


Abstract
Despite the identification of many factors that facilitate ribosome assembly, the molecular mechanisms by which they drive ribosome biogenesis are poorly understood. Here, we analyze the late stages of assembly of the 50S subunit using Bacillus subtilis cells depleted of RbgA, a highly conserved GTPase. We found that RbgA-depleted cells accumulate late assembly intermediates bearing sub-stoichiometric quantities of ribosomal proteins L16, L27, L28, L33a, L35 and L36. Using a novel pulse labeling/quantitative mass spectrometry technique, we show that this particle is physiologically relevant and is capable of maturing into a complete 50S particle. Cryo-electron microscopy and chemical probing revealed that the central protuberance, the GTPase associating region and tRNA-binding sites in this intermediate are unstructured. These findings demonstrate that key functional sites of the 50S subunit remain unstructured until late stages of maturation, preventing the incomplete subunit from prematurely engaging in translation. Finally, structural and biochemical analysis of a ribosome particle depleted of L16 indicate that L16 binding is necessary for the stimulation of RbgA GTPase activity and, in turn, release of this co-factor, and for conversion of the intermediate to a complete 50S subunit.
INTRODUCTION
In bacteria, ribosome biogenesis requires the synthesis, folding, chemical modification and assembly of 3 large RNAs and 55 proteins. This complex process is completed rapidly and efficiently with bacterial cells synthesizing up to 50 000 ribosomes per generation (1). Several decades ago, the groups of Nomura (2–4) and Nierhaus (5,6) determined the thermodynamic binding dependencies for ribosomal proteins (r-proteins) in the 30S and 50S bacterial subunits and used these data to reconstruct assembly maps depicting r-protein binding. These assembly maps suggest that assembly proceeds in stages with some proteins binding directly to the RNA while others require the pre-binding of other r-proteins.
More recently, work from the Williamson (7) and Woodson (8) laboratories established that assembly of the 30S subunit does not flow through a single pathway but, instead, utilizes multiple alternative pathways, each exhibiting hierarchical binding as predicted by Nomura et al. Although similar in vitro studies have not been reported for the 50S subunit, the existence of multiple parallel pathways has also been suggested from in vivo studies (9). Overall, the assembly of the 50S subunit is less understood than that of the 30S subunit due to its larger number of components, more intricate structure and the lack of simple, highly efficient in vitro reconstruction protocol.
In the cell, 50S ribosomal subunit assembly is assisted by a variety of protein factors including a critically important class of GTPases (10). While the precise roles of many of these factors are still unknown, genetic studies suggest that these GTPases act late in 50S assembly (11–14). It is hypothesized that during ribosomal assembly these GTPases may facilitate protein–RNA interactions, participate in rRNA processing or otherwise ensure that the assembly process progresses quickly and efficiently by limiting the rRNA folding landscape and preventing the assembling subunit from entering kinetic traps common in the folding of large RNA molecules. By coupling their enzymatic activity to guanine nucleotide concentrations in the cell, these enzymes may also provide cells with a rapid and direct mechanism to shut down ribosome biogenesis in response to decreases in cellular energy levels (10,11).
The focus of this study was to gain insights into the events occurring during the late stages of 50S subunit assembly and to understand the role of the essential maturation factor RbgA in this process. This protein (also known as YlqF) is a widely conserved GTPase found in all three kingdoms of life. Although Escherichia coli lacks an RbgA homolog, this protein is found in most Gram-positive and Gram-negative bacteria as well as in all eukaryotes (11).
Cells depleted of RbgA grow at a significantly decreased rate, exhibit dramatically reduced levels of 70S ribosomes and completely lack 50S particles. Instead, they accumulate a large subunit intermediate that migrates as a 45S particle in sucrose gradients (11,15). The 45S particle exhibits disordered functional centers as visualized by electron microscopy and is severely depleted of the tertiary-binding r-proteins L16, L27 and L36 (11,15,16) suggesting it may be a late stage assembly intermediate of the 50S subunit. Depletion of other highly conserved GTPases, including ObgE, YsxC (YihA) and YphC (EngA or Der) (12–14,17) also causes the accumulation of immature large ribosomal subunits. However, it is presently unknown whether any of these ribosomal particles are physiological assembly intermediates, competent for assembly or instead simply terminal dead-end products.
We hypothesized that if the 45S particles from RbgA-depleted Bacillus subtilis cells are in fact competent for assembly, their analysis could elucidate the conformational changes occurring during the late stages of large ribosomal subunit maturation. Such an analysis could also reveal how RbgA assists this process and would constitute an important first step toward characterizing any functional interplay between RbgA and the other assembly factor GTPases.
To this end, we performed pulse-labeling experiments and determined that the 45S particle could mature into functional 70S particles. Quantitative mass spectrometry (qMS) and cryo-EM experiments revealed that the 45S particles lacked several r-proteins and, structurally, exhibited substantial distortion in multiple key functional sites of the ribosome. These results suggest that the functional core is the last region of the 50S subunit to mature during the assembly process and provide a mechanistic explanation to how the cell ensures that assembly is complete before large ribosomal particles are allowed to initiate translation. We additionally provide evidence that L16 plays a key role in this late structural rearrangement. Our results indicate that the entry of this r-protein directly or indirectly requires RbgA activity and its binding triggers most of the conformational changes that the 45S particle undergoes to become a mature 50S subunit. In addition, binding of L16 to the maturing large ribosomal subunit also triggers the GTPase activity of RbgA and the subsequent release of this assembly factor.
MATERIALS AND METHODS
Ribosome and 50S subunit purifications
The 50S and 45S subunits from Bacillus subtilis were purified from IF2-depleted (RB419) and RbgA-depleted (RB301) strains, respectively. Construction of these two strains has been previously described (11). In these cells transcription of infB (encoding IF2) and ylqF (encoding RbgA) is under the control of an isopropyl-beta-d-thiogalactopyranoside (IPTG) inducible Pspank promotor. To minimize the probability of genetic suppressors arising, strains were first grown at 37°C on LB agar plates containing 5 µg/ml of chloramphenicol and 1 mM IPTG. To initiate depletion, cells were then scraped off the plate and resuspended in 1 ml of LB media, which was then used to inoculate 50 ml of pre-warmed LB media lacking IPTG, and cultures were grown to an OD600 of 0.05. Cultures were incubated at 37°C with agitation until they reached ∼150 min doubling time. Cells were not allowed to grow beyond an OD600 of 0.5, thus if at this point cells were still exhibiting doubling times ≤150 min, then the culture was diluted again in 50 ml of new fresh pre-warmed LB media to an OD600 of 0.05. Typically, two cycles of growth dilution were required before the cells reached ∼150 min doubling time. At that point, cultures were used to inoculate 1 l of pre-warmed media to an OD600 of 0.05. Full depletion of the IF2 or RbgA proteins was obtained in cells with a doubling time of ∼120–150 min. At OD600 0.4–0.5, these cells were harvested via centrifugation at 8500 g for 15 min. Cell pellets were processed as previously described (18) to obtain the crude ribosomes with the only exception that buffers contained 15 mM magnesium acetate (versus 10 mM). To obtain the 50S or 45S fraction, the crude ribosome pellet was resuspended in buffer containing 10 mM Tris–HCl at pH 7.5, 15 mM magnesium acetate, 60 mM NH4Cl and 2 mM 2-mercaptoethanol (non-dissociating conditions). About 50–60 A260 units of the subunit suspension were layered onto a 32-ml 10–30% (wt/vol) sucrose gradient made up in the same buffer and centrifuged at 43 000 g for 16 h using a Beckman SW32 Ti rotor. Gradients were fractionated using a Brandel fractionator apparatus and an AKTAprime FPLC system (GE Healthcare). The elution profile was monitored by UV absorbance at A260, and fractions corresponding to the 50S or 45S subunit peaks were collected and pooled. Subunits were removed from the sucrose buffer by centrifugation at 100 000 g for 16 h and the pellet resuspended in storage buffer (10 mM Tris–HCl pH 7.5, 10 mM magnesium acetate, 60 mM NH4Cl, 3 mM 2-mercaptoethanol) and kept at −80°C.
L16-depleted 50S subunits were obtained by performing the cell lysis step in higher volume. In this case, cell pellets from 200 ml cultures were first washed with buffer containing 10 mM Tris–HCl pH 7.5, 10 mM MgCl2 and 60 mM KCl. Subsequently, cell pellets were resuspended in 10 ml of lysis buffer (versus 2.5 ml for control 50S subunit) containing 10 mM Tris–HCl pH 7.5, 10 mM MgCl2, 60 mM KCl, 1 mM DTT, 0.5% Tween 20 (v/v), protease inhibitor cocktail (Roche) and 27 units of RNAse free DNAse (Qiagen). Cell lysis was performed by three consecutive passes of the cells suspension through a cell pressure cell at 1200–1400 psi. Lysate was clarified by centrifuging at 16 000 g for 20 min. Crude ribosomes were pelleted by centrifugation at 166 880 g for 2 h and 25 min, and the pellet was dissolved in 1 ml of HMA 06 buffer (20 mM Tris–HCl pH 7.5, 10 mM MgCl2, 30 mM NH4Cl and 1 mM DTT). Subsequently, 1 ml of high salt HMA 08 buffer (20 mM Tris–HCl pH 7.5, 10 mM MgCl2, 800 mM NH4Cl and 1 mM DTT) was added, and the mixture was incubated on ice for 1 h. Then, ribosomes were pelleted again by centrifugation at 166 880 g for 2 h and 25 min and resuspended in 500 µl of HMA06 buffer before loading them onto a 35-ml 18–43% sucrose gradient made in buffer 10 mM Tris–HCl pH 7.5, 10 mM MgCl2, 50 mM NH4Cl and 1mM DTT. Gradients were centrifuged at 81 991 g for 14 h, and fractionation was monitored by absorbance at A260. The peak corresponding to 50S subunit was concentrated, and buffer was exchanged to buffer containing 10 mM Tris–HCl pH 7.5, 10 mM MgCl2, 60 mM KCl using Amicon Ultra-15 centrifugal concentrators (Millipore).
Quantitative mass spectrometry
15N-labeled 70S ribosomes were prepared by growing B. subtilis ATCC 6051 strain aerobically in 1 l MSpitz9 medium (8.0 mM K2HPO4, 4.4 mM KH2PO4, 0.39 mM Na3-citrate, 10 mM MgCl2, 10 mM MgSO4, 5.6 mM glucose, 50 µM Na3·EDTA, 25 mM CaCl2, 50 µM FeCl3, 0.5 µM ZnSO4, 0.5 µM CuSO4, 0.5 µM MnSO4, 0.5 µM CoCl2, 0.04 µM d-biotin, 0.02 µM folic acid, 0.08 µM vitamin B1, 0.11 µM calcium pantothenate, 0.4 nM vitamin B12, 0.2 µM nicotinamide, 0.07 µM riboflavin and 7.6 mM (15NH4)2SO4) at 37°C. Cells were harvested at OD600 ∼ 0.4 by directly adding the culture to 1 l of ice. After centrifugation at 6000 g for 20 min, cells were resuspended in buffer containing 10 mM Tris (pH 7.8), 20 mM MgCl2, 100 mM NH4Cl, 6 mM 2-mercaptoethanol and lysed using a BioSpec mini-bead beater. The lysate was clarified by centrifugation (30 000 g for 40 min) and loaded onto a 10–40% (w/v) sucrose gradient made up in the same buffer. The gradient was then centrifuged at 4°C in a Beckman SW32 Ti rotor at 83 000 g for 28 h. Fractions were collected as described above and those containing 70S particles were pooled and stored at 4°C.
14N-labeled 50S subunits and 45S particles produced and purified as described above were prepared for LC/MS analysis using published protocols (19). Briefly, 20 pmol of 15N-labeled 70S spike was mixed with each experimental sample (20 pmol) before addition of trichloroacetic acid (TCA) to a final concentration of 13% (v/v). Samples were incubated on ice overnight, and precipitated material was isolated by centrifugation, washed with 10% TCA followed by acetone and finally resuspended in 40 µl buffer B (100 mM NH4CO3, 5% acetonitrile and 5 mM dithiotreitol). After incubation for 10 min at 65°C, 10 mM iodoacetamide was added, and samples were incubated at 30°C for 30 min. Samples were then digested using 0.2 µg trypsin at 37°C overnight. Peptides were injected onto a C18 column and were eluted over 105 min across a concave 5–50% acetonitrile gradient and detected on an Agilent G1969A ESI-TOF mass spectrometer using a detection range set at 250–1300 m/z. Unique ribosomal peptides were identified in the raw data using a custom suite of software designed to identify 14N/15N peak pairs given a theoretical digest (20). For each peptide, the quantities of 14N or 15N species were calculated by fitting the isotope distribution using a Least Square Fourier Transform Convolution algorithm (21). The relative protein level for each r-protein was calculated as the 14N fitted isotope distribution amplitude divided by the sum of the 14N and 15N fitted isotope distribution amplitudes [14N/(14N + 15N)]. Each isotope distribution and its local chromatographic contour map were carefully examined and fits with low signal-to-noise ratios were excluded. To account for differences in the total amount of each experimental sample analyzed, each relative protein level was normalized to that of L24, a primary binding r-protein shown to be present in stoichiometric quantities in each sample analyzed.
To improve protein coverage and quantification accuracy, samples were run multiple times and averaged across runs. Because proteins that are completely absent from an experimental sample cannot be identified by the 14N/15N peak pair, each experimental sample was also analyzed on a AB/Sciex 5600 Triple-TOF run in MS2 mode. Peptides were eluted from analytical C18 nano-column across a 120-min linear 5–40% acetonitrile gradient at 300 nl/min. Precursor ions were detected in a 400–1250 m/z mass window, and product ions were detected with a 100–1800 m/z mass window. Precursor peptides were identified from the fragmentation data, using Mascot (Matrix Science) and relevant MS1 scans were inspected to quantify the 14N/15N species as described above.
Pulse-labeling experiments and quantitation of r-protein labeling kinetics
In these experiments, the RbgA-depleted cells were grown exponentially in 14N-labeled minimal media in the presence of limiting inducer (10 µM IPTG). Control cells were grown in the presence of 1 mM IPTG. During mid-log phase growth, the cells were pulsed by the addition of an equal volume of 15N-labeled media. Samples were taken at multiple time points over a 80-min period and rapidly frozen for storage. After lysis, either 45S or 70S particles were purified on a sucrose gradient, mixed with a 15N-labeled 70S internal standard and the mixture of proteins were prepared for mass spectrometry analysis as described above. For each peptide, three isotope envelopes were observed corresponding to proteins synthesized before the pulse (0% 14N; lowest m/z), after the pulse (50% 15N; middle m/z) or the internal standard (100% 15N; highest m/z). The fraction of material labeled, which was calculated as [50% 15N/(50% 15N + 100% 14N)], increased monotonically as a function of time, consistent with proteins synthesized after the pulse constituting a greater fraction of the 70S particles as time progresses. To ease comparison between the 1 mM and 10 µM IPTG conditions, each time course was scaled by the corresponding growth rate. For each protein, the pulse-labeling time course was fit to Equation (1) as described in Chen et al. (22).
| (1) |
where P is the pool size, t is the time, k is the growth rate and fr is the fraction labeled, and P is the only free parameter. The max lab curves were calculated using Equation (2):
| (2) |
Cryo-electron microscopy
Ribosomal subunits were diluted to a concentration of 16 nM in storage buffer, and 3.6 µl of this dilution was applied in freshly coated holey carbon grids with an additional layer (5–10 nm) of thin carbon. Before the sample was applied the grid was glow discharged at 5 mA for 15 s. Vitrification of the specimen was performed in a Vitrobot (FEI) by blotting the grids twice, 7 s each time, before they were plunged into liquid ethane. Grids were transferred to a JEOL 2010F electron microscope operated at 200 kV using a Gatan 914 cryo-holder. Micrographs were collected at a nominal magnification of 50 000× and a range of defocus from −1.5 to −3.9 µm and digitalized with a step size of 12.7 µm in a Nikon Supercool Scan 9000. The sampling of the images was 2.54 Å/pixel.
Image processing
Projection images of the ribosomal subunits were picked manually using Boxer (23). The total of number of particles picked for the mature 50S subunit, 45S particles and L16-depleted 50S subunit were 53 053, 76 499 and 51 016 particles, respectively. The contrast transfer function of the micrographs was estimated using CTFFIND software (24) and corrected using the Xmipp software package (25).
The three-dimensional (3D) reconstructions of the mature 50S subunit were calculated using 3D projection matching alignment procedures as implemented in the Xmipp software package (25). The reference map used to refine all three reconstructions was the X-ray structure of the E. coli 50S subunit (PDB ID: 2AW4) (26) low-pass filtered to 25 Å resolution. The resulting map was sharpened using a B-factor calculated using a previously described method (27) implemented in the Xmipp software package (28).
The dataset of immature 45S subunits was first subjected to a maximum likelihood-based classification approach following previously published protocols (29–32). The 3D reconstruction of the mature 50S subunit low-pass filtered to 70 Å was used as an initial reference to produce four seeds from random subsets of experimental images by performing one iteration of maximum likelihood optimization. Using these four seeds, we performed 30 iterations of maximum likelihood-based classification. This approach rendered four homogeneous subpopulations of particles. Increasing the number of classes in the ML3D classifications produced classes with similar features. The total number of particles in each class was 20 269, 10 990, 21 178 and 24 059. The 3D reconstructions of the four conformational populations of the 45S particle were obtained from these groups of particles using projection matching approaches. The reference maps for the projection matching refinements were produced from the particles in each subclass using a maximum likelihood-based refinement approach (30,33) to prevent model bias.
In the case of the L16-depleted 50S subunit, particle images in the dataset were first used to produce an initial reference map using a maximum likelihood based refinement approach (30,33). The initial seed for this procedure was the cryo-EM map of B. subtilis IF2-depleted 50S subunit low-pass filtered to 50 Å. The 3D structure was refined against this initial model using a projection matching approach (25).
To estimate the resolution of the 3D reconstructions, even and odd-numbered particles from the last cycle of refinement were used to produce two 3D maps from which the Fourier Shell Correlation plot was calculated. Resolution values were obtained from this plot using the 0.5 criteria, and these values were used to low-pass filter the corresponding 3D reconstructions.
Visualization and docking of X-ray structures into the cryo-EM maps was done using the Chimera program (34).
Chemical probing
Chemical probing of the 23S rRNA was performed with dimethyl sulfate (DMS) and kethoxal. DMS methylates specifically unpaired and exposed adenine and cytosine in RNA, while kethoxal methylates gunanine. The site of methylation is subsequently identified by reverse transcription of the RNA using fluorescent primers. A minimum cutoff of at least a 2-fold difference in chemical reactivity was employed to select bases that had a significant change in structure when comparing the 45S particle and 50S subunit. We also required that these changes were observed in a minimum of three biological replicates of the experiment and report the arithmetic mean and standard error for each base. To statistically test the significance of the data, we log transformed the ratios and calculated the geometric mean and standard deviation and performed a one-sample two-tailed Student’s t-test.
Approximately 100 pmol of 50S subunits or 45S particles were incubated with 4 µl of 0.88 M DMS solution prepared in 95% ethanol. Reactions were incubated on ice for 1 h and stopped by adding DMS stop solution containing 1 M Tris–HCl at pH 7.5, 0.1 M EDTA and 1 M 2-mercaptoethanol. DMS modified subunits were precipitated by addition of subunit precipitation buffer (85% ethanol, 0.1 M Na-acetate and 25 µg/µl glycogen). For kethoxal modification, 100 pmol of ribosomal subunits were incubated with 2.5 µl of a 2.5 M kethoxal solution on ice for 1 h. In this case, reactions were terminated by addition of subunit precipitation buffer (85% ethanol, 0.1 M Na-acetate and 25 µg/µl glycogen) supplemented with 25 mM potassium borate. All further solution used for kethoxal treated rRNA had 25 mM potassium borate to maintain the stable modification The volume of all reactions was 100 µl. Subsequent to the chemical modification, the 23S rRNA from the ribosomal subunits was prepared by ethanol precipitation and phenol extraction. Precipitation was obtained by centrifugation at 16 000 g for 20 min after addition of 1 ml of subunit precipitation buffer. Pellets containing the ribosomal subunits were dissolved in RNA extraction buffer (0.5% SDS, 5 mM EDTA pH 8, 0.3 M Na-acetate). Proteins were removed by two rounds of PCI solution (phenol: chloroform: Isoamylalcohol: 25:24:1), treatment and traces of phenol were removed by chloroform treatment. RNA was precipitated from the aqueous layer by using 100% ethanol. The RNA pellet was air dried and dissolved in water.
The reverse transcriptase reaction was carried out using 5′ 6-carboxyfluorescein (6-FAM) labeled primers listed in Supplementary Table S3 that were complementary to a portion of the 23S rRNA sequence. Reactions containing 5 µl of 0.3 µM modified rRNA, 2 µl of 1 µM 5′ 6-FAM labeled primer and 2 µl of 4.5× hybridization buffer (225 mM HEPES, pH 7, 450 mM KCl) were incubated for 5 min at 75°C. A volume of 11 µl of Master mix containing 0.5 µl (100 units) Reverse transcriptase (Superscript III reverse transcriptase from Life technologies), 0.5 µl of RNase Inhibitor (Roche), 1 µl 10 mM dNTP, 4 µl 5× buffer, 1 µl 100 mM DTT and 4 µl water was added to the reaction. Reactions were incubated at 55°C for 1 h. Then, 2 µl chase solution (1 mM dNTP) was added to the reaction and incubated for another 30 min. Then, 2 units of RNase H was added to the reactions and incubated at 37°C for 1 h. Reactions were cleaned using DTR edgebio ultra gel filtration cartridge and incubated with 5 µl of formamide for 2 min at 70°C. Then, they were rapidly transferred to ice. Sequencing was done on ABI PRISM® 3130 Genetic Analyzer (Applied Biosystems), and sequence analysis was carried out using ABI peak-scanner software (Applied Biosystems). More than 2-fold difference in modification between the 50S subunit and 45S particle was considered for further analysis. To eliminate peaks due to spontaneous reverse transcriptase stops, the difference cut off between control and modified |45S R-50S R| versus |45S C-50SC| > 5-fold was considered as genuine modification. The modifications reported here were observed in a minimum of three biological replicates of the experiment. To find the position of a modified base, it was compared with di-deoxysequencing reaction carried out using cDNA of 23S rRNA using Thermo Sequenase Cycle Sequencing Kit (Affymetrix) using fluorescent primer, and positions were determined within three bases. Nucleotide bases were mapped on the 23S rRNA secondary structure map of B. subtilis which was obtained from comparative RNA Web (CRW) Site and project (http://www.rna.ccbb.utexas.edu/). Corresponding bases in B. subtilis were mapped in the X-ray structure of the 50S subunit from E. coli (PDB ID 2AW4) using Chimera (34).
GTPase activity assays
The GTPase activity of RbgA in the presence of mature 50S subunit, 45S particle and L16-depleted 50S subunits was determined by incubating the protein with GTP and then measuring released free phosphate by the malachite green/ammonium molybdate colorimetric assay (BioAssays Systems) according to manufacturer protocols. RbgA protein for these experiments was expressed and purified as described (11). Mixtures contained 0.5 µM of RbgA and the same concentration of ribosomal subunits in reaction buffer (50 mM Tris–HCl pH 7.5, 250 mM KCl and 5 mM MgCl2) were incubated at 37°C for 30 min. Reaction was started by adding GTP to a final concentration of 200 µM. At this GTP concentration, the obtained values are within the linear range of the assay (35). The GTPase activity obtained for the RgbA protein by itself was defined as 1 and the GTPase activity for RbgA in the presence of each ribosomal subunits was expressed and plotted with respect to this standard.
RESULTS
The 45S particle from RbgA-depleted B. subtilis cells lacks several late-binding r-proteins
The 45S particles analyzed in this study were obtained by depleting B. subtilis cells of RbgA using a strain in which the rbgA gene was under the control of the IPTG-inducible Pspank promoter (RB301) (11). In the presence of 1 mM IPTG, these cells produced a ribosome profile indistinguishable from wild-type cells (Supplementary Figure S1; bottom panel). In contrast, withholding IPTG caused depletion of RbgA and led to a decrease in the levels of 70S ribosomes and the accumulation of free 45S particles (Supplementary Figure S1; top panel) (11,15).
We analyzed the protein complement of the 45S particles by qMS. Cells were grown in 14N-labeled media, and 45S particles were purified under non-dissociating conditions (see Materials and Methods) that prevented contamination with mature 50S subunits (Supplementary Figure S1; top panel). These purified 45S particles were then spiked with a fixed concentration of 70S particles purified from cells grown in 15N-labeled media. Due to the isotope shift that results from growth in 15N-labeled media, we could independently quantify the r-protein levels in the experimental sample (45S from RbgA-depleted cells) and the reference sample (70S particles from wild-type cells).
We also quantified the protein composition of 50S subunits purified under identical conditions from IF-2 depleted cells (RB419) to allow for a direct comparison between immature (45S) and mature particles (50S). We found that multiple proteins were completely absent (L16, L28 and L36) or dramatically depleted (L27, L33a and L35) from 45S particles (Figure 1A and B; red). All six of these proteins displayed normal levels in the 50S particle isolated from IF-2 depleted cells, confirming that the observed effect was specific to the 45S particle and not a result of our mass spectrometry based quantification procedure (Figure 1A and B; blue).
Figure 1.

Protein complement of the 45S particle. (A) Relative protein abundance in 14N-labeled 50S subunits (blue) and 45S particles (red) with respect to 15N-labeled 50S subunits from functional 70S ribosomes. The 50S subunits and 45S particles were purified from IF2-depleted and RbgA-depleted cells, respectively. The 70S subunits used as reference were purified from wild-type cells. Each marker represents a unique measurement of a peptide resulting from a tryptic digest of the parent protein. Each sample was analyzed in quadruplicate, and datasets were merged to improve proteomic coverage. (B) Representative mass chromatograms of proteins present stochiometrically (left), completely absent (middle) or depleted (right) from the 45S particle are shown in red, with mass chromatograms from the reference particles in brown. Equivalent chromatograms for the 50S subunits are shown in blue. (C) Transparent surface representation of the control cryo-EM map obtained for the mature 50S subunit. The X-ray structure from T. thermophilus (PDB ID 2Y11) was docked into the cryo-EM to show the r-proteins that were found absent or depleted in the 45S particles. Individual proteins are shown as a surface. The central protuberance is labeled as CP.
Depletion of L16, L27 and L36 from the 45S particles has been reported in earlier biochemical studies (11,15). However, the higher sensitivity of these qMS experiments identified additional r-proteins, including L28, L33a and L35 are depleted from the 45S particle. All these r-proteins (except for L28) cluster to the base of the central protuberance (CP) (36,37) (Figure 1C) and bind late in 50S subunit reconstitution experiments (Supplementary Figure S2) (5,6,19,38,39) indicating that the 45S particles isolated from RbgA-depleted strains represent a late assembly intermediate of the 50S subunit.
The 45S particle can mature into a complete ribosome
We performed in vivo pulse-labeling experiments to determine whether the 45S particles that accumulate in RbgA-depleted cells can mature into functional subunits or if they are simply dead-end assembly products (Figure 2A and B). In these experiments, RB301 cells (Pspank-rbgA) were grown in 14N-labeled media either in the presence of 1 mM or 10 µM IPTG. When grown with 1 mM IPTG, cells displayed a wild-type growth rate (48-min doubling time) and ribosome profile (Figure 2C; blue). In contrast, when grown with 10 µM IPTG they grew slower (85-min doubling time) and their ribosome profile contained significant quantities of 45S particles (Figure 2C; red). At mid-log phase, cells were pulsed with 15N-labeled media and, at various times post-pulse, 70S ribosomes were purified on a sucrose gradient. Using qMS, we determined the fraction of label incorporated as a function of time for each r-protein by quantifying the isotope distributions of the 14N (protein synthesized before the pulse) and 50% 15N material (proteins synthesized after the pulse) (Supplementary Figure S3A and C). The resulting label incorporation time course was then fit to a previously described theoretical pulse-labeling model (22), which allowed us to accurately calculate a ‘relative precursor pool size’ (P) for each r-protein (Figure 2D; Supplementary Figure S3D). Here, the parameter (P) reports the quantities of particles in the precursor pool relative to those in the fully mature ribosomal pool.
Figure 2.

The 45S particle can mature into a complete ribosome. Cells were pulsed with a stable isotope label and, at various times post-pulse, cells were harvested, 70S particles were purified on a sucrose gradient, and the degree of label incorporation was quantified using qMS. The pulse’s arrival time in 70S particles is a function of the size of the precursor pools through which the label must transit. (A) Dead-end intermediates do not alter the pulse transit time as the label is able to directly progress via the green arrows to the 70S particle. 15N-labeled amino acids that are incorporated in the dead-end intermediate (red arrows) are never detected as, according to the off-pathway model, these particles do not mature. (B) Intermediates competent for maturation will increase the pulse transit time as the labeled material must wash through the large 45S precursor pool in this model. (C) Sucrose gradient profiles corresponding to cells in which transcription of rbgA is under the control of an IPTG inducible Pspank promoter. Cells were grown in either the presence of 1 mM (blue) or 10 µM IPTG (red). To calculate the abundance of the precursor relative to that of the 70S particles, 45S and 50S peaks were integrated, corrected using the appropriate rRNA extinction coefficient and normalized by the integrated 70S peak intensity (shading and inset text). (D) Representative pulse-labeling kinetics for various proteins for cells grown with RbgA induced (blue; 1 mM IPTG) or RbgA limited (red; 10 µM IPTG). Cellular doubling times were 48 and 85 min for the RbgA-induced and RbgA-limited conditions, respectively. To ease comparison, the time domain has been normalized by the doubling rate for each condition. The maximum labeling rate in the absence of protein turnover is strictly a function of the growth rate and is depicted by the gray line. Each experimental time course was fit (dashed lines) to a previously described pulse-labeling model (22) resulting in a ‘relative precursor pool size’ (P) for the r-protein. (E) Relationship between precursor pool size and protein abundance in the 45S particle. With the exception of exchangeable proteins L10 and L12, proteins present in the 45S particle also have large precursor pools upon RbgA-limitation (group II). With the sole exception of L36, proteins depleted from the 45S particle (group I) exhibit no pronounced increase in precursor pool size upon RbgA-limitation.
The conceptual framework of this model is presented in Figure 2A and B. If the 45S particle is a dead-end for assembly, labeled nitrogen accumulates in 45S particles but is never incorporated in 70S subunits (Figure 2A) and therefore does not contribute to the measured (P) value. Conversely, if the generated 45S particles are competent for maturation, then labeled material must wash through this 45S precursor pool, which will delay label incorporation into 70S ribosomes (Figure 2B). Thus, if the 45S particles are competent for maturation, the model predicts that all of the r-proteins incorporated into this particle will show increased (P) values in the RbgA-limiting condition.
Using the described pulse-labeling time courses, we determined the precursor pool size for each r-protein under each condition (Supplementary Figure S3D). The data fit well using this theoretical model and most r-proteins present in the 45S particle, clearly showed an increase in the precursor pool under RbgA-limiting conditions [Figure 2D (L3, L21, L22)]. Small subunit proteins did not exhibit appreciable pools in either condition (data not shown). These data are inconsistent with a dead-end intermediate, instead confirming that the 45S particle can mature into a functional 70S particle and is likely a genuine on-pathway intermediate (Supplementary Figure S4A). However, these results cannot distinguish between several similar mechanistic models in which the 45S particle is competent for maturation (Supplementary Figure S4B).
For proteins absent from the 45S particle (except L36), we observed very small pools [Figure 2D (L16, L35, L33a)] and, in some cases (i.e. L28, L33a), we observed ‘over-labeling’, which is consistent with cellular turnover of these proteins [Figure 2D (L33a)]. Because our pool parameter (P) measures free proteins as well as all intermediates upstream of the 70S, this result indicates that the biosynthesis and/or degradation of these proteins must be tightly regulated. Were it not, large quantities of free proteins would accumulate in the cell and contribute to the observed precursor pool.
To determine the relationship between protein occupancy in the 45S and precursor pool size upon RbgA depletion, we plotted these quantities against one another (Figure 2E). This treatment generally divides the r-proteins into two groups: those absent from the 45S, which exhibit small precursor pools, and those present in the 45S, which exhibit large precursor pools. The stalk proteins, L10 and L7/L12 are known to exchange during cellular growth and translation, likely explaining the observed small precursor pools (22,40). Unregulated synthesis of L36 may account for its relatively large precursor pool.
Our pulse-labeling model also makes clear predictions for the labeling kinetics of the intermediate itself. In particular, if the 45S particle is competent for maturation, we expect significant over-labeling of material purified from the 45S peak. Alternatively, if this material is a ‘dead-end’ particle that is not turned-over, we expect proteins purified from this peak to label at the cellular growth rate (see Chen et al. for a theoretical treatment of these predictions). After pulse-labeling cells as described above, we purified material from the 45S peak and analyzed label incorporation as a function of time. Consistent with conversion to a mature subunit, we observe significant over-labeling of the 45S particle (Supplementary Figure S3B).
Because our model reports pool sizes as a fraction of completed ribosomes, we can estimate the fraction of the 45S peak that is competent for maturation by integrating the 45S and 70S peaks in the sucrose profile and comparing this ratio to the fit pool size. For proteins in the 45S, the average pool size (1.1; Figure 2E) is similar to the 45S:70S integrated peaks ratio observed in the sucrose gradients (1.4; Figure 2C), implying that the vast majority of the 45S particles purified from the sucrose gradient eventually form mature 50S subunits. Notably, the measured pool size upon RbgA-depletion (1.1) represents a ∼40-fold increase relative to the average pool size of cells bearing RbgA (0.03).
The central protuberance in the 45S particle is unstructured
To visualize the large structural changes that occur in the 50S subunit during the late stages of maturation, we obtained cryo-EM structures of the mature 50S subunit and 45S particle and we complemented this analysis by measuring each particle’s reactivity to chemical probes.
The cryo-EM structure of the 50S subunit from IF2-depleted cells was similar to that from E. coli or Thermus thermophilus (Figure 3A). As expected, our cryo-EM maps lacked density corresponding to the stress specific L25/Ctc protein, consistent with having harvested cells growing exponentially (41). In analyzing the 45S particles, we classified (18,30,31,33) the entire dataset and found four predominant conformations of the purified 45S particles (Figure 3B). The 3D structures of the 45S particle conformers showed that most of the subunit domains resembled the fully mature state, consistent with a late-stage intermediate. However, the CP displayed significant conformational alterations from the native structure, with deviations and missing densities gradually increasing from class 1 to class 4 (Figure 3B). Consequently, we believe these maps illustrate the progressive evolution of the CP toward the mature state during the late stages of assembly.
Figure 3.

Cryo-EM structure of the 45S particle. (A) Crown view of the cryo-EM structure of the mature 50S subunits refined to 11 Å (Supplementary Figure S5) represented as a semitransparent surface. The X-ray structure of the 50S subunit from T. thermophilus (PDB ID 2Y11) is shown docked into the cryo-EM map. The central protuberance is labeled as CP. L25/Ctc r-protein for which a corresponding density was not found in the cryo-EM map is labeled. The rRNA is represented as a blue ribbon. The r-proteins are shown in red except for those that were found missing or depleted in the 45S particle that are colored as in Figure 1C. The six domains of the 23S rRNA are indicated in the structure. (B) Crown view of the cryo-EM maps obtained for the four conformational subpopulations (labeled from CL1 to CL4) of the 45S particles refined to a resolution that ranged between 13 and 15 Å (Supplementary Figure S5). The particle distribution between the CL1, CL2, CL3 and CL4 classes was 27%, 14%, 28% and 31%, respectively. Landmarks of the subunit including the CP, L1 and L7/L12 stalks are indicated. (C) The cryo-EM map for one of the conformational subpopulations of the 45S particle (CL1) is shown with the X-ray structure of the 50S subunit from T. thermophilus (PDB ID 2Y11) being docked. Labeled r-proteins and in a color different from red are those for which a corresponding density was not found in the cryo-EM map of the 45S particle and were also found either depleted or absent in the qMS analysis. Color coding of these proteins is as in Figure 1C.
To identify the specific components in the CP that were either missing or in a different conformation, the X-ray structure of the 50S subunit from T. thermophilus (PDB IDs 2Y11) was docked as a rigid body into the cryo-EM maps obtained for the four conformational subpopulations of the 45S particle (Figure 3C; Supplementary Figure S6). We found that proteins located at the base of the CP including L16, L27, L33a, L35 and L36 lacked corresponding density in the cryo-EM maps obtained for the four conformational subpopulations of the 45S particle (Figure 4; Supplementary Figure S7). This finding was consistent with our qMS analysis (Figure 1A). Careful inspection revealed that the amount of density missing for these r-proteins varied among the four conformational subpopulations of the 45S particle again suggesting that these maps represent various stages of CP maturation (Supplementary Figure S7).
Figure 4.

Late binding r-proteins missing from the 45S subunit. Close-up view of the densities representing the r-proteins that qMS found depleted or absent in the cryo-EM map of the 45S particles (right panels). The panels in the left are from the control cryo-EM map for the mature 50S subunit. The map shown for the 45S particle corresponds to conformation subpopulation ‘CL1’. The X-ray structure of the 50S subunit from T. thermophilus (PDB ID 2Y11) was docked into the cryo-EM maps. Proteins are colored as indicated by the color code, and 23S rRNA is shown as a blue ribbon.
Density corresponding to protein L5 was missing from maps for classes 3 and 4 (Figure 5A; Supplementary Figure S8) despite the observed stoichiometric occupancy by qMS (Figure 1A). This discrepancy suggested enhanced flexibility of L5 in these two classes. L5 is a central organizer of the CP, assisting the entry of all other r-proteins in this domain and promoting the association of the 5S rRNA (42). Our maps showed that while most of the 5S rRNA density is present, its location deviated slightly from that observed in the mature 50S subunit (Figure 5A; Supplementary Figure S8; top panels). Similarly, helices 81, 84 and 85 located at the core of the CP adopted conformations distinct from the mature structure (Figure 5A; Supplementary Figure S8). These results suggested that in addition to L5, the r-proteins at the base of the CP also stabilize the mature conformation of the rRNA at the CP. More importantly, this structural analysis revealed that the CP is one of the last structural motifs to mature.
Figure 5.
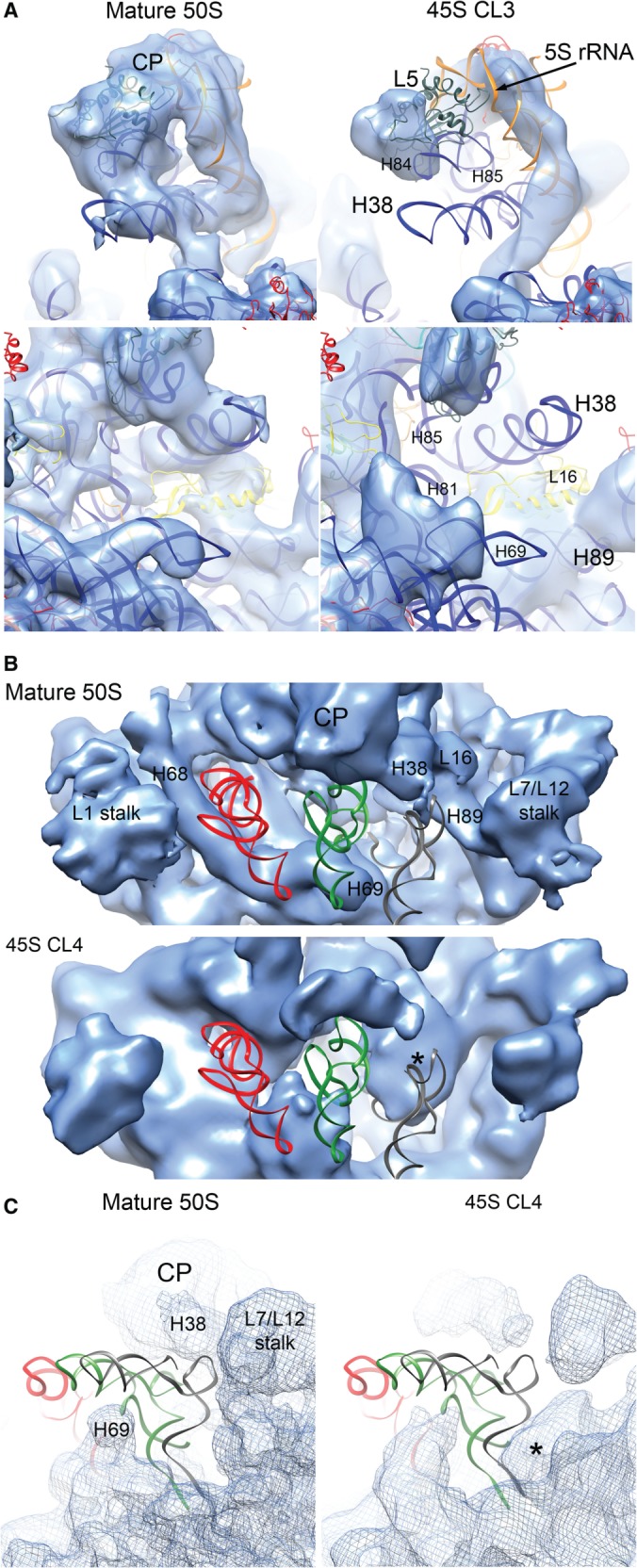
The CP and tRNA-binding sites in the 45S particle are distorted. (A) Side view from the L7/12 stalk (top panels) and front view (bottom panels) of the CP of the mature 50S subunit (left panels) and the 45S particle (right panels). The cryo-EM map of the 45S particle displayed in this figure corresponds to the conformational subpopulation ‘CL3’. Helices from the rRNA for which a corresponded density was not found or the existing density deviated from the mature 50S structure are labeled. The X-ray structure shown docked into the map corresponds to the 50S subunit from T. thermophilus (PDB ID 2Y11). (B) Top view of the canyon containing the A, P and E tRNA-binding sites in the cryo-EM map of the mature 50S subunit and 45S particle (conformational subpopulation ‘CL4’). The tRNA molecules in the A (gray), P (green) and E (red) sites are placed for reference in the two maps. Location of the tRNA molecules was determined by docking the X-ray structure of the 50S subunit from T. thermophilus (PDB ID 1GIY). Important landmarks and rRNA helices that showed distortions in the 45S particles are labeled. A prominent density occupying part of the A site is labeled with an asterisk. (C) A side view of the A tRNA-binding site in the 45S particle (conformational subpopulation ‘CL4’) partially occupied with the prominent density is shown in the right panel and compared to a similar view of the 50S subunit cryo-EM map (left panel).
The structure of the 45S particle is incompatible with tRNA binding
Helices 38, 69 and 89 in the 23S rRNA stabilize tRNA binding to the A and P sites of the mature 50S subunit. Helix 38 (A-site finger) contacts the elbow, D and T loops of the A-tRNA, whereas helix 89 interacts with its acceptor arm. In addition, helix 69 interacts with the D arm of the A and P-tRNA. Helices 38 and 89 also create a binding cleft for L16, which makes direct contacts with the A-site tRNA (Figure 5B; top panel). These helices all displayed severe distortions in the 45S particle structures with their corresponding densities in the EM maps either absent or shifted from their positions in the mature 50S subunit (Figure 5B; Supplementary Figure S9). In addition, a prominent density likely resulting from displacement of rRNA in this region occupied part of the A site (Figure 5B; asterisk, Figure 5C; Supplementary Figure S9; asterisk).
The acceptor stem of the E-site tRNA binds in a pocket formed by helices 68, 74, 75, 76 and 88 and the two r-proteins, L28 and L35 (43). These two proteins were depleted from the 45S particles according to qMS analysis (Figure 1A) and the cryo-EM maps for two of the conformational populations of the 45S particle (CL1 and CL4) lacked density corresponding to these proteins (Figure 4; Supplementary Figure S7). In addition, the ∼50 Å-long helix 68 found at the cleft between the L1 stalk and the CP was missing (Figure 5B). Taken together, these distortions are incompatible with tRNA molecules binding at the A, P or E sites of the 45S particle.
To probe for nucleotide-level structural differences between the 45S particle and the mature 50S subunit, we performed chemical modifications of the two particles using DMS or kethoxal followed by primer extension. Consistent with the cryo-EM maps, five bases in helix 38, G911(864), G916(869), G953(906), C955(908) and C961(914) (number in brackets correspond to E. coli numbering of the 23S rRNA) all displayed dramatically increased reactivity in the 45S particle (Supplementary Figure S10; Supplementary Tables S1 and S2). Four residues that exhibited significantly increased modification in the 45S (4- to 11-fold) make direct contact with L16 (Figure 6; Supplementary Tables S1 and S2). We hypothesized that the increased reactivity in the 45S particle was due to the absence of L16 and/or local conformational differences in the central part of helix 38. Nucleotide A2281 (2252) of helix 80, which makes a direct contact with the P-site tRNA (Figure 6), was less reactive (58-fold) in the 45S particle (Supplementary Figure S11; Supplementary Table S2). This indicates that this base pair is less available for contact with the P-site tRNA. Finally, 14 bp in helix 68 had enhanced modifications (5 - to 34-fold) in the immature particle (Figure 6; Supplementary Figure S11; Supplementary Tables S1 and S2). Therefore, the chemical probing analysis confirmed the severe structural limitations of the 45S particle to bind tRNA in the A, P and E sites.
Figure 6.

Bases with increased and decreased chemical reactivity in the 45S particle. The center of the figure shows a top view of the 50S subunit cryo-EM map. The X-ray structure of the 50S subunit from E. coli (PDB ID 2AW4) is shown docked into the cryo-EM maps. The r-proteins from the X-ray structure have been removed for clarity, except L16 that is displayed as a yellow ribbon. Helices 68, 38 and GAR (helices 42–44) that showed the highest levels of chemical modification are colored in red. Bases with increased and decreased reactivity to DMS and kethoxal in these helices are displayed in the 3D structure as red and blue spheres, respectively. They are also mapped into the secondary structure of the 23S rRNA. Detailed views of the framed areas are shown at both sides. In these views the cryo-EM map displayed is that from the 45S particle conformation subpopulation ‘CL2’ for easier comparison between the chemical modification data and the structural distortions observed in the immature ribosomal subunit. Landmarks of the 50S structure including the CP, and L1 stalk are indicated. A, P and E tRNA molecules are also shown for orientation purposes.
Key intersubunit bridges are not properly structured in the 45S particle
Previous work has shown that 45S particles do not associate with 30S subunits in RbgA-depleted cells (11,15), however, the detailed structural underpinnings of these results have not been explored. Crystal structures of the 70S particles show that near the CP, three intersubunit bridges mediate the association of the large and small subunits. Bridge B1a utilizes helix 38 and connects the CP to protein S13 in the head of the 30S subunit; bridge B2a, an essential interaction for 70S stability, connects helix 69 and the decoding site of the 30S subunit; and bridge B7a links helix 68 with helix 23 in the 30S subunit (26,44). In E. coli, A1848 in helix 68 interacts with A702 in helix 23 of the 16S rRNA to form this last intersubunit bridge (Figure 6). As described earlier, helices 38, 68 and 69 in the 23S rRNA were significantly distorted in the cryo-EM maps of the 45S structure (Figure 5A and B). Moreover, chemical probing showed significantly increased reactivity for multiple nucleotides in helices 38 and 68 as well as a 17-fold reactivity increase for residue A1877 (1848 in E. coli) in the 45S particle (Figure 6; Supplementary Figures S11 and S12; Supplementary Tables S1 and S2). The severe structural distortion observed in these critical regions likely explains the inability of the immature 45S to associate with 30S subunits.
The GTPase associated region is not properly structured in the 45S particles
Given the distortions observed in the CP and intersubunit bridges, we used our highly sensitive chemical probing approach to analyze whether other smaller structural differences were present in the 45S using primers covering the entire 23S rRNA (Supplementary Table S3). This analysis revealed altered reactivity in five bases of helix 43 in the 23S rRNA (Figure 6; Supplementary Figure S10; Supplementary Tables S1 and S2). This helix, together with helix 44, forms the GTPase associating region (GAR) (Figure 6), which serves as an interaction site for translation factors (i.e. EF-Tu and EF-G). Three of the bases with altered reactivity, G1105(1059), C1110(1064) and C1118(1072) were less reactive when compared to the 50S subunit, while the other two bases, G1102(1056) and G1114(1068) were more reactive. In addition, we found significantly increased modification of nucleotides C1098(1052) and G1166(1120) in helix 42 (Figure 6, top right panel; Supplementary Tables S1 and S2), which is proposed to provide flexibility and orientation specificity to the GAR. While these structural changes are more subtle than those observed in the CP, these data indicates that the GAR is also malformed in this immature particle.
L16 enables the 50S subunit to stimulate the GTPase activity of RbgA
Mature 50S subunits, but not the 45S particles, significantly stimulate RbgA GTPase activity (35). Given the numerous differences between the 45S and 50S particles described above, we attempted to isolate subtly perturbed 50S-like particles that might illuminate the specific structural features required for RbgA GTPase stimulation. Fortuitously, we found that purification of the 50S subunit from a dilute cellular lysate caused partial dissociation of L16 from the subunit. Analysis of these ‘L16-depleted 50S subunits’ by qMS (Figure 7A) showed that all other r-proteins were present in nearly stoichiometric amounts, except for L28 and L7/L12, which were also partially depleted in the control 50S subunits purified from a more concentrated lysate. Despite the depletion of L16, we found that these subunits co-migrated with mature 50S particles (Figure 7B).
Figure 7.

Depletion of L16 from the mature 50S subunit partially reverts the subunit to an immature state. (A) Relative protein abundance in 14N-labeled 50S subunit purified using standard (2.5 ml; blue) or higher (10 ml; red) lysis volumes with respect to 15N-labeled 50S subunits from functional 70S ribosomes. The 50S subunits were purified from IF2-depleted cells (see methods). The 70S subunits used as reference were purified from wild-type cells. Each marker represents a unique measurement of a peptide resulting from a tryptic digest of the parent r-protein. Each sample was analyzed in duplicate and datasets were merged to improve proteomic coverage. 50S subunits purified using a higher lysis volume showed a specific depletion of L16, and these particles were called ‘L16-depleted 50S subunits’. (B) Sedimentation profiles of L16-depleted 50S subunits in sucrose gradients. Cells grown as described in (B) were lysed either in 10 ml buffer (top panel) or in 2.5 ml of buffer (bottom panel). Analysis of sedimentation profiles in a sucrose gradient revealed that in each instance, large subunit particles co-migrated at 50S. (C) GTPase activity of RbgA in the presence of L16-depleted 50S subunit, 50S subunits purified using standard lysis volume and 45S particles. In each case, the GTPase activity was normalized to that of the RbgA protein in isolation. Plotted values represent the average and standard deviation obtained from three replicas of each reaction. (D) Cryo-EM structure of the L16-depleted 50S subunit refined to 13 Å resolution (Supplementary Figure S12). The X-ray structure of the 50S subunit from T. thermophilus (PDB ID 2Y11) was docked in the cryo-EM map. L1 and L25/Ctc have been removed for clarity. L16, L6 and the rRNA helices that exhibited conformational deviations from the mature 50S subunit are labeled. The central protuberance is labeled as CP. (E) Model for the functional interplay between RgbA and L16 during the last stages of maturation of the 50S subunit. Under this model, RbgA directly or indirectly mediates binding of L16 to the 45S particle (red), which in turn releases RbgA (upon GTP hydrolysis). Binding of L16 and release of RbgA induce conformational changes that transition the assembling particle toward the mature structure (blue).
Surprisingly, these nearly mature 50S-like particles only weakly stimulated RbgA GTPase activity, behaving much like the immature 45S subunits (Figure 7C). 50S particles purified from the more concentrated lysate exhibited stoichiometric L16 incorporation and maximally stimulated RbgA GTPase activity, suggesting L16 incorporation is necessary for this activity. Notably, L16 is the largest of the r-proteins depleted from the 45S particles and has been implicated in driving major conformational changes in the 50S subunit during in vitro assembly (45).
To inspect structural changes in the L16-depleted 50S subunit we performed a cryo-EM reconstruction (Figure 7D). Structurally, these particles were similar to the least distorted conformational subpopulation of the 45S particle (CL1) (Figure 3B). Consistent with the qMS analysis, we could not observe density corresponding to L16, L28 (not shown) and L7/L12 and, much like the 45S structures, part of the density corresponding to L6 was missing, suggesting enhanced flexibility of this r-protein. Like in the 45S structure, we also noted partial displacement of helices 38, 68 and 89, which form the binding pocket for L16, suggesting that L16 affords stabilizing contacts to these RNA structures.
These results suggested that the presence of L16 enabled the large ribosomal subunit to stimulate RbgA GTPase activity. In addition, our finding that the loss of L16 (and perhaps L28) caused a partial reversion of the structure of the mature 50S subunit to that of the immature 45S particle suggested that L16 contributes substantially to the conformational changes occurring during the late stages of large subunit maturation. Taken together, these results show that L16 binding either directly or indirectly requires RbgA activity and that GTPase stimulation and release of RbgA are directly or indirectly dependent on L16 binding to the 50S subunit. We hypothesize that together, these two events (L16 binding and RbgA release) then result in substantial conformational changes that structure the tRNA-binding sites and intersubunit bridges, effectively transitioning the assembling particle into the mature structure (Figure 7E).
DISCUSSION
In this study, we found that the 45S particle from RbgA-depleted cells is a physiologically relevant assembly intermediate that is able to mature into the 50S ribosomal subunit. Structural analysis revealed the conformational changes that the large ribosomal subunit undergoes during the late stages of maturation. We found that the 45S particle contains an incomplete protein complement and presents severe distortions in the CP and tRNA binding sites by cryo-EM and chemical probing. In contrast, few nucleotides in the body of the subunit showed significantly altered reactivity in chemical probing experiments (Supplementary Tables S1 and S2). This finding was consistent with our cryo-EM structures, which show that the body of the immature 45S particle is folded in a conformation closely resembling the structure of the mature 50S subunit. Overall, these results indicate that the functional core of the 50S subunit is the last region to become structured, and that RbgA activity is required for the maturation of these motifs and for the incorporation of late binding proteins. Because homologs of RbgA are involved in mitochondrial, chloroplast and eukaryotic ribosome assembly (10) it is likely our studies have relevance for the assembly of these ribosomes as well.
Interesting parallels are revealed by comparison of the 45S structures reported here with recently described structures of 30S subunit assembly intermediates (18,32,46). In each case the main structural domains of the immature particles are fully formed in late intermediates, whereas the functional core (the decoding center in the 30S, the tRNA-binding sites in the 50S) is severely distorted. These structural distortions likely prevent the immature 30S and 50S ribosomal subunits from assembling into 70S ribosomes and prematurely engaging in translation. Evidence for terminal maturation of ribosomal particle’s functional elements can also be found in cryo-EM structures of the late cytoplasmic 40S ribosome assembly intermediate from Saccharomyces cerevisiae, which bears a distorted decoding center (47). Further highlighting the need to keep inactive ribosome particles out of the translating pool, recent work has identified quality control checkpoints in yeast that ensure that newly formed ribosome subunits are fully functional before engaging in translation (48,49). That this general assembly principle is conserved for both ribosomal subunits and between bacteria and eukaryotes suggests that allowing immature particles to initiate translation results in a major stress for the cell.
The structural rearrangement that the 45S particle undergoes to become a mature subunit seems to be induced by the combined effect of RbgA binding and the entrance of late binding r-proteins into the assembly. Our structural and biochemical results in the L16-depleted 50S subunits suggest that L16 is a key player in the structural rearrangements that the 45S particles undergoes on the pathway to become a mature subunit. L16 was previously shown to be important for subunit association (50) and incorporation of L16 during in vitro 50S assembly causes a large-scale conformational change (45). In addition, the yeast homolog of L16, Rpl10p, is one of the last r-proteins to be incorporated into the 60S subunit and its binding is controlled by the RbgA homolog Lsg1p (51). Thus, it appears that this late stage incorporation of L16 is an evolutionarily conserved process that may control the level of functional ribosomes in both bacteria and eukaryotes.
The lack of L27, L28 and L36 also has significant consequences for ribosome structure and function. Removal of L27 impairs peptidyl transferase activity and P-site tRNA binding (52) and elimination of L28 in E. coli cells results in accumulation of a 47S particle (53). Finally, E. coli 50S subunits lacking L36 contain distortions that extend from the L36 binding site ∼60 Å to the peptidyl transferase center (54). When combined with results from our L16-depleted 50S subunits, which contain normal quantities of L27 and L36, these studies suggest that the distortions observed in the 45S particle are caused by the absence of L16 and, possibly to a lesser extent, L28.
While this paper was in preparation, a study reporting the cryo-EM reconstruction of a 45S particle also purified from RbgA-depleted B. subtilis cells was published (16). The structural distortions exhibited by these structures are similar to those observed in our cryo-EM reconstructions of the 45S particles. The work presented here significantly adds to this initial characterization of the 45S particles. We show that the 45S particle is indeed a physiologically relevant assembly intermediate competent to become a mature 50S subunit. In addition, we have also extended the structural analysis of the 45S particle to nucleotide resolution with chemical probing experiments. Finally, we found that the presence of L16 in the large ribosomal subunit is required for the stimulation of RbgA GTPase activity either through direct contacts between L16 and RbgA, or indirectly through the large conformational changes induced upon L16 binding.
The main question that remains to be answered for RbgA and other essential GTPase ribosome assembly factors is precisely how they facilitate late stages of ribosomal subunit assembly. Li et al. (16) assigns RbgA the role of an rRNA chaperone that likely facilitates a structural remodeling of the large subunit contributing to the re-orientation of helix 38 to its native position. However, intriguingly one-third of the 45S particles in their RbgA-depleted cells displayed helix 38 in a native-like conformation (state I). This finding is difficult to reconcile with the essential role of RbgA being involved in positioning helix 38 during 50S subunit maturation. They further propose that this re-orientation of helix 38 is a rate-limiting prerequisite for the conformational maturation of the CP and tRNA binding sites. In contrast, our structural analysis of the L16-depleted 50S subunits demonstrated that despite helix 38 adopting a proper overall orientation, some helices in the CP (helices 81, 85 and 89) and tRNA-binding sites (helix 68) still adopt non-mature conformations (Figure 7D). These data indicate that re-orientation of helix 38 alone is not sufficient for maturation of these functional sites. While the orientation of helix 38 may play an important role in a late stage of assembly, at this time we feel there is not enough evidence to distinguish between this activity and a model in which RbgA aids in the incorporation or conformational positioning of late-binding proteins such as L16.
Despite differences in our proposed mechanistic models, the presented data and that from Li et al. (16) firmly establish RbgA’s role as as key player in the structural maturation of the 50S subunit’s functional core and also suggest that bacteria could use this assembly factor to control the number of translationally competent ribosomes as a function of cellular GTP concentrations. Indeed, RbgA binds the 45S subunit tightly only in the presence of GTP (35) and thus a drop in cellular GTP concentration could be used to inhibit RbgA’s activity and halt maturation of the functional core of the ribosomal subunits (10). Because cellular GTP levels are linked to nutrient availability, such a mechanism could provide a relatively early checkpoint and limit the number of ribosomes entering the translational pool during conditions of nutrient deprivation.
ACCESSION NUMBERS
EMDB IDs for the conformational subpopulations CL1, CL2, CL3 and CL4 of the 45S particle are 5789, 5790, 5791, 5792, respectively. The cryo-EM map of the mature 50S subunit and the L16-depleted 50S subunit has been assigned the EMDB IDs 5787, 5788, respectively.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Canadian Institutes of Health Research [MOP-82930 to J.O.]; CAREER Award from the National Science Foundation [0643565 to R.A.B.]; National Institutes of Health [R37-GM-053757 to J.R.W.]; Canadian Institutes of Health Research Doctoral Research Award supported (to A.J.); Jane Coffin Childs Fund Postdoctoral Fellowship supported (to J.H.D.); The funders had no role in study design, data collection and analysis, decision to publish or preparation of the manuscript. Funding for open access charge: Canadian Institute of Health Research.
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
We are grateful to Vivian Leong for technical assistance and to the staff at the EM Facility of the Faculty of Health Sciences and at Canadian Centre for Electron Microscopy at McMaster. We are also thankful to Richard Fekete for suggestions about the primer extension method. We also acknowledge Brian King and Christopher Radek for early work in the project.
REFERENCES
- 1.Bremer H, Dennis PD. Modulation of chemical composition and other parameters of the cell by growth rate. In: Neidhardt FC, editor. Escherichia coli and Salmonella. Washington D.C: ASM Press; 1996. pp. 1553–1569. [Google Scholar]
- 2.Traub P, Nomura M. Structure and function of Escherichia coli ribosomes. I. Partial fractionation of the functionally active ribosomal proteins and reconstitution of artificial subribosomal particles. J. Mol. Biol. 1968;34:575–593. doi: 10.1016/0022-2836(68)90182-4. [DOI] [PubMed] [Google Scholar]
- 3.Traub P, Nomura M. Structure and function of E. coli ribosomes. V. Reconstitution of functionally active 30S ribosomal particles from RNA and proteins. Proc. Natl Acad. Sci. U.S.A. 1968;59:777–784. doi: 10.1073/pnas.59.3.777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Traub P, Nomura M. Studies on the assembly of ribosomes in vitro. Cold Spring Harb. Symp. Quant. Biol. 1969;34:63–67. doi: 10.1101/sqb.1969.034.01.010. [DOI] [PubMed] [Google Scholar]
- 5.Rohl R, Nierhaus KH. Assembly map of the large subunit (50S) of Escherichia coli ribosomes. Proc. Natl Acad. Sci. U.S.A. 1982;79:729–733. doi: 10.1073/pnas.79.3.729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Herold M, Nierhaus KH. Incorporation of six additional proteins to complete the assembly map of the 50 S subunit from Escherichia coli ribosomes. J. Biol. Chem. 1987;262:8826–8833. [PubMed] [Google Scholar]
- 7.Talkington MW, Siuzdak G, Williamson JR. An assembly landscape for the 30S ribosomal subunit. Nature. 2005;438:628–632. doi: 10.1038/nature04261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Adilakshmi T, Bellur DL, Woodson SA. Concurrent nucleation of 16S folding and induced fit in 30S ribosome assembly. Nature. 2008;455:1268–1272. doi: 10.1038/nature07298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sykes MT, Williamson JR. A complex assembly landscape for the 30S ribosomal subunit. Annu. Rev. Biophys. 2009;38:197–215. doi: 10.1146/annurev.biophys.050708.133615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Britton RA. Role of GTPases in bacterial ribosome assembly. Annu. Rev. Microbiol. 2009;63:155–176. doi: 10.1146/annurev.micro.091208.073225. [DOI] [PubMed] [Google Scholar]
- 11.Uicker WC, Schaefer L, Britton RA. The essential GTPase RbgA (YlqF) is required for 50S ribosome assembly in Bacillus subtilis. Mol. Microbiol. 2006;59:528–540. doi: 10.1111/j.1365-2958.2005.04948.x. [DOI] [PubMed] [Google Scholar]
- 12.Schaefer L, Uicker WC, Wicker-Planquart C, Foucher AE, Jault JM, Britton RA. Multiple GTPases participate in the assembly of the large ribosomal subunit in Bacillus subtilis. J. Bacteriol. 2006;188:8252–8258. doi: 10.1128/JB.01213-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hwang J, Inouye M. The tandem GTPase, Der, is essential for the biogenesis of 50S ribosomal subunits in Escherichia coli. Mol. Microbiol. 2006;61:1660–1672. doi: 10.1111/j.1365-2958.2006.05348.x. [DOI] [PubMed] [Google Scholar]
- 14.Jiang M, Datta K, Walker A, Strahler J, Bagamasbad P, Andrews PC, Maddock JR. The Escherichia coli GTPase CgtAE is involved in late steps of large ribosome assembly. J. Bacteriol. 2006;188:6757–6770. doi: 10.1128/JB.00444-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Matsuo Y, Morimoto T, Kuwano M, Loh PC, Oshima T, Ogasawara N. The GTP-binding protein YlqF participates in the late step of 50 S ribosomal subunit assembly in Bacillus subtilis. J. Biol. Chem. 2006;281:8110–8117. doi: 10.1074/jbc.M512556200. [DOI] [PubMed] [Google Scholar]
- 16.Li N, Chen Y, Guo Q, Zhang Y, Yuan Y, Ma C, Deng H, Lei J, Gao N. Cryo-EM structures of the late-stage assembly intermediates of the bacterial 50S ribosomal subunit. Nucleic Acids Res. 2013;41:7073–7083. doi: 10.1093/nar/gkt423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Morimoto T, Loh PC, Hirai T, Asai K, Kobayashi K, Moriya S, Ogasawara N. Six GTP-binding proteins of the Era/Obg family are essential for cell growth in Bacillus subtilis. Microbiology. 2002;148:3539–3552. doi: 10.1099/00221287-148-11-3539. [DOI] [PubMed] [Google Scholar]
- 18.Jomaa A, Stewart G, Martin-Benito J, Zielke R, Campbell TL, Maddock JR, Brown ED, Ortega J. Understanding ribosome assembly: the structure of in vivo assembled immature 30S subunits revealed by cryo-electron microscopy. RNA. 2011;17:697–709. doi: 10.1261/rna.2509811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen SS, Williamson JR. Characterization of the ribosome biogenesis landscape in E. coli using quantitative mass spectrometry. J. Mol. Biol. 2013;425:767–779. doi: 10.1016/j.jmb.2012.11.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bunner AE, Trauger SA, Siuzdak G, Williamson JR. Quantitative ESI-TOF analysis of macromolecular assembly kinetics. Anal. Chem. 2008;80:9379–9386. doi: 10.1021/ac8020505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sperling E, Bunner AE, Sykes MT, Williamson JR. Quantitative analysis of isotope distributions in proteomic mass spectrometry using least-squares Fourier transform convolution. Anal. Chem. 2008;80:4906–4917. doi: 10.1021/ac800080v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen SS, Sperling E, Silverman JM, Davis JH, Williamson JR. Measuring the dynamics of E. coli ribosome biogenesis using pulse-labeling and quantitative mass spectrometry. Mol. Biosyst. 2012;8:3325–3334. doi: 10.1039/c2mb25310k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ludtke SJ, Baldwin PR, Chiu W. EMAN: semiautomated software for high-resolution single-particle reconstructions. J. Struct. Biol. 1999;128:82–97. doi: 10.1006/jsbi.1999.4174. [DOI] [PubMed] [Google Scholar]
- 24.Mindell JA, Grigorieff N. Accurate determination of local defocus and specimen tilt in electron microscopy. J. Struct. Biol. 2003;142:334–347. doi: 10.1016/s1047-8477(03)00069-8. [DOI] [PubMed] [Google Scholar]
- 25.Scheres SH, Nunez-Ramirez R, Sorzano CO, Carazo JM, Marabini R. Image processing for electron microscopy single-particle analysis using XMIPP. Nat. Protoc. 2008;3:977–990. doi: 10.1038/nprot.2008.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schuwirth BS, Borovinskaya MA, Hau CW, Zhang W, Vila-Sanjurjo A, Holton JM, Cate JH. Structures of the bacterial ribosome at 3.5 A resolution. Science. 2005;310:827–834. doi: 10.1126/science.1117230. [DOI] [PubMed] [Google Scholar]
- 27.Rosenthal PB, Henderson R. Optimal determination of particle orientation, absolute hand, and contrast loss in single-particle electron cryomicroscopy. J. Mol. Biol. 2003;333:721–745. doi: 10.1016/j.jmb.2003.07.013. [DOI] [PubMed] [Google Scholar]
- 28.Fernandez JJ, Luque D, Caston JR, Carrascosa JL. Sharpening high resolution information in single particle electron cryomicroscopy. J. Struct. Biol. 2008;164:170–175. doi: 10.1016/j.jsb.2008.05.010. [DOI] [PubMed] [Google Scholar]
- 29.Scheres SH, Marabini R, Lanzavecchia S, Cantele F, Rutten T, Fuller SD, Carazo JM, Burnett RM, San Martin C. Classification of single-projection reconstructions for cryo-electron microscopy data of icosahedral viruses. J. Struct. Biol. 2005;151:79–91. doi: 10.1016/j.jsb.2005.04.003. [DOI] [PubMed] [Google Scholar]
- 30.Scheres SH, Valle M, Nunez R, Sorzano CO, Marabini R, Herman GT, Carazo JM. Maximum-likelihood multi-reference refinement for electron microscopy images. J. Mol. Biol. 2005;348:139–149. doi: 10.1016/j.jmb.2005.02.031. [DOI] [PubMed] [Google Scholar]
- 31.Scheres SH, Gao H, Valle M, Herman GT, Eggermont PP, Frank J, Carazo JM. Disentangling conformational states of macromolecules in 3D-EM through likelihood optimization. Nat. Methods. 2007;4:27–29. doi: 10.1038/nmeth992. [DOI] [PubMed] [Google Scholar]
- 32.Leong V, Kent M, Jomaa A, Ortega J. Escherichia coli rimM and yjeQ null strains accumulate immature 30S subunits of similar structure and protein complement. RNA. 2013;19:789–802. doi: 10.1261/rna.037523.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Scheres SH, Valle M, Carazo JM. Fast maximum-likelihood refinement of electron microscopy images. Bioinformatics. 2005;21(Suppl. 2):ii243–ii244. doi: 10.1093/bioinformatics/bti1140. [DOI] [PubMed] [Google Scholar]
- 34.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 35.Achila D, Gulati M, Jain N, Britton RA. Biochemical characterization of ribosome assembly GTPase RbgA in Bacillus subtilis. J. Biol. Chem. 2012;287:8417–8423. doi: 10.1074/jbc.M111.331322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schmeing TM, Voorhees RM, Kelley AC, Ramakrishnan V. How mutations in tRNA distant from the anticodon affect the fidelity of decoding. Nat. Struct. Mol. Biol. 2011;18:432–436. doi: 10.1038/nsmb.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dunkle JA, Xiong L, Mankin AS, Cate JH. Structures of the Escherichia coli ribosome with antibiotics bound near the peptidyl transferase center explain spectra of drug action. Proc. Natl Acad. Sci. U.S.A. 2010;107:17152–17157. doi: 10.1073/pnas.1007988107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nomura M, Erdmann VA. Reconstitution of 50S ribosomal subunits from dissociated molecular components. Nature. 1970;228:744–748. doi: 10.1038/228744a0. [DOI] [PubMed] [Google Scholar]
- 39.Fahnestock S, Held W, Nomura M. The assembly of bacterial ribosomes. In: Markham R, Bancroft JB, Davies DR, Hopwood DE, Horne RW, editors. Generation of Sub-cellular Structures. Amsterdam: North-Holland; 1973. pp. 179–217. [Google Scholar]
- 40.Deroo S, Hyung SJ, Marcoux J, Gordiyenko Y, Koripella RK, Sanyal S, Robinson CV. Mechanism and rates of exchange of L7/L12 between ribosomes and the effects of binding EF-G. ACS Chem. Biol. 2012;7:1120–1127. doi: 10.1021/cb300081s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schmalisch M, Langbein I, Stulke J. The general stress protein Ctc of Bacillus subtilis is a ribosomal protein. J. Mol. Microbiol. Biotechnol. 2002;4:495–501. [PubMed] [Google Scholar]
- 42.Korepanov AP, Korobeinikova AV, Shestakov SA, Garber MB, Gongadze GM. Protein L5 is crucial for in vivo assembly of the bacterial 50S ribosomal subunit central protuberance. Nucleic Acids Res. 2012;40:9153–9159. doi: 10.1093/nar/gks676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wilson DN, Nierhaus KH. The E-site story: the importance of maintaining two tRNAs on the ribosome during protein synthesis. Cell Mol. Life Sci. 2006;63:2725–2737. doi: 10.1007/s00018-006-6125-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yusupov MM, Yusupova GZ, Baucom A, Lieberman K, Earnest TN, Cate JH, Noller HF. Crystal structure of the ribosome at 5.5 A resolution. Science. 2001;292:883–896. doi: 10.1126/science.1060089. [DOI] [PubMed] [Google Scholar]
- 45.Teraoka H, Nierhaus KH. Protein L16 induces a conformational change when incorporated into a L16-deficient core derived from Escherichia coli ribosomes. FEBS Lett. 1978;88:223–226. doi: 10.1016/0014-5793(78)80179-3. [DOI] [PubMed] [Google Scholar]
- 46.Guo Q, Goto S, Chen Y, Feng B, Xu Y, Muto A, Himeno H, Deng H, Lei J, Gao N. Dissecting the in vivo assembly of the 30S ribosomal subunit reveals the role of RimM and general features of the assembly process. Nucleic Acids Res. 2013;41:2609–2620. doi: 10.1093/nar/gks1256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Strunk BS, Loucks CR, Su M, Vashisth H, Cheng S, Schilling J, Brooks CL, 3rd, Karbstein K, Skiniotis G. Ribosome assembly factors prevent premature translation initiation by 40S assembly intermediates. Science. 2011;333:1449–1453. doi: 10.1126/science.1208245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Strunk BS, Novak MN, Young CL, Karbstein K. A translation-like cycle is a quality control checkpoint for maturing 40S ribosome subunits. Cell. 2012;150:111–121. doi: 10.1016/j.cell.2012.04.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Karbstein K. Quality control mechanisms during ribosome maturation. Trends Cell Biol. 2013;23:242–250. doi: 10.1016/j.tcb.2013.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kazemie M. The importance of Escherichia coli ribosomal proteins L1, L11 and L16 for the association of ribosomal subunits and the formation of the 70-S initiation complex. Eur. J. Biochem. 1975;58:501–510. doi: 10.1111/j.1432-1033.1975.tb02398.x. [DOI] [PubMed] [Google Scholar]
- 51.West M, Hedges JB, Chen A, Johnson AW. Defining the order in which Nmd3p and Rpl10p load onto nascent 60S ribosomal subunits. Mol. Cell. Biol. 2005;25:3802–3813. doi: 10.1128/MCB.25.9.3802-3813.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Maguire BA, Beniaminov AD, Ramu H, Mankin AS, Zimmermann RA. A protein component at the heart of an RNA machine: the importance of protein l27 for the function of the bacterial ribosome. Mol. Cell. 2005;20:427–435. doi: 10.1016/j.molcel.2005.09.009. [DOI] [PubMed] [Google Scholar]
- 53.Maguire BA, Wild DG. The roles of proteins L28 and L33 in the assembly and function of Escherichia coli ribosomes in vivo. Mol. Microbiol. 1997;23:237–245. doi: 10.1046/j.1365-2958.1997.2131578.x. [DOI] [PubMed] [Google Scholar]
- 54.Maeder C, Draper DE. A small protein unique to bacteria organizes rRNA tertiary structure over an extensive region of the 50 S ribosomal subunit. J. Mol. Biol. 2005;354:436–446. doi: 10.1016/j.jmb.2005.09.072. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.