

Abstract
For many fungi the number of known secondary metabolites is surprisingly small compared to the astonishingly large number of terpene cyclase, polyketide synthase (PKS), and non-ribosomal peptide synthetase (NRPS) secondary metabolite gene clusters found in their genomes. Correspondingly, the majority of fungal secondary metabolite genes have not yet been associated with the biosynthesis of any known small molecules, and it seems likely that for many more PKS and NRPS known small molecule products represent but a fraction of the entire spectrum of metabolites produced by the associated pathways. Comparative metabolomics based on differential analysis by 2D NMR spectroscopy (DANS) in conjunction with LC–MS analyses is emerging as a highly effective tool for pursuing small molecule structures and biosynthetic pathways associated with orphan PKS and NRPS gene clusters. Here we describe the use of DANS paired with LC–MS analyses for the comparison of the metabolomes of various fungal strains including wild-type (WT), PKS/NRPS overexpressing, and/or corresponding PKS/NRPS knock-out (KO) strains.
Keywords: DANS, NMR spectroscopy, Nonribosomal peptide synthetase, Polyketide synthase, Metabolomics, Mass spectrometry, Biosynthesis
1. Introduction
A number of recent examples have demonstrated the utility of comparative metabolomics using 2D NMR spectroscopy and mass spectrometry for identifying the biogenic small molecules (1) whose biosynthesis is dependent on a specific genetic background (2, 3) or environmental conditions (4, 5). In fungi, biosynthesis of many if not most secondary metabolites appears to depend on an integrated network of regulatory processes that responds to specific environmental stimuli (6, 7). As a result, most fungal PKS and NRPS genes are not active under standard laboratory conditions and correspondingly their small molecule products have not been identified. Variation of fungal growth conditions can induce activation of additional biosynthetic pathways, and it was shown that 2D NMR-based comparative metabolomics can be used to detect and identify associated changes in secondary metabolite production (4). This first example for DANS was based on comparison of a specific type of high-resolution 2D NMR spectra (a well-known version of 1H, 1H correlation referred to as dqfCOSY or 2qfCOSY) of entire fungal metabolite extracts. Whereas previously 2D NMR spectra had been used almost exclusively for the characterization of isolated, pure natural products, this example demonstrated that differential comparison of such spectra provides a highly effective means to map and characterize changes in fungal metabolism. dqfCOSY spectra are particularly well-suited for this type of analysis because of their good dynamic range and information-rich peak fine structure (8) which enables interpretation even in cases of significant peak overlap and includes detailed proton–proton coupling information (see Note 1) (9).
Subsequent studies established DANS as a general method for correlating secondary metabolite production with genetic changes. The first example where DANS was utilized to link secondary metabolite gene cluster expression and specific metabolic output came from the study of the Bacillus subtilis pksX gene cluster, a very large hybrid polyketide/nonribosomal peptide synthase. The antibiotic metabolites produced by the pksX-encoded pathway had eluded structure elucidation due to their rapid degradation under ambient laboratory conditions and upon chromatography (10). Therefore a strategy was developed in which 2D NMR spectra of metabolite extracts derived from a pksX-expressing strain (pksX+) and a pksX knock-out strain (pksX−) were compared. Initial comparison of dqfCOSY spectra corresponding to extracts derived from pksX+ and pksX− led to the identification of partial structures (i.e., fragments of a larger chemical structure) belonging to the sought-after pksX-dependent metabolites. Additional 2D NMR spectroscopic analysis including: (1H, 13C)-HMQC, (1H, 13C)-HMBC, (1H, 15N)-HMBC, and ROESY spectra, in conjunction with high-resolution positive-ion electrospray MS of the crude extracts, enabled the identification of the highly labile hexaene bacillaene and several-related metabolites as the products of the PksX megacomplex.
DANS methodology has also been applied to higher organisms. In pursuit of endogenous small molecule signals used by the model organism Caenorhabditis elegans, Pungaliya et al.'s DANS comparison of WT worms and daf-22 mutant worms, a strain known to be defective in the production of the signaling molecules of interest, led to the characterization of a family of glycosides of the dideoxy sugar ascarylose (ascarosides) with potent biological activity (11). In this study the use of chemically synthesized ascarosides served as standards for parallel LC–MS analysis of crude worm extracts, which confirmed the identity of the daf-22-dependent C. elegans metabolites identified via DANS.
A recent study of the Aspergillus fumigatus metabolome demonstrated that DANS can reveal large numbers of new metabolites even for previously well-studied pathways (3). Gliotoxin (1, Fig. 1b), a major product of the gli non-ribosomal peptide synthetase (NRPS) gene cluster, is strongly associated with virulence of A. fumigatus. Despite identification of all genes of the gli cluster, the pathway of gliotoxin biosynthesis had remained elusive, in part because few potential intermediates have been characterized. DANS-based comparison of the WT metabolome with that of ΔgliZ (Fig. 1), a knockout strain devoid of the gene encoding the transcriptional regulator of the gli cluster, revealed nine novel gliZ-dependent metabolites including unexpected structural motifs. Their identification provided detailed insight into the biosynthesis of gliotoxin and related compounds and may benefit studies of the role of the gli cluster in A. fumigatus virulence.
Fig. 1.
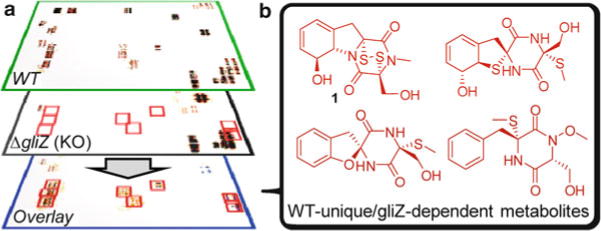
Schematic overview for the use of DANS to identify gliZ-dependent metabolites in A. fumigatus (3). (a) Spectroscopic signals present in both WT and ΔgliZ (KO) spectra, representing metabolites not strongly affected by the ΔgliZ mutation, are eliminated in the overlay, whereas signals only present in WT (i.e. they are gliZ-dependent) remain (red boxes). (b) Further analysis of the COSY signals highlighted by DANS, with additional 2D NMR and HPLC–MS analysis, established full chemical structures of gliZ-dependent metabolites, including both known and novel compounds.
DANS-based comparative metabolomics paired with HPLC–MS analyses promises to greatly improve detection of changes in fungal metabolite production and to simplify identification of the structures of differentially produced metabolites. Potential applications include comparison of fungi grown under different culture conditions as well as comparison of different genotypes or ecotypes. However, great care must be taken when designing DANS studies to ensure that metabolite extracts are obtained using strictly reproducible conditions and remain uncontaminated. Below we outline a detailed protocol and note some pitfalls for DANS analysis of fungal samples, discussing specific aspects of extract preparation, sample enrichment, data acquisition, processing and interpretation, and replication. As a specific example, we present a general outline for DANS/LC–MS comparison of a wild-type strain with an NRPS or PKS knock-out strain.
2. Materials
2.1. Solvents and Filters
Extraction and chromatography: Solvents must be of highest available purity. Use HPLC-grade solvents with purity ≥99.9% (see Note 2).
Wash Celite or cotton to be used for extract filtration by placing into a glass frit and rinsing with 20 ml of extraction solvent (methanol or acetonitrile/water mixtures) per 1 ml of filtration material.
2.2. NMR Spectroscopy
NMR solvents: Use methanol-d4 (see Note 3), acetonitrile-d3, acetone-d6, or DMSO-d6 of 99.8% or greater purity.
NMR tubes: Use high quality 5 mm tubes (see Note 4).
NMR spectrometer and probes: Use of NMR spectrometers with a 1H resonance frequency of 600 MHz or higher is recommended; however, high-resolution dqfCOSY spectra acquired at 900 MHz can provide more than twice the resolution of spectra acquired at 600 MHz. Use inverse-detection or HCN triple resonance probes (see Note 5).
2.3. Fractionation and HPLC–MS
Fractionation of extracts for DANS: Use a programmable, fully automated flash chromatography system (e.g., Teledyne ISCO CombiFlash), equipped with either normal-phase silica gel or reverse-phase C18 (octadecyl-functionalized) silica columns.
LC–MS: Use a standard reversed phase column (e.g., 4.6 × 150 mm, 5 μm particle diameter) and acetonitrile–water (alternatively methanol–water) solvent gradients (see Note 3). Addition of 0.1% acetic acid can improve chromatographic separation for some compound classes. Analyze all samples under carefully standardized conditions, using both positive- and negative-ion electrospray ionization.
Use a rotary evaporator with vacuum control and temperature-controlled water bath for evaporation of extracts and fractions.
3. Methods
Fungal secondary metabolism can be affected by minor, often unnoted changes in culture conditions. Therefore, we recommend that all analyses are carried out using samples from at least two independent biological replicates.
3.1. Preparation of Fungal Cultures
Wild-type and knock-out strains (as well as any additional strains to be compared, for example a PKS/NRPS overexpressor) must be cultured at the same time under identical conditions. Culture parameters must be carefully controlled in order to ensure comparability with later replicates. These include temperature, humidity, light levels, media (make one batch and partition out for individual strains), incubation time, and inoculum concentration, in addition to other parameters specific to the type of culture (e.g., liquid/solid).
The number of individual culture replicates for each strain required for DANS varies depending on media type (rich/poor) and fungal species. If possible, enough cultures should be set up, pooled, and processed to produce at least 50–100 mg of metabolite extract for each strain.
3.2. Culture Extraction
Lyophilize fungal cultures (liquid culture or homogenized solid/agar based), break up the residue into a fine powder, and extract with organic solvent. Suitable solvent systems for extraction of compounds of a wide polarity range include (from least to most polar): a mixture of ethyl acetate and methanol (9:1, v:v), acetonitrile (3), methanol, or a mixture of acetonitrile and water (9:1, v:v) (see Note 3). Use 100 ml solvent per 2 g of lyophilized culture. Extract for at least 2 h with effective stirring. Process all cultures on the same day, using equivalent volumes of the same batch of extraction solvents.
Filter the extract over pre-washed Celite (5 g per 100 ml extract) or cotton (see Note 6). Rinse filter with 100 ml of extraction solvent.
Evaporate filtered extracts in vacuo using a rotary evaporator equipped with a room temperature water bath. Do not heat the extracts above room temperature. Use the same size of evaporation flask and the same vacuum settings for all extracts.
3.3. Optional Pre-fractionation/Polarity Segregation
Crude fungal extracts usually represent highly complex mixtures of small molecules (complexity can be assessed by acquiring a dqfCOSY spectrum for a crude fungal extract, see below) and contain compounds of a wide polarity range. Some compounds in such crude extracts may not be well soluble in the NMR solvent, thus preventing their detection. Furthermore, the presence of poorly soluble components in metabolite samples often degrades the quality of the NMR spectra. The procedures described below employ reproducible chromatographic enrichment to yield samples of reduced chemical complexity and increased polarity uniformity, helping to prevent precipitate formation, and improving NMR spectral line shapes as well as dynamic range. Two alternative procedures are described, for samples derived from extraction procedures that use either relatively polar or nonpolar solvents. These protocols separate the wild-type and knock-out mutant extracts (as well as any additional samples) into sets of three “pools” each.
3.3.1. Samples from Polar Extraction
Dissolve wild-type fungal sample completely in suitable organic solvent (e.g., the solvent used for extraction) in a round-bottomed flask. Use at least 5 ml of solvent per 100 mg of extract or more if needed for complete dissolution.
Add 1 g (per 100 mg of extract dissolved) of Celite or octadecyl-functionalized silica gel to the solution.
To adsorb the solute extract onto the solid phase, remove solvent in vacuo using rotary evaporation. To ensure reproducible chromatography amongst sets of fungal extracts, remove residual solvent after rotary evaporation by subjecting the sample to reduced pressure (0.1 mmHg, ∼1 h).
Remove the extract-imbued solid phase from flask and set aside. Add 10 ml of additional solvent and 1 g of Celite or octadecyl-functionalized silica gel to flask and repeat step 3 to collect residual extract stuck to the sides of the flask.
Condition a 100 g reverse-phase C-18 flash chromatography column (for 80–500 mg of extracts) by changing solvent composition from 100% organic solvent to 1–5% organic solvent/99–95% water (or water with 0.1% acetic acid) over 10 column volumes, holding at 99–95% water for five additional column volumes.
Load the solid phase adsorbed sample onto the chromatography system as described by the manufacturer.
Fractionate the sample using a solvent-gradient based chromatography method. A reverse-phase gradient (acetonitrile–water or methanol–water) that works well for fungal extracts was recently described (3).
Combine individual fractions to give three pools corresponding to high (e.g., 1–5% organic solvent), medium (e.g., 6–54% organic solvent), and low (e.g., 55–100% organic solvent) polarities. UV chromatograms acquired by the chromatography system during a test separation run can assist with determining which fractions to combine.
Repeat steps 1–8 for knock-out extract (and any additional samples to be compared via DANS) using identical solvents, media, and fractionation parameters. Comparison of UV chromatograms acquired for different extracts can be utilized to assess reproducibility of elution times, helping to determine which fractions should be included in a given pool.
3.3.2. Samples from Nonpolar Extraction
Dissolve wild-type fungal sample completely in suitable organic solvent (e.g., the solvent used for extraction) in a round-bottomed flask. Use at least 5 ml of solvent per 100 mg of extract or more if needed for complete dissolution.
Add 2 g (per 100 mg of extract dissolved) of normal-phase silica gel to solution.
Same as polar extract enrichment procedures above.
Same as polar extract enrichment procedures above.
Condition a 40 g Normal-Phase Silica Gel flash chromatography column (for extracts 100–1,000 mg) by changing solvent gradient composition from 50% more polar solvent (e.g., methanol, acetonitrile, ethyl acetate, with optional 0.1% acetic acid) to 100% less polar organic solvent (e.g., dichloromethane, hexanes) over 8 column volumes, followed by conditioning with the same solvent for 4 additional column volumes.
Same as polar extract enrichment procedures above.
Fractionate the sample using a solvent-gradient based chromatography method. Example, chromatographic method for fractionation of Aspergillus flavus ethyl acetate:methanol (9:1) extracts: start with 100% dichloromethane for 2.5 column volumes, followed by a linear increase of methanol over 20 column volumes up to 30% methanol, which is held for an additional 10 column volumes.
Combine individual fractions to give three pools. For example when using the chromatographic method in step 7, combine fractions with 0–8% methanol as pool#1 (least polar), fractions with 8–16% methanol as pool#2 (medium polar), and 17–30% methanol as pool#3 (most polar). UV chromatograms acquired by the chromatography system during a test separation run can help guide pooling.
Same as polar extract enrichment procedures above.
3.4. NMR Sample Preparation
The following steps assume use of a 5 mm sample tube and 0.6 ml of NMR solvent. If using smaller samples volumes, sample and solvent amounts must be adjusted proportionally.
Suspend 50 mg of extract in 0.4 ml of deuterated NMR solvent in a 1 ml vial and place on a shaker for 30 min. Centrifuge to remove insoluble material and transfer supernatant into a fresh 1 ml vial (see Note 7).
Evaporate all solvent in vacuo using a rotary evaporator (using a proper adaptor for the 1 ml vial). Redissolve the residue in 0.6 ml of NMR solvent (see Note 8). If any turbidity remains at this stage, reevaporate all solvent and repeat step 1. Treat all samples to be compared in exactly the same way.
Place NMR samples into NMR tubes, taking care that solvent amount for all samples is identical. Store samples in the dark at room temperature for at least 12 h prior to NMR spectroscopy.
Acquire 1H spectra for all samples. Inspect lines shape and peak position for known polar components (e.g., media components such as nicotinamide). If the spectra show considerable variation in proton chemical shift values, especially for signals representing chemical species with acidic or basic functional groups, the pH of the sample should be adjusted such that peaks corresponding to the same chemical species in the various samples have the same chemical shift values (see Note 9).
3.5. Acquisition and Processing of 2D NMR Spectra
Acquisition parameters: For acquisition of dqfCOSY spectra at 600 MHz, use 0.8 s acquisition time, 80 complex increments per 1 ppm of sweep width, a relaxation delay of 1.6 s, and 8–32 scans per increment (see Note 10). Phase cycled dqfCOSY sequences must be used, not gradient-selected versions (see Note 11). For field strengths greater than 600 MHz, proportionally increase the number of complex increments per 1 ppm of sweep width.
Processing of NMR spectra: All spectra are processed in two different ways: (1) for comparative analysis (spectral overlay) and (2) for detailed follow-up analysis. Method 1 (for spectral overlay): shorten time domain to 0.25 s, zero-fill to 4096 data points in F2, apply a squared sine bell window function, and perform first Fourier transform. Next, zero-fill to 4096 data points in F1, apply a squared sine bell window function, and apply second Fourier transform. Switch display to absolute value-mode and use real part of the spectrum for overlay (see below). Method 2 (for detailed follow-up analysis): use fulltime domain, zero-fill to 8192 data points in F2 (see Note 12), apply cosine window function, and perform first Fourier transform. Next, zero-fill to 4096 data points in F1, apply a cosine window function, and apply second Fourier transform. Phase correct spectra as appropriate.
DANS overlay of dqfCOSY spectra using graphics software: Overlays of large dqfCOSY spectra are possible with several NMR processing software packages, e.g., Bruker's Topspin. However, differences between spectra can often be visualized better using overlays of spectral images using graphics software such as Adobe Photoshop. For the purpose of detecting differences between a wild-type and a knock-out strain, the dqfCOSY spectra are arranged as follows. Display spectra as color maps in the NMR processing software (e.g., MNOVA, Topspin) using a color scheme that correlates color saturation and/or brightness with signal intensity. Place bitmaps from the spectra to be compared (e.g., spectra of wild-type and knock-out strain) into two sets of layers in a Photoshop document as follows. On top of a white background layer, two layers are placed containing absolute value-mode dqfCOSY spectra for wild-type and knockout strain. For these two layers, the blending mode is set to “difference.” On top of this stack of layers, another two layers are placed, again containing the wild-type and knock-out spectra. For these three layers, “multiply” is used as the blending mode. Finally, a “curves” adjustment layer is placed as the topmost layer. This curves layer is used to invert colors and to adjust gamma to increase or decrease contrast.
Comparative analysis: In order to compare spectra, visibility of layers is toggled off and on as needed. For example, to visualize all signals from compounds present in wild-type, the layer containing the wild-type spectrum in “difference” mode is turned on and visibility of all other layers containing spectra is turned off. An example for resulting spectral image is in Fig. 2a. Additionally turning on visibility of the layer containing the knock-out spectrum in “multiply” mode results in suppression or color changes for all signals, except those only present in wild-type and absent in the knock-out spectrum (Fig. 2b) (see Note 13).
Construct partial structures based on interpretation of the identified differential dqfCOSY cross peaks, using the phase-sensitive processing of the spectra (see step 2) (8, 12). Compare partial structures with known metabolites of the investigated fungal strains, and use HPLC–MS to confirm presence of any known metabolites. For the identification of potentially novel metabolites that are present in high concentrations (>1% of total sample), acquire supplemental (1H, 13C)-HSQC and (1H, 13C)-HMBC spectra (12) of wild-type and mutant sample for additional comparative analysis. If the abundance of the metabolites of interest is lower (<1% of the total) or in cases of extensive overlap in the dqfCOSY spectra, additional chromatographic fractionation will be required to obtain satisfactory dqfCOSY, (1H, 13C)-HSQC, and (1H, 13C)-HMBC data. If the presence of new metabolites is confirmed, isolate compounds using additional chromatography (see Note 14). Confirm structures of novel metabolites by high-resolution mass spectrometry.
Fig. 2.
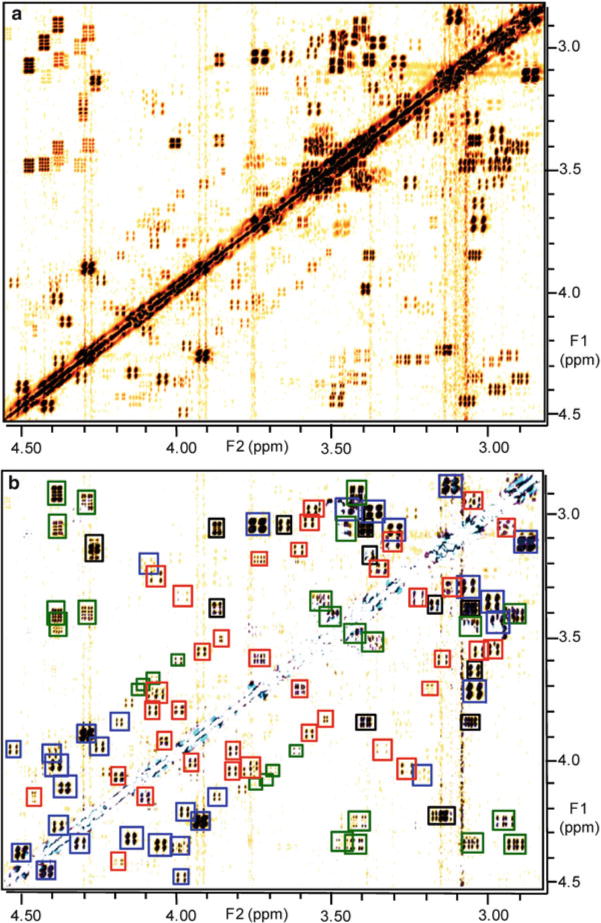
Actual DANS spectra from analysis of A. fumigatus gliZ-dependent metabolites (3) (a) Section of the WT dqfCOSY spectrum in absolute-value mode. (b) DANS overlay of WT and ΔgliZ (KO) spectra, showing signals representing known gliotoxin derivatives (blue), novel gli Z-dependent compounds (red), and previously described fungal metabolites (black). Several crosspeaks representing metabolites present in both WT and ΔgliZ were not fully suppressed in the overlay due to chemical shift variation (green).
Acknowledgments
This work was supported by the National Institutes of Health (Grant No. GM088290 and GM008500) and DuPont Crop Protection.
Footnotes
Instead of dqfCOSY spectra, several other types of two-dimensional NMR spectra could be used for the differential analysis of complex small molecule mixtures, for example TOCSY spectra with short mixing times. Phase-sensitive TOCSY spectra exhibit good peak shape and excellent sensitivity, and short mixing times could be used to prevent over-crowding of the spectra. However, for the differential analysis of fungal extracts, which usually are not mass-limited, the slightly higher sensitivity of the TOCSY experiment does not confer any significant advantage. We chose dqfCOSY spectra over TOCSY spectra because the fine structure of the dqfCOSY cross peaks allows for the extraction of more structural information (coupling constant information) than would be possible to derive from TOCSY cross peaks. This difference becomes relevant in the second step of the differential analysis, in which signals, identified as representing compounds of interest, are analyzed in greater detail. For this second step, the dqfCOSY method thus provides a clear advantage. Nonetheless, for mass-limited samples, differential analysis based on TOCSY spectra could be useful.
Care should be taken to avoid or reduce contact of the sample and solvents to plastic materials/surfaces, as many plastics contain soluble components that can result in significant contamination of samples and degradation of NMR spectral quality.
Methanol (when used as extraction, chromatography, or NMR solvent) can react with labile metabolites (e.g., methylsulfanyl derivatives, carboxylic acids, unsaturated aldehydes, and ketones), resulting in artifact formation. Acetonitrile/water mixtures should be used if the presence of methanol-sensitive compounds is suspected. Additionally, methanol and acetonitrile/water mixtures will extract media carbon sources such as glycerol, glucose, or other mono- and disaccharides. Signals corresponding to these components will dominate NMR spectra acquired for crude methanol extracts, requiring their eventual removal from the sample via chromatography (see Subheading 3.3).
The use of 5 mm NMR tubes and corresponding NMR probe heads offers excellent dynamic range at good sensitivity, facilitating detection of even very minor components of a metabolome. In cases where extract quantity is limited, smaller sample tubes/probe heads should be used.
The use of cryogenic (cold) probes can be advantageous for mass-limited samples; however, in cases where sample size is not limiting, room temperature probes may provide better results with fewer artifacts when acquiring dqfCOSY spectra. For (1H, 13C)-HSQC and (1H, 13C)-HMBC, cryogenic probes are generally preferable (8).
Of the two filtration materials, Celite is more effective at removing proteins and other macromolecular components from the sample, which often cause line shape problems and/or precipitate formation during NMR acquisition. However, cotton tends to be less chemically abrasive than Celite and should be used when labile chemical species are suspected.
Appropriate choice of NMR solvent is critical for spectral quality and dynamic range. Methanol-d4 dissolves most organic compounds, but leads to deuterium exchange of many types of protons (hydroxyl, amine, amide, and also β-keto carbonyl) and may cause problems for compounds susceptible to nucleophilic addition or substitution (see Note 3). DMSO-d6 or acetonitrile-d3 with 5–10% D2O can be used alternatively, but may not dissolve very lipophilic compounds. Any insoluble small molecules will be retained in the centrifugation pellet.
The use of 0.4 ml of solvent in the centrifugation step ensures that the actual NMR sample, prepared using a slightly larger solvent volume of 0.6 ml, will not be completely saturated. This is necessary to avoid formation of precipitates during NMR spectroscopic analysis.
The proton chemical shifts of small molecules containing acidic or basic functional groups can vary greatly (>1 ppm) depending on sample pH; this variation can complicate the interpretation of DANS spectra. To adjust the pH of an NMR sample, make a solution of 5% acetic acid-d4 in an NMR solvent of choice (acidified solution). Choose an internal standard 1H signal to monitor during the process of adjusting the sample pH; for example, the well resolved singlet corresponding to the pH-sensitive proton at position 2 of nicotinic acid or nicotinamide (8.6–8.7 ppm). Titrate the sample with the acid solution (∼10 μl/aliquot), monitoring how sample chemical shifts respond to the addition of the acidified solution. Continue the process iteratively until internal standard 1H signal and overall peak shift landscape is consistent for all samples that will be compared in the DANS analysis (11).
Use of 8 scans per increment may be sufficient for concentrated samples; however, on most spectrometers artifact suppression is better at 16 scans per increment. For high dynamic range requirements or dilute samples 32 (or more) scans per increment may be required.
Use of gradient-selected dqfCOSY sequences (i.e. sequences that use gradients for coherence selection) results in spectra with significantly worse line shape characteristics and lower signal-to-noise and dynamic range. The use of homospoil pulses at the beginning of the relaxation delay d1 is recommended.
Digital resolution in F2 in the Fourier-transformed spectrum should be no lower than 1 Hz/data point, and preferably closer to 0.5 Hz/data point. Spectra with large sweep widths, or spectra acquired at high field strength, may thus require zero-filling to 16384 data points in F2 prior to Fourier transformation.
The graphical comparison of 2D-NMR spectra as described here can be regarded as a “weighing” of one spectrum against one or more related spectra. Essentially, the “multiply” function weighs the RGB values of a given data point against the RGB values of corresponding data points in the overlaid spectra. The use of the “multiply” blending mode as a simple out-of-the-box weighing mode is convenient; however, other blending modes may in some situations provide better results. For example, simply subtracting one spectrum from another would create a mask that could be used to completely block out any signals occurring in both spectra. However, in our experience color shifts of signals as they result from the “multiply” blending mode are generally better to work with than complete signal extinction, because the preserved multiplicity structures of partially suppressed peaks allows one to confirm that they indeed represent superposition(s) of equivalent cross peaks in the compared spectra, as opposed to mutual extinction of unrelated cross peaks that accidentally occur at similar chemical shift values in the compared spectra.
Isolation is not a requirement for structure determination of novel compounds; however, full structural characterization of pure compounds (or significantly enriched samples) is often easier than their identification from spectra of complex mixtures. Nonetheless, there is small but increasing number of metabolites that have never been isolated in pure form and, therefore, were identified based on spectra of complex metabolite mixtures (1).
References
- 1.Forseth RR, Schroeder FC. NMR-spectroscopic analysis of mixtures: from structure to function. Curr Opin Chem Biol. 2011;15:38–47. doi: 10.1016/j.cbpa.2010.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Challis GL. Genome mining for novel natural product discovery. J Med Chem. 2008;51:2618–2628. doi: 10.1021/jm700948z. [DOI] [PubMed] [Google Scholar]
- 3.Forseth RR, Fox EM, Chung D, Howlett BJ, Keller NP, Schroeder FC. Identification of cryptic products of the gliotoxin gene cluster using NMR-based comparative metabolomics and a model for gliotoxin biosynthesis. J Am Chem Soc. 2011;133:9678–9681. doi: 10.1021/ja2029987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schroeder FC, Gibson DM, Churchill AC, Sojikul P, Wursthorn EJ, Krasnoff SB, Clardy J. Differential analysis of 2D NMR spectra: new natural products from a pilot-scale fungal extract library. Angew Chem Int Ed Engl. 2007;46:901–904. doi: 10.1002/anie.200603821. [DOI] [PubMed] [Google Scholar]
- 5.Shiloach J, Reshamwala S, Noronha SB, Negrete A. Analyzing metabolic variations in different bacterial strains, historical perspectives and current trends-example E. coli. Curr Opin Biotechnol. 2010;21:21–26. doi: 10.1016/j.copbio.2010.01.001. [DOI] [PubMed] [Google Scholar]
- 6.Georgianna DR, Fedorova ND, Burroughs JL, Dolezal AL, Bok JW, Horowitz-Brown S, et al. Beyond aflatoxin: four distinct expression patterns and functional roles associated with Aspergillus flavus secondary metabolism gene clusters. Mol Plant Pathol. 2010;11:213–226. doi: 10.1111/j.1364-3703.2009.00594.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yin W, Keller NP. Transcriptional regulatory elements in fungal secondary metabolism. J Microbiol. 2011;49:329–339. doi: 10.1007/s12275-011-1009-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Claridge TDW. High-resolution NMR techniques in organic chemistry. Elsevier; Amsterdam, Boston: 2009. pp. xiv–383. [Google Scholar]
- 9.Taggi AE, Meinwald J, Schroeder FC. A new approach to natural products discovery exemplified by the identification of sulfated nucleosides in spider venom. J Am Chem Soc. 2004;126:10364–10369. doi: 10.1021/ja047416n. [DOI] [PubMed] [Google Scholar]
- 10.Butcher RA, Schroeder FC, Fischbach MA, Straight PD, Kolter R, Walsh CT, et al. The identification of bacillaene, the product of the PksX megacomplex in Bacillus subtilis. Proc Natl Acad Sci USA. 2007;104:1506–1509. doi: 10.1073/pnas.0610503104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pungaliya C, Srinivasan J, Fox BW, Malik RU, Ludewig AH, Sternberg PW, et al. A shortcut to identifying small molecule signals that regulate behavior and development in Caenorhabditis elegans. Proc Natl Acad Sci USA. 2009;106:7708–7713. doi: 10.1073/pnas.0811918106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bross-Walch N, Kuhn T, Moskau D, Zerbe O. Strategies and tools for structure determination of natural products using modern methods of NMR spectroscopy. Chem Biodivers. 2005;2:147–177. doi: 10.1002/cbdv.200590000. [DOI] [PubMed] [Google Scholar]