 0.5; 2 µg/ml tamoxifen–0.25 µg/ml AMB). The combination of either triphenylethylene with FC was additive.
One approach to improving the efficacy of FLU is to identify molecules that yield a fungicidal combination with FLU (21). We therefore further characterized the combination of FLU with toremifene/tamoxifen by a time-kill assay. The combination of subinhibitory concentrations of toremifene or tamoxifen (2 µg/ml) and FLU (2 µg/ml) decreased the initial inoculum by >2 log10, the definition of a fungicidal combination (the time-kill curve for toremifene is shown in Fig. 1B). Importantly, the combination of toremifene or tamoxifen with FLU is fungicidal at concentrations of toremifene (22) and tamoxifen (23) that have been safely achieved in serum or tissue in humans with oral doses in the setting of clinical trials for the treatment of malignancies other than breast cancer.
Toremifene and tamoxifen are active against intraphagocytic C. neoformans.
An important facet of C. neoformans pathogenesis is its ability to replicate within the phagolysosomes of macrophages (17). Consequently, the phagolysosome represents an important host niche for C. neoformans; however, neither AMB nor FLU is active against intraphagocytic C. neoformans (18). To test the activity of tamoxifen and toremifene against intraphagocytic C. neoformans, we infected mouse J774 macrophage-like cells with opsonized C. neoformans; this system has been widely used to study C. neoformans-macrophage interaction (24). The cells were washed to remove extracellular organisms and then incubated for 24 h in medium containing toremifene or tamoxifen (8 µg/ml) or dimethyl sulfoxide (DMSO) (1%). The macrophages were lysed, and the number of viable C. neoformans organisms was determined using standard plating assays. Treatment of the infected J774 cells with 8 µg/ml of toremifene or tamoxifen led to a 0.5- to 1-log10 reduction in the number of viable organisms within the macrophage relative to DMSO-treated J774 cells (Fig. 1C). Importantly, these drug concentrations had no effect on the viability of J774 by standard cytotoxicity assays, nor did they affect the morphology or appearance of the cells (>90% survival of uninfected macrophages treated with drug by CytoToxOne assay). The anti-malarial chloroquine is also active against intraphagocytic C. neoformans by virtue of its ability to alkalinize the phagolysosome (25). Tamoxifen also alkalinizes macrophage compartments but does so at concentrations (0.5 µg/ml) well below that which affects C. neoformans replication (26). Thus, it appears that direct antifungal activity is likely to play an important role in the activity of triphenylethylenes for C. neoformans within macrophages.
The combination of tamoxifen or toremifene and fluconazole is more active than fluconazole alone in a murine model of cryptococcosis.
To further test triphenylethylenes as potential anti-cryptococcal agents, we examined the activities of tamoxifen and toremifene both alone and in combination with FLU in a murine model of cryptococcosis. The pharmacology of tamoxifen has been well characterized in mice and differs significantly from that in humans with respect to the steady-state levels achieved in the serum and tissues (27). In mice, much higher doses of tamoxifen (200 mg/kg/day) are required to establish serum and tissue levels comparable to standard doses of tamoxifen in humans (25 to 60 mg/day). Although this dose in mice leads to serum levels below the MIC of tamoxifen against C. neoformans, the hydrophobic nature of the molecule leads to tissue levels that are up to 100-fold higher than those in serum and are comparable to the MIC of tamoxifen and the FIC of tamoxifen combined with FLU (28, 29).
To account for the differences in pharmacology and humans, male AJ/Cr mice were treated with tamoxifen (200 mg/kg/day) or carrier daily for 3 days prior to inoculation to allow tamoxifen to accumulate in tissue to levels approximately equal to that seen with standard dosing in humans. On the day of inoculation, mice were infected with the C. neoformans serotype A parent strain H99 by tail-vein injection. The infected mice were treated with fluconazole alone (5 mg/kg/day), tamoxifen alone (200 mg/kg/day), or tamoxifen in combination with fluconazole for 7 days, after which the brain fungal burdens from these mice were determined. As shown in Fig. 1D, neither tamoxifen nor FLU at this dose alone yielded a statistically significant reduction in fungal brain burden. However, the combination of tamoxifen and FLU reduced brain burden >1 log10 relative to that in untreated mice and 0.7 log10 compared to FLU-treated mice. Similar experiments with toremifene were complicated by the fact that AJ/Cr mice were much less tolerant of toremifene than the strains of mice reported in the literature (30). These results indicate that tamoxifen doses that give serum and tissue levels comparable to those achievable in humans improve the in vivo activity of FLU against C. neoformans.
Triphenylethylenes directly bind to C. neoformans calmodulin and inhibit calmodulin-mediated calcineurin activation in vitro.
The antifungal activity of triphenylethylenes was first described over 30 years ago, but their fungal target has not been defined at the molecular level. We and others have reported genetic data obtained with the model yeast Saccharomyces cerevisiae indicating that tamoxifen interferes with calcium homeostasis and calmodulin function (13, 31). In mammalian systems, tamoxifen has been shown to directly bind calmodulin (32). Consistent with previous work in S. cerevisiae (13), expression of the C. neoformans gene CAM1 (CnCAM1) from the strong GPD1 promoter led to decreased toremifene susceptibility compared to a strain expressing CnCAM1 from the endogenous promoter (Fig. 2A). We therefore hypothesized that calmodulin is a direct molecular target of the triphenylethylenes in C. neoformans.
FIG 2 .
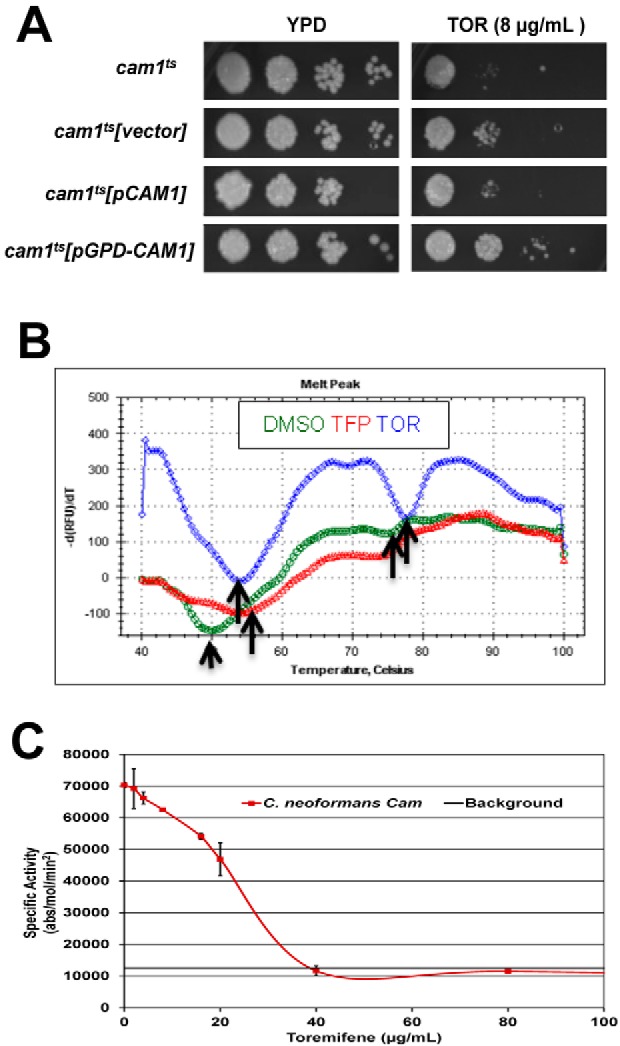
Toremifene binds and inhibits the function of C. neoformans calmodulin in vitro. (A) C. neoformans strain (cam1Ts) with reduced CAM1 expression (38) was transformed with empty vector, a vector expressing CAM1 from its endogenous promoter (pCAM1), or a vector expressing CAM1 from the strong constitutive promoter GPD1 (pGPD-CAM1). Serial dilutions (10-fold) of a suspension (1 OD600 unit/ml) suspension of cells for the indicated strains were spotted on yeast peptone dextrose (YPD) plates containing DMSO solvent (1%) or toremifene (TOR; 8 µg/ml). The plates were incubated at 30°C for 3 days and photographed. (B) Thermal denaturation curves for CnCam1 in the presence of DMSO (green), trifluopromazine (TFP; red), and toremifene (TOR; blue), as determined by differential scanning fluorimetry. The y axis shows the negative derivative of the fluorescence with respect to change in temperature. The arrows indicate low-temperature and high-temperature inflection points corresponding to the Tm for N-terminal-domain and C-terminal-domain denaturation, respectively. (C) CnCam1-mediated activation of human calcineurin is inhibited by TOR. The specific activity (absorbance [abs]/mol calcineurin/min) of human calcineurin is plotted on the y axis, and the no-drug (DMSO-only) control indicates activation. The dose-response curve was fitted by linear analysis (R = 0.986) to give an IC50 of 25 µg/ml. Three independent experiments were performed with similar results, and results of a representative single experiment are shown. Error bars indicate the standard deviations for technical replicates of each drug concentration.
To test whether triphenylethylenes directly bind C. neoformans calmodulin, we used a thermal shift assay with CnCam1 purified from Escherichia coli by affinity chromatography. As shown in Fig. 2B, CnCam1 shows two denaturation inflection points at 50°C and 78°C. These curves are consistent with isothermal calorimetry experiments indicating that the C-terminal domain of mammalian calmodulin denatures at a lower temperature than the N terminus. Addition of the well-characterized calmodulin antagonist trifluopromazine (TFP) shifts the melting temperature (Tm) of CnCam1 by 4°C; a ΔTm greater than 2°C indicates significant binding (33). The addition of either tamoxifen or toremifene stabilized CnCam1 at 8°C and 10°C, respectively (see Fig. S2A in the supplemental material). In addition, toremifene and tamoxifen shifted the Tm of both the low- and high-temperature inflection points (Fig. 2B), suggesting that triphenylethylenes bind to two sites on CnCam1. The hydrophobic drug FLU did not alter the Tm of CnCam1, indicating that the Tm shift is unlikely to be due to nonspecific hydrophobic interactions (see Fig. S2A). These data indicate that toremifene and tamoxifen directly bind to C. neoformans calmodulin and strongly support calmodulin as a molecular target of triphenylethylenes in Cryptococcus.
We next tested whether triphenylethylenes interfered with CnCam1 function in vitro. Calmodulin binds to and activates calcineurin, a protein involved in stress response, antifungal tolerance, and virulence in C. neoformans as well as other fungi (34). We had previously shown that CnCam1 activates human calcineurin using a standard in vitro assay (11). Toremifene inhibits CnCam1-mediated calcineurin activity in a dose-dependent manner with an apparent 50% inhibitory concentration (IC50) of 25 µg/ml based on linear regression analysis of the dose-response curve shown in Fig. 2C (R = 0.98). The well-characterized calmodulin inhibitor trifluopromazine also inhibited the reaction (data not shown). Consistent with these results, similar concentrations of toremifene (20 µg/ml) reduced the amount of green fluorescent protein (GFP)-tagged C. neoformans calcineurin (Cna1-GFP) (35) bound by CnCam1 immobilized on beads by 45% (standard deviation, 5%; three replicates) (a representative blot is shown in Fig. S2B in the supplemental material). Taken together, these in vitro data indicate that triphenylethylenes bind C. neoformans calmodulin and interfere with its function in vitro.
Triphenylethylenes phenocopy C. neoformans calmodulin mutants and inhibit nuclear localization of the calcineurin-dependent transcription factor Crz1/SP-1.
Calmodulin plays a crucial role in vacuolar function and morphology in S. cerevisiae (36). Specifically, calmodulin is required for vacuolar fusion in a Ca2+-dependent process, and accordingly, temperature-sensitive S. cerevisiae calmodulin mutants show both fragmented and enlarged vacuoles (37). If triphenylethylenes inhibit calmodulin, C. neoformans cells exposed to the drug should also show vacuolar fusion defects. To test this hypothesis, we used the vacuolar membrane marker MDY-64 to characterize the vacuolar morphology of C. neoformans calmodulin mutants and triphenylethylene-treated cells. Wild-type C. neoformans cells show one or two intermediately sized vacuolar structures (Fig. 3A). In contrast, the vacuolar structures displayed by the cam1Ts mutant of C. neoformans, which expresses 5% of the wild-type levels of calmodulin (38), are either enlarged or very small (see Fig. S3 in the supplemental material) and, thus, phenocopy the vacuolar fusion defects of S. cerevisiae calmodulin mutants (37). Consistent with the Ca2+ dependence of calmodulin’s role in vacuolar fusion, the cam1-4DA mutant, which cannot bind Ca2+ (38), also shows vacuolar fusion defects (see Fig. S3). Exposure of wild-type C. neoformans cells to subinhibitory concentrations of toremifene caused vacuolar defects matching those of the calmodulin mutants (Fig. 3A). Furthermore, treatment of C. neoformans with fluspirilene, a structurally distinct calmodulin antagonist (11), also induces fragmented/altered vacuoles (Fig. 4B). Fluconazole-treated cells do not display this phenotype, suggesting that this phenotype is not related to a generalized stress response induced by antifungal agents (data not shown). Toremifene-treated C. neoformans cells, therefore, show vacuole morphology phenotypes consistent with altered calmodulin function.
FIG 3 .
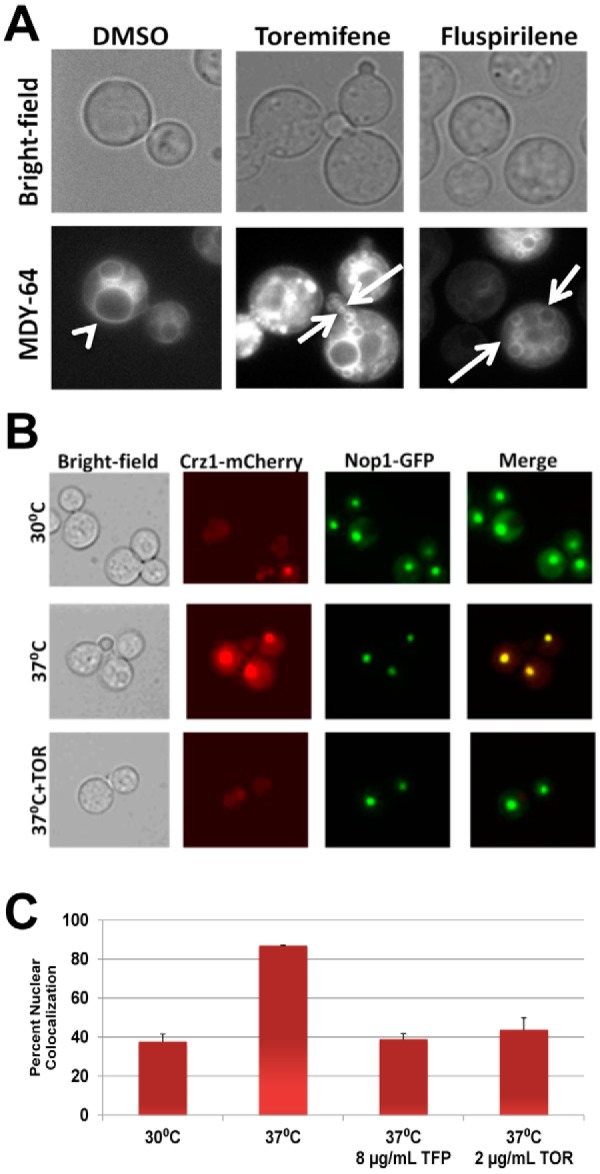
Toremifene phenocopies vacuolar defects of calmodulin mutants and inhibits stress-induced nuclear localization of Crz1. (A) Logarithmic-phase C. neoformans H99 cells were treated with subinhibitory toremifene (2 µg/ml) and the calmodulin antagonist fluspirilene (32 µg/ml), stained with the vacuolar marker MDY-64, and photographed in bright-field or fluorescence channels. The arrowhead indicates a normal, moderate-sized vacuole in DMSO-treated cells, and arrows indicate fragmented, small vacuoles indicative of vacuole fusion defects. (B) C. neoformans strains containing fluorescently tagged Nop1-GFP and Crz1-mCherry were cultivated to logarithmic phase at 30°C, 37°C, and 37°C with a subinhibitory concentration of toremifene (2 µg/ml). Photomicrographs from the appropriate channels are shown, as well as a merged fluorescence image. (C) The percentage of cells with colocalized Nop1-GFP and Crz1-mCherry under the indicated conditions is graphed. Cells were also treated with subinhibitory concentrations of the known calmodulin inhibitor trifluopromazine (8 µg/ml; 1/4 MIC) as a positive control. Bars indicate means from three independent experiments (>100 cells counted per condition per experiment), and error bars indicate standard deviations.
FIG 4 .
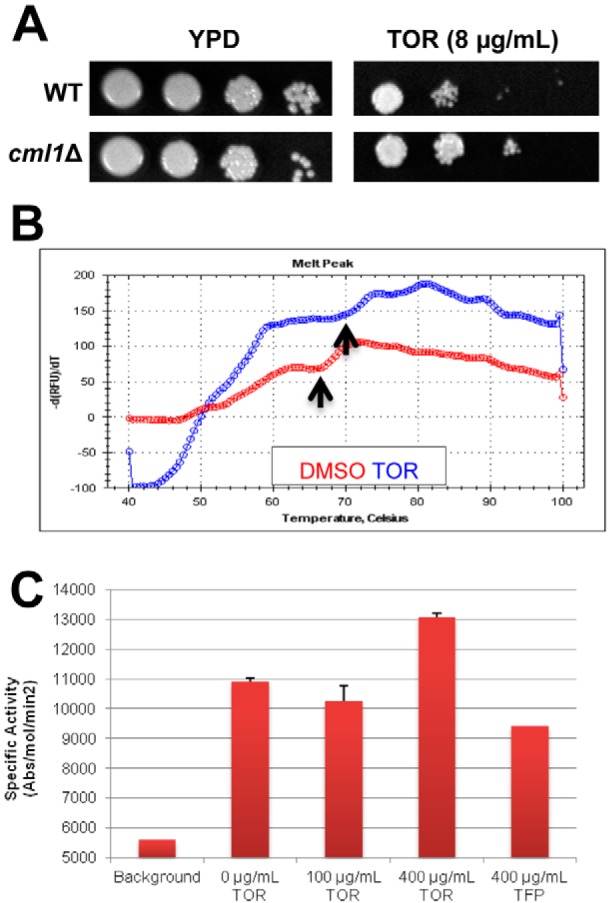
Toremifene binds the EF hand protein Cml1 and modulates its ability to activate calcineurin. (A) Serial dilutions (10-fold) of suspensions (1 OD600 unit/ml) of cells for wild-type (CM018) and cml1Δ strains were spotted on yeast-peptone-dextrose (YPD) plates containing DMSO solvent (1%) or toremifene (TOR; 8 µg/ml). The plates were incubated at 30°C for 3 days and photographed. (B) TOR (blue trace) shifts the Tm for denaturation of Cml1 compared to solvent control (DMSO, red trace). Differential scanning fluorimetry was performed as described in the legend to Fig. 2 and in Materials and Methods. Arrows indicate the Tm for the two conditions, which were determined as described in Materials and Methods. (C) TOR increases the activity of Cml1-mediated calcineurin activity. The specific activity (absorbance [abs]/mol calcineurin/min) of human calcineurin is plotted on the y axis, and the no-drug (DMSO-only) control indicates activation.
To more specifically test whether triphenylethylenes interfere with calmodulin function in C. neoformans cells, we took advantage of the fact that the calcineurin substrate Crz1 was recently identified in C. neoformans (39). Crz1 is a zinc finger transcription factor that localizes to the nucleus in a calcineurin-dependent fashion during stress in fungi (40). Lev et al. have shown that in C. neoformans, CnCrz1 localizes to the nucleus upon exposure to heat stress and high extracellular calcium, consistent with the behavior of Crz1 of S. cerevisiae (ScCrz1). Importantly, addition of the calcineurin inhibitor FK506 prevented the nuclear localization of CnCrz1 during heat stress, indicating that the process is calcineurin dependent (39).
We utilized a C. neoformans strain containing Crz1 tagged with mCherry and the nucleolus-localized protein Nop1 tagged with GFP to test the effect of toremifene on Crz1 localization. At 30°C in the presence of DMSO (1%), the nuclei of 38% of cells contained Crz1; upon shifting to 37°C, the percentage increased to 87% (Fig. 3B and C). Treatment with a subinhibitory concentration of toremifene (2 µg/ml; >90% of cells were viable by propidium iodide staining) at the time of the temperature shift reduced the number of cells with nucleus-localized Crz1 to 39% (P < 0.005; Student’s t test). Treatment with a subinhibitory concentration of the known calmodulin inhibitor trifluopromazine also reduced Crz1 nuclear localization (Fig. 3C), while treatment with fluconazole did not block localization (data not shown). Our data indicate that triphenylethylenes not only interfere with calmodulin function in vitro but also disrupt the function of calmodulin in C. neoformans cells, strongly indicating that calmodulin antagonism contributes to the antifungal activity of this scaffold.
A large-scale chemical genetic screen identifies a second EF hand protein as a triphenylethylene target.
Our observations indicate that calmodulin is a target of triphenylethylenes, but, as with all small molecules, other targets may exist. Chemical genetic screens with large collections of yeast mutants have emerged as a powerful tool for identifying targets and pathways related to small-molecule mechanisms of action (41). Accordingly, we screened a collection of 1,450 C. neoformans deletion mutants for altered growth relative to wild-type strains on agar plates containing toremifene using an automated procedure to evaluate colony size. From this collection, a total of 86 genes with increased or decreased susceptibility to toremifene were identified (see Table S2 in the supplemental material). GO term analysis of genes annotated in the Broad Institute C. neoformans database or identified as homologous to genes in the S. cerevisiae database (P < 1 × 10−3) showed that genes encoding transmembrane transporters (e.g., hexose transporters) were the predominant functional type of gene whose deletion altered toremifene susceptibility (14/86).
Of the mutants identified in the screen, the most likely additional direct target was the EF hand motif-containing protein CNAG_05655, the deletion of which resulted in decreased toremifene susceptibility (see Table S2 in the supplemental material). Retesting of an independent isolate confirmed that deletion of CNAG_05655 decreases susceptibility to toremifene (Fig. 4A). CNAG_05655 is annotated in the Broad Institute database as an EF hand-containing protein, and BLAST searches of other fungal genomes identified homology to S. cerevisiae centrin Cdc31 and to other fungal centrin-like proteins. Like calmodulin, both CNAG_05655 and S. cerevisiae Cdc31 contain 4 EF hand calcium-binding motifs. Indeed, overexpression of CDC31 has been shown previously to suppress the lethality of calmodulin mutants in S. cerevisiae (42). Alignment of CNAG_05655 and calmodulin indicates that, except for an N-terminal extension, CNAG_05655 is very similar to calmodulin (74% identity) (see Fig. S4A in the supplemental material). Based on the strong homology to calmodulin, we refer to this gene as CML1, for calmodulin-like protein 1.
To test the hypothesis that Cml1 is an additional target of the triphenylethylenes, we expressed and purified codon-optimized CML1 from E. coli as described above for CnCam1 and determined whether toremifene bound to it using thermal shift assays. Toremifene induced a 6°C shift in the Tm of Cml1 (Fig. 4B). Proteins with homology to calmodulin such as ScCdc31 have been shown to bind to calmodulin target proteins in yeast (42). Consistent with those observations, Cml1 activates human calcineurin as determined using the assay described above (Fig. 4C). Interestingly, addition of toremifene to Cml1-mediated calcineurin assays increased the activity of enzyme 40% (Fig. 4C). Consistent with this result, Cml1 immobilized on beads pulled down Cna1-GFP more effectively in the presence of toremifene than in its absence (87% increase relative to DMSO; for a representative blot, see Fig. S4B in the supplemental material). Thus, our data indicate that triphenylethylenes bind to and modulate the in vitro activity of a second EF hand-containing C. neoformans protein. Because there are multiple EF hand proteins in eukaryotic genomes, the triphenylethylenes may interfere with the function of multiple EF hand proteins as part of their mechanism of antifungal activity.
Tamoxifen analogs with increased calmodulin antagonist activity have increased anti-cryptococcal activity.
As discussed above, many drugs have multiple targets within the cell, and hence, the crucial question in the setting of the SOSA strategy is whether optimizing the interactions of a scaffold with a given molecular target will lead to improved activity. The activity of the triphenylethylene scaffold as a calmodulin antagonist in mammalian systems was linked to the scaffold’s estrogen receptor-independent anti-tumor activity (43). Rowlands et al. (44) and Hardcastle et al. (45) synthesized tamoxifen analogs designed to optimize calmodulin antagonism. Based on their data, we synthesized two tamoxifen analogs with increased calmodulin activity: idoxifene (5-fold-greater calmodulin activity) and the chain-extended analog of idoxifene, which has 6-fold-greater calmodulin activity than tamoxifen. Consistent with our model, the MICs of both idoxifene (4 µg/ml) and methylene-idoxifene (2 µg/ml) were lower than that of tamoxifen/toremifene (8 µg/ml). Importantly, both idoxifene and methylene-idoxifene blocked temperature-induced Crz1 nuclear localization more effectively than toremifene at the same drug concentrations (Fig. 5). Importantly, these data strongly support the hypothesis that the anti-cryptococcal activity of tamoxifen analogs correlates with anti-calmodulin activity and provide a mechanistic basis for additional structure-activity-based optimization.
FIG 5 .
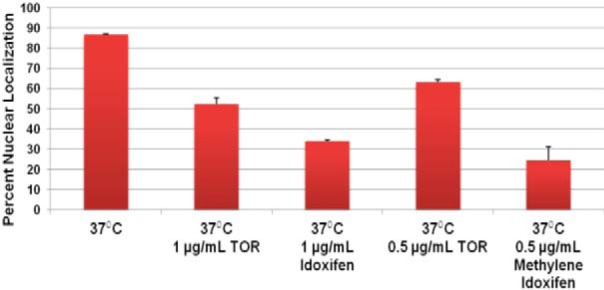
Anti-cryptococcal activity of triphenylethylenes correlates with calmodulin antagonism. C. neoformans strain containing fluorescently tagged Nop1-GFP and Crz1-mCherry were cultivated to logarithmic phase at 37°C with the indicated concentrations of toremifene (TOR), idoxifene, and methylene-idoxifene. As in Fig. 3B, the percentage of cells with colocalized Nop1-GFP and Crz1-mCherry under the indicated conditions is graphed. Bars show the means from three independent experiments (>100 cells counted per condition per experiment), and error bars indicate standard deviations.
DISCUSSION
An effective, widely available treatment for cryptococcal meningitis is an unmet clinical need of global importance (1, 6). The current gold standard therapy is based on medications over 50 years old (AMB/FC) and is not available in the regions of the world with the highest burden of disease (6, 8). To meet this clinical need, a new anti-cryptococcal drug or drug combination must satisfy a very stringent set of criteria. First, the drug(s) must be fungicidal for C. neoformans. Second, the drug(s) must be able to pass the blood-brain barrier and achieve active concentrations within the central nervous system. Third, the regimen must be orally administered so that it can be readily implemented in regions where resources are limited. The triphenylethylenes satisfy all three of these criteria and thus appear to be well-suited for optimization as an anti-cryptococcal scaffold.
Our characterization of the in vitro and in vivo activities of tamoxifen-like molecules has revealed a number of additional features of the scaffold that further support its potential as an anti-cryptococcal agent. First, the antifungal activity appears to be a characteristic of the class in general, since we have shown that seven derivatives are active. In contrast, the activity does not appear to be characteristic of all estrogen receptor antagonists, because non-triphenylethylene-based estrogen receptor antagonists, such as raloxifene, are not active against C. neoformans in our hands (L. DiDone and D. J. Krysan, unpublished results). Second, the major metabolites of tamoxifen retain activity and, indeed, are slightly more active than the parent drug. Third, tamoxifen and toremifene are active toward C. neoformans within macrophages, a niche not accessed by either FLU or AMB (18). Fourth, triphenylethylenes have either synergistic or additive in vitro activity with the drugs (AMB, FLU, and FC) currently used to treat cryptococcosis. While our work was in progress, Spitzer et al. (14) reported that tamoxifen was synergistic with FLU using a C. neoformans strain that had a very high FLU MIC (64 µg/ml). Our characterization has extended these findings in two important ways.
First, we have shown that the combination of tamoxifen/toremifene and FLU is fungicidal by time-kill experiments. FLU is fungistatic, and its inability to directly kill Cryptococcus is widely accepted as one of the most important reasons for its decreased efficacy relative to fungicidal AMB/FC (7). Despite its decreased efficacy, FLU has a number of attractive properties as a therapy in regions where resources are limited. It is safe, orally bioavailable, and provided free of charge to many regions through a donation by its manufacturer. Consequently, an attractive strategy to improve the treatment of cryptococcosis in regions where resources are limited would be to identify a drug that could be combined with FLU to yield a fungicidal cocktail. Triphenylethylenes appear to be promising candidates in that regard.
Second, we tested the combination of FLU and triphenylethylenes in a mouse model of cryptococcosis. Both tamoxifen and toremifene show synergistic activity with FLU in vivo, but neither was active as a single agent at the concentrations tested. Since the pharmacokinetics of triphenylethylenes differ significantly between mice and humans, we used a dose of tamoxifen in the animal experiments that has been shown by a number of studies to give serum and tissue concentrations similar to those obtained by standard dosing (25 to 60 mg/day) in humans (15, 27). The brain concentrations established in mice (and humans) by this dosing are similar to the concentration of tamoxifen that is synergistic with FLU in vitro but is below the MIC of tamoxifen as a single agent against C. neoformans. Thus, it is not surprising that tamoxifen did not have a significant effect on cryptococcal brain burden when used as a single agent, whereas the combination of tamoxifen and FLU led to a reasonable 1-log10 reduction. Although tamoxifen and toremifene did not have significant activity alone in the mouse model, it is important to consider that much higher serum and tissue levels have been achieved in humans as part of early-phase clinical trials. For example, doses of 300 to 400 mg/day have been studied as part of a regimen for multiple myeloma and glioblastoma; these doses lead to micromolar concentrations of tamoxifen in the serum and 10- to 50-fold-higher concentrations in tissue (23). As such, it is theoretically possible that high-dose tamoxifen could achieve sufficient brain levels to be active as a single agent or more active in combination with FLU. Irrespective of whether tamoxifen ultimately emerges as a potential therapy, these results indicate that triphenylethylene-based molecules warrant additional study and optimization.
The systematic optimization of the biological activity of candidate scaffold is greatly facilitated by the knowledge of its molecular target(s). Although triphenylethylenes were developed as estrogen receptor antagonists and are used for the treatment of breast cancer and osteoporosis, a number of off-target effects on eukaryotic cells have been attributed to these molecules. One of the best characterized targets of tamoxifen-related molecules in eukaryotes is calmodulin (31). Our in vitro and cellular assays indicate that triphenylethylenes (i) directly bind CnCam1 by thermal shift assay, (ii) prevent CnCam1 from binding and activating calcineurin in vitro, (iii) cause vacuolar fusion defects that phenocopy those displayed by calmodulin mutants, and (iv) inhibit the stress-induced localization of the calcineurin-dependent transcription factor CnCrz1 in a dose-dependent fashion.
In addition to our previous work (13), two S. cerevisiae-based genome-wide chemical genetic studies have investigated the possible mechanisms for the antifungal activity of tamoxifen (14, 31). Parsons et al. found that the chemical genetic profile of tamoxifen was similar to that of amiodarone (31), a drug that interferes with calcium homeostasis in yeast (46). In addition, tamoxifen clustered with chlorpromazine and trifluoperazine, two well-characterized calmodulin inhibitors. Spitzer et al. (14) performed a similar analysis and also found that tamoxifen was similar to trifluoperazine in a chemical-genetic profile. Calmodulin is an essential protein which binds to and modulates the activity of a wide range of targets, including proteins required for secretory pathway functions (e.g., Myo2) and membrane fusion (36). The previous large-scale studies, however, did not identify a specific molecular target for tamoxifen. Our data strongly support calmodulin as an important target for triphenylethylenes and provide a model that is consistent with the finding of the previous genome-wide chemical studies in S. cerevisiae. Furthermore, Edlind et al. showed that genetic or pharmacological interference with calmodulin function increases S. cerevisiae sensitivity to azole antifungal drugs (47). In addition, one of our laboratories has demonstrated that calcineurin inhibitors are synergistic with FLU and combine to yield to a fungicidal cocktail (21). Thus, inhibition of the calmodulin-calcineurin axis provides an explanation for the synergistic interaction with FLU.
Through the large-scale chemical genetic screen, we also found an additional EF hand-containing protein, Cml1 (CNAG_05655), to which triphenylethylenes bind. The S. cerevisiae centrin Cdc31 and centrin/caltractins from other fungal species are the closest orthologs to Cml1 (42). Similar to other centrins and caltractins, the homology between CNAG_05655 and Cdc31 is primarily due to the EF hand motifs in the C terminus. Other yeast centrins are essential, while CNAG_0565 does not appear to be essential in C. neoformans. Cml1 binds to both toremifene and the known calmodulin antagonist TFP, suggesting that it is also a target of these drugs. Cml1 also binds to calcineurin and activates it. Interestingly, the addition of toremifene increases the affinity of Cml1 for calcineurin and increases its ability to activate calcineurin. These data suggest that the presence of toremifene may increase the affinity of Cml1 for calmodulin targets, leading to inappropriate activation that is detrimental to the organism, a hypothesis that would explain the toremifene resistance of the Cml1 deletion strain. There are a number of other sequences corresponding to EF hand-containing proteins within the genome of Cryptococcus, and thus, interaction with these proteins may also contribute to the antifungal activity of triphenylethylenes.
The main goal of identifying the target of a molecule is that one can then use it to guide optimization of the desired activity. We found that two analogs of tamoxifen, idoxifene and its chain-extended derivative methylene-idoxifene, which were specifically designed by Rowlands et al. (44) and Hardcastle et al. (45) to have increased anti-calmodulin activity, also have increased anti-cryptococcal activity. Further supporting the correlation between calmodulin activity and anti-cryptococcal activity, we also found that the analogs were more effective at blocking calmodulin-calcineurin-dependent nuclear localization of Crz1. These data not only provide more evidence for calmodulin as an important target for triphenylethylenes in Cryptococcus but also provide a mechanistic basis for future studies designed to optimize the anti-cryptococcal activity of this scaffold.
In summary, we have found that the triphenylethylene-based estrogen receptor antagonists related to tamoxifen represent an attractive scaffold for development as anti-cryptococcal agents. This class of molecules has a number of features relevant to the treatment of cryptococcosis, including fungicidal activity, concentration within the CNS to levels well above the MIC, activity within macrophages, synergy with existing therapies, and oral bioavailability. We have also shown that this class of molecules interferes with calmodulin and calmodulin-like proteins as part of its mechanism of action and that molecules with improved activity for these targets are more effective against Cryptococcus. This provides us with the opportunity to embark on a target-based, systematic optimization of the anti-cryptococcal activity of this scaffold. Although calmodulin is essential in all eukaryotes, fungal and parasite calmodulins are among the most divergent of examples of this important class of proteins and have emerged as a potential target for the treatment of parasitic diseases (48). Our results indicate that calmodulin has potential as an anti-cryptococcal target and that triphenylethylenes represent a potentially important scaffold for the development of new therapies for cryptococcosis.
MATERIALS AND METHODS
Strains, plasmid construction, media, and growth conditions.
Cryptococcus neoformans var. grubii strain H99 and Cryptococcus neoformans var. neoformans strain JEC21 are from Heitman lab collections. The deletion collection strains are derivatives of the H99 Cryptococcus neoformans var. grubii isolate CM018 as described by Liu et al. (49). The genotypes of all strains used are described in Table S1 in the supplemental material. C. neoformans were cultivated from frozen stocks on yeast–peptone–2% dextrose (YPD) agar plates at 30°C. Liquid cultures (YPD) were incubated at 30°C unless otherwise noted. YPD medium and plates were prepared using standard recipes (50). Toremifene-containing plates were prepared according to the standard YPD recipe with the addition of MOPS (morpholinepropanesulfonic acid; 0.165 M). The molten agar was adjusted to pH 7 after autoclaving followed by the addition of toremifene citrate dissolved in DMSO (<1%). All small molecules and drugs were obtained from Sigma except for toremifene citrate, which was purchased from Tecoland for in vitro experiments or from the University of Rochester Medical Center Pharmacy for in vivo experiments. E. coli strains were grown in LB medium containing 1% (wt/vol) Bacto-tryptone, 0.5% (wt/vol) yeast extract, and 1% (wt/vol) NaCl adjusted to pH 7.0. LB agar plates contained 1.5% (wt/vol) Bacto-agar.
pRSET A-JEC21:CAM1 was described previously (38). H99:CAM1 and H99:CML1 were synthesized by Biomatik using the coding sequences provided by the Broad Institute C. neoformans var. grubii H99 database (CNAG_01557 and CNAG_05655, respectively). These genes were subcloned into the pRSET A vector (Invitrogen) by Biomatik. Sequences were verified by Biomatik, and expression yielded proteins of the appropriate sizes, as determined by SDS-PAGE.
In vitro antifungal susceptibility assays.
All MICs were determined using CSLI standard methods (24). The fractional inhibitory concentration index (FICI) was determined using the checkerboard method and formulae as described elsewhere (26). Time-kill assays and spot dilution assays were performed using previously published methods (11, 13). Intracellular antifungal activity was determined using freshly split murine J774 phagocyte-like cells as described previously (11). The cytotoxicity of drugs toward J774 cells was determined by measuring lactate dehydrogenase (LDH) activity in the supernatants of J774 cells treated with drugs in the absence of H99 cells (CytoToxOne; Promega) with the manufacturer’s protocol. LDH release triggered by either concentration of toremifene/tamoxifen or by DMSO (1%) treatment was not distinguishable from background and was <5% of that generated by cells lysed with the manufacturer’s lysis reagent.
Mouse model of disseminated cryptococcosis.
All experimental procedures were approved by the University of Rochester University Committee on Animal Resources (IACUC). Male AJ/Cr mice (20 to 25 g) were purchased from the Frederick National Laboratory for Cancer Research (NCI, Frederick, MD). Animals were housed in the University of Rochester Medical Center vivarium and allowed food ad libitum. Beginning on day −3, mice were treated daily with tamoxifen (200 mg/kg) suspended in peanut oil (40 mg/ml) by oral gavage. Control groups received peanut oil alone. On day 0, mice were inoculated via lateral tail vein injection with 4.5 × 104 CFU of C. neoformans H99 in 100 µl of saline. Beginning 2 to 4 h after inoculation and continuing daily, mice were sham treated or treated with fluconazole (200 µg; approximately 5 mg/kg) by intraperitoneal injection; toremefine or tamoxifen and carrier treatments were continued daily. On day 7, all mice were euthanized, after which the brains were removed and homogenized in YPD (2 ml). To determine organism burden, serial dilutions of the homogenates were plated on YPD plates containing vancomycin (10 µg/ml) and gentamicin (100 µg/ml). The number of CFU per gram of brain tissue was calculated and transformed into log10 units, and differences between groups were analyzed by analysis of variance (ANOVA); statistical significance was set at a P value of <0.05 (SigmaPlot Software).
Microscopy.
To evaluate vacuolar morphology, overnight cultures of JEC21, PK7, and PK50 were diluted 1:25 into fresh YPD and mock treated or treated with 2 µg/ml toremifene. After growth for an additional 4 h, 1 ml of each culture was washed with DPBS (Dulbecco's phosphate-buffered saline) and resuspended in 10 µM MDY-64 (Invitrogen), 10 mM HEPES (pH 7.4), 5% glucose. Cells were incubated in the dark at room temperature for 1 h and then placed on ice until imaging. To determine the effect of small molecules on Crz1 nuclear localization, overnight cultures of strain XW252 were diluted 1:50 in fresh YPD and treated with 1/4 the MIC of the test compound or carrier. The resulting cultures were incubated for an additional 4 h at either 30°C or 37°C. Samples were washed and resuspended in DPBS for imaging. Images were collected with a Nikon ES80 epifluorescence microscope equipped with a CoolSnap charge-coupled device (CCD) camera using NIS-Elements software and processed in PhotoShop. For quantitative analysis, at least 100 cells with clearly visible signals for both fluorophores were evaluated by eye for colocalization of Crz1 and Nop1. All imaging results were performed on multiple days from independent cultures.
Differential scanning fluorimetry.
Assays were performed on a CFX96 real-time PCR detection system (Bio-Rad) using Cam1 and Cml1 purified according to previously published protocol (11). All melting curves were generated in DPBS with calcium chloride (total volume of 25 µl; 5× SYPRO dye [Invitrogen], 0.25 mg/ml protein, and 4% DMSO).
In vitro calcineurin assay.
A colorimetric calcineurin activity assay kit (catalog no. 207005; Calbiochem) was used following the manufacturer’s recommendations. Reactions were performed at 30°C. Each well contained calcineurin (40 U), RII phosphopeptide substrate (0.15 mM), 10 µl distilled water (dH2O; 10 µl), in a volume of 50 µl. Cam1 and Cml1 were added to yield a final concentration of ~0.25 µM. Reactions progressed for 10 min. Control experiments indicated that the reaction was linear with time over 30 min. The reactions were quenched with the addition of malachite green (100 µl), and the mixtures were incubated at room temperature for an additional 10 min. Absorbance (620 nm) was measured with a SpectraMax M5 (Molecular Devices). The apparent IC50 for toremifene was determined by linear regression analysis using Sigma Plot software: the data were fitted to the following equation: x = (39.948 × y) − (0.00000000534 × y2).
In vitro calmodulin pulldown assays.
An overnight culture of LK214 (42) was diluted in fresh YPD (OD600, 0.1; 25 ml) and incubated for 8 h. Cells were collected and resuspended in calmodulin-binding buffer (180 µl; 25 mM Tris-HCl [pH 8.0], 150 mM NaCl, 1 mM magnesium acetate, 1 mM imidazole, 2 mM CaCl2, 1 mM β-mercaptoethanol supplemented with EDTA-free protease cocktail [Roche]). Cells were lysed using glass beads (0.5 mm; BioSpec Products, Inc.) with ten 20-s bursts of vortexing in a bead beater, each followed by 60 s on ice. Lysates was clarified by centrifugation at 3,000 × g for 10 min at 4°C. Cam1 or Cml1 was expressed and purified as previously described (11) except that the proteins were not eluted from the affinity resin. Cell lysates were pretreated with empty beads to reduce nonspecific binding. Binding reactions were performed by combining Cam1 or Cml1 resin, cell lysate and drug or DMSO and incubating at 4°C for 2 h. The resin was then washed three times with calmodulin binding buffer, resuspended in 2× Laemmli sample buffer and boiled for 5 min, after which samples were directly loaded onto a gradient polyacrylamide gel. Fractionated protein was transferred to a nitrocellulose membrane and blocked overnight with 5% nonfat dried milk. The membrane was probed with Living Colors anti-GFP antibody for 2 h, washed, and detected with horseradish peroxidase-conjugated secondary antibody. Blots were developed using an Amersham ECL Plus Western blotting detection reagent kit (GE Healthcare Life Sciences).
Chemical genetic screen of Cryptococcus neoformans deletion mutant library.
A C. neoformans deletion mutant library (1,450 strains) was arrayed using a RoTor high-density colony pinning robot (Singer Instruments). Each library plate was pinned in duplicate to three test plates each of yeast nitrogen base (YNB) and YNB plus 4 µg/ml toremifene (n = 6 for each knockout strain for each condition). Plates were incubated plates for 24 h at 30°C and then scanned them using an Epson V350 Photo scanner with autofocus. Colony size data were extracted from scanned plate images using the publically available software ScreenMill (51). We adjusted for edge effects and knockout mutant growth rate variations using the S-score method developed by Collins et al. (52) and then performed quartile normalization for each screening plate to adjust for test colony position on the plate (53). Deletion mutants with colony size scores two standard deviations from the mean were considered significant. Independent isolates of selected mutants were retested to confirm results, and all showed the phenotype observed in the large-scale screen.
SUPPLEMENTAL MATERIAL
Chemical structures of all triphenylethylenes and their MICs for C. neoformans. ChewDraw-generated structures for all triphenylethylenes are shown. MICs were determined by CLSI methods, as described in Materials and Methods. Download
Toremifene binds to CnCam1 and interferes with its ability to bind CnCna1. (A) ΔTm of CnCam1 in the presence of dimethyl sulfoxide (DMSO), trifluopromazine (TFP), fluconazole (FLU), tamoxifen (TAM), and toremifene (TOR). Values are means from at least three independent experiments performed in triplicate; error bars indicate standard deviations. (B) CnCam1 containing a histidine tag was purified from E. coli, immobilized on beads containing cobalt, and incubated with cellular lysates of a C. neoformans strain containing a fusion of Cna1 to green fluorescent protein (Cna1-GFP) in the presence of DMSO or toremifene (TOR; 20 µg/ml). The blots of input and beads alone are shown as controls. The experiment was performed three times, and a representative blot is shown. Download
C. neoformans calmodulin mutants have fragmented vacuolar morphology. Logarithmic-phase C. neoformans H99 cells from the indicated strains were stained with the vacuolar marker MDY-64 and photographed in bright-field or fluorescence channels. The arrowhead indicates a normal, moderate-sized vacuole in DMSO-treated cells, and arrows indicate fragmented, small vacuoles indicative of vacuole fusion defects. Download
Cml1 is an EF hand protein that binds Cna1 more efficiently in the presence of toremifene. (A) Alignment of Cm11 (CNAG_05655, red) with Cam1 (blue). Bars indicate EF hand motifs. (B) CnCam1 containing a histidine tag was purified from E. coli, immobilized on beads containing cobalt, and incubated with cellular lysates of a C. neoformans strain containing a fusion of Cna1 to green fluorescent protein (Cna1-GFP) in the presence of DMSO or toremifene (TOR; 20 µg/ml). The blots of input and beads alone are shown as controls. The experiment was performed twice, and a representative blot is shown. The lanes are from the same gel, but for technical reasons the image was spliced for clarity. Download
Strain table listing Cryptococcus neoformans strains used in the study, along with the source of each strain and references for each source.
C. neoformans deletion mutants with altered susceptibility to toremifene. As described in Materials and Methods, a collection of 1,450 single-gene deletion mutants was screened for alteration in growth on solid medium containing either 0, 4, or 8 µg/ml toremifene. − and + indicate mutants with colony sizes smaller and larger, respectively, than that of the wild type by >2 standard deviations. The Broad ID number is the identifier assigned to the open reading frame (ORF) on the Broad Institute database (http://www.broadinstitute.org/annotation/genome/cryptococcus_neoformans/MultiHome.html). A gene name was assigned to an ORF if it was homologous to a characterized S. cerevisiae gene with a P value of >10−6. Function is based on the known function of the S. cerevisiae homolog or as notated in the Broad Institute database.
ACKNOWLEDGMENTS
We thank Joe Chen (Duke) and Jennifer Martin for their contributions to this project. We thank Arturo Casadevall (Albert Einstein) for antibody samples.
This work was supported by National Institute of Allergy and Infectious Disease grants R01AI091422 (D.J.K.), 2T32AI007464-16 (Y.C.-R.), R01AI099206 (H.D.M.), and R01 AI50438-10 (J.H.).
Footnotes
Citation Butts A, Koselny K, Chabrier-Roselló Y, Semighini CP, Brown JCS, Wang X, Annadurai S, DiDone L, Tabroff J, Childers WE, Jr., Abou-Gharbia M, Wellington M, Cardenas ME, Madhani HD, Heitman J, Krysan DJ. 2014. Estrogen receptor antagonists are anti-cryptococcal agents that directly bind EF hand proteins and synergize with fluconazole in vivo. mBio 5(1):e00765-13. doi:10.1128/mBio.00765-13.
REFERENCES
- 1. Park BJ, Wannemuehler KA, Marston BJ, Govender N, Pappas PG, Chiller TM. 2009. Estimation of the current global burden of cryptococcal meningitis among persons living with HIV/AIDS. AIDS 23:525–530. 10.1097/QAD.0b013e328322ffac [DOI] [PubMed] [Google Scholar]
- 2. Chayakulkeeree M, Perfect JR. 2006. Cryptococcosis. Infect. Dis. Clin. North Am. 20:507–544. 10.1016/j.idc.2006.07.001 [DOI] [PubMed] [Google Scholar]
- 3. Perfect JR. 2012. The triple threat of cryptococcosis: it’s the body site, the strain and/or the host. mBio 3:e100165-12. 10.1128/mBio.00165-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Perfect JR, Dismukes WE, Dromer F, Goldman DL, Graybill JR, Hamill RJ, Harrison TS, Larsen RA, Lortholary O, Nguyen MH, Pappas PG, Powderly WG, Singh N, Sobel JD, Sorrell TC. 2010. Clinical practice guidelines for the management of cryptococcal disease: 2010 update by the Infectious Diseases Society of America. Clin. Infect. Dis. 50:291–322. 10.1086/649858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Day JN, Chau TT, Wolbers M, Mai PP, Dung NT, Mai NH, Phu NH, Nghia HD, Phong ND, Thai CQ, Thai, le H, Chuong LV, Sinh DX, Duong VA, Hoang TN, Diep PT, Campbell JI, Sieu TP, Baker SG, Chau NV, Hien TT, Lalloo DG, Farrar JJ. 2013. Combination therapy for cryptococcal meningitis. N. Engl. J. Med. 368:1291–1302. 10.1056/NEJMoa1110404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sloan DJ, Dedicoat MJ, Lalloo DG. 2009. Treatment of cryptococcal meningitis in resource-limited settings. Curr. Opin. Infect. Dis. 22:455–463. 10.1097/QCO.0b013e32832fa214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bicanic T, Muzoora C, Brouwer AE, Meintjes G, Longley N, Taseera K, Rebe K, Loyse A, Jarvis J, Bekker LG, Wood R, Limmathurotsakul D, Chierakul W, Stepniewska K, White NJ, Jaffar S, Harrison TS. 2009. Independent association of clearance of infection and clinical outcome of HIV-associated cryptococcal meningitis: analysis of combined cohort of 262 patients. Clin. Infect. Dis. 49:702–709. 10.1086/604716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Butts A, Krysan DJ. 2012. Antifungal drug discovery: something old and something new. PLoS Pathog. 8:e1002870. 10.1371/journal.ppat.1002870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wermuth CG. 2004. Selective optimization of side activities: another way for drug discovery. J. Med. Chem. 47:1303–1314. 10.1021/jm030480f [DOI] [PubMed] [Google Scholar]
- 10. Bisson WH. 2012. Drug repurposing in chemical genomics: can we learn from the past to improve the future? Curr. Top. Med. Chem. 12:1867–1868. 10.2174/156802612804547399 [DOI] [PubMed] [Google Scholar]
- 11. Butts A, DiDone L, Koselny K, Baxter BK, Chabrier-Rosello Y, Wellington M, Krysan DJ. 2013. A repurposing approach identifies off-patent drugs with fungicidal cryptococcal activity, a common structural chemotype, and pharmacological properties relevant to the treatment of cryptococcosis. Eukaryot. Cell 12:278–287. 10.1128/EC.00314-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wiseman H, Cannon M, Arnstein HR. 1989. Observation and significance of growth inhibition of Saccharomyces cerevisiae (A224A) by the anti-oestrogen drug tamoxifen. Biochem. Soc. Trans. 17:1038–1039 [DOI] [PubMed] [Google Scholar]
- 13. Dolan K, Montgomery S, Buchheit B, DiDone L, Wellington M, Krysan DJ. 2009. The antifungal activity of tamoxifen: in vitro, in vivo, and mechanistic characterization. Antimicrob. Agents Chemother. 53:3337–3346. 10.1128/AAC.01564-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Spitzer M, Griffiths E, Blakely KM, Wildenhain J, Ejim L, Rossi L, De Pascale G, Curak J, Brown E, Tyers M, Wright GD. 2011. Cross-species discovery of syncretic drug combinations that potentiate the antifungal fluconazole. Mol. Syst. Biol. 7:499. 10.1038/msb.2011.31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Morello KC, Wurz GT, DeGregorio MW. 2003. Pharmacokinetics of selective estrogen receptor modulators. Clin. Pharmacokinet. 42:361–372. 10.2165/00003088-200342040-00004 [DOI] [PubMed] [Google Scholar]
- 16. Zhang L, Yu J, Pan H, Hu P, Hao Y, Cai W, Zhu H, Yu AD, Xie X, Ma D, Yuan J. 2007. Small molecule regulators of autophagy identified by an image-based high-throughput screen. Proc. Natl. Acad. Sci. U. S. A. 104:19023–19028. 10.1073/pnas.0709695104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Voelz K, May RC. 2010. Cryptococcal interactions with the host immune system. Eukaryot. Cell 9:835–846. 10.1128/EC.00039-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Herrmann JL, Dubois N, Fourgeaud M, Basset D, Lagrange PH. 1994. Synergic activity of amphotericin-B and gamma interferon against intracellular Cryptococcus neoformans in macrophages. J. Antimicrob. Chemother. 34:1051–1058. 10.1093/jac/34.6.1051 [DOI] [PubMed] [Google Scholar]
- 19. National Committee for Clinical Laboratory Standards 2002. Reference method for broth dilution antifungal susceptibility testing of yeasts. Approved standard M27–MA2. National Committee for Clinical Laboratory Standards, Wayne, PA [Google Scholar]
- 20. Mukherjee PK, Sheehan DJ, Hitchcock CA, Ghannoum MA. 2005. Combination treatment of invasive fungal infections. Clin. Microbiol. Rev. 18:163–194. 10.1128/CMR.18.1.163-194.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Del Poeta M, Cruz MC, Cardenas ME, Perfect JR, Heitman J. 2000. Synergistic antifungal activities of bafilomycin A(1), fluconazole, and the pneumocandin MK0991/caspofungin acetate (L-743,873) with calcineurin inhibitors FK506 and L-685,818 against Cryptococcus neoformans. Antimicrob. Agents Chemother. 44:739–746. 10.1128/AAC.44.3.739-746.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lara PN, Jr, Gandara DR, Wurz GT, Lau D, Uhrich M, Turrell C, Raschko J, Edelman MJ, Synold T, Doroshow J, Muggia F, Perez EA, DeGregorio M. 1998. High-dose toremifene as a cisplatin modulator in metastatic non-small cell lung cancer: targeted plasma levels are achievable clinically. Cancer Chemother. Pharmacol. 42:504–508. 10.1007/s002800050852 [DOI] [PubMed] [Google Scholar]
- 23. Decaudin D, Etienne MC, De Cremoux P, Maciorowski Z, Vantelon JM, Voog E, Urien S, Tran-Perennou C, Renée N, Vielh P, Némati F, Pouillart P. 2004. Multicenter phase II feasibility trial of high dose tamoxifen in patients with refractory or relapsed multiple myeloma. J. Natl. Cancer Inst. 96:636–637. 10.1093/jnci/djh108 [DOI] [PubMed] [Google Scholar]
- 24. Alanio A, Desnos-Ollivier M, Dromer F. 2011. Dynamics of Cryptococcus neoformans-macrophage interactions reveal that fungal background influences outcome during cryptococcal meningoencephalitis in humans. mBio 2:e00158–e00159. 10.1128/mBio.00158-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Harrison TS, Griffin GE, Levitz SM. 2000. Conditional lethality of the diprotic weak bases chloroquine and quinacrine against Cryptococcus neoformans. J. Infect. Dis. 182:283–289. 10.1086/315649 [DOI] [PubMed] [Google Scholar]
- 26. Miguel DC, Yodoyama-Yasunaka JKU, Andreioli WK, Moratara RA, Uliana SRB. 2007. Tamoxifen is effective against Leishmania and induces a rapid alkalization of the parasiophorous vacuoles harbouring Leishmania (Leishmania) amazonensis amastigotes. J. Antimicrob. Ther. 60:526–534. 10.1093/jac/dkm219 [DOI] [PubMed] [Google Scholar]
- 27. Robinson SP, Langan-Fahey SM, Johnson DA, Jordan VC. 1991. Metabolites, pharmacodynamics, and pharmacokinetics of tamoxifen in rats and mice compared to the breast cancer patient. Drug Metab. Dispos. 19:36–43 [PubMed] [Google Scholar]
- 28. Iusuf D, Teunissen SF, Wagenaar E, Rosing H, Beijnen JH, Schinkel AH. 2011. P-glycoprotein (ABCB1) transports the primary active tamoxifen metabolites endoxifen and 4-hydroxytamoxifen and restricts their brain penetration. J. Pharmacol. Exp. Ther. 337:710–717. 10.1124/jpet.110.178301 [DOI] [PubMed] [Google Scholar]
- 29. Lien EA, Wester K, Lønning PE, Solheim E, Ueland PM. 1991. Distribution of tamoxifen and metabolites into brain tissue and brain metastases in breast cancer patients. Br. J. Cancer 63:641–645. 10.1038/bjc.1991.147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kuroiwa S, Koyama M, Watanabe N, Ekimoto H, Ohnishi Y, Saito M, Maruo K, Inaba M, Tashiro T, Yamori T, et al. 1993. Antitumor activity of a new antiestrogenic drug, toremifene (NK622) against human breast cancer xenografts in nude mice. Gan To Kagaku Ryoho 20:617–623 [PubMed] [Google Scholar]
- 31. Parsons AB, Lopez A, Givoni TE, Williams DE, Gray CA, Porter J, Chua G, Sopko R, Brost RL, Ho CH, Wang J, Ketela T, Brenner C, Brill JA, Fernandez GE, Lorenz TC, Payne GS, Ishihara S, Ohya Y, Andrews B, Hughes TR, Frey BJ, Graham TR, Andersen RJ, Boone C. 2006. Exploring the mode-of-action of bioactive compounds by chemical-genetic profiling in yeast. Cell 126:611–625. 10.1016/j.cell.2006.06.040 [DOI] [PubMed] [Google Scholar]
- 32. Lopes MC, Vale MG, Carvalho AP. 1990. Ca2(+)-dependent binding of tamoxifen to calmodulin isolated from bovine brain. Cancer Res. 50:2753–2758 [PubMed] [Google Scholar]
- 33. Senistenra G, Chau I, Vedad M. 2011. Thermal denaturation assays in chemical biology. Assay Drug Dev. Technol. 10:128–136 [DOI] [PubMed] [Google Scholar]
- 34. Kraus PR, Heitman J. 2003. Coping with stress: calmodulin and calcineurin in model and pathogenic fungi. Biochem. Biophys. Res. Commun. 311:1151–1157. 10.1016/S0006-291X(03)01528-6 [DOI] [PubMed] [Google Scholar]
- 35. Kozubowski L, Thompson JW, Cardenas ME, Moseley MA, Heitman J. 2011. Association of calcineurin with the COPI protein Sec28 and the COPII protein Sec13 revealed by quantitative proteomics. PLoS One 6:e25280. 10.1371/journal.pone.0025280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Cyert MS. 2001. Genetic analysis of calmodulin and its targets in S. cerevisiae. Annu. Rev. Genet. 35:647–672. 10.1146/annurev.genet.35.102401.091302 [DOI] [PubMed] [Google Scholar]
- 37. Peters C, Mayer A. 1998. Ca2+/calmodulin signals completion of docking and triggers a late step of vacuole fusion. Nature 396:575–580. 10.1038/25133 [DOI] [PubMed] [Google Scholar]
- 38. Kraus PR, Nichols CB, Heitman J. 2005. Calcium- and calcineurin-independent roles for calmodulin in Cryptococcus neoformans morphogenesis and high-temperature growth. Eukaryot. Cell 4:1079–1087. 10.1128/EC.4.6.1079-1087.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lev S, Desmarini D, Chayakulkeeree M, Sorrell TC, Djordjevic JT. 2012. The Crz1/Sp1 transcription factor of Cryptococcus neoformans is activated by calcineurin and regulates cell wall integrity. PLoS One 7:e51403. 10.1371/journal.pone.0051403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Stathopoulos AM, Cyert MS. 1997. Calcineurin acts through the CRZ1/TCN1-encoded transcription factor to regulate gene expression in yeast. Genes Dev. 11:3432–3444. 10.1101/gad.11.24.3432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Schenone M, Dančík V, Wagner BK, Clemons PA. 2013. Target identification and mechanism of action in chemical biology and drug discovery. Nat. Chem. Biol. 9:232–240. 10.1038/nchembio.1199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Greier BM, Wiech H, Schiebel E. 1996. Binding of centrins and yeast calmodulin to synthetic peptides corresponding to binding sites in the spindle pole body components Kar1p and Spc110p. J. Biol. Chem. 45:28366–28374 [DOI] [PubMed] [Google Scholar]
- 43. Gulino A, Barrera G, Vacca A, Farina A, Ferretti C, Screpanti I, Dianzani MU, Frati L. 1986. Calmodulin antagonism and growth-inhibiting activity of triphenylethylene antiestrogens in MCF-7 human breast cancer cells. Cancer Res. 46:6274–6278 [PubMed] [Google Scholar]
- 44. Rowlands MG, Budworth J, Jarman M, Hardcastle IR, McGague R, Gescher A. 1995. Comparison between protein kinase C inhibition and antagonism of calmodulin by tamoxifen analogues. Biochem. Pharmacol. 50:723–726. 10.1016/0006-2952(95)00186-4 [DOI] [PubMed] [Google Scholar]
- 45. Hardcastle IR, Rowlands MG, Grimshaw RM, Houghton J, Jarman M. 1996. Homologs of idoxifene: variations of estrogen receptor binding and calmodulin antagonism with chain length. J. Med. Chem. 39:999–1004. 10.1021/jm9505472 [DOI] [PubMed] [Google Scholar]
- 46. Gupta SS, Ton VK, Beaudry V, Rulli S, Cunningham K, Rao R. 2003. Antifungal activity of amiodarone is mediated by disruption of calcium homeostasis. J. Biol. Chem. 278:28831–28839. 10.1074/jbc.M303300200 [DOI] [PubMed] [Google Scholar]
- 47. Edlind T, Smith L, Henry K, Katiyar S, Nickels J. 2002. Antifungal activity in Saccharomyces cerevisiae is modulated by calcium signaling. Mol. Microbiol. 46:257–268. 10.1046/j.1365-2958.2002.03165.x [DOI] [PubMed] [Google Scholar]
- 48. Benaim B, Garcia CR. 2011. Targeting calcium homeostasis as a therapy of Chagas’ disease and leishmanisis—a review. Trop. Biomed. 28:471–481 [PubMed] [Google Scholar]
- 49. Liu OW, Chun CD, Chow ED, Chen C, Madhani HD, Noble SM. 2008. Systematic genetic analysis of virulence in the human fungal pathogen Cryptococcus neoformans. Cell 135:174–188. 10.1016/j.cell.2008.07.046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Burke DJ, Dawson D, Stearns T. 2000. Methods in yeast genetics. CSHL Press, Woodbury, NY [Google Scholar]
- 51. Dittmar JC, Reid RJ, Rothstein R. 2010. ScreenMill: A freely available software suite for growth measurement, analysis and visualization of high-throughput screen data. BMC Bioinformatics 11:353–363. 10.1186/1471-2105-11-353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Collins SR, Schuldiner M, Krogan NJ, Weissman JS. 2006. A strategy for extracting and analyzing large-scale quantitative epistatic interaction data. Genome Biol. 7:R63. 10.1186/gb-2006-7-7-r63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Bolstad BM, Irizarry RA, Astrand M, Speed TP. 2003. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics 19:185–193. 10.1093/bioinformatics/19.2.185 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Chemical structures of all triphenylethylenes and their MICs for C. neoformans. ChewDraw-generated structures for all triphenylethylenes are shown. MICs were determined by CLSI methods, as described in Materials and Methods. Download
Toremifene binds to CnCam1 and interferes with its ability to bind CnCna1. (A) ΔTm of CnCam1 in the presence of dimethyl sulfoxide (DMSO), trifluopromazine (TFP), fluconazole (FLU), tamoxifen (TAM), and toremifene (TOR). Values are means from at least three independent experiments performed in triplicate; error bars indicate standard deviations. (B) CnCam1 containing a histidine tag was purified from E. coli, immobilized on beads containing cobalt, and incubated with cellular lysates of a C. neoformans strain containing a fusion of Cna1 to green fluorescent protein (Cna1-GFP) in the presence of DMSO or toremifene (TOR; 20 µg/ml). The blots of input and beads alone are shown as controls. The experiment was performed three times, and a representative blot is shown. Download
C. neoformans calmodulin mutants have fragmented vacuolar morphology. Logarithmic-phase C. neoformans H99 cells from the indicated strains were stained with the vacuolar marker MDY-64 and photographed in bright-field or fluorescence channels. The arrowhead indicates a normal, moderate-sized vacuole in DMSO-treated cells, and arrows indicate fragmented, small vacuoles indicative of vacuole fusion defects. Download
Cml1 is an EF hand protein that binds Cna1 more efficiently in the presence of toremifene. (A) Alignment of Cm11 (CNAG_05655, red) with Cam1 (blue). Bars indicate EF hand motifs. (B) CnCam1 containing a histidine tag was purified from E. coli, immobilized on beads containing cobalt, and incubated with cellular lysates of a C. neoformans strain containing a fusion of Cna1 to green fluorescent protein (Cna1-GFP) in the presence of DMSO or toremifene (TOR; 20 µg/ml). The blots of input and beads alone are shown as controls. The experiment was performed twice, and a representative blot is shown. The lanes are from the same gel, but for technical reasons the image was spliced for clarity. Download
Strain table listing Cryptococcus neoformans strains used in the study, along with the source of each strain and references for each source.
C. neoformans deletion mutants with altered susceptibility to toremifene. As described in Materials and Methods, a collection of 1,450 single-gene deletion mutants was screened for alteration in growth on solid medium containing either 0, 4, or 8 µg/ml toremifene. − and + indicate mutants with colony sizes smaller and larger, respectively, than that of the wild type by >2 standard deviations. The Broad ID number is the identifier assigned to the open reading frame (ORF) on the Broad Institute database (http://www.broadinstitute.org/annotation/genome/cryptococcus_neoformans/MultiHome.html). A gene name was assigned to an ORF if it was homologous to a characterized S. cerevisiae gene with a P value of >10−6. Function is based on the known function of the S. cerevisiae homolog or as notated in the Broad Institute database.