

Abstract
In the current study zidovudine loaded PLGA nanoparticles were prepared, coated and further investigated for its effectiveness in brain targeting. IR and DSC studies were performed to determine the interaction between excipients used and to find out the nature of drug in the formulation. Formulations were prepared by adopting 23 factorial designs to evaluate the effects of process and formulation variables. The prepared formulations were subjected for in vitro and in vivo evaluations. In vitro evaluations showed particle size below 100 nm, entrapment efficiency of formulations ranges of 28–57%, process yield of 60–76% was achieved and drug release for the formulations were in the range of 50–85%. The drug release from the formulations was found to follow Higuchi release pattern, n–value obtained after Korsemeyer plot was in the range of 0.56–0.78. In vivo evaluations were performed in mice after intraperitoneal administration of zidovudine drug solution, uncoated and coated formulation. Formulation when coated with Tween 80 achieved a higher concentration in the brain than that of the drug in solution and of the uncoated formulation. Stability studies indicated that there was no degradation of the drug in the formulation after 90 days of preparation when stored in refrigerated condition.
Keywords: Zidovudine, Brain targeting, Tween 80, Coated nanoparticles, Factorial design
1. Introduction
HIV reaches and infects the central nervous system soon after entering the host followed by the establishment of a viral reservoir which produces complex neurological problems, latent infections and resistance (Wong et al., 2010). Main reason for Human Immuno Deficiency Virus (HIV) to infect and replicate in the brain is these strains have the tendency to reside and replicate at sites where there is low concentration of circulating antibodies as these virus strains are susceptible for neutralization by host antibodies (Kolchinsky et al., 2001). Usually HIV and other infectious organisms gain access to the brain by adopting two pathways Transcellular and paracellular pathway and/or Trojan horse mechanism Micro organisms use any one of these approaches or a combination of these approaches to access CNS which mainly depends on the nature of the organism (Huang and Jong, 2001). HIV invades the central nervous system (CNS) after peripheral infection, Highly Active Anti Retroviral Therapy (HAART) is unlikely to prevent the entry of HIV into the CNS. HIV-infected monocytes in the circulatory system can permeate the blood–brain barrier and thus cause infection in the CNS. It has been observed from previous studies that the pathological features of HIV-1 infection in the brain lead to widespread activation of macrophages, microglial nodules, activated resident microglia, astrocytes, myelin pallor, multinucleated giant cells and blood-derived macrophages (Fischer-Smith and Rappaport, 2005).
The drug used in the study zidovudine was classified under the antiretroviral category and these classes of drugs are active against a anti retro virus such as HIV. These drugs prolong and improve the quality of life by suppressing many complications associated with AIDS. Zidovudine increases CD4 counts and reduces HIV RNA to undetectable levels and this results in good immune status, reduces opportunistic infections, also reduces mortality rate, suppresses the occurrence of new Kaposis lesions, and reduces neurological manifestations of AIDS. It is also found to slowdown the progress of the disease (Tripathi, 2008).
PLGA possess ES good CNS biocompatibility, which was proved in many studies (Emerich et al., 1999; Menei et al., 1993). Devices and formulations made of PLGA nanoparticles are available in Europe, Japan, and the US (Ueda and Tabata, 2003). The importance of incorporating Quality by Design is recognized and implemented now days, by USFDA and other regulatory agencies worldwide, ICH has issued guidance that describes the implementation of Quality by Design in various stages of product development. ICH Q8 has given a full description about the use of some tools for the design of the experiment and its implementation for various applications in pharmaceutical manufacturing (ICH, 2008). The objective of the study is to develop tween 80 coated zidovudine loaded PLGA nanoparticles to achieve good concentration of the drug in the brain. To obtain this objective formulations were prepared using nanoprecipitation technique. 23 factorial design was adopted in preparing formulations. The coating of tween 80 on prepared formulations was achieved by the adsorption method. The formulations were evaluated in vitro to evaluate all physico chemical and release parameters. Brain availability and tissue distribution studies for the prepared formulations were studied by using mice.
2. Materials and methods
2.1. Materials
Zidovudine was gifted by Alembic Pharma Gujarat, Poly (Lactic-co-glycolic acid) PLGA
50:50 Ratio Molecular weight 7000–17,000, was purchased from Sigma Aldrich chemicals Pvt. Ltd., USA, Tween 80 was purchased from Loba Chemie Pvt. Ltd., Mumbai, Poloxamer 407 was purchased from Sigma Aldrich chemicals Pvt. Ltd., USA, Dialysis membrane was purchased from HiMedia Laboratories Pvt. Ltd. Mumbai, Methanol HPLC Grade was purchased from Thermo Fisher Scientific Ind. Pvt. Ltd. Mumbai, Acetonitrile HPLC Grade was purchased from Ranbaxy Fine Chemicals Ltd., Mumbai, Water HPLC Grade was purchased from Ranbaxy Fine Chemicals Ltd., Mumbai.
2.2. Methods
2.2.1. Experimental design
Formulation design adopted in the study was the 23 factorial design. The dependant and independent variables of the design are shown below.
| Independent variable | Levels | Dependent variable | |
| Low | High | ||
| Polymer | 100 | 200 | Particle size |
| Poloxamer | 50 | 100 | Entrapment efficiency |
| pH of aqueous phase | 1.2 | Neutral | In vitro release |
The formulation design was generated by using Design Expert Software (Version 8.0).
2.2.2. Differential scanning calorimetry studies for zidovudine formulations
DSC studies were performed to assess the presence and nature of the encapsulated drug in formulations and also to study the interaction between excipients used, DSC studies were carried out in NETZSCH DSC 204. The samples were loaded in the chamber and gradually heated till 300 °C with a heating rate of 10 °C/min.
2.2.3. Preparation of zidovudine loaded PLGA nanoparticles by nanoprecipitation technique
Organic phase was prepared by dissolving weighed quantities of drug and polymer in 4 ml acetone. The aqueous phase was prepared by dissolving a weighed quantity of poloxamer in water. Then the organic phase was poured into the aqueous phase and stirred constantly using a magnetic stirrer at 100 RPM. The stirring is continued for 4 h till complete evaporation of the organic solvent (Fessi et al., 1989). The formulation composition is summarized in Table 1.
Table 1.
Formulation table showing the composition of zidovudine, PLGA, and poloxamer 407.
| Formulation code | Zidovudine (mg) | PLGA (mg) | Poloxamer 407 (mg) | Aqueous medium |
|---|---|---|---|---|
| F1 | 100 | 100 | 50 | PH 1.2 |
| F2 | 100 | 200 | 50 | PH 1.2 |
| F3 | 100 | 100 | 100 | PH 1.2 |
| F4 | 100 | 200 | 100 | PH 1.2 |
| F5 | 100 | 100 | 50 | Distilled water |
| F6 | 100 | 200 | 50 | Distilled water |
| F7 | 100 | 100 | 100 | Distilled water |
| F8 | 100 | 200 | 100 | Distilled water |
The prepared nanoparticles were suspended in PBS 7.4 and tween 80 was added to the suspended nanoparticles to give a concentration of 1% and incubated for 1 h (Wilson et al., 2008).
2.2.4. Evaluation of nanoparticles
2.2.4.1. Particle size determination
Mean particle size of the prepared nanoparticles was determined by phase contrast microscope (PCM). The prepared samples were placed on a slide and images were taken by using a Leica Inverted Phase Contrast Microscope (Leica S 40, 230 ± 10v, 5 W). Average particle sizes of 50 particles were measured and mean particle size was calculated.
2.2.4.2. Surface morphology
Surface characteristics of the formulation F9 were analyzed by performing HR TEM analysis. Samples for the analysis were prepared on carbon coated copper grid, and a drop of the prepared formulation is placed on the grid and dried. The dried samples were placed in HR TEM analyzer JEOL TEM 2100 and images were taken.
2.2.4.3. Surface charge
Surface charge of the coated formulation F9 was determined by using Malvern Zeta sizer ZS90. The formulations were placed on the instrument and the surface charge was determined.
2.2.4.4. Entrapment efficiency
The nanoformulations were centrifuged and the supernatant containing free drug was collected which was further analyzed by UV at 266 nm. This gives the amount of drug that is unentrapped in the nanoparticles. Amount of drug found in the supernatant was subtracted from the total amount of drug added to the formulation which gives the amount of drug entrapped in the nanoparticles (Govender et al., 1999). The formulations were evaluated for entrapment efficiency by the following formula
2.2.4.5. Process yield
The yield of nanoparticles were calculated using the following equation
2.2.4.6. In vitro release study
The release studies were carried out by using the dialysis membrane technique where the dialysis bag with a molecular weight cut off of 12,000–14,000 was used. The membrane was pretreated with warm phosphate buffer saline for 10 min. Then 50 mg of the prepared formulation and 1 ml of PBS were added into the dialysis bag and then immersed into 50 ml of phosphate buffered saline medium (pH 7.4). Stirring was performed at 100 RPM by using a magnetic stirrer at 37 °C. Samples (2 ml) were withdrawn at various time points and equal volume of fresh buffer is replaced and the absorbance was measured by a UV spectrophotometer at 266 nm3.
2.2.4.7. In vitro release kinetics
The data obtained from release studies were fitted into kinetic models like zero order, first order, Higuchi, Hixson Crowell, Korsemeyer-Peppas, to study the mechanism of drug release from the prepared nanoparticles.
2.2.4.8. In vivo studies
In vivo studies were performed to determine the efficiency of prepared nanoparticles for effective brain targeting. The study was performed after obtaining the approval from PSG Institutional Animal Ethics Committee Registration No.: 158/99/CPCSEA. In vivo studies were performed on male mice with weights of 30–35 g. The animals were divided into four groups with three animals in each group. Group 1 received the drug solution, group 2 received formulation without any surface modification, group 3 received formulation with surface modification using tween 80, and group 4 was kept as control. The doses were calculated based on body surface area.
After 60 min the animals were anesthetized and sacrificed by using chloroform. The organs like brain, liver, kidney, spleen were removed and washed with phosphate buffer saline pH 7.4 and homogenized using tissue homogenizer for 15 min by using 5 ml of phosphate buffered saline (pH 7.4). The homogenates were stored in a deep freezer at −80 °C until use (Schaffazick et al., 2008).
2.2.4.9. Tissue sample preparation
50 μl of tissue homogenate was transferred to a clean test tube and 5 μl of the internal standard was added (10 μg/ml of lamivudine) into tissue homogenate and vortexed for 5 min. To each sample, 0.8 ml of ethyl acetate was added and vortex-mixed for 5 min and centrifuged at 10,000 RPM for 3 min at 4 °C. 0.6 ml of organic layer was separated and placed into a clean glass test tube and evaporated to dryness under vacuum at 45 °C in a concentrator. The residue was reconstituted in 100 μl of mobile phase and vortex-mixed for 3 min. The reconstituted samples were injected for analysis (Tan and Douglas Boudinot, 2000).
2.2.4.10. HPLC conditions
HPLC was performed using waters 515 system equipped with a binary solvent delivery pump, Rheodyne manual injector and UV detector (Waters 2489). The chromatographic separation was performed on a C18 column: Luna C18, 100A (150 × 3.0 mm, 5 μ) (Phenomenex) and the absorbance was monitored at 266 nm. The system was run in the isocratic mode with a mobile phase consisting of methanol: potassium dihydrogen phosphate buffer (pH 3.0, 5 mM) in the ratio of 40:60 (v/v) and was delivered at a flow rate of 0.3 mL/min. The mobile phase was filtered through a 0.22 μm Nylon filter (Pall Scientific, Ville St. Laurent, QC, Canada) and degassed in an ultrasonic bath for 30 min prior to use. The injection volume was 20 μl while the column was maintained at room temperature during the run.
3. Results and discussion
3.1. DSC studies
DSC studies were performed to assess the nature of drug present in the formulations and also to study the interaction between excipients used, DSC studies were performed to find out the interaction between excipients used in the formulation and also to determine the form in which the drug is entrapped in the nanoparticles. Fig. 1(A) and (B) depicts the DSC Thermogram for zidovudine and zidovudine loaded nanoparticles respectively.
Figure 1.
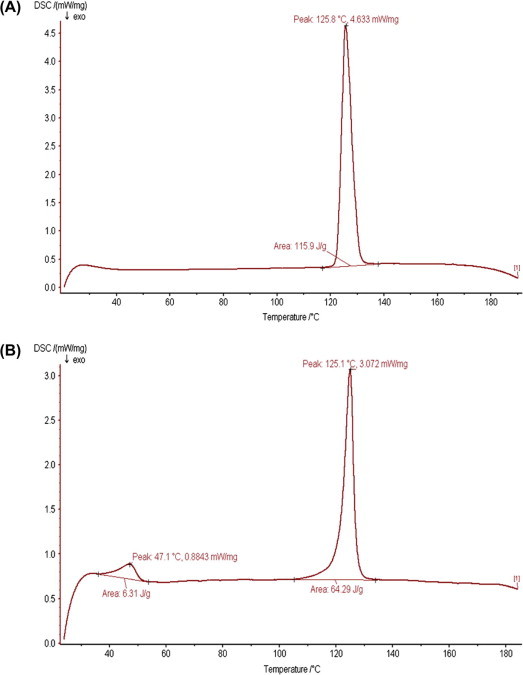
DSC Thermograms of (A) zidovudine and (B) formulation F9.
The drug zidovudine displayed a characteristic endothermic peak at 125.8 °C which is the melting point/transition temperature for the drug clearly depicted in Fig. 1(A). DSC thermogram of the drug loaded nanoparticles depicted in Fig. 1(B) showed two endothermic peaks at 47.1 °C and 125.1 °C. The peak at 47.1 °C corresponds to the PLGA glass transition temperature (Tg) and the peak at 125 °C corresponds to the endothermic peak of zidovudine respectively. The slight change in drug endotherm was due to morphological changes attained when formulated (Sunil et al., 2005).
3.2. Particle size
Particle size which is an important parameter for brain targeting has to be considered because particles lesser than 200 nm are required for good targeting to the brain. Studies have showed particles lesser than 100 nm are more effective in delivering drugs to the brain (Wilson et al., 2008).
The average mean particle size of the prepared nanoparticles is summarized in Table 2, the mean particle size was found to be in the range of 56 nm to 93 nm. The particle size in this range is essential for the particle to reach the brain by evading phagocytosis.
Table 2.
In vitro evaluation results of prepared formulations F1–F9.
| F code | Mean particle size (nm) | Entrapment efficiency (%) | Process yield (%) | % drug release |
|---|---|---|---|---|
| F1 | 81 | 28.00 | 75.80 | 85.28 ± 0.80 |
| F2 | 93 | 41.50 | 65.71 | 79.29 ± 0.35 |
| F3 | 68 | 30.25 | 65.33 | 85.67 ± 0.67 |
| F4 | 74 | 46.00 | 69.75 | 76.49 ± 0.28 |
| F5 | 71 | 50.50 | 73.20 | 82.84 ± 1.20 |
| F6 | 79 | 59.50 | 76.00 | 74.17 ± 0.67 |
| F7 | 56 | 48.25 | 64.00 | 85.30 ± 1.03 |
| F8 | 69 | 52.75 | 60.75 | 70.67 ± 1.73 |
| F9 | 83 | 57.27 | 74.84 | 82.38 ± 1.34 |
Formulation F9 was prepared by coating formulation F6 with tween 80 for the purpose of targeting, mean particle size of F9 was found to be 83 nm. Smaller particles with a size range below 100 nm with the hydrophilic surface were found to have longer circulation in the blood, thus prolonging the duration of therapeutic effect and improving targeting to the desired sites (Banerjee et al., 2002).
The concentration of poloxamer 407 was found to influence the particle size of the prepared nanoparticles which was in accordance with the results obtained which described that nanoparticles with a smaller size range and narrow size distribution are obtained only when there is sufficient surfactant concentration (Vauthier and Bouchemal, 2009).
In formulations with a low level of poloxamer F1, F2, F5 and F6, the particle size was found to be higher than preparations containing high concentrations F3, F4, F7 and F8 of the poloxamer in both aqueous and acidic pH. This shows that the particle size of the prepared nanoparticles was influenced by the concentration of the poloxamer.
Formulation F7 comprising 100 mg of drug, 100 mg of polymer and 100 mg of poloxamer was found to possess a low mean particle size of 56 nm and formulation F2 with 100 mg drug, 200 mg polymer and 50 mg poloxamer has the larger size of 93 nm. When PBS pH 7.4 was used as aqueous phase it resulted in the aggregation of particles which could not be measured.
The surface plot shown in Fig. 2 indicates the effect of poloxamer and PLGA on the particle size of nanoparticles. This proves that the polymer and stabilizer combination selected have good suitability to use in the nanoprecipitation technique to produce nanoparticles with a particle size below 100.
Figure 2.

Surface plot illustrating the influence of PLGA and Poloxamer on particle size of the prepared formulations.
3.3. Surface morphology
Surface morphology of the prepared formulations was evaluated by obtaining TEM images. TEM images were taken for formulation F9 and the images obtained proved the spherical shape of the nanoparticles as shown in Fig. 3.
Figure 3.
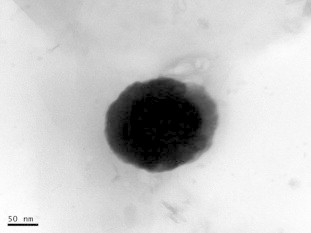
TEM image of tween 80 coated formulation F9.
3.4. Zeta potential
Samples were diluted appropriately using Millipore water before the measurement of zeta potential. Zeta potential value of formulation F9 was found to be – 20 mV. Zeta potential value is an important parameter which influences formulation stability. Stability is very important to prevent settling of nanoparticles.
3.5. Entrapment efficiency
Entrapment efficiency of formulations was in the range of 28–59.5%. Entrapment efficiency was found to be influenced by the amount of polymer and the pH of the aqueous phase. Table 2 depicts the results obtained for the prepared formulations. Formulation F6 containing 100 mg drug, 200 mg polymers and 50 mg poloxamer was found to possess high entrapment efficiency.
The formulations containing 200 mg of polymer in composition show higher entrapment than the formulations containing 100 mg of polymer. Formulations F1–F4 were found to possess low entrapment efficiency and this may be due to acidic pH of the aqueous medium (Govender et al., 1999). Formulations prepared with distilled water as external aqueous phase show high entrapment than formulations prepared with acidic medium as external aqueous phase. This shows the influence of pH on the entrapment efficiency. Changes in aqueous phase pH can improve the entrapment efficiency of nanoparticles (Govender et al., 1999; Peltonen et al., 2004). The response surface plot illustrating the effect of polymer PLGA and aqueous medium on the entrapment efficiency of nanoparticles is shown in Fig. 4.
Figure 4.
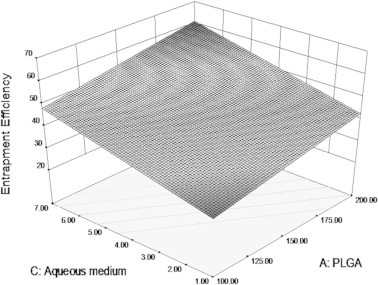
Surface plot depicting the influence of PLGA and aqueous medium on entrapment efficiency of PLGA nanoparticles.
Change in pH of the medium results in diffusion of the drug from the inner phase to outer phase which results in low entrapment. This is due to the ionization of the drug at a particular pH which results in high solubility and this enhances the drug to stay in aqueous phase while the molecular form of drug stays in the hydrophobic polymer phase (Govender et al., 1999).
3.6. Process yield
The process yield values for the prepared formulations are summarized in Table 2. Process yield of the prepared formulations ranged from 60.75% to 76%. Results are summarized in Table 2. The yield was very low for F8 and F6 shows a good process yield of 76%. Good yield shows good efficiency of the preparation method. This shows the suitability of the method to prepare formulations as the process yield was found to be above 50% (Govender et al., 1999).
3.7. In vitro drug release
The cumulative percentage release of formulations F1–F8 ranges from 70.67 ± 1.73 to 85.67 ± 0.67, the values are presented in Table 2.
All formulations showed burst release initially clearly seen in Fig. 5, followed by sustained release, burst release was due to drug molecules adsorbed in the surface of nanoparticles. These drug particles at the surface instantaneously dissolve when it comes into contact with the medium (Agnihotri et al., 2004).
Figure 5.
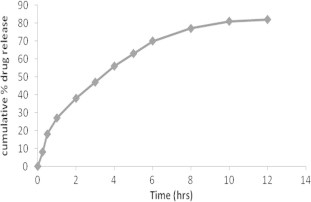
Cumulative mean % release of zidovudine from PLGA nanoparticles containing poloxamer 407 in PBS pH 7.4.
Surface plot in Fig. 6 clearly shows the effect of formulation variables PLGA and poloxamer on drug release. Amount of PLGA affected the drug release, as the concentration of the polymer increases, release decreases. Retardation of release was more pronounced in formulations with 200 mg of polymer than formulations containing 100 mg of polymer. This was clearly seen in formulations F2, F4, F6, and F8. The surface plot clearly depicts the decrease in release with increase in the amount of polymer. Poloxamer when used in high levels delays release than its presence in low levels.
Figure 6.
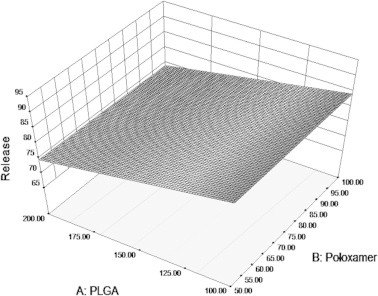
Surface plot depicting the influence of PLGA and poloxamer on drug release from PLGA nanoparticles.
Release of drug from nano carriers is influenced by parameters that are drug related, polymer related and environmentally related. The drug related parameters that influence release are its molecular weight, concentration, interaction with carrier, diffusion, ion exchange, desorption from surface, position, and physicochemical properties.
The polymer related parameters are size and density of the particle, amount and type of matrix material, denaturation of polymerization, nature and extent of cross linking, presence of excipients, surface erosion, total disintegration of particles and surface erosion.
Environmentally related parameters are polarity, ionic strength, presence of enzymes, light, magnetism, microwave and temperature (Tomlinson, 1983).
The data obtained from in vitro release studies were fitted into release kinetic models like zero order, first order, Higuchi, Hixson Crowell and Korsemeyer Peppas. The values are presented in Table 3. The formulations were found to have higher r2 values for the first order model which shows that the release of drug from the nanoparticles was concentration dependent. Higher r2 values were seen for the Higuchi model indicating diffusion controlled release (Dhanalekshmi et al., 2010). The n-value obtained after the Korsemeyer plot was in the range of 0.56–0.76 indicating that release followed anomalous non Fickian transport.
Table 3.
Determination coefficient (r2) and diffusion exponent (n) after fitting the release data.
| F code | Zero order | First order | Hixson Crowell | Higuchi | Korsmeyer Peppas | |
|---|---|---|---|---|---|---|
| r2 | r2 | r2 | r2 | r2 | n | |
| F1 | 0.8652 | 0.9787 | 0.9566 | 0.9813 | 0.8666 | 0.56 |
| F2 | 0.8844 | 0.9743 | 0.9483 | 0.9783 | 0.8701 | 0.58 |
| F3 | 0.8913 | 0.9416 | 0.9115 | 0.9799 | 0.8975 | 0.60 |
| F4 | 0.8409 | 0.9869 | 0.965 | 0.9686 | 0.9057 | 0.65 |
| F5 | 0.8852 | 0.9546 | 0.9398 | 0.9618 | 0.9189 | 0.68 |
| F6 | 0.9104 | 0.9528 | 0.9271 | 0.9872 | 0.9626 | 0.76 |
| F7 | 0.9123 | 0.9782 | 0.9518 | 0.9813 | 0.8958 | 0.66 |
| F8 | 0.9056 | 0.9916 | 0.9747 | 0.9877 | 0.9391 | 0.67 |
| F9 | 0.8589 | 0.8977 | 0.8747 | 0.9803 | 0.9304 | 0.53 |
3.8. In vivo studies
Nanoparticles with sizes above 100 nm will tend to restrict their biodistribution, they are cleared from systemic circulation by Kupffer cells or other phagocytic cell populations within the mononuclear phagocytic system. Table 4 shows concentration of zidovudine in tissues.
Table 4.
Zidovudine concentration (ng/ml) in different organs after the administration of formulation F9, uncoated formulation F6 and zidovudine drug in solution.
| S.No | Organ | F9 | F6 | Zidovudine |
|---|---|---|---|---|
| 1. | Brain | 2464.76 ± 43.22 | 913.63 ± 11.45 | 352.33 ± 25.96 |
| 2. | Plasma | 11943.33 ± 34.26 | 12635.67 ± 14 | 16286.33 ± 38.52 |
| 3. | Liver | 7328.27 ± 30.53 | 8044.97 ± 76.43 | 5423.36 ± 67.25 |
| 4. | Spleen | 7904.61 ± 57.50 | 9508.31 ± 34.35 | 8584.96 ± 20.01 |
| 5. | Kidney | 5035 ± 57.64 | 5680.81 ± 44.86 | 7679.3 ± 51.7 |
Higher concentration of zidovudine 2464.76 ± 43.22 was observed in the brain when nanoparticles coated with tween 80 (formulation F9) were administered whereas the concentration of uncoated nanoparticles formulation F6 in the brain was 913.63 ± 11.45. The concentration of zidovudine achieved in the brain when administered as plain drug solution was 352.33 ± 25.96.
These results show that more drug reaches the brain when the formulation coated with tween 80 is administered which was in accordance with results reported for doxorubicin and loperamide by means of polysorbate 80 coated nanoparticles and for dalargin (Gelperina et al., 2010; Kreuter et al., 1997). The mechanism behind high transport of drugs to the brain after coating with polysorbate 80 according to that it was due to the adsorption of apolipoprotein B and E from blood over the polysorbate 80 coated nanoparticles which further enhances endocytosis mediated transport into the brain (Kreuter et al., 2002).
The reason for the animal sacrifice and excision of organs 1 h after dosing was based on the result that maximum concentration of zidovudine was at 1 h after the dose administration (Lobenberg et al., 1998). When compared to plain drug solution high concentration of the drug is seen in the brain with formulation F6 containing uncoated nanoparticles and this may be due to effect of particle size. Fig. 7 depicts the distribution of zidovudine to various organs.
Figure 7.
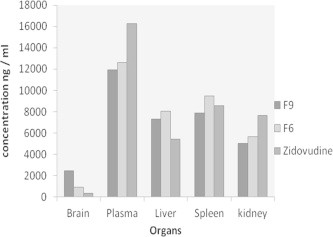
Concentration of zidovudine (ng/ml) in various organs on administration of the drug solution, F6 and F9.
Plasma concentration of coated formulation F9 was 11,943.33 ± 34.26 1 h after administration to animals and for formulation F6 it was 12,635.67 ± 14, for the zidovudine drug in solution it was 16,286.33 ± 38.52. Zidovudine plasma concentration was less after the administration of formulations F9 and F6 than the drug in solution and this may be due to the slow release of the drug from the nanoparticles.
Concentration achieved in the liver after the administration of Formulation F9, F6 and drug in solution was, 7328.27 ± 30.53, 8044.97 ± 76.43, and 5423.36 ± 67.25 respectively. Higher concentration of drug is seen in the liver when formulations F9 and F6 were administered. This was due to the uptake of nanoparticles by the reticuloendothelial system especially by the kupffer cells (Plard and Bazile, 1999).
The concentrations of drug in the spleen were, 7904.61 ± 57.50, 9508.31 ± 34.35, and 8584.96 ± 20.01 for coated formulation F9, uncoated formulation F6, and plain drug in solution. The distribution of zidovudine to the spleen reduced after coating with polysorbate 80 solution.
The drug concentration achieved in the kidney after the administration of coated formulation F9 was 5035 ± 57.64, for uncoated formulation it was 5680.81 ± 44.86 and for drug in solution it was higher than formulations 7679.3 ± 51.7. The diagramatic representation of the concentration of zidovudine achieved in different organs is depicted in Fig. 7.
3.9. Stability determination of formulation F9 after 90 days
Stability of the prepared formulations was evaluated at room temparature and in refrigerated condition for a period of 90 days. The formulations were evaluated for pH, particle size, and appearance. The results proved that the formulations were stable.
4. Conclusion
Zidovudine loaded PLGA nanoparticles were prepared successfully by nanoprecipitation technique and coated with tween 80 to achieve good concentration in the brain. The prepared formulations were evaluated in vitro and in vivo. The results revealed that formulations were found to be spherical in shape with good size distribution. It also possesses good entrapment efficiency and prolonged release of the drug. The in vivo results indicated that the formulations possess required qualities to achieve good concentration in the brain. In vivo studies in mice revealed that formulation coated with tween 80 achieved higher concentrations in the brain than formulation with uncoated nanoparticles and drug in solution. Further studies are required to improve the qualities of the formulation and make it an efficient drug delivery system.
Footnotes
Peer review under responsibility of King Saud University.

Reference
- Agnihotri S.A., Mallikarjuna N.N., Aminabhavi T.M. Recent advances on chitosan-based micro- and nanoparticles in drug delivery. J. Controlled Release. 2004;100:5–28. doi: 10.1016/j.jconrel.2004.08.010. [DOI] [PubMed] [Google Scholar]
- Banerjee T., Mitra S., Singh K., Sharma R.K., Maitra A. Preparation and biodistribution of ultrafine chitosan nanoparticles. Int. J. Pharm. 2002;243:93–105. doi: 10.1016/s0378-5173(02)00267-3. [DOI] [PubMed] [Google Scholar]
- Dhanalekshmi U.M., Poovi G., Kishore Narra, Neelakanta Reddy P. In vitro characterization and in vivo toxicity study of repaglinide loadedpoly (methyl methacrylate) nanoparticles. Int. J. Pharm. 2010;396:194–203. doi: 10.1016/j.ijpharm.2010.06.023. [DOI] [PubMed] [Google Scholar]
- Emerich D.F., Tracy M.A., Ward K.L., Figueiredo M., Qian R., Henschel C. Biocompatibility of poly (DL-lactide-co-glycolide) microspheres implanted into the brain. Cell Transplant. 1999;8:47–58. doi: 10.1177/096368979900800114. [DOI] [PubMed] [Google Scholar]
- Fessi H., Puisieux F., Devissaguet J.P., Ammoury N., Benita S. Nanocapsule formation by interfacial polymer deposition following solvent displacement. Int. J. Pharm. 1989;55:1–4. [Google Scholar]
- Fischer-Smith T., Rappaport J. Evolving paradigms in the pathogenesis of HIV-1-associated dementia. Expert Rev. Mol. Med. 2005;7 doi: 10.1017/S1462399405010239. 1e26. [DOI] [PubMed] [Google Scholar]
- Gelperina Svetlana, Maksimenko Olga. Drug delivery to the brain using surfactant-coated poly(lactide-co-glycolide) nanoparticles: influence of the formulation parameters. Eur. J. Pharm. Biopharm. 2010;74:157–163. doi: 10.1016/j.ejpb.2009.09.003. [DOI] [PubMed] [Google Scholar]
- Govender Thirumala, Stolnik Snjezana, Garnett Martin C., Illum Lisbeth, Davis Stanley S. PLGA nanoparticles prepared by nanoprecipitation: drug loading and release studies of a water soluble drug. J. Controlled Release. 1999;57:171–185. doi: 10.1016/s0168-3659(98)00116-3. [DOI] [PubMed] [Google Scholar]
- Huang S.-H., Jong A.Y. Cellular mechanisms of microbial protein contributing to invasion of the blood–brain barrier. Micro Rev. Cell Microbiol. 2001;3:277–287. doi: 10.1046/j.1462-5822.2001.00116.x. [DOI] [PubMed] [Google Scholar]
- ICH. Q8(R1), Pharmaceutical Development. Geneva, Switzerland; 2008 Nov.
- Kolchinsky P., Kiprilov E., Sodroski J. Increased neutralization sensitivity of CD4 independent human immunodeficiency virus variants. J. Virol. 2001;75:2041–2050. doi: 10.1128/JVI.75.5.2041-2050.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreuter J., Petrov V.E., Kharkevich D.A., Alyautdin R.N. Influence of the type of surfactant on the analgesic effects induced by the peptide dalargin after its delivery across the blood–brain barrier using surfactant-coated nanoparticles. J. Controlled Release. 1997;49(1):81–87. [Google Scholar]
- Kreuter J., Shamenkov D., Petrov V., Ramge P., Cychutek K., Koch-Brandt C., Alyautdin R. Apolipoprotein-mediated transport of nanoparticle-bound drugs across the blood–brain barrier. J. Drug Targeting. 2002;10:317–325. doi: 10.1080/10611860290031877. [DOI] [PubMed] [Google Scholar]
- Lobenberg R., Araujo L., von Brisen H., Rodgers E., Kreuter J. Body distribution of azidothymidine bound to hexyl-cyanoacrylate nanoparticles after i.v. injection to rats. J. Controlled Release. 1998;50:21–30. doi: 10.1016/s0168-3659(97)00105-3. [DOI] [PubMed] [Google Scholar]
- Menei P., Daniel V., Montero-Menei C., Brouillard M., Pouplard-Barthelaix A., Benoit J.P. Biodegradation and brain tissue reaction to poly(D, L-lactide-co-glycolide) microspheres. Biomaterials. 1993;14:470–478. doi: 10.1016/0142-9612(93)90151-q. [DOI] [PubMed] [Google Scholar]
- Peltonen Leena, Aitta Johanna, Hyvönen Samuli, Karjalainen Milja, Hirvonen Jouni. Improved entrapment efficiency of hydrophilic drug substance during nanoprecipitation of poly(l)lactide nanoparticles. AAPS PharmSciTech. 2004;5:1–6. doi: 10.1208/pt050116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plard J.P., Bazile D. Comparison of the safety profiles of PLA and Me PEG-PLA nanoparticles after single dose intravenous administration to rat. Colloids Surf. B. 1999;16:173–183. [Google Scholar]
- Schaffazick Scheila R., Siqueira Ionara R. Incorporation in polymeric nanocapsules improves the antioxidant effect of melatonin against lipid peroxidation in mice brain and liver. Eur. J. Pharm. Biopharm. 2008;69:64–71. doi: 10.1016/j.ejpb.2007.11.010. [DOI] [PubMed] [Google Scholar]
- Sunil K.J., Awasthi A.M., Jain N.K., Agarwal G.P. Calcium silicate based micro-spheres of repaglinide for gastro retentive floating drug delivery: preparation and in vitro characterization. J. Controlled Release. 2005;107:300–309. doi: 10.1016/j.jconrel.2005.06.007. [DOI] [PubMed] [Google Scholar]
- Tan Xiaolei, Douglas Boudinot F. Simultaneous determination of zidovudine and its monophosphate in mouse plasma and peripheral red blood cells by high-performance liquid chromatography. J. Chromatogr. B. 2000;740:281–287. doi: 10.1016/s0378-4347(00)00109-2. [DOI] [PubMed] [Google Scholar]
- Tomlinson E. Microsphere delivery systems for drug targeting and controlled release. Int. J. Pharm. Technol. Prod. Manuf. 1983;4:49. [Google Scholar]
- Tripathi K.D. 6th ed. Jaypee Brothers Medical Publishers; New Delhi, India: 2008. Essentials of Medical Pharmacology. 770–771. [Google Scholar]
- Ueda H., Tabata Y. Polyhydroxyalkanonate derivatives in current clinical applications and trials. Adv. Drug Delivery Rev. 2003;55:501–518. doi: 10.1016/s0169-409x(03)00037-1. [DOI] [PubMed] [Google Scholar]
- Vauthier C., Bouchemal K. Methods for the preparation and manufacture of polymeric nanoparticles. Pharm. Res. 2009;26:1025–1058. doi: 10.1007/s11095-008-9800-3. [DOI] [PubMed] [Google Scholar]
- Wilson Barnabas, Samanta Malay Kumar. Targeted delivery of tacrine into the brain with polysorbate 80-coated poly(n-butylcyanoacrylate) nanoparticles. Eur. J. Pharm. Biopharm. 2008;70:75–84. doi: 10.1016/j.ejpb.2008.03.009. [DOI] [PubMed] [Google Scholar]
- Wong Ho Lun, Chattopadhyay Niladri, Wu Xiao Yu, Bendayan Reina. Nanotechnology applications for improved delivery of antiretroviral drugs to the brain. Adv. Drug Delivery Rev. 2010;62:503–517. doi: 10.1016/j.addr.2009.11.020. [DOI] [PubMed] [Google Scholar]