


Abstract
Eukaryotic translation termination results from the complex functional interplay between two release factors, eRF1 and eRF3, in which GTP hydrolysis by eRF3 couples codon recognition with peptidyl-tRNA hydrolysis by eRF1. Here, we present a cryo-electron microscopy structure of pre-termination complexes associated with eRF1•eRF3•GDPNP at 9.7 -Å resolution, which corresponds to the initial pre-GTP hydrolysis stage of factor attachment and stop codon recognition. It reveals the ribosomal positions of eRFs and provides insights into the mechanisms of stop codon recognition and triggering of eRF3’s GTPase activity.
INTRODUCTION
Termination occurs when a stop codon enters the ribosomal A site and consists of stop codon recognition followed by peptide release, which involves the nucleophilic attack of a water molecule on the P-site peptidyl-tRNA in the ribosomal peptidyl transferase center (PTC). Eukaryotic termination is mediated by two directly interacting release factors: eRF1, which is responsible for stop codon recognition and triggering peptide release, and eRF3, a GTPase that strongly stimulates peptide release by eRF1 in a GTP-dependent manner (1–3). eRF1, in turn, stabilizes binding of GTP to eRF3 so that they form a stable ternary complex (4,5), and is required for eRF3’s ribosome-dependent GTPase activity (6). eRF1 has omnipotent decoding capacity and recognizes all three stop codons.
It comprises N-terminal (N), middle (M) and C-terminal (C) domains (7). The rigid core of domain C contains a flexible insertion forming a mini-domain (8). Domain N is involved in stop codon recognition. Although the mechanism by which eRF1 responds to all three stop codons is not clear, extensive mutational and genetic analyses identified the essential role in this process of GTS31–33 (human numbering), TASNIKS58–64 and YxCxxxF125–131 motifs located at the apex of the N-domain (7,9–18). eRF1’s domain M contains the universal GGQ loop, whose placement into the PTC causes rearrangement of rRNA, allowing a water molecule to enter and induce peptide release (1,7,19–22).
eRF3 consists of the essential C-terminal region comprising GTP-binding (G) domain and β-barrel domains 2 and 3, which are homologous to elongation factors EF-Tu and eEF1A (23), and a non-conserved N-terminal region that is not essential for termination (24). eRF1’s C and M domains interact with eRF3 (15,25). eRF1 and eRF3 bind to the pre-termination complexes (pre-TCs) as an eRF1•eRF3•GTP ternary complex, but peptide release does not occur until eRF3 hydrolyzes GTP. GTP hydrolysis releases eRF1’s domain M from eRF3, which enables its GGQ loop to enter the PTC and trigger peptide release.
Although the structures of individual eRFs have been determined (7,15,23), the structural basis for key steps in termination, such as stop codon recognition or triggering of eRF3’s GTPase activity, remains unresolved. Our previous cryo-EM structure of the eRF1•eRF3•GDPNP-bound pre-TC had a resolution of 18 Å (26), which did not allow accurate modeling of the ribosome-bound factors and therefore could not address such questions. The advent of a more powerful classification algorithm, RELION (27), prompted us to re-examine the original data set (26), resulting in a much higher resolution (9.7 Å), which enabled us to propose a more detailed model of the interactions pivotal for peptide release.
MATERIALS AND METHODS
Data processing
Mammalian pre-TCs were assembled in vitro on a derivative of β-globin messenger RNA (mRNA) encoding MVHL tetrapeptide followed by a UAA stop codon, using purified rabbit 40S and 60S ribosomal subunits, initiation (2,3,1,1A,4A,4B,4G,5 and 5B) and elongation (1A and 2) factors and aminoacylated tRNAs (26). eRF1•eRF3•GDPNP-bound pre-TCs were formed with full-length eRF1 and eRF3 lacking the non-essential N-terminal 138aa (26). A cryo-EM data set of the eRF1•eRF3•GDPNP-bound pre-TCs comprising 195 432 particles (26) was aligned and refined using the RELION autorefine procedure (27). It resulted in a 9.1-Å structure [gold standard Fourier shell correlation (FSC) = 0.143] showing fragmented densities in the intersubunit space, indicative of heterogeneity. The data set was then subjected to RELION classification (28) starting with 10 classes (k = 10). Of the 10 classes, class 8 (Figure 1) was well populated (48 973 particles, 25.1%), with well-defined additional masses of density in the intersubunit space corresponding to eRF1, eRF3 and P-site tRNA as previously described (26). The particles from this class were isolated, and the structure was first refined with RELION autorefine procedure. This yielded a structure at 9.7 Å (gold standard FSC = 0.143), but the density exhibited strong stretching artifacts when filtered at the measured resolution, probably due to preferred orientations of the particles on the cryo-EM grid. The alignment parameters were further refined using RELION classification with k = 1 and a T factor of four for three iterations at an angular spacing of 1.8°, followed by three additional iterations at 0.9°. The resulting reconstruction displayed the same resolution of 9.7 Å (FSC = 0.143), but the quality of the structure was very much improved and the stretching artifacts were no longer visible. The statistics of alignment precision estimated by RELION were also significantly improved [improvement in terms of overall accuracy of rotations, 41%; overall accuracy of translations, 43% and average Pmax at 1.8° angular step, 180% (Supplementary Table S1)].
Figure 1.

Unsupervised 3D classification. The 195 432 particles were first refined with RELION (27) to give a 9.1-Å ribosome reconstruction having low occupancy for the factors and tRNAs (top). The particles were then classified with RELION (27) starting with 10 seeds. The 10 resulting maps of the ribosome (bottom) are shown from the P stalk side (top vignette), with a close-up on the eRF1-eRF3 binding site (middle vignette), and from the 40S solvent side (bottom vignette). Bottom table: strength of the electron density for each domain of eRF1, eRF3 and tRNAs, as well as position of the L1 stalk.
Modeling of eRF1, eRF3, mRNA and P-site tRNALeu
The cryo-EM map segmentation was performed using the Segger (29) plug-in in UCSF Chimera (30). Models of human eRF1 and eRF3 were built based on several experimental structures. Atomic coordinates of eRF1 and eRF3 domains 2 and 3 were taken from their crystal structure (15) (PDBID: 3E1Y), and the C-terminal mini-domain structure of eRF1 (residues 326–373) was modeled based on its nuclear magnetic resonance (NMR) structure (8) (PDBID: 2KTU). Most of eRF3 G-domain (residues 317–439, 207–216) was modeled by homology based on the crystal structure of Saccharomyces pombe eRF3•GDP (23) (PDBID: 1R5B) because of its significantly higher resolution over the S. pombe eRF3•GMPPNP crystal structure (23) (2.35 versus 3.20 Å, respectively). Only the GTP-binding pocket of the G-domain (residues 217–231) was modeled based on the crystal structure of eRF3•GMPPNP (23) (PDBID: 1R5O). Missing residues and the switch region (residues 232–316) in eRF3 G-domain were modeled by homology based on the archeal elongation factor 1α (aEF1α) taken from the eRF1•GTP-bound aEF1α crystal structure (31) (PDBID: 3VMF). The arrangement between eRF3’s domains II and III and G-domain was derived from the latter crystal structure as well. The P-site tRNALeu was modeled from its crystal structure in complex with leucyl-tRNA synthetase from Pyrococcus horikoshii (32) (PDBID: 1WZ2). The mRNA was modeled based on the mRNA of the crystal structure of the Thermus thermophilus 70S ribosome bound with the Q253P mutant form of release factor 2 (22) (PDBID: 4KFK). All homology modeling was done using SWISS-MODEL (33,34).
Molecular dynamics flexible fitting
The derived model was first rigid body-fitted into its corresponding segmented cryo-EM density map using UCSF Chimera (30). Then, using the molecular graphics software VMD (35), the system was prepared for molecular dynamics flexible fitting (MDFF) (36) in explicit solvent, a procedure which applies the cryo-EM map as an additional potential to the system—in this study the eRF1•eRF3•tRNA•mRNA cryo-EM segmented map—thus comprising only the molecules to be simulated: eRF1, eRF3, mRNA and P-site tRNALeu. The MDFF system was embedded in a box of TIP3P water molecules with an extra 12-Å padding in each direction. The system was neutralized by potassium ions, and an excess of ∼0.2 M KCl was added. The simulated system was prepared using CHARMM force field parameters [Combined CHARMM All-hydrogen topology file for CHARMM22 proteins and CHARMM27 lipids (37)]. The system was energy-minimized in 600 steps in the molecular dynamics simulation package NAMD (38). After minimization, the fitting trajectories were run for 400 ps, once the root-mean-square deviation and cross-correlation coefficient had stabilized.
RESULTS AND DISCUSSION
Structure determination of the mammalian eRF1•eRF3•GDPNP-bound pre-TC
Classification of the cryo-EM data set comprising 195 432 particles of the mammalian eRF1•eRF3•GDPNP-bound pre-TC formed on mRNA encoding MVHL tetrapeptide, followed by a UAA stop codon, yielded 10 classes with different factor and tRNA occupancies (Figure 1). One of these classes (25% of the particles) had P-site tRNA along with well-defined eRF1 and eRF3 densities. The characteristic long variable loop of tRNALeu was apparent in the P-site tRNA, indicating that it was correctly programmed with an A-site stop codon. The eRF1•eRF3•GDPNP-bound pre-TC class yielded a 9.7-Å reconstruction (Figure 2a), which allowed precise docking of each factor's domains. eRF1 and eRF3 were modeled (Figures 2b–d) based on the crystal structures of eRF1, eRF3, eRF1-eRF32-3 and aRF1-aEF1α (7,15,23,31) and the NMR structure of eRF1’s C-domain (8). The model was refined using MDFF (36) with secondary structure constraints.
Figure 2.
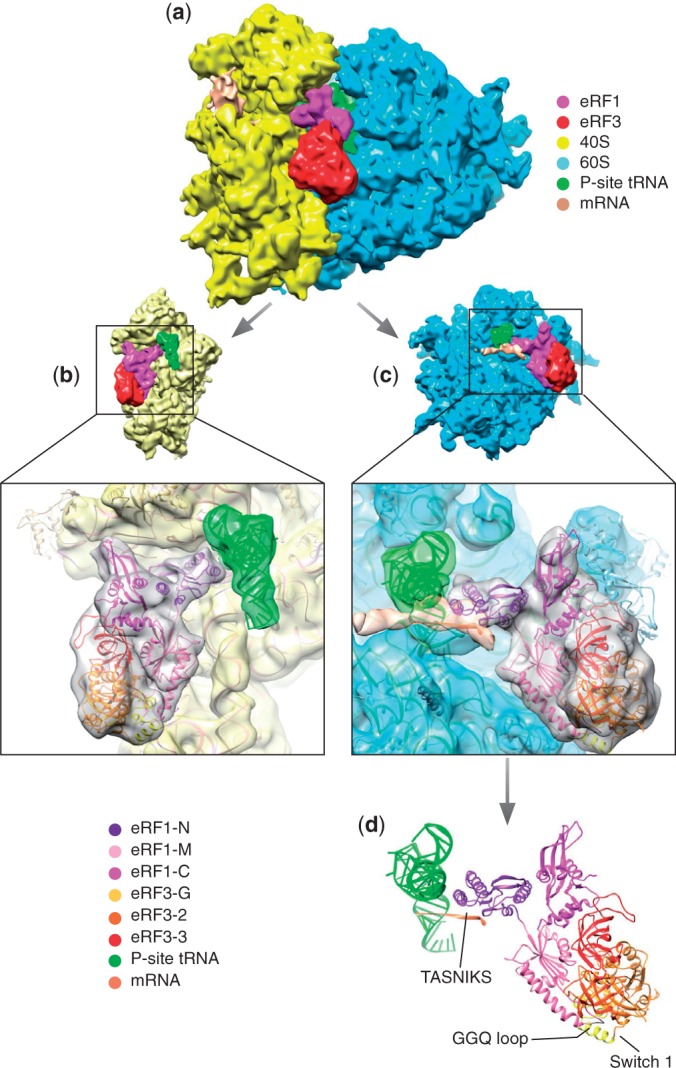
Overview of the complex. (a) Overview of the map showing the 40S (yellow), 60S (blue), eRF1 (Magenta), eRF3 (red), P-site tRNA (green) and mRNA (coral). (b) Atomic model fitted into its density viewed from the 60S side with the 60S density removed. (c) Atomic model fitted into its density viewed from the 40S side with the 40S density removed. (d) Atomic model in the same orientation as in c, showing eRF1, eRF3, the P-site tRNA and mRNA path.
Overview of the complex
The position and conformation of ribosome-bound eRF1/eRF3 are similar to those of aa-tRNA/EF-Tu (39) and the eRF1/eRF3 paralogs Dom34/Hbs1 (40) involved in the dissociation of stalled elongation complexes (41,42), with eRF1’s domain N binding to the decoding center, and the rigid core and mini-domain of eRF1’s domain C forming a bridge between the P stalk and the beak of the 40S subunit. eRF1 interacts with eRF3’s domain 3 via its domain C, whereas eRF1’s extended domain M containing the GGQ motif is tucked between eRF3’s domains 2 and G. eRF3 is bound to the universal GTPase-associated center (GAC) of the ribosome, between the sarcin–ricin loop (SRL) on the 60S subunit, and helices (h) 5 and 14 of 18S rRNA on the 40S subunit. No additional conformational changes were observed on the ribosome at the improved resolution, compared with those reported previously (26): it is in a non-rotated state, the L1 stalk in the open position (Figure 1) and the P stalk base shifted inward. However, what was previously interpreted as the result of a ribosomal rearrangement at the mRNA entrance (26) now appears more likely to be an mRNA bundle (Figure 2 and Supplementary Figure S1).
Interaction between eRF1-N and the stop codon
eRF1’s N-domain reaches deep into the decoding center (Figure 2), establishing multiple contacts with the 40S subunit, including h18, h30, h31, h34 and h44 of 18S rRNA and ribosomal proteins rpS30e and rpS31e. R65/R68 in the long α3 helix are in proximity to nucleotides 1330–1331 of h34, consistent with these residues’ strong influence on eRF1’s ribosomal binding (11). In our structure, mRNA occupies its normal position in the decoding center, and eRF1’s N-domain interacts directly with the stop codon (Figure 3), primarily via the TASNIKS58–64 motif, consistent with ultraviolet cross-linking of the stop codon to this element (10). The positions of the mRNA bases and amino acid side-chains cannot be determined precisely at the present resolution. However, the observed proximity of particular residues and bases in the fitted structures makes it likely that they interact with one another. The TASN residues appear to be close to the first position of the stop codon, I to the second and KS to the third. The proximity of K to the nucleotide immediately downstream of the stop codon could potentially contribute to the mechanism by which this base influences termination efficiency (43,44). In addition to TASNIKS, three residues from GTS31–33 and YxCxxxF125–131 motifs are also positioned within interacting distance of the stop codon: G31 and T32 are close to the third and second base, respectively, and C127 to the bases in positions 1 and 2. The positions of stop codon nucleotides relative to these three residues are consistent with the experimentally determined influence of T32 and C127 on the specificity of recognition of the nucleotide in the second position of the stop codon (14,15,17). Y125 and F131 are farther away, and their influence on termination (9,12,15,17) may therefore reflect roles in stabilizing the domain structure. Additional contacts with the pre-TC may be important for proper placement of the N-domain into the decoding site. Thus, the β-sheet supporting the GTS and YCF motifs interacts with H69 of 28S rRNA, whereas the continuous density between helix α2 and the P-site tRNA (which is particularly strong between S46 at the tip of α2 and nucleotide 30 of the tRNALeu anticodon stem-loop) indicates likely contact between eRF1 and P-site tRNA. Functional interaction between them has been proposed, and preferential binding to specific tRNAs could contribute to the observed bias for certain codons preceding stop codons (45,46). Interestingly, the characteristic long variable loop of the P-site tRNALeu is also in proximity to the highly conserved P-site loop of rpL11 on the 60S subunit and to h30 and rpS18 of the 40S subunit (Supplementary Figure S2). The P-site loop of rpL11 (L5 in bacteria) contacts the T-loop of tRNA in the P/P, as well as in neighboring intermediate states, and was therefore proposed to escort tRNA through the P site during translocation (47–50). Notably, residues in the P-site loop, which are seen in proximity to the variable loop of tRNALeu in the present reconstruction, were shown to play a key role in binding of the P-site tRNA (51). The potential strengthening of the contact with the P-site loop of L11 and additional specific contacts with h30 and rpS18 may contribute to the known higher affinity of tRNALeu to the P/P state, accounting for the increased stability of ribosomal complexes containing P-site tRNALeu after peptide release (52).
Figure 3.

Close-up of the eRF1 N-domain and mRNA showing the TASNIKS amino-acids (blue), GTS loop (red) and YxCxxxF motif (green), and their positions relative to the approximate location of the stop codon bases represented as slabs. Codon positions are indicated in orange: first position, yellow: second position and blue: third position.
Additional stabilization of eRF1’s interaction with the 40S subunit appears to be provided by its mini-domain, which protrudes toward the beak where it interacts via its flexible loop with h33/h34 of 18S rRNA, or possibly with the N-terminal tail of rpS31e (Figure 4).
Figure 4.
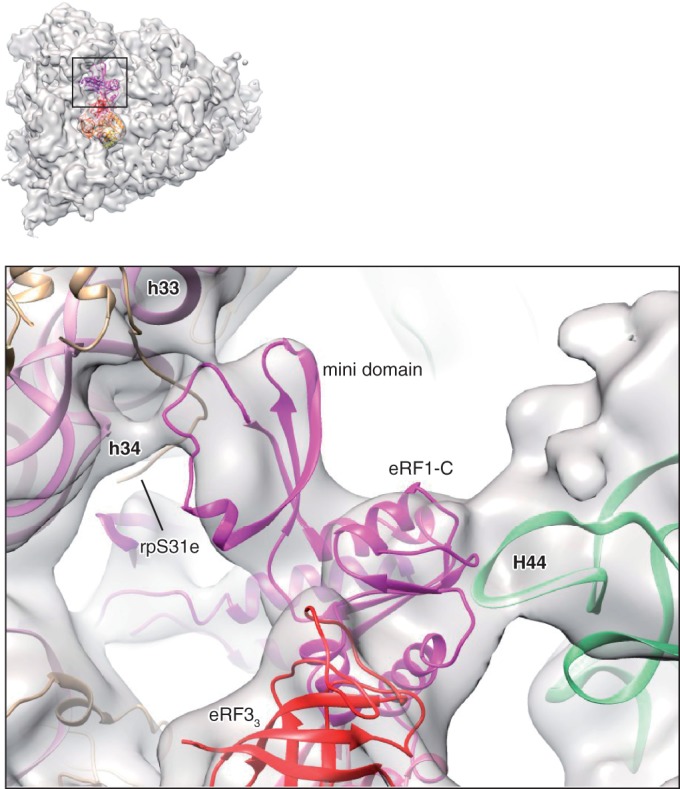
Close-up of eRF1-C and the mini-domain, showing the interaction of eRF1-C with H44 of the P stalk, and the interaction of the mini-domain with the 40S beak. rpL12 and rpLP0 not shown.
Interaction between eRF1 and eRF3
The rigid core of eRF1’s C-domain interacts with eRF3’s domain 3 as observed in the eRF1-eRF32–3 crystal structure (15), and with H44 of the P stalk (Figures 2 and 4). rpL12 is in proximity to eRF1’s C-domain and domain 3 of eRF3 and may contact them directly, but its scattered density suggests that they do not interact stably in this configuration. The inward rotation of the stalk base (26) is similar to that observed in Dom34/Hbs1-associated ribosomes (40).
The topology of eRF1’s M domain is now clearly visible, folded back onto eRF3, with the GGQ loop close to the switch I region, similarly to the interaction between Dom34’s central domain with Hbs1 (40) and aRF1 domain B with aEF1α (31) (Supplementary Figure S3), whereas α-helix 8 interacts with eRF3’s domain 3, close to the conserved GRFTLRD613–619 motif (23) (Figure 5a).
Figure 5.

(a) Close-up of eRF3 showing the eRF1-eRF3 model fitted in the electron density together with the human 80S ribosome model (PDBID: 3J3A/B/D/F) [18S rRNA (purple), small subunit ribosomal proteins (beige), 28S rRNA (green), large subunit ribosomal proteins (light blue)]. (b) The 70S ribosome/EF-Tu/tRNA/GDPNP from (39) compared with the 80S ribosome-eRF1-eRF3 in (a).
Structure of eRF3 bound to the ribosome
eRF3’s domains are well resolved: domains 2 and 3 have the same orientation relative to eRF1 as in the eRF1-eRF32–3 complex (15), whereas the G-domain is positioned similarly to that of aEF1α in the aRF1-aEF1α complex (31) and to G-domains in other GTPases, such as EF-Tu (39), and contacts the GAC in a similar manner (Figure 5). The switch I region of eRF3's G-domain is well ordered and is tucked between the SRL, h14 of 18S rRNA and the GGQ loop of eRF1-M. The SRL nt 4599–4600 and 4601–4603 are in proximity to DK273–274 in the switch I and GE326–327 in helix α5, respectively (Figure 5a).
GTPase-activation mechanism
There are numerous similarities in the functioning of eRF1•eRF3•GTP and aa-tRNA•EF-Tu•GTP complexes that suggest likely conservation of the GTPase-activation mechanism: hydrolysis of EF-Tu/eRF3-bound GTP is triggered by binding to the A site of aa-tRNA or of eRF1, respectively, followed by dissociation of the factor and accommodation of either the aa-tRNA acceptor end or the apical loop of eRF1’s M domain in the PTC. In the case of EF-Tu, binding of aa-tRNA•EF-Tu•GTP to the ribosome induces a shift in domain 2, resulting in the interaction of a highly conserved β-turn with h5 and disruption of the interaction of the 3'-end of tRNA with the switch I loop, which leads to GTPase activation in a process that also involves direct participation of the SRL (39,53). A similar contact is established between h5 on the 40S subunit and the conserved KDxGT449–453 β-turn in eRF3’s domain 2 (Figure 5a), and a C3302U substitution in the Saccharomyces cerevisiae SRL (equivalent to human C4603U) caused a termination defect (54). One notable difference is that there is additional density between h14 and eRF1’s M domain (Figure 5a), which involves the conserved R192 that is important for GTP hydrolysis, but not for GTP binding or the eRF1/eRF3 interaction (15). A hypothetical mechanism of GTPase activation based on the presumed similarity with EF-Tu would be that off the ribosome, eRF1’s M domain is too tightly packed onto the switch I region to allow GTP hydrolysis, but after eRF1•eRF3•GTP binds to the ribosome, the R192/h14 and β-turn/h5 interactions together pull the M-domain from eRF3, loosening the switch I region. The P stalk may act as a gatekeeper in this regard, impairing the eRF1-M/h14 and eRF3/h5 interactions until eRF1’s binding to a bona fide stop codon is strong enough to pull the P stalk inward. Although eRF1 can stimulate ribosome-dependent GTPase activity in the absence of an A-site stop codon (6), it is likely that GTP hydrolysis would be accelerated by the interaction between eRF1 and a stop codon.
In eRF1•eRF3•GDPNP-bound pre-TCs, the GGQ loop of eRF1 is positioned far away from the PTC, and its accommodation in the PTC would require substantial rearrangement of eRF1’s domain M. Superimposing the structure of eRF1 in the eRF1•eRF3•GDPNP-bound pre-TC onto the crystal structures of individual or eRF32–3-bound eRF1 (7,15) shows that domain M can undergo large hinge movements (Figure 6). If eRF3-bound eRF1 adopts a strained conformation on binding to the ribosome similar to that of the A/T state of EF-Tu-bound tRNA (55,56), then dissociation of eRF3 following GTP hydrolysis would initiate spontaneous relaxation of eRF1 resulting in accommodation of the GGQ loop in the PTC. Full entry of the GGQ loop into the PTC would then likely be facilitated by the high mobility of the apical GGQ loop in the thermal environment (57), the positive charge of this region promoting its retention in the PTC following entry.
Figure 6.

(a) Superimposition onto domain C of eRF1 in the crystal structure (7) (PDBID: 1DT9) and bound to the pre-TC ribosome. The mini-domain is not displayed for clarity. (b) Comparison between the positions of eRF1-M in the eRF1-eRF3-ribosome structure, the eRF1-eRF32–3 (15) (PDBID: 3E1Y) and the eRF1 (7) (PDBID: 1DT9) crystal structures with their domain C superimposed. Position of the P-site tRNA in the context of the pre-termination complex is shown to compare the distance necessary to be travelled for the GGQ loop to reach the peptidyl transfer center (PTC) with the amplitude of movement between the different conformations of eRF1 observed. Only the N domain of eRF1 in the pre-TC cryo-EM structure is shown for clarity.
CONCLUSION
Our structure revealed the topology of the interaction between eRF1’s N-domain and the UAA stop codon, as well as the intricate network of interactions between eRF3’s switch I region, eRF1’s GGQ loop and the GAC. It suggests that GTP hydrolysis may be triggered once eRF1 is properly bound to the STOP codon in a manner similar to that of EF-Tu. Now, atomic resolution structures of this complex bound to the three different stop codons may be required to achieve complete understanding of how a single factor can recognize them all.
ACCESSION NUMBERS
The Cryo-EM map is deposited in the EMDataBank with accession code EMD-5801. The atomic model of eRF1-eRF3 bound to the pre-TC is deposited in the Protein Data Bank with the accession code 3J5Y.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Howard Hughes Medical Institute and the National Institute of Health [R01 GM29169 to J.F., R01 GM80623 to T.V.P.]. Funding for open access charge: National Institutes of Health.
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENT
The authors thank Melissa Thomas for assistance in the preparation of the figures and Harry Kao for assistance with computer hardware.
REFERENCES
- 1.Jackson RJ, Hellen CU, Pestova TV. Termination and post-termination events in eukaryotic translation. Adv. Protein Chem. Struct. Biol. 2012;86:45–93. doi: 10.1016/B978-0-12-386497-0.00002-5. [DOI] [PubMed] [Google Scholar]
- 2.Alkalaeva EZ, Pisarev AV, Frolova LY, Kisselev LL, Pestova TV. In vitro reconstitution of eukaryotic translation reveals cooperativity between release factors eRF1 and eRF3. Cell. 2006;125:1125–1136. doi: 10.1016/j.cell.2006.04.035. [DOI] [PubMed] [Google Scholar]
- 3.Salas-Marco J, Bedwell DM. GTP hydrolysis by eRF3 facilitates stop codon decoding during eukaryotic translation termination. Mol. Cell. Biol. 2004;24:7769–7778. doi: 10.1128/MCB.24.17.7769-7778.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pisareva VP, Pisarev AV, Hellen CU, Rodnina MV, Pestova TV. Kinetic analysis of interaction of eukaryotic release factor 3 with guanine nucleotides. J. Biol. Chem. 2006;281:40224–40235. doi: 10.1074/jbc.M607461200. [DOI] [PubMed] [Google Scholar]
- 5.Mitkevich VA, Kononenko AV, Petrushanko IY, Yanvarev DV, Makarov AA, Kisselev LL. Termination of translation in eukaryotes is mediated by the quaternary eRF1*eRF3*GTP*Mg2+ complex. The biological roles of eRF3 and prokaryotic RF3 are profoundly distinct. Nucleic Acids Res. 2008;34:3947–3954. doi: 10.1093/nar/gkl549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Frolova L, Le Goff X, Zhouravleva G, Davydova E, Philippe M, Kisselev L. Eukaryotic polypeptide chain release factor eRF3 is an eRF1-and ribosome-dependent guanosine triphosphatase. RNA. 1996;2:334–341. [PMC free article] [PubMed] [Google Scholar]
- 7.Song H, Mugnier P, Das AK, Webb HM, Evans DR, Tuite MF, Hemmings BA, Barford D. The crystal structure of human eukaryotic release factor eRF1—mechanism of stop codon recognition and peptidyl-tRNA hydrolysis. Cell. 2000;100:311–321. doi: 10.1016/s0092-8674(00)80667-4. [DOI] [PubMed] [Google Scholar]
- 8.Mantsyzov AB, Ivanova EV, Birdsall B, Alkalaeva EZ, Kryuchkova PN, Kelly G, Frolova LY, Polshakov VI. NMR solution structure and function of the C-terminal domain of eukaryotic class 1 polypeptide chain release factor. FEBS J. 2010;277:2611–2627. doi: 10.1111/j.1742-4658.2010.07672.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bertram G, Bell HA, Ritchie DW, Fullerton G, Stansfield I. Terminating eukaryote translation: domain 1 of release factor eRF1 functions in stop codon recognition. RNA. 2000;6:1236–1247. doi: 10.1017/s1355838200000777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chavatte L, Seit-Nebi A, Dubovaya V, Favre A. The invariant uridine of stop codons contacts the conserved NIKSR loop of human eRF1 in the ribosome. EMBO J. 2002;21:5302–5311. doi: 10.1093/emboj/cdf484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Frolova L, Seit-Nebi A, Kisselev L. Highly conserved NIKS tetrapeptide is functionally essential in eukaryotic translation termination factor eRF1. RNA. 2002;8:129–136. doi: 10.1017/s1355838202013262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Seit-Nebi A, Frolova L, Kisselev L. Conversion of omnipotent translation termination factor eRF1 into ciliate-like UGA-only unipotent eRF1. EMBO Rep. 2002;3:881–886. doi: 10.1093/embo-reports/kvf178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ito K, Frolova L, Seit-Nebi A, Karamyshev A, Kisselev L, Nakamura Y. Omnipotent decoding potential resides in eukaryotic translation termination factor eRF1 of variant-code organisms and is modulated by the interactions of amino acid sequences within domain 1. Proc. Natl Acad. Sci. USA. 2002;99:8494–8499. doi: 10.1073/pnas.142690099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fan-Minogue H, Du M, Pisarev AV, Kallmeyer AK, Salas-Marco J, Keeling KM, Thompson SR, Pestova TV, Bedwell DM. Distinct eRF3 requirements suggest alternate eRF1 conformations mediate peptide release during eukaryotic translation termination. Mol. Cell. 2008;30:599–609. doi: 10.1016/j.molcel.2008.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cheng Z, Saito K, Pisarev AV, Wada M, Pisareva VP, Pestova TV, Gajda M, Round A, Kong C, Lim M, et al. Structural insights into eRF3 and stop codon recognition by eRF1. Genes Dev. 2009;23:1106–1118. doi: 10.1101/gad.1770109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Conard SE, Buckley J, Dang M, Bedwell GJ, Carter RL, Khass M, Bedwell DM. Identification of eRF1 residues that play critical and complementary roles in stop codon recognition. RNA. 2012;18:1210–1221. doi: 10.1261/rna.031997.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kryuchkova P, Grishin A, Eliseev B, Karyagina A, Frolova L, Alkalaeva E. Two-step model of stop codon recognition by eukaryotic release factor eRF1. Nucleic Acids Res. 2013;41:4573–4586. doi: 10.1093/nar/gkt113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Merritt GH, Naemi WR, Mugnier P, Webb HM, Tuite MF, von der Haar T. Decoding accuracy in eRF1 mutants and its correlation with pleiotropic quantitative traits in yeast. Nucleic Acids Res. 2010;38:5479–5492. doi: 10.1093/nar/gkq338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Frolova LY, Tsivkovskii RY, Sivolobova GF, Oparina NY, Serpinsky OI, Blinov VM, Tatkov SI, Kisselev LL. Mutations in the highly conserved GGQ motif of class 1 polypeptide release factors abolish ability of human eRF1 to trigger peptidyl-tRNA hydrolysis. RNA. 1999;5:1014–1020. doi: 10.1017/s135583829999043x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Laurberg M, Asahara H, Korostelev A, Zhu J, Trakhanov S, Noller HF. Structural basis for translation termination on the 70S ribosome. Nature. 2008;454:852–857. doi: 10.1038/nature07115. [DOI] [PubMed] [Google Scholar]
- 21.Weixlbaumer A, Jin H, Neubauer C, Voorhees RM, Petry S, Kelley AC, Ramakrishnan V. Insights into translational termination from the structure of RF2 bound to the ribosome. Science. 2008;322:953–956. doi: 10.1126/science.1164840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Santos N, Zhu J, Donohue JP, Korostelev AA, Noller HF. Crystal structure of the 70S ribosome bound with the Q253P mutant form of release factor RF2. Structure. 2013;21:1258–1263. doi: 10.1016/j.str.2013.04.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kong C, Ito K, Walsh MA, Wada M, Liu Y, Kumar S, Barford D, Nakamura Y, Song H. Crystal structure and functional analysis of the eukaryotic class II release factor eRF3 from S. pombe. Mol. Cell. 2004;14:233–245. doi: 10.1016/s1097-2765(04)00206-0. [DOI] [PubMed] [Google Scholar]
- 24.Kushnirov VV, Ter-Avanesyan MD, Telckov MV, Surguchov AP, Smirnov VN, Inge-Vechtomov SG. Nucleotide sequence of the SUP2 (SUP35) gene of Saccharomyces cerevisiae. Gene. 1988;66:45–54. doi: 10.1016/0378-1119(88)90223-5. [DOI] [PubMed] [Google Scholar]
- 25.Kononenko AV, Mitkevich VA, Dubovaya VI, Kolosov PM, Makarov AA, Kisselev LL. Role of the individual domains of translation termination factor eRF1 in GTP binding to eRF3. Proteins. 2008;70:388–393. doi: 10.1002/prot.21544. [DOI] [PubMed] [Google Scholar]
- 26.Taylor D, Unbehaun A, Li W, Das S, Lei J, Liao HY, Grassucci RA, Pestova TV, Frank J. Cryo-EM structure of the mammalian eukaryotic release factor eRF1-eRF3-associated termination complex. Proc. Natl Acad. Sci. USA. 2012;109:18413–18418. doi: 10.1073/pnas.1216730109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Scheres SH. RELION: implementation of a Bayesian approach to cryo-EM structure determination. J. Struct. Biol. 2012;180:519–530. doi: 10.1016/j.jsb.2012.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Scheres SH. A Bayesian view on cryo-EM structure determination. J. Mol. Biol. 2012;415:406–418. doi: 10.1016/j.jmb.2011.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pintilie GD, Zhang J, Goddard TD, Chiu W, Gossard DC. Quantitative analysis of cryo-EM density map segmentation by watershed and scale-space filtering, and fitting of structures by alignment to regions. J. Struct. Biol. 2010;3:427–438. doi: 10.1016/j.jsb.2010.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 2004;13:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 31.Kobayashi K, Saito K, Ishitani R, Ito K, Nureki O. Structural basis for translation termination by archaeal RF1 and GTP-bound EF1α complex. Nucleic Acids Res. 2012;40:9319–9328. doi: 10.1093/nar/gks660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fukunaga R, Yokoyama S. Aminoacylation complex structures of leucyl-tRNA synthetase and tRNALeu reveal two modes of discriminator-base recognition. Nat. Struct. Mol. Biol. 2005;12:915–922. doi: 10.1038/nsmb985. [DOI] [PubMed] [Google Scholar]
- 33.Bordoli L, Kiefer F, Arnold K, Benkert P, Battey J, Schwede T. Protein structure homology modeling using SWISS-MODEL workspace. Nat. Protoc. 2009;1:1–13. doi: 10.1038/nprot.2008.197. [DOI] [PubMed] [Google Scholar]
- 34.Schwede T, Kopp J, Guex N, Peitsch MC. SWISS-MODEL: an automated protein homology-modeling server. Nucleic Acids Res. 2003;13:3381–3385. doi: 10.1093/nar/gkg520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Humphrey W, Dalke A, Schulten KJ. VMD: visual molecular dynamics. Mol. Graph. 1996;1:33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 36.Trabuco LG, Villa E, Mitra K, Frank J, Schulten K. Flexible fitting of atomic structures into electron microscopy maps using molecular dynamics. Structure. 2008;16:673–683. doi: 10.1016/j.str.2008.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.MacKerell AD, Bashford D, Bellott M, Dunbrack RL, Evanseck JD, Field MJ, Fischer S, Gao J, Guo H, Ha S, et al. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B. 1998;18:3616. doi: 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- 38.Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kale L, Schulten K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005;16:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Voorhees RM, Schmeing TM, Kelley AC, Ramakrishnan V. The mechanism for activation of GTP hydrolysis on the ribosome. Science. 2010;330:835–838. doi: 10.1126/science.1194460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Becker T, Armache JP, Jarasch A, Anger AM, Villa E, Sieber H, Motaal BA, Mielke T, Berninghausen O, Beckmann R. Structure of the no-go mRNA decay complex Dom34-Hbs1 bound to a stalled 80S ribosome. Nat. Struct. Mol. Biol. 2011;18:715–720. doi: 10.1038/nsmb.2057. [DOI] [PubMed] [Google Scholar]
- 41.Pisareva VP, Skabkin MA, Hellen CU, Pestova TV, Pisarev AV. Dissociation by Pelota, Hbs1 and ABCE1 of mammalian vacant 80S ribosomes and stalled elongation complexes. EMBO J. 2011;30:1804–1817. doi: 10.1038/emboj.2011.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shoemaker CJ, Eyler DE, Green R. Dom34:Hbs1 promotes subunit dissociation.and peptidyl-tRNA drop-off to initiate no-go decay. Science. 2010;330:369–372. doi: 10.1126/science.1192430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bonetti B, Fu L, Moon J, Bedwell DM. The efficiency of translation termination is determined by a synergistic interplay between upstream and downstream sequences in Saccharomyces cerevisiae. J. Mol. Biol. 1995;251:334–345. doi: 10.1006/jmbi.1995.0438. [DOI] [PubMed] [Google Scholar]
- 44.McCaughan KK, Brown CM, Dalphin ME, Berry MJ, Tate WP. Translational termination efficiency in mammals is influenced by the base following the stop codon. Proc. Natl Acad. Sci. USA. 1995;92:5431–5435. doi: 10.1073/pnas.92.12.5431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mottagui-Tabar S, Tuite MF, Isaksson LA. The influence of 5′ codon context on translation termination in Saccharomyces cerevisiae. Eur. J. Biochem. 1998;257:249–254. doi: 10.1046/j.1432-1327.1998.2570249.x. [DOI] [PubMed] [Google Scholar]
- 46.Cridge AG, Major LL, Mahagaonkar AA, Poole ES, Isaksson LA, Tate WP. Comparison of characteristics and function of translation termination signals between and within prokaryotic and eukaryotic organisms. Nucleic Acids Res. 2006;34:1959–1973. doi: 10.1093/nar/gkl074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Spahn CM, Beckmann R, Eswar N, Penczek PA, Sali A, Blobel G, Frank J. Structure of the 80S ribosome from Saccharomyces cerevisiae—tRNA-ribosome and subunit-subunit interactions. Cell. 2001;107:373–386. doi: 10.1016/s0092-8674(01)00539-6. [DOI] [PubMed] [Google Scholar]
- 48.Yusupov MM, Yusupova GZ, Baucom A, Lieberman K, Earnest TN, Cate JH, Noller HF. Crystal structure of the ribosome at 5.5 A resolution. Science. 2001;292:883–896. doi: 10.1126/science.1060089. [DOI] [PubMed] [Google Scholar]
- 49.Fischer N, Konevega AL, Wintermeyer W, Rodnina MV, Stark H. Ribosome dynamics and tRNA movement by time-resolved electron cryomicroscopy. Nature. 2010;466:329–333. doi: 10.1038/nature09206. [DOI] [PubMed] [Google Scholar]
- 50.Bock LV, Blau C, Schröder GF, Davydov II, Fischer N, Stark H, Rodnina MV, Vaiana AC, Grubmüller H. Energy barriers and driving forces in tRNA translocation through the ribosome. Nat. Struct. Mol. Biol. 2013;20: 1390–1396. doi: 10.1038/nsmb.2690. [DOI] [PubMed] [Google Scholar]
- 51.Rhodin MH, Dinman JD. A flexible loop in yeast ribosomal protein L11 coordinates P-site tRNA binding. Nucleic Acids Res. 2010;38:8377–8389. doi: 10.1093/nar/gkq711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Skabkin MA, Skabkina OV, Hellen CU, Pestova TV. Reinitiation and other unconventional posttermination events during eukaryotic translation. Mol. Cell. 2013;51:249–264. doi: 10.1016/j.molcel.2013.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Vorstenbosch E, Pape T, Rodnina MV, Kraal B, Wintermeyer W. The G222D mutation in elongation factor Tu inhibits the codon-induced conformational changes leading to GTPase activation on the ribosome. EMBO J. 1996;15:6766–6774. [PMC free article] [PubMed] [Google Scholar]
- 54.Liu R, Liebman SW. A translational fidelity mutation in the universally conserved sarcin/ricin domain of 25S yeast ribosomal RNA. RNA. 1996;2:254–263. [PMC free article] [PubMed] [Google Scholar]
- 55.Valle M, Sengupta J, Swami NK, Grassucci RA, Burkhardt N, Nierhaus KH, Agrawal RK, Frank J. Cryo-EM reveals an active role for aminoacyl-tRNA in the accommodation process. EMBO J. 2002;21:3557–3567. doi: 10.1093/emboj/cdf326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Schmeing TM, Voorhees RM, Kelley AC, Gao YG, Murphy FV, IV, Weir JR, Ramakrishnan V. The crystal structure of the ribosome bound to EF-Tu and aminoacyl-tRNA. Science. 2009;326:688–694. doi: 10.1126/science.1179700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ivanova EV, Kolosov PM, Birdsall B, Kelly G, Pastore A, Kisselev LL, Polshakov VI. Eukaryotic class 1 translation termination factor eRF1—the NMR structure and dynamics of the middle domain involved in triggering ribosome-dependent peptidyl-tRNA hydrolysis. FEBS J. 2007;274:4223–4237. doi: 10.1111/j.1742-4658.2007.05949.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.