

Abstract
The first-principles method and the evolutionary algorithm are used to identify stable high pressure phases of potassium azide (KN3). It has been verified that the stable phase with space group I4/mcm below 22 GPa, which is consistent with the experimental result, will transform into the C2/m phase with pressure increasing. These two phases are insulator with 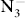 anions. A metallic phase with P6/mmm symmetry is preferred above 40 GPa, and the N atoms in this structure form six-membered rings which are important for understanding the pressure effect on
anions. A metallic phase with P6/mmm symmetry is preferred above 40 GPa, and the N atoms in this structure form six-membered rings which are important for understanding the pressure effect on 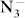 anions and phase transitions of KN3. Above the studied pressure (100 GPa), a polymerization of N6 rings may be obtained as the result of the increasing compactness.
anions and phase transitions of KN3. Above the studied pressure (100 GPa), a polymerization of N6 rings may be obtained as the result of the increasing compactness.
Metal azides have drawn considerable attention for their interesting chemical and physical properties. Under external influences (impact, heat, irradiation, etc), they become unstable and decompose into metal and nitrogen1. Their practical applications include explosives, pure nitrogen sources and photographic materials. Additionally, metal azides are structurally simple among solids that deflagrate or detonate, so they are potentially model systems for theories of the fast reactions2. Being different from the extensively studied alkali halides, metal azides are ionic compounds containing internal molecular structure which makes them the candidates for understanding the complex nature of chemical bonding. On the other hand, the polymerization of nitrogen may form a high-energy-density material because the transformation from the N–N single bond (160 kJ/mol) to the N≡N triple bond (954 kJ/mol) is accompanied by a large energy release. The non-molecular nitrogen under high pressure was first predicted theoretically by A. K. McMahan et al.3. A single-bonded cubic gauche form of N2 (cg-N) was also successfully synthesized by M. I. Eremets et al.4,5. It is suggested that cg-N can be stabilized in compounds with other elements or by introducing impurities4. Recently, metal azides have been proposed to be a precursor in the formation of polymeric nitrogen. It could be expected that the 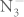 anion will create polymeric single-covalent-bond networks more easily than diatomic nitrogen because the
anion will create polymeric single-covalent-bond networks more easily than diatomic nitrogen because the 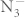 anion is more weakly bonded than the diatomic triple-bonded nitrogen6. Alkali azides are one class of compound among metal azides. Pressure-induced phase transitions in alkali azides have been reported by both experimental6,7,8,9,10,11 and theoretical investigations12,13,14,15,16. Previous study reported that sodium azide undergoes a set of phase transitions6,8, and the
anion is more weakly bonded than the diatomic triple-bonded nitrogen6. Alkali azides are one class of compound among metal azides. Pressure-induced phase transitions in alkali azides have been reported by both experimental6,7,8,9,10,11 and theoretical investigations12,13,14,15,16. Previous study reported that sodium azide undergoes a set of phase transitions6,8, and the 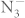 anions in NaN3 transform to polymeric nitrogen net above 120 GPa6. Lithium azide, which is isostructural to the low-temperature phase of NaN3 at ambient condition, is stable up to the pressure of 60 GPa at room temperature7. Recent high-pressure study of CsN3 up to 55.4 GPa reveals three phase transitions approximately at 0.5, 4.4, and 15.4 GPa11. Strikingly, theoretical studies have predicted polymerization of nitrogen in LiN314,15 and NaN316. Thus, a study of the high-pressure behavior of KN3 would provide more insights into the mechanism of pressure-induced rearrangement of azide anions. It is helpful to investigate theoretically the pressure effect on potassium azide which might result in the formation of polymeric nitrogen.
anions in NaN3 transform to polymeric nitrogen net above 120 GPa6. Lithium azide, which is isostructural to the low-temperature phase of NaN3 at ambient condition, is stable up to the pressure of 60 GPa at room temperature7. Recent high-pressure study of CsN3 up to 55.4 GPa reveals three phase transitions approximately at 0.5, 4.4, and 15.4 GPa11. Strikingly, theoretical studies have predicted polymerization of nitrogen in LiN314,15 and NaN316. Thus, a study of the high-pressure behavior of KN3 would provide more insights into the mechanism of pressure-induced rearrangement of azide anions. It is helpful to investigate theoretically the pressure effect on potassium azide which might result in the formation of polymeric nitrogen.
At ambient condition, potassium azide crystallizes in a body-centered tetragonal lattice with I4/mcm symmetry, and the 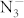 ion in the lattice is a straight chain of three nitrogen atoms. Raman scattering up to 4.0 GPa17 and single-crystal x-ray diffraction up to 7.0 GPa18 show that no phase transition is found at these measured pressures. Recently, a structural phase transition at 15.5 GPa is revealed by X-ray diffraction study9,10. A Raman scattering study of KN3 up to 55.0 GPa suggests that a first-order phase transition starts at 13.6 GPa and completes at 32.2 GPa10. However, a detailed structure of solid KN3 under high pressure has been unsolved yet. In this paper, we employ the first-principles study to understand the phase transitions and properties of KN3 under high pressure.
ion in the lattice is a straight chain of three nitrogen atoms. Raman scattering up to 4.0 GPa17 and single-crystal x-ray diffraction up to 7.0 GPa18 show that no phase transition is found at these measured pressures. Recently, a structural phase transition at 15.5 GPa is revealed by X-ray diffraction study9,10. A Raman scattering study of KN3 up to 55.0 GPa suggests that a first-order phase transition starts at 13.6 GPa and completes at 32.2 GPa10. However, a detailed structure of solid KN3 under high pressure has been unsolved yet. In this paper, we employ the first-principles study to understand the phase transitions and properties of KN3 under high pressure.
In the present work, KN3 is compressed up to 100 GPa. The phase transition from I4/mcm to C2/m is determined at a pressure about 22 GPa. In addition, we have defined another high-pressure phase with P6/mmm symmetry in which the 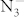 ions transform to N6 rings, and the hexagonal phase is metallic. We have just noticed the discussion on the P6/mmm phase by Li et al. recently19. The three phases of this crystal are stable against decomposition of KN3 into
ions transform to N6 rings, and the hexagonal phase is metallic. We have just noticed the discussion on the P6/mmm phase by Li et al. recently19. The three phases of this crystal are stable against decomposition of KN3 into 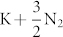 under pressure up to 100 GPa. The results provide an insight into the formation of polymeric nitrogen in metal azides.
under pressure up to 100 GPa. The results provide an insight into the formation of polymeric nitrogen in metal azides.
Computational details
To find the lowest energy structures of KN3 under extreme conditions, the USPEX code based on the evolutionary algorithm20,21 is employed for the search where the VASP code22 is used as an external ab initio code for the underlying structural optimizations. Evolutionary variable-cell structure prediction simulations are performed at 20, 60, and 100 GPa with 1, 2, and 4 KN3 formula units per unit cell. The first generation is produced randomly. All newly generated structures are relaxed at constant pressure and ranked by their enthalpy value. The lowest-enthalpy 60% structures of each generation are used to produce the next generation through heredity (70%), atomic permutation (10%), atom position mutation (10%) and lattice mutation (10%). The structures are relaxed by using density-functional theory with the Perdew-Burke-Ernzerhof (PBE) exchange correlation functional23, and projector augmented wave (PAW) method24 is also adopted. The 3s23p64s1 for K and 2s22p3 for N are treated as valence electrons. Having selected the lower enthalpy structures, we recalculate their enthalpies with increased accuracy between 0 and 100 GPa. An energy cutoff of 520 eV is used for the plane-wave basis sets, and the total energy is converged to 1.0 × 10−6 eV in the self-consistent loop. The atomic positions, lattice parameters, and cell volume are fully optimized by using a conjugate-gradient algorithm. The iterative relaxation of the atomic positions stop when all forces are smaller than 0.001 eV/Å, and the total stress tensor is reduced to the order of 0.01 GPa. The phonon calculations are performed using the Quantum ESPRESSO code based on density functional perturbation theory25, where vanderbilt ultrasoft pseudopotentials are used for K and N. Before the application, the pseudopotentials are tested by comparing the relaxed structural parameters and electronic structure with the results obtained from VASP code. All cases concerned are zero-temperature ground state.
Results and discussion
The analysis of the predicted structures gives us a list of candidate structure with space groups I4/mcm, C2/m, P21, Cmc21, P6/mmm, 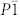 , and Imma, which are depicted in Fig. 1. In order to exam the difference between theoretical results and experimental data, the theoretical lattice constants and unit cell volume are compared with experimental I4/mcm structure at ambient condition (Table I). The LDA calculations underestimate a, c, and V by 3.3%, 5.2%, and 11.4%, whereas GGA calculations overestimate a, c, and V by 1.5%, 2.0%, and 5.2%, respectively. The GGA results are closer to the experimental results than the LDA ones. Therefore the GGA exchange-correlation functional is adopted for the further calculations. The C2/m structure detected from the simulations has the same space group as in LiN37, NaN326 and CsN311. The I4/mcm, C2/m, P21 and Cmc21 structures possess linear molecular
, and Imma, which are depicted in Fig. 1. In order to exam the difference between theoretical results and experimental data, the theoretical lattice constants and unit cell volume are compared with experimental I4/mcm structure at ambient condition (Table I). The LDA calculations underestimate a, c, and V by 3.3%, 5.2%, and 11.4%, whereas GGA calculations overestimate a, c, and V by 1.5%, 2.0%, and 5.2%, respectively. The GGA results are closer to the experimental results than the LDA ones. Therefore the GGA exchange-correlation functional is adopted for the further calculations. The C2/m structure detected from the simulations has the same space group as in LiN37, NaN326 and CsN311. The I4/mcm, C2/m, P21 and Cmc21 structures possess linear molecular 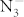 anions, while the structures with
anions, while the structures with 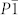 and Imma symmetries display the characteristic of the N-atom chains and are radically different from the structures containing
and Imma symmetries display the characteristic of the N-atom chains and are radically different from the structures containing 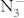 anions (see Fig. 1 and supplementary Table S1). In variable-cell simulations at 100 GPa, the P6/mmm structure is clearly identified as the most stable one and it is assigned to a new structure type for alkali metal azides. Interestingly, this is a layer-like structure with six N atoms forming a planar N6 ring. Additionally, polynitrogen molecules as clean high energy density materials have been extensively explored for several years27,28,29. A lot of theoretical work on N6 has shown that the planar hexagonal (D6h) ring is not minimum30,31,32. However, the six-membered nitrogen rings can be stabilized by coordinate covalent to oxygen33 and by the incorporation of metal atom in metal-N6 molecules34. We now obtain the N6 rings in bulk materials. It reveals that extra atoms play an important role in stabilizing N6 ring. More recently, a P6/m structure containing N6 ring has been also predicted in LiN313,14,15 and NaN316. Thus, the P6/m structure is considered in KN3. The results show that the P6/m structure becomes P6/mmm phase after structure optimization. Parameters describing the C2/m and P6/mmm structures are listed in Table II.
anions (see Fig. 1 and supplementary Table S1). In variable-cell simulations at 100 GPa, the P6/mmm structure is clearly identified as the most stable one and it is assigned to a new structure type for alkali metal azides. Interestingly, this is a layer-like structure with six N atoms forming a planar N6 ring. Additionally, polynitrogen molecules as clean high energy density materials have been extensively explored for several years27,28,29. A lot of theoretical work on N6 has shown that the planar hexagonal (D6h) ring is not minimum30,31,32. However, the six-membered nitrogen rings can be stabilized by coordinate covalent to oxygen33 and by the incorporation of metal atom in metal-N6 molecules34. We now obtain the N6 rings in bulk materials. It reveals that extra atoms play an important role in stabilizing N6 ring. More recently, a P6/m structure containing N6 ring has been also predicted in LiN313,14,15 and NaN316. Thus, the P6/m structure is considered in KN3. The results show that the P6/m structure becomes P6/mmm phase after structure optimization. Parameters describing the C2/m and P6/mmm structures are listed in Table II.
Figure 1. The structures computed for KN3 in the pressure range 0–100 GPa.
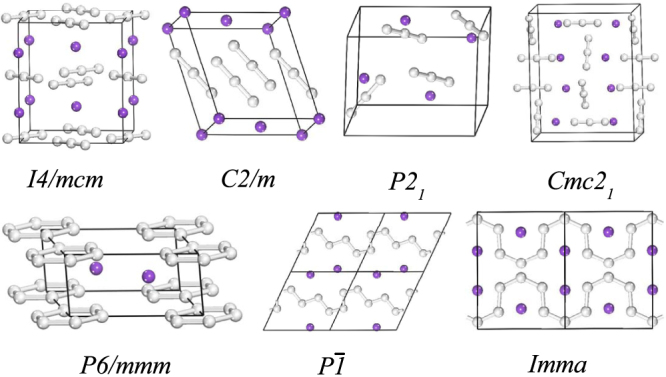
The purple spheres are potassium atoms and grey are nitrogen atoms. The 2 × 1 × 2 supercell of the 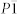 structure and the 2 × 1 × 1 supercell of the Imma structure are viewed along b-axis.
structure and the 2 × 1 × 1 supercell of the Imma structure are viewed along b-axis.
Table 1. Theoretical lattice constants and unit cell volumes V at ambient pressure compared with experimental data for I4/mcm structure.
| Present work | |||
|---|---|---|---|
| Parameter | Experiment (ref. 9) | LDA | GGA |
| a = b(Å) | 6.11094 | 5.90883 | 6.20556 |
| c(Å) | 7.09755 | 6.72696 | 7.24091 |
| V(Å3) | 265.043 | 234.867 | 278.841 |
Table 2. Optimized structural parameters of C2/m phase at 30 GPa and P6/mmm phase at 100 GPa from the first-principles calculations.
| Pressure(GPa) | Space group | Lattice parameters(Å,°) | Atomic coordinates(fractional) |
|---|---|---|---|
| 30 | C2/m | a = 4.293, b = 4.360, c = 4.767 | K 2a (0.0000, 0.0000, 0.0000) |
| α = 90, β = 107.969, γ = 90 | N 2d (0.0000, 0.5000, 0.5000) | ||
| N 4i (0.6272, 0.0000, 0.3151) | |||
| 100 | P6/mmm | a = b = 5.376, c = 2.366 | K 2d (0.6667, 0.3333, 0.5000) |
| α = β = 90, γ = 120 | N 6j (0.0000, 0.2396, 0.0000) |
The enthalpies of the most energetically competitive structures are compared over the pressure range 0-100 GPa as shown in Fig. 2. The most stable structure is a tetragonal phase with I4/mcm symmetry from ambient pressure up to 22 GPa, which is then replaced by a lower-enthalpy C2/m structure. Actually, at 13.6 GPa, Raman spectra10 have identified a phase transition that completes at 32.2 GPa, which is in agreement with the C2/m phase being stable above 22 GPa. The I4/mcm-C2/m sequence has also been observed in CsN311. During I4/mcm-C2/m transition, the tetragonal lattice is distorted under pressure, and the orientation of 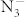 anions changes between the two K layers. The
anions changes between the two K layers. The  anion in the C2/m structure is parallel to one another. Moreover, compression induces a symmetry reduction and
anion in the C2/m structure is parallel to one another. Moreover, compression induces a symmetry reduction and 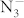 anions are still in a molecular state in this transition, which are consistent with the experimental results10. Above 40 GPa, a hexagonal structure with P6/mmm symmetry is favored over other structures and remains the lowest-enthalpy phase up to 100 GPa. We find that the crystal structures of KN3 containing the
anions are still in a molecular state in this transition, which are consistent with the experimental results10. Above 40 GPa, a hexagonal structure with P6/mmm symmetry is favored over other structures and remains the lowest-enthalpy phase up to 100 GPa. We find that the crystal structures of KN3 containing the 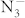 ions are energetically favorable at lower pressure, while at higher pressure, there is a tendency to the N chains or rings. Although pressure induces the rearrangement of azide ions, the formation of N–N single bond needs further compression. The dependence of volume on pressure is shown in the inset of Fig. 2. The volume reductions of ~2.3% and ~9.5% are found for the I4/mcm–C2/m and C2/m–P6/mmm transitions, respectively. The discontinuous change in volume indicates that the two phase transitions are first order. As the transformation from C2/m to P6/mmm involves the forming of N-N bonds and is reconstructive, there is a large kinetic barrier. In addition, the possibility of decomposition formula of
ions are energetically favorable at lower pressure, while at higher pressure, there is a tendency to the N chains or rings. Although pressure induces the rearrangement of azide ions, the formation of N–N single bond needs further compression. The dependence of volume on pressure is shown in the inset of Fig. 2. The volume reductions of ~2.3% and ~9.5% are found for the I4/mcm–C2/m and C2/m–P6/mmm transitions, respectively. The discontinuous change in volume indicates that the two phase transitions are first order. As the transformation from C2/m to P6/mmm involves the forming of N-N bonds and is reconstructive, there is a large kinetic barrier. In addition, the possibility of decomposition formula of 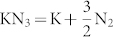 is checked by the enthalpies of decomposition, where we consider
is checked by the enthalpies of decomposition, where we consider 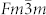 , Pnma, I41/amd, and Cmca structures for K35,36 and the cubic gauche (I213) structure for the N24,37. It turns out that the enthalpies of
, Pnma, I41/amd, and Cmca structures for K35,36 and the cubic gauche (I213) structure for the N24,37. It turns out that the enthalpies of 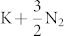 are much higher than that of KN3 at the concerned pressure range (>3.2 eV). Thus the KN3 crystal keeps stable against decomposition over the 0–100 GPa pressure range.
are much higher than that of KN3 at the concerned pressure range (>3.2 eV). Thus the KN3 crystal keeps stable against decomposition over the 0–100 GPa pressure range.
Figure 2. Calculated enthalpies per KN3 unit as the function of pressure.

The enthalpies are referenced to that of C2/m. Inset: the pressure dependence of volume for I4/mcm, C2/m and P6/mmm phases of KN3.
Since the high-pressure P6/mmm phase is novel for alkali metal azides, it is essential to investigate the properties of P6/mmm structure in detail. The ratios of a/a0 and c/c0 for the P6/mmm structure seen in Fig. 3a exhibit that the compression is anisotropic with the reduction of lattice parameter a by 13.97% over the pressure studied, while lattice parameter c decreases by 26.73%. This implies that it is more compressive along interlayer direction than intralayer direction for the layer-like P6/mmm phase. The N6 ring with D6h symmetry possesses benzene-like characteristics. The charge density of N6 ring in Fig. 3c suggests that each nitrogen atom forms two σ bonds with its two nearest neighbors by overlapping two sp2 hybrid orbitals, which is similar to the C-C bonds of C6H6. Additionally, for covalent bonds, there is a general trend in that the shorter the bond length, the stronger the bond strength. As shown in Fig. 3b, the N-N bond length of N6 ring is longer than that of N = N double bond (1.25Å) but shorter than that of N–N single bond (1.45Å). This means that the σ bond of N6 ring is weaker than the N = N double bond but stronger than the N–N single bond. In general, pressure induces the destabilization of intramolecular bonds. The structure with N2 and N6 units has been reported around 60 GPa by first-principles simulations for pure nitrogen38. Recently, a diamondoid structure of the polymeric nitrogen is predicted above 263 GPa39. These findings also provide a basis for understanding the high-pressure behavior of nitrogen-related materials. As pressure increases, there is a competition among the σ bond, ionic bond, and van der waals. Thus, it is probably fair to say that N6 rings in P6/mmm phase will transform to polymeric nitrogen networks under further compression.
Figure 3.

(a) Variation of normalized lattice parameters of P6/mmm structure with pressure. a0 and c0 are the lattice parameters of the equilibrium volume structure at zero pressure. (b) Pressure dependence of the N-N bond length of N6 ring. (c) Total charge density plotted in the (001) plane for P6/mmm phase at 100 GPa.
The ambient-pressure phase I4/mcm is insulating with a calculated band gap of 4.2 eV at 0 GPa (see supplementary Fig. S1), and the C2/m structure is also found to be an insulator with a band gap of 4.1 eV at 30 GPa (see supplementary Fig. S2). On further compression, KN3 transforms to a metallic state with P6/mmm symmetry at 40 GPa. Besides, the  and Imma structures are metastable above 60 GPa. The Imma structure is metal (see supplementary Fig. S3), and the
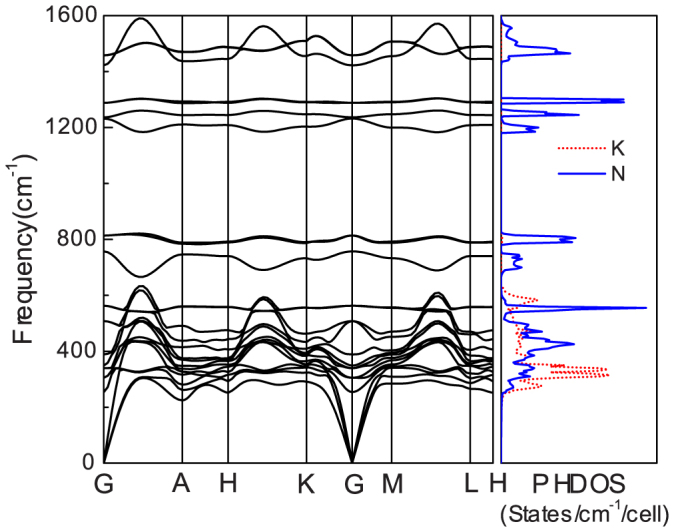
and Imma structures are metastable above 60 GPa. The Imma structure is metal (see supplementary Fig. S3), and the  structure is a semiconductor with narrow band gap (0.78 eV, see supplementary Fig. S4). Fig. 4 presents the band structure and the density of states of P6/mmm phase at 100 GPa. It reveals that P6/mmm structure is a weak metal with a small density of states at the Fermi level (0.07 states/eV/cell) which are associated with the N-2p electrons. The bands across the Fermi level are highly dispersive along the c-axis (G → A, H → K, and M → L). In addition, the Bader method40 is chosen to analyze the charge transfer as implemented in the algorithm developed by Henkelman et al.41. The Bader charges of the P6/mmm phase are around +0.73 and −0.24 for potassium and nitrogen, respectively, which suggests that the high-pressure P6/mmm phase has ionic characteristics for K-N chemical bonds. It means that the N6 anion in P6/mmm phase has nearly 8 π-electrons. Six Pz orbitals form three bonding π orbitals and three antibonding π* orbitals. Thus, the π* orbitals are partially occupied by two electrons, accompanied by two conduction bands crossing the Fermi level. Furthermore, the dynamical stability of the P6/mmm structure is established from the phonon calculations. As shown in Fig. 5, the absence of any imaginary frequency confirms the stability of the P6/mmm phase. The lower bands, ranging up to 620 cm−1, are formed by a significant mixing of K and N vibrations, while the higher bands are mostly attributed due to N-atom.
structure is a semiconductor with narrow band gap (0.78 eV, see supplementary Fig. S4). Fig. 4 presents the band structure and the density of states of P6/mmm phase at 100 GPa. It reveals that P6/mmm structure is a weak metal with a small density of states at the Fermi level (0.07 states/eV/cell) which are associated with the N-2p electrons. The bands across the Fermi level are highly dispersive along the c-axis (G → A, H → K, and M → L). In addition, the Bader method40 is chosen to analyze the charge transfer as implemented in the algorithm developed by Henkelman et al.41. The Bader charges of the P6/mmm phase are around +0.73 and −0.24 for potassium and nitrogen, respectively, which suggests that the high-pressure P6/mmm phase has ionic characteristics for K-N chemical bonds. It means that the N6 anion in P6/mmm phase has nearly 8 π-electrons. Six Pz orbitals form three bonding π orbitals and three antibonding π* orbitals. Thus, the π* orbitals are partially occupied by two electrons, accompanied by two conduction bands crossing the Fermi level. Furthermore, the dynamical stability of the P6/mmm structure is established from the phonon calculations. As shown in Fig. 5, the absence of any imaginary frequency confirms the stability of the P6/mmm phase. The lower bands, ranging up to 620 cm−1, are formed by a significant mixing of K and N vibrations, while the higher bands are mostly attributed due to N-atom.
Figure 4. Electronic band structure and projected density of states (PDOS) for P6/mmm phase at 100 GPa.
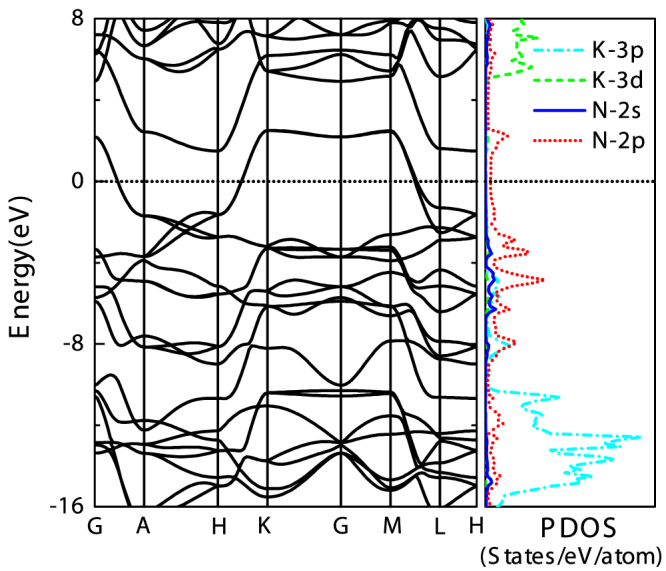
Figure 5. Phonon dispersion curve and phonon density of states (PHDOS) for P6/mmm phase at 100 GPa.
Conclusion
In summary, an evolutionary algorithm in conjunction with first-principles electronic structure computations has been used to predict the stable high-pressure phases of potassium azide. Our calculations indicate that the experimental I4/mcm phase of KN3 transforms to C2/m structure at 22 GPa and then to a hexagonal P6/mmm structure at 40 GPa. The planar N6 ring is formed in the metallic P6/mmm phase. This phase of KN3 is dynamically stable. Further compression could lead to the polymerization of N6 rings, which can be used as a potential high-energy-density material.
Author Contributions
J.Z. and Z.Z. conceived the research. J.Z. carried out the calculations. J.Z., Z.Z., H.-Q.L. and Y.-L.L. analyzed the data. J.Z. and Z.Z. wrote the paper.
Supplementary Material
supplementary information
Acknowledgments
This work was supported by the National Science Foundation of China under Grants Nos. 11174284 and NSAF U1230202, the special Funds for Major State Basic Research Project of China (973) under Grant No. 2012CB933702, Hefei Center for Physical Science and Technology under Grant no. 2012FXZY004, and Director Grants of CASHIPS. Y.-L.L. is supported by the National Science Foundation of China under Grants Nos. 11047013 and 11347007. The calculations were performed in Center for Computational Science of CASHIPS and on the ScGrid of Supercomputing Center, Computer Network Information Center of Chinese Academy of Sciences.
References
- Evans B. L., Yoffe A. D. & Gray P. Physics and chemistry of the inorganic azides. Chem. Rev. 59, 515–568 (1959). [Google Scholar]
- Bowden F. P. & Yoffe A. D. Fast Reactions in Solids (Butterworths Scientific Publications, London, 1958). [Google Scholar]
- McMahan A. K. & LeSar R. Pressure dissociation of solid nitrogen under 1 Mbar. Phys. Rev. Lett. 54, 1929–1932 (1985). [DOI] [PubMed] [Google Scholar]
- Eremets M. I., Gavriliuk A. G., Trojan I. A., Dzivenko D. A. & Boehler R. Single-bonded cubic form of nitrogen. Nat. Mater. 3, 558–563 (2004). [DOI] [PubMed] [Google Scholar]
- Eremets M. I., Gavriliuk A. G. & Trojan I. A. Single-crystalline polymeric nitrogen. Appl. Phys. Lett. 90, 171904 (2007). [Google Scholar]
- Eremets M. I. et al. Polymerization of nitrogen in sodium azide. J. Chem. Phys. 120, 10618–10623 (2004). [DOI] [PubMed] [Google Scholar]
- Medvedev S. A. et al. Phase stability of lithium azide at pressures up to 60 GPa. J. Phys.: Condens. Matter 21, 195404 (2009). [DOI] [PubMed] [Google Scholar]
- Zhu H. et al. Pressure-induced series of phase transitions in sodium azide. J. Appl. Phys. 113, 033511 (2013). [Google Scholar]
- Ji C. et al. High pressure X-ray diffraction study of potassium azide. J. Phys. Chem. Solids 72, 736–739 (2011). [Google Scholar]
- Ji C. et al. Pressure-induced phase transiton in potassium azide up to 55 GPa. J. Appl. Phys. 111, 112613 (2012). [Google Scholar]
- Hou D. et al. Series of phase transition in cesium azide under high pressure studied by in situ x-ray diffraction. Phys. Rev. B 84, 064127 (2011). [Google Scholar]
- Babu K. R., Lingam Ch. B., Tewari S. P. & Vaitheeswaran G. High-pressure study of lithium azide from density-functional calculations. J. Phys. Chem. A 115, 4521–4529 (2011). [DOI] [PubMed] [Google Scholar]
- Zhang M., Yan H., Wei Q., Wang H. & Wu Z. Novel high-pressure phase with pseudo-benzene “N6” molecule of LiN3. Europhys. Lett. 101, 26004 (2013). [Google Scholar]
- Prasad D. L. V. K., Ashcroft N. W. & Hoffmann R. Evolving structural diversity and metallicity in compressed lithium azide. J. Phys. Chem. C 117, 20838–20846 (2013). [Google Scholar]
- Wang X. et al. Polymerization of nitrogen in lithium azide. J. Chem. Phys. 139, 164710 (2013). [DOI] [PubMed] [Google Scholar]
- Zhang M. et al. Structural and electronic properties of sodium azide at high pressure: A first principles study. Solid State Commun. 161, 13–18 (2013). [Google Scholar]
- Christoe C. W. & Iqbal Z. Raman scattering in alkali azides at high pressures. Chem. Phys. Lett. 39, 511–514 (1976). [Google Scholar]
- Weir C. E., Block S. & Piermarini G. J. Compressibility of inorganic azides. J. Chem. Phys. 53, 4265–4269 (1970). [Google Scholar]
- Li J. et al. Pressure-induced polymerization of nitrogen in potassium azides. Europhys. Lett. 104, 16005 (2013). [Google Scholar]
- Oganov A. R. & Glass C. W. Crystal structure prediction using ab initio evolutionary techniques: Principles and applications. J. Chem. Phys. 124, 244704 (2006). [DOI] [PubMed] [Google Scholar]
- Glass C. W., Oganov A. R. & Hansen N. USPEX–Evolutionary crystal structure prediction. Comp. Phys. Comm. 175, 713–720 (2006). [Google Scholar]
- Kresse G. & Furthmüller J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 54, 11169–11186 (1996). [DOI] [PubMed] [Google Scholar]
- Perdew J. P., Burke K. & Ernzerhof M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868 (1996). [DOI] [PubMed] [Google Scholar]
- Kresse G. & Joubert D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 59, 1758–1775 (1999). [Google Scholar]
- Baroni S., Gironcoli S. d., Corso A. D. & Giannozzi P. Phonons and related crystal properties from density-functional perturbation theory. Rev. Mod. Phys. 73, 515–562 (2001). [Google Scholar]
- Iqbal Z. Temperature dependence of ramanactive phonons and nature of the phase transition in lithium and sodium azide. J. Chem. Phys. 59, 1769–1774 (1973). [Google Scholar]
- Cacace F., de Petris G. & Troiani A. Experimental detection of tetranitrogen. Science 295, 480–481 (2002). [DOI] [PubMed] [Google Scholar]
- Vij A. et al. Polynitrogen Chemistry. Synthesis, chatacterization, and crystal structure of surprisingly stable fluoroantimonate salts of N5+. J. Am. Chem. Soc. 123, 6308–6313 (2001). [DOI] [PubMed] [Google Scholar]
- Saxe P. & Schaefer H. F. Cyclic D6h hexaazabenzeneA relative minimum on the N6 potential energy hypersurface? J. Am. Chem. Soc. 105, 1760–1764 (1983). [Google Scholar]
- Glukhovtsev M. N. & Schleyer P. v. R. Structures, bonding and energies of N6 isomers. Chem. Phys. Lett. 198, 547–554 (1992). [Google Scholar]
- Tobita M. & Bartlett R. J. Structures and stability of N6 isomers and their spectroscopic characteristics. J. Phys. Chem. A 105, 4107–4113 (2001). [Google Scholar]
- Raczyńska E. D. On the basicity and π-electron delocalization of ‘hexaazabenzene' N6 - Quantum-chemical studies. Comp. Theor. Chem. 971, 38–41 (2011). [Google Scholar]
- Wilson K. J., Perera S. A., Bartlett R. J. & Watts J. D. Stabilization of the pseudo-benzene N6 ring with oxygen. J. Phys. Chem. A 105, 7693–7699 (2001). [Google Scholar]
- Duan H.-X. & Li Q.-S. A series of novel aromatic compounds with a planar N6 ring. Chem. Phys. Lett. 432, 331–335 (2006). [Google Scholar]
- Ma Y., Oganov A. R. & Xie Y. High-pressure structures of lithium, potassium, and rubidium predicted by an ab initio evolutionary algorithm. Phys. Rev. B 78, 014102 (2008). [Google Scholar]
- Marqués M. et al. Potassium under pressure: A pseudobinary ionic compound. Phys. Rev. Lett. 103, 115501 (2009). [DOI] [PubMed] [Google Scholar]
- Kotakoski J. & Albe K. First-principles calculations on solid nitrogen: A comparative study of high-pressure phases. Phys. Rev. B 77, 144109 (2008). [Google Scholar]
- Mattson W. D., Sanchez-Portal D., Chiesa S. & Martin R. M. Prediction of new phases of nitrogen at high pressure from first-principles simulations. Phys. Rev. Lett. 93, 125501 (2004). [DOI] [PubMed] [Google Scholar]
- Wang X. et al. Cagelike diamondoid nitrogen at high pressures. Phys. Rev. Lett. 109, 175502 (2012). [DOI] [PubMed] [Google Scholar]
- Bader R. F. W. Atoms in Molecules: A Quantum Theory (Oxford University Press, New York, 1990). [Google Scholar]
- Henkelman G., Arnaldsson A. & Jónsson H. A fast and robust algorithm for Bader decomposition of charge density. Comput. Mater. Sci. 36, 354–360 (2006). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
supplementary information