

Abstract
Monoallelic expression within a gene family is found in pathogens exhibiting antigenic variation and in mammalian olfactory neurons. Trypanosoma brucei, a lethal parasite living in the human bloodstream, expresses variant surface glycoprotein (VSG) from 1 of 15 bloodstream expression sites (BESs) by virtue of a multifunctional RNA polymerase I. The active BES is transcribed in an extranucleolar compartment termed the expression site body (ESB), whereas silent BESs, located elsewhere within the nucleus, are repressed epigenetically. The regulatory mechanisms, however, are poorly understood. Here we show that two essential subunits of the basal class I transcription factor A (CITFA) predominantly occupied the promoter of the active BES relative to that of a silent BES, a phenotype that was maintained after switching BESs in situ. In these experiments, high promoter occupancy of CITFA was coupled to high levels of both promoter-proximal RNA abundance and RNA polymerase I occupancy. Accordingly, fluorescently tagged CITFA-7 was concentrated in the nucleolus and the ESB. Because a ChIP-seq analysis found that along the entire BES, CITFA-7 is specifically enriched only at the promoter, our data strongly indicate that monoallelic BES transcription is activated by a mechanism that functions at the level of transcription initiation.
INTRODUCTION
Monoallelic expression of a large gene family is a phenomenon found in parasites that exhibit antigenic variation of cell surface proteins to evade the mammalian host’s immune system (1). It also occurs in mammalian olfactory sensory neurons, which express a single member of the large odorant receptor (OR) gene family (2). While monoallelic gene expression of a gene family is typically regulated at the level of transcription, the mechanisms enabling transcription of a single gene/allele are not well understood.
Trypanosoma brucei is a vector-borne lethal parasite in sub-Saharan Africa that lives freely in the human bloodstream by virtue of a dense coat of variant surface glycoprotein (VSG) expressed from a single gene. The uniform coat shields invariant membrane proteins from immune recognition (3) and is periodically altered by switching VSG expression to a different gene. While the parasite harbors more than 2000 VSG genes and pseudogenes, VSG expression is restricted to 1 of 15 telomeric bloodstream expression sites (BESs) (4). A BES is a tandem array of several expression site-associated genes (ESAGs) followed by a single VSG gene, all of which are polycistronically transcribed by a multifunctional RNA polymerase (pol) I from a promoter that is located 40–60 kb upstream of the VSG gene (5). The active BES is transcribed outside of the nucleolus (6) in the extranucleolar expression site body (ESB), a DNase-resistant compartment that appears to limit productive transcription to a single BES (7).
Silencing of gene family members is mediated by epigenetic mechanisms. Silent OR genes in mice are marked with histone methylations H3K9me3 and H4K20me3 that are characteristic of constitutive heterochromatin (8). Plasmodium falciparum, the parasite causing cerebral malaria, exhibits monoallelic expression of the var gene family, which encodes variants of the P. falciparum erythrocyte membrane protein 1 (9). This protein is expressed on the surface of an infected erythrocyte and mediates cytoadherence to endothelial host receptors keeping the infected cell in the peripheral vascular system. Deficiency of the histone deacetylase Sir2 in P. falciparum led to activation of silent var genes and loss of monoallelic var gene expression (10,11). Accordingly, knockdown of T. brucei RAP1, encoding a telomeric protein known to induce epigenetic gene silencing, led to derepression of all known BESs, detection of additional extranucleolar RNA pol I foci and formation of mixed VSG cell surface coats (12). In addition, several other factors have been implicated in BES silencing because their gene knockdowns resulted in derepression of silent BESs; these comprise the methyltransferase DOT1B (13), the chromatin remodelers TbISWI (14) and FACT (15), the histone deacetylase DAC3 (16), the nucleoplasmin-like protein NLP (17), histone H1 (18), the origin recognition complex (19), histone H3 and the histone chaperones CAF-1b and ASF1A (20) as well as the mini-chromosome maintenance-binding protein MCM-BP (21). Moreover, nucleosomes are enriched in the chromatin of silent BESs but are depleted in that of the active BES (22,23).
While these studies demonstrated the importance of epigenetic factors in restricting VSG expression to a single gene, the mechanism of transcriptional silencing is yet to be determined. Importantly, in the cited studies, mature VSG mRNA levels from derepressed BESs remained ∼10–10 000-fold lower than that of the active VSG. BES derepression by RAP1 and DOT1B silencing did not affect the high expression level of the active VSG (12,13), indicating that derepressed BESs cannot effectively compete with VSG expression from the active BES. On the other hand, silencing either SPT16, encoding a FACT subunit, or NLP did reduce the abundance of active VSG transcript (15,17). However, as shown in the NLP study, promoter-proximal gene expression of the active site was much less affected, suggesting again that in the derepressed state, BES promoters cannot efficiently compete for transcription factors from the active promoter.
We have recently characterized the multi-subunit class I transcription factor A (CITFA) as an essential factor for RNA pol I transcription of the active BES, the large ribosomal gene unit (RRNA) and the procyclin gene units, which encode the major cell surface proteins of insect-stage, procyclic form (PF) trypanosomes (24). Characterization of CITFA revealed seven apparently trypanosomatid-specific proteins, termed CITFA-1 to -7, and the dynein light chain DYNLL1 (24,25). The purified complex bound to the 67-bp-long BES promoter in a gel shift assay requiring the integrity of both known promoter elements for efficient binding. Moreover, depletion of CITFA-2 or CITFA-7 from extract or inactivation of CITFA-2 by an anti-CITFA-2 antibody abolished RNA pol I transcription in vitro from BES, rRNA gene (RRNA) and procyclin gene promoters, as determined by 121–146-bp-long primer extension products. This identified CITFA as a basal promoter-binding transcription initiation factor for RNA pol I in trypanosomes (24,25). Accordingly, silencing of CITFA-2 and of CITFA-7 resulted in rapid loss of rRNA and mRNA of the active VSG gene in bloodstream forms (BFs) and in trypanosome death in culture. Here, anti-CITFA-2 and anti-CITFA-7 chromatin immunoprecipitation (ChIP) assays showed that CITFA predominantly occupied the active BES promoter relative to that of a silent BES, a phenotype maintained after switching BESs in situ. High CITFA occupancy at the active BES promoter correlated with high promoter occupancy by RNA pol I and with a high promoter-proximal RNA level. Accordingly, as shown for the RRNA loci, CITFA-7 silencing considerably reduced both promoter-proximal RNA pol I occupancy and RNA abundance. Together these data strongly indicate that monoallelic VSG transcription is regulated at the level of transcription initiation. Moreover, fluorescently tagged CITFA-7 was found to be concentrated in the nucleolus and the ESB supporting the notion that CITFA has a role in BES activation. Because a ChIP-seq analysis revealed that among BES sequences high CITFA-7 occupancy is restricted to the promoter, our results raise the possibility that the sequestration of CITFA limits productive transcription initiation by RNA pol I to the nucleolus and the ESB.
MATERIALS AND METHODS
DNAs
pCITFA-7-PTP-NEO and the RNAi stem-loop vector pCITFA-7-T7-stl were described previously (25). pCITFA-7-eYFP-NEO was generated by inserting 353 bp of the CITFA-7 coding region (position 56 to position 408) into the ApaI and NotI sites of pC-eYFP-NEO (26). pCITFA-7-PTP-BLA was made by replacing the neomycin phosphotransferase with the blasticidin S deaminase ORF, pBLA-PTP-CITFA-2 and pBLA-PTP-RPB6z were likewise generated from pPURO-PTP-CITFA-2 (24) and pPURO-PTP-RPB6z (27). pPURO-mCherry-RPB6z was obtained by replacing the sequence of the composite PTP tag (protein C epitope, tobacco etch virus [TEV] protease site, tandem protein A domain) with the mCherry fruit fluorescent protein coding region. Allele deletions were achieved by transfecting polymerase chain reaction (PCR) products in which 100 bp of 5′ and 3′ gene flanks were fused to the hygromycin phosphotransferase coding region. DNA oligonucleotides that were used in competitive and quantitative PCR (qPCR) are listed in Supplementary Table S1. Semiquantitative reverse transcriptase-PCR (RT-PCR) was performed using cycle numbers which were empirically determined to be within the linear amplification range for each oligonucleotide pair. To demonstrate that consensus oligonucleotides co-amplify promoter sequences from different BESs and RRNA repeats, we sequenced PCR amplification products of genomic DNA and detected 11 out of 14 and 2 out of 3 known single-nucleotide polymorphisms of BES (4) and RRNA promoters, respectively (Supplementary Figure S1 and data not shown).
Cells
Culturing and stable transfection of BFs and PFs were carried out as previously described (25,28). BF cells were grown with either G418, puromycin, hygromycin, phleomycin or blasticidin at final concentrations of 2.5, 0.2, 1, 1 and 2 µg/ml, respectively, unless specified otherwise. Double-strand CITFA-7 RNA expression was induced with 2 µg/ml doxycycline. For in situ BES switching, 1.8 × 104 Np/nP cells were washed three times with prewarmed medium, resuspended in medium to 2 × 103 cells/ml, divided into six aliquots and each aliquot grown in antibiotic-free medium to a 13-ml culture of 1 × 106 cells/ml. After dilution with an equal volume of medium, cells were plated in 1 ml aliquots of either 10 µg/ml puromycin or 100 µg/ml G418. Stable clonal cell lines were maintained at either 1 µg/ml puromycin or 10 µg/ml G418. Assays of drug sensitivity of cells following BES switchings were as described in (13), namely, 10 ml of 1 × 103 cells/ml were grown in either 1 µg/ml puromycin or 100 µg/ml G418 for 48 h, growth was then monitored for 72 h.
Fluorescence microscopy
Microscope slides and coverslips (2.5 × 2.5 cm) were washed in methanol in a heated ultrasonic water bath for 20 min, rinsed thoroughly with deionized water and washed with 100% ethanol for 20 min. Following a final rinse in water, the slides and coverslips were air-dried. A square chamber was made on each slide using strips of labeling tape. Cells in exponential growth were centrifuged for 4 min at 800g and room temperature, washed in cold medium and resuspended in cold BBSG buffer (50 mM Bicine, 77 mM glucose, 50 mM NaCl and 5 mM KCl, pH 8.0) to a density of 1 × 105 cells/µl. Cells were fixed by adding 4% paraformaldehyde to a final concentration of 0.05% for 1 min after which they were pelleted at 800g for 45 s. Following two washes with BBSG buffer, the cell pellet was resuspended in BBSG buffer to a final concentration of 4 × 105 cells/µl and 4,6-diamidino-2-phenylindol (DAPI) was added to a final concentration of 0.04 µg/ml. Twenty-five microliters of cell suspension were spread on the chamber and covered with 125 µl of 1% low gelling agarose kept warm at 37°C (Sigma, St. Louis, MO, USA), and a coverslip was placed on top to close the chamber. Slides were stored at 4°C for ∼16 h for complete cell fixation and pictures were acquired within 24 h from the time of embedding, using an Axiovert 200 microscope (Zeiss, Thornwood, NY, USA). Images were processed with Axiovision software (Zeiss) and ImageJ. Whole cell and ESB enhanced yellow fluorescent protein (eYFP) signals were quantified using ImageJ and corrected by subtracting the mean pixel intensity of the surrounding region of each cell analyzed. Individual cell analysis was restricted to cells with only one nucleus and one kinetoplast.
ChIP and ChIP-seq analyses
ChIP experiments were carried out as previously described (29), resulting in average chromatin fragments of 200–400 bp. Analysis of Np/nP cell lines required larger chromatin fragments to accommodate amplification of the selectable marker regions downstream of the transcription initiation site (TIS). DNA fragments of ∼600 bp were obtained with medium sonication power using a Bioruptor UCD-200 (Diagenode, New York, NY, USA) and a reduced sonication time from 25 to 8 min (30 s on/30 s off). PTP-tagged CITFA-7, CITFA-2 and RPB6z were immunoprecipitated with an anti-ProtA rabbit polyclonal serum (Sigma) and untagged RPA31 with a rat anti-RPA31 immune serum (27). Negative control precipitations were conducted with either rabbit or rat nonspecific immune sera. The percent IP was calculated relative to the input material and corrected by subtracting the percent IP of the negative control assays. The fold enrichment was obtained by dividing the amount of precipitated DNA by the amount of DNA from control precipitations. To generate the library for ChIP-seq analysis, 1 × 109 cells were processed to produce 10 ng of precipitated DNA. In this case, chromatin was sonicated for 100 min (ten 10 min of 30 s on/30 s off with 5 min cooling on ice in between) on high setting to obtain smaller fragments ranging from 100 to 250 bp in size. qPCR quality controls and setup were analyzed as described previously (29). For the preparation of ChIP-seq libraries, 10 ng of input DNA and 10 ng of DNA precipitated with CIFTA-7 were end-repaired by incubating the DNA with 1 × T4 ligase buffer (NEB), 0.4 mM dNTPs, 0.5 µl of End Repair Enzyme Mix (NEB, 30 µl of 3 U/µl T4 DNA polymerase, 6 µl of 1 U/µl Klenow Fragment, 30 µl of 10 U/µl T4 DNA Polynucleotide Kinase) in a total volume of 50 µl for 30 min at 20°C. The end-repaired DNA fragments were cleaned up using AmPure XP beads (Beckman Coulter) as suggested by Illumina. To generate a protruding 3′A base for adapter ligation, the end-repaired DNA was mixed with 2 µl of 10 × NEB buffer 2, 1 µl of 4 mM dATP and 0.5 µl of Klenow 3′ to 5′ exo minus enzyme (5 U/µl, NEB) and incubated for 30 min at 37°C. Next, the DNA was ligated to 1 µl of forked DNA adapters (0.2 µM) using the Quick Ligation Kit (NEB) according to the manufacturer’s specifications. To prepare the forked adapters, forward (AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTTCCGATC*T) and reverse DNA oligonucleotides (/5Phos/GATCGGAAGAGCACACGTCTGAACTCCAGTCACGCCAATAT CTCGTATGCCGTCTTCTGCTT*G or/5Phos/GATCGGAAGAGCACACGTCTGAACTCCAGTCACCTT GTAATCTCGTATGCCGTCTTCTGCTT*G) were suspended at 100 µM in 10 mM Tris–HCl, pH 7.8, 0.1 mM EDTA, pH 8.0, 50 mM NaCl, mixed at equal concentrations, heated to 95°C and annealed by slowly cooling the mix to room temperature (asterisk denotes a phosphorothioate bond). Adaptor-annealed DNA was cleaned up using AmPure XP beads and amplified using the KAPA HIFI HotStart Ready Mix (Kapabiosystems) and PCR primers 5′-AATGATACGGCGACCACCGA*G-3′ (forward primer) and 5′-CAAGCAGAAGACGGCATACGA*G-3′ (reverse primer) for five cycles as suggested by Kapabiosystems. The PCR product was gel-purified and, again, PCR-amplified (15 cycles). The final PCR product was cleaned up using the MinElute PCR Purification Kit (Qiagen) and gel purified on a 1% agarose gel followed by a cleanup using the MinElute Gel extraction Kit (Qiagen). The quality and concentration of the final libraries was determined using a BioAnalyzer 2100 (Agilent).
Libraries from input and immunoprecipitated DNA were sequenced using a HiSeq2000 sequencer (Illumina). For the input DNA, we obtained 75 167 347 sequence reads of which 73.6% could be mapped (45.6% mapped to exactly 1 site). For the immunoprecipitated DNA, we obtained 16 722 060 sequence reads of which 59.8% could be mapped (39.8% mapped to exactly one site). The sequence reads (100 bp) were annotated based on the T. brucei 427 genome (version 4.1) to which we added the sequences of the 14 BESs available at http://www.sanger.ac.uk/resources/downloads/protozoa/trypanosoma-brucei.html. Standard and fast gapped read alignments were performed using Bowtie version 2 (30). To normalize the read coverage between the libraries, the read coverage was divided by the total number of aligned reads in each library and multiplied by the total number of aligned reads of the smaller library.
RNA analysis
Total RNA was prepared by the hot phenol method as specified previously (27). For pre-mRNA analysis, total RNA was reverse transcribed with random hexanucleotides (Roche) as primers and SuperScript II reverse transcriptase (Invitrogen) following the manufacturer’s recommendations, and cDNA was amplified with at least one oligonucleotide hybridizing outside the sequence of the mature mRNA.
RESULTS
CITFA-7 occupancy of BES and RRNA promoters
To further investigate the transcriptional roles of the CITFA complex in RNA pol I transcription, we analyzed CITFA-7 occupancy of RRNA and BES promoters in BFs. Previously, we have generated the BF cell line TbC7ee that expresses CITFA-7 with a C-terminal PTP tag and no untagged CITFA-7 (Figure 1) (25). The large composite PTP tag which consists of a protein C epitope, a TEV protease site and dual protein A (ProtA) domains (31) did not affect the functionality of CITFA-7 because CITFA-7 is essential for trypanosome viability and TbC7ee cells proliferated as efficiently as wild-type cells (25). Using a ChIP-grade, polyclonal anti-ProtA antibody (32) and a nonspecific rabbit immune serum for normalization, we precipitated chromatin from TbC7ee cells. According to qPCR analysis using consensus oligonucleotides for BES and for RRNA promoters, both promoter sequences were clearly enriched in the precipitate in comparison with the β−α tubulin intergenic region, which served as a negative control (Figure 1). DNA fragments within the ESAG7 and 18S rRNA coding regions, which extended to ∼1.6 and ∼1.2 kb downstream of the BES and RRNA TISs, respectively, precipitated much less efficiently than their promoters. Because we have previously found that RNA pol I cross-linked equally to the RRNA promoter and to the 18S rRNA coding region (29), these results strongly indicated that CITFA-7 does not associate with elongating RNA pol I and that it binds only to the promoter region, thus substantiating the role of CITFA as a transcription initiation factor. Most interestingly, RRNA promoter DNA was consistently ∼5-fold more enriched than BES promoter DNA. Because the trypanosome harbors multiple copies of these promoters, this result suggested that CITFA-7 occupied relatively more RRNA than BES promoters and, therefore, raised the possibility that among BES promoters it primarily bound the promoter of the active BES.
Figure 1.

CITFA-7 occupies specifically RNA pol I promoters. Top panel, schematic drawing of the CITFA-7 locus in cell line TbC7ee, which exclusively expresses CITFA-7 with a C-terminal PTP fusion. The open reading frames of CITFA-7, of HYGR and NEOR, and the PTP tag are indicated by an open box, striped boxes and a black box, respectively. Smaller gray boxes represent gene flanks for RNA processing signals. The PTP tag sequence was fused to an endogenous CITFA-7 allele by targeted integration of pCITFA-7-PTP as indicated. Bottom panel, anti-ProtA ChIP with TbC7ee cells expressing CITFA-7-PTP. Precipitated DNA was analyzed by qPCR of the consensus BES promoter (BES prom), a conserved region of the ESAG7 coding region (ESAG7 ORF), the consensus RRNA promoter (RRNA prom), 18S rRNA coding region (18S rDNA) and the β − α tubulin intergenic region (β − α tub). The precipitation percentage relative to the input material was corrected by subtracting corresponding values from negative control precipitations that were carried out with a comparable nonspecific antiserum.
CITFA-7 predominantly occupies the active BES promoter relative to a silent BES promoter
The previously described BF cell line PN13 in which the silent BES1 and the active BES17 were marked with a puromycin N-acetyltransferase gene (PUROR) and a neomycin phosphotransferase (NEOR) gene, respectively (13), was used to directly test the binding of CITFA-7 to promoters of active and silent BESs. Both genes were introduced at corresponding positions 240 bp downstream of the TIS, locations that allowed for the selection of in situ switches between these two BESs and for specific DNA amplification downstream of the BES1 and BES17 promoters. To enable anti-CITFA-7 ChIP experiments in these cells, we stably integrated plasmid CITFA-7-PTP-BLA into one CITFA-7 allele and termed the corresponding line C7-Np1, indicating that these trypanosomes are resistant to neomycin (upper case N), sensitive to puromycin (lower case p) and represent the basal line from which nP2 and Np3 cell lines were directly derived by consecutive in situ BES switching experiments (Figure 2A).
Figure 2.

BES transcription is activated at the level of transcription initiation. (A) Schematic outline of genomic modifications in cell line C7-Np1 in which NEOR and PUROR were inserted into the active BES17 and silent BES1, respectively, and the PTP sequence fused to the CITFA-7 coding sequence in one CITFA-7 allele. Arrows labeled 1–3 represent primers used in the subsequent competitive PCR analysis. (B) Anti-CITFA-7-PTP ChIP assays, carried out with C7-Np1 and one C7-nP2 cell line, in which BES1 was activated and BES17 silenced, were analyzed by competitive PCR amplifying NEOR- and PUROR-specific sequences in total DNA (Inp), control IP (ctrl) and anti-ProtA IP (IP). (C) qPCR analysis of anti-CITFA-7-PTP ChIP assays, carried out with clonal cell lines obtained from in situ switching events between BES17 and BES1, using NEOR- or PUROR-specific primer pairs. The difference between BES17 and BES1 occupancies were statistically analyzed by paired two-tailed student’s t-tests. One, two and three asterisks indicate P-values that are <0.05, 0.01 and 0.001, respectively. (D) Calculation of the fold enrichment over negative control precipitations for the ChIP experiments shown in the bottom panel of Figure 1 and the C7-Np1 cell line shown in Figure 2C (BES17 NEOR and BES1 PUROR). (E) Reverse transcription-qPCR analysis of NEOR or PUROR pre-mRNA abundances in C7-Np/nP cell lines. The values for these relative abundances were standardized with the tubulin pre-mRNA level, which was set to 1. (F) qPCR analysis of anti-PTP-RPB6z ChIP with R6z-Np1 cells. Results shown in (E) and (F) were statistically analyzed as described above.
Anti-ProtA IP of chromatin prepared from C7-Np1 cells was first analyzed by a competitive PCR assay in which a common sense primer was paired with two antisense oligonucleotides that were complementary to either the NEOR or the PUROR coding region (Figure 2A). While the NEOR band was only slightly stronger than the PUROR band in DNA isolated from total chromatin or from the control IP, anti-CITFA-7 precipitated chromatin was strongly enriched in NEOR DNA derived from the active BES17 (Figure 2B). Incubation of C7-Np1 cells without antibiotics and subsequent selection with puromycin combined with limiting dilution, revealed independent cell lines that had switched to the expression of BES1. A corresponding ChIP analysis of one of these lines (C7-nP2) showed that now the BES1-derived PUROR band was highly enriched over the NEOR band, demonstrating that high CITFA-7 occupancy correlated with BES activation (Figure 2B).
To quantitatively assess CITFA-7 occupancy at BES1/BES17 promoters in these cell lines, we carried out qPCR analysis of precipitated DNA. To ensure specific amplification of BES1 and BES17 sequences, we chose oligonucleotide pairs that hybridized to the PUROR and NEOR coding regions, respectively. As shown for the original C7-Np1 cell line and six independently derived C7-nP2 clones and C7-Np3 clones, CITFA-7 occupied predominantly the active BES promoter relative to that of the silenced BES (Figure 2C). This pattern was not due to continuous selective antibiotic pressure because it remained unchanged when the antibiotics were removed for ten generations before the ChIP experiment. We did, however, consistently detect above background levels of CITFA-7 at the promoter of the silenced BES, indicating that the transcription factor does interact with silent BES promoters to some extent.
Although we specifically amplified the active BES in these experiments, the precipitation percentage did not increase over that of the assays of the consensus BES promoter (compare Figure 2C with Figure 1). This has two likely explanations: firstly, TbC7ee cells exclusively expressed tagged CITFA-7, whereas C7-Np/nP lines co-expressed wild-type and tagged CITFA-7. Secondly, in the first experiment (Figure 1), we amplified the BES promoter, which is the CITFA binding site, whereas the specific NEOR and PUROR amplification products of the subsequent experiment (Figure 2A) extended to positions 482 and 545 downstream of the TIS, respectively, thus amplifying only a fraction of the precipitated DNA. Accordingly, when we calculated the fold enrichment over negative control precipitations, the active BES17 in C7-Np1 cells exhibited a similarly high enrichment as the consensus RRNA promoter, a nearly 8-fold higher enrichment than that of the inactive BES1 promoter, and an approximately four times higher enrichment than that of the consensus BES promoter in TbC7ee cells, confirming high CITFA-7 occupancy of the active BES promoter (Figure 2D).
BES activation is regulated at the level of transcription initiation
Our previous in vitro transcription results strongly indicated that CITFA is a basal, DNA-binding transcription initiation factor that mediates RNA pol I recruitment to its cognate promoters. The purified CITFA complex required both BES promoter elements for efficient DNA binding and depletion of CITFA from extract resulted in the loss of RRNA and BES promoter transcripts that were only 127 and 146 nucleotides long, respectively (24;25). Hence, high CITFA occupancy should lead to high promoter-proximal transcription rates and to high RNA pol I occupancy at the active BES promoter. To assess promoter-proximal transcription levels at the active versus the silenced BES, we quantified the abundances of pre-NEOR and pre-PUROR mRNA relative to tubulin pre-mRNA in C7-Np1 cells as well as in one C7-nP2 and one C7-Np3 cell line (Figure 2E). Because in these cell lines NEOR and PUROR RNA processing is directed by aldolase gene flanks (13), we paired a sense oligonucleotide that targets the aldolase 5′ gene flank upstream of the known aldolase splice site with either a NEOR- or PUROR-specific antisense oligonucleotide to amplify random hexamer-derived cDNA. As expected, pre-mRNA of the selectable marker from the active site was 16 (Np1), 40 (nP2) and 24 (Np3) times more abundant than tubulin pre-mRNA and exceeded the selectable marker pre-mRNA from the inactive BES 59- to 79-fold (Figure 2E).
Because this finding suggested a higher transcription initiation rate from the active BES promoter than from the inactive BES promoter, we wanted to determine the promoter-proximal occupancy of RNA pol I at these sites. We generated analogous to the C7-Np1 cell line (Figure 2A), an R6z-Np1 cell line in which we introduced the functional N-terminal PTP fusion (27) to the RNA pol I-specific subunit RPB6z in PN13 cells. The corresponding ChIP experiment demonstrated that RNA pol I occupancy was ∼17-fold higher at the active BES17 promoter than at the silent BES1 promoter (Figure 2F).
Finally, we confirmed that CITFA-7 silencing is functionally linked to the loss of promoter-proximal RNA pol I occupancy and the reduced abundance of promoter-proximal pre-RNA. Because BESs in our extant single marker cells for gene knockdowns were unmarked, we were unable to specifically investigate the active BES. Instead, we focused our analysis on the RRNA promoter region because CITFA-7 is equally important for RRNA and BES transcription (25), and it is likely that most RRNA repeats are active in rapidly dividing, cultured trypanosomes. For these ChIP experiments we used a polyclonal antibody directed against the essential RNA pol I-specific subunit RPA31 (27). This antibody effectively precipitated the RRNA promoter and the 18S rRNA coding region in noninduced cells, while the β−α tubulin intergenic region, which is transcribed by RNA pol II, was not precipitated above the control precipitation with a comparable nonspecific polyclonal antibody (Figure 3A). On silencing CITFA-7 for 42 h, RNA pol I occupancy decreased, on average, by 83% at the RRNA promoter and by 84% in the 18S rRNA coding region. Similarly, promoter-proximal 18S pre-rRNA abundances were reduced, on average, by 91.5% in CITFA-7-silenced cells as analyzed by qPCR of random hexamer-derived cDNA. The corresponding semiquantitative qPCR analysis shows the drop of pre-rRNA abundance and the concomitant increase of tubulin pre-mRNA (Figure 3B). The latter finding is consistent with our previous observation that the massive loss of rRNA in CITFA-7-silenced cells led to an increase of all nonaffected RNAs in total RNA preparations of equal amounts (25). These results confirmed the pivotal role of CITFA in RNA pol I transcription initiation.
Figure 3.

CITFA-7 silencing affects RRNA promoter-proximal RNA pol I occupancy and pre-18S rRNA abundance. (A) Anti-RPA31 ChIP assays were conducted with noninduced bloodstream trypanosomes (BFs) and with BFs in which CITFA-7 was silenced for 42 h. Precipitated chromatin was analyzed by qPCR, amplifying regions of the RRNA promoter, the 18S rDNA and, as a control, the β − α tubulin intergenic region. The experiments were repeated three times and analyzed by an unpaired two-tailed Student’s t-test assuming unequal variances. (B) Semiquantitative RT-PCR analysis of 18S pre-rRNA and tubulin pre-mRNA (pre-tub) in noninduced cells (n.i.) and CITFA-7-silenced cells.
Taken together, we have shown here that high CITFA-7 occupancy of the active BES promoter coincides with high levels of promoter-proximal RNA from this BES and high RNA pol I occupancy in the promoter region. In addition, we demonstrated for the RRNA gene array that depletion of CITFA-7 strongly reduces RNA pol I occupancy in the promoter region and causes significant decreases of promoter-proximal RNA abundance. Therefore, we concluded that monoallelic BES transcription in T. brucei is regulated at the level of transcription initiation.
BES promoter occupancy by CITFA-2 parallels that of CITFA-7
The high occupancy of the active BES promoter by CITFA-7 suggested that the whole CITFA complex is bound there. To confirm this notion, we repeated the ChIP analysis with the essential CITFA-2 subunit. Similar to the CITFA-7 analysis, we generated cell line C2-Np1 by tagging CITFA-2 N-terminally with PTP in PN13 cells. We previously showed that PTP-CITFA-2 is fully functional (24). The competitive PCR analysis demonstrated even more strikingly than in the CITFA-7 analysis the predominant association of CITFA-2 with the active BES promoter (Figure 4A). qPCR analyses of anti-PTP-CITFA-2 ChIP assays conducted in C2-Np1 cells and in independently derived, switched C2-nP2 and C2-Np3 lines showed that CITFA-2 was even more distinctly associated with the active BES than CITFA-7 (Figure 4B). While in case of CITFA-7, on average 3.47 times more DNA was precipitated from the active versus the silent BES promoter, this fold difference in CITFA-2 occupancy was, at 6.24, significantly higher (P = 2.4 × 10−5, two-tailed, unpaired student’s t-test assuming unequal variance). Previously, by sedimenting tandem affinity-purified CITFA complex through a linear sucrose gradient, we obtained evidence that trypanosomes possess two CITFA complexes: a more abundant complex that lacks CITFA-2 and DYNLL1 and a slightly faster sedimenting full complex (25). Because CITFA-2 is indispensable for CITFA function in RNA pol I transcription from BES, RRNA and GPEET procyclin gene promoters, it appears that binding of CITFA-2 to the complex activates CITFA. Therefore, the more distinct association of CITFA-2 with the active BES promoter may be directly related to the fact that precipitating CITFA-2 specifically pulled down the active CITFA complex. Together, these data strongly indicated that, among BES promoters, CITFA is predominantly bound to the active BES promoter.
Figure 4.

High CITFA-2 occupancy of the active BES promoter. (A) Anti-PTP-CITFA-2 ChIP assays with C2-Np1 and C2-nP2 cell lines analyzed by competitive PCR of NEOR and PUROR sequences. (B) Corresponding qPCR analysis of C2-Np/pN cell lines. Statistical analysis was by paired two-tailed Student’s t-tests.
CITFA-7 is concentrated in the nucleolus and the ESB
The efficiency of the RNA pol I transcription system is due to its high initiation rate, which, as was shown for the yeast ribosomal gene unit (33), can lead to an RNA pol I density of one actively transcribing enzyme complex for every ∼100 bp. Accordingly, detection of RNA pol I in the ESB has been straightforward [reviewed in (34)]. On the other hand, it was demonstrated in the human system that the basal, promoter–binding RNA pol I transcription initiation factors SL1 and UBF can support multiple rounds of transcription in vitro without dissociating from the promoter (35), which suggests that relatively few transcription factors are required to ensure maximal RNA pol I transcription. Because the ESB appears to harbor a single BES with either one or two BES promoters (4), it was not certain whether CITFA would be detectable in this compartment. However, in a previous study, when we used indirect immunofluorescence microscopy to co-localize the PTP-tagged RNA pol I subunit RPB6z and HA-tagged CITFA-7, we occasionally detected a CITFA-7 signal outside the nucleolus that co-localized with RPB6z, suggesting that the ESB contains detectable amounts of the transcription factor (25). To enhance the sensitivity of the RPB6z/CITFA-7 co-localization, we created a BF cell line in which, by targeted plasmid integrations into the RPB6z and CITFA-7 loci, the mCherry protein and the eYFP were N- and C-terminally fused to RPB6z and CITFA-7, respectively (Figure 5A). Fluorescence microscopy showed that in 39% of cells (n = 155) mCherry-RPB6z was detected only in the nucleolus, which is apparent in DAPI stains as a spherical structure of low DNA density (Figure 5B, long arrow). In 41% of cells, mCherry-RPB6z localized to the nucleolus as well as to a single extranucleolar spot that was clearly separate from the nucleolus and, therefore, represented the ESB. Like the nucleolus, the ESB consistently exhibited a reduced DAPI stain (Figure 5B, short arrow). In the remaining 20% of cells, two extranucleolar mCherry-RPB6z signals were detectable. These numbers were in accordance with previous RNA pol I-based ESB identifications (7,13). Although the nucleolar staining patterns of CITFA-7-eYFP and mCherry-RPB6z were not completely congruent, which could be expected from a transcription initiation factor and an RNA pol, these proteins exhibited extensive co-localization in the nucleolus of each cell analyzed. Most importantly, in 91% of the cells with one or two extranucleolar spots, we could detect co-localization of CITFA-7-eYFP in all these spots (Figures 5B, C, two additional examples are shown in Supplementary Figure S2A). Although after our previous study we had anticipated that CITFA-7 would be detectable in the ESB, the rather robust signal of CITFA-7-eYFP in this compartment was nevertheless surprising because it suggested a much larger accumulation of the transcription factor in the ESB than is required for RNA pol I transcription initiation from a single BES. Quantification of eYFP fluorescence revealed that the ESB signal contributed 4.5% to that of a whole cell (standard deviation of 1.3%, n = 10). Because an immunoblot comparison of recombinant CITFA-7 with endogenous CITFA-7 of whole cell lysates uncovered that a BF trypanosome harbors ∼4000 CITFA-7 molecules (Supplementary Figure S2B), we calculated that the ESB contains ∼180 CITFA-7 molecules. Together, these data show that CITFA-7 is concentrated in both the nucleolus and the ESB, which strongly supports our finding of predominant CITFA occupancy of the active BES promoter.
Figure 5.
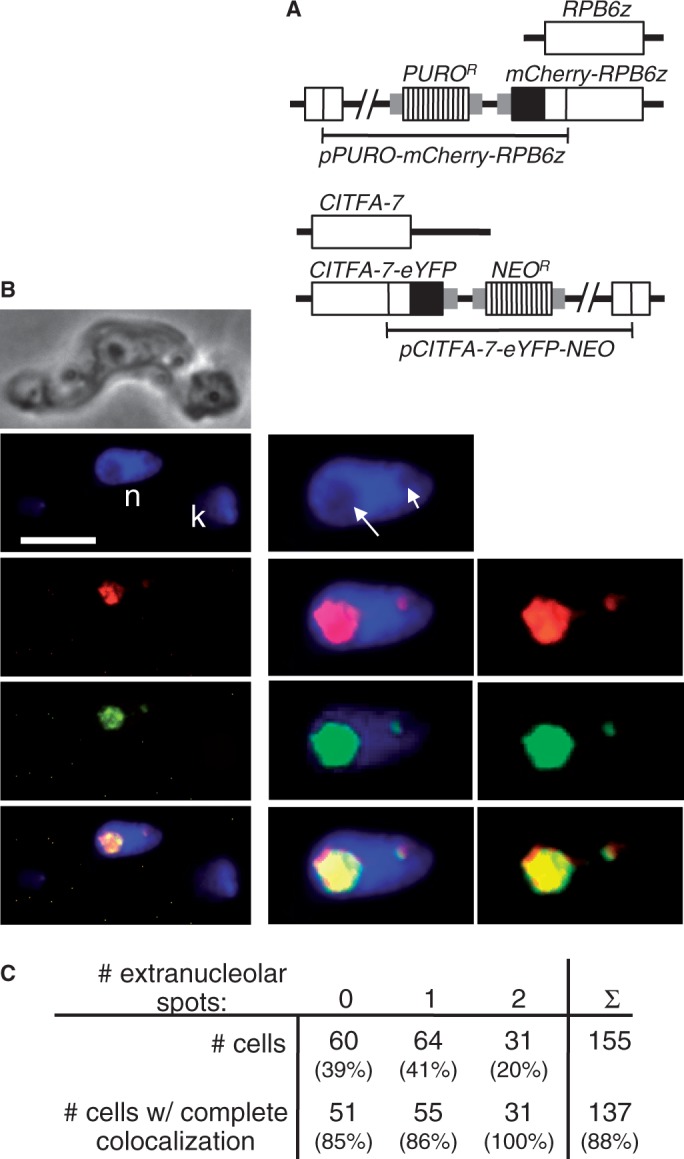
Co-localization of mCherry-RPB6z and CITFA-7-eYFP. (A) Schematic of the RPB6z and CITFA-7 loci showing the targeted pPURO-mCherry-RPB6z and pCITFA-7-eYFP-NEO integrations in the BF cell line used here. (B) Images from top to bottom show phase contrast, DAPI stain (bar: 10 µm; n, nucleus; k, kinetoplast; long and short arrows indicate nucleolus and ESB, respectively), mCherry-RPB6z, CITFA-7-eYFP (pseudo-colored in green) and an image merge. Images on the left are unmodified, whereas red and green signals in the enlarged images (panels in the middle and on the right) were recolored and enhanced in brightness and contrast. (C) Statistical analysis of 155 individual cells. Cells were categorized by the number of extranucleolar RNA pol I (mCherry-RPB6z) spots. Cells in which mCherry-RPB6z and CITFA-7-eYFP exhibited clear co-localization in the nucleolus and all detectable extranucleolar spots were scored as cells with complete co-localization.
Peak(s) of CITFA-7 occupancy within a BES is restricted to its promoter region(s)
Our findings of high CITFA occupancy of the active BES promoter and the presence of ∼200 CITFA-7 molecules in the ESB raised the question of how this factor is concentrated in this compartment. We have previously shown in vitro that the CITFA complex is a sequence-specific DNA binding factor that requires both BES promoter elements for efficient binding (24). However, it was possible that BESs harbor multiple CITFA binding sites or that CITFA-7 remains bound to RNA pol I during transcription elongation, which would lead to an enrichment of CITFA-7 along the entire BES. To determine the presence of CITFA along the entire BES, we conducted a genome-wide CITFA-7-PTP ChIP-seq analysis and aligned the reads to the genome of T. brucei brucei 427 including the 14 BES sequences, which were available for this strain (4). To account for sequencing biases and alignment artifacts, the number of sequencing reads obtained from the IP was normalized with the number of sequencing reads obtained from the input material. Reads (100 nt) were aligned to the most similar genomic sequence or, if the target site was not unique, randomly distributed among all possible target sites. Thus, the analysis was limited by two constraints: firstly, BES promoter regions are nearly identical, differing only in a few single nucleotide polymorphisms, which made unambiguous alignment of most reads to particular BES promoters impossible. Hence, reads from the active site were presumably distributed among most BESs. Secondly, for our analysis, we used cells that originated from the Tc221 clone of the 427 strain (36) that had switched expression from the original VSG221 gene to an unknown VSG gene (kind gift from Dr Peter Overath, MPI Tübingen, Germany). As we found out during our analysis, the BES promoter regions of this strain differed in a few positions from the published 427 strain (4), preventing the unambiguous identification of individual BESs according to the known promoter sequences (data not shown). Nevertheless, sequence reads could be aligned along all BESs, indicating that the Tc221 BES sequences still resembled the published ones enough to enable a meaningful analysis.
Figure 6 shows the analysis of BES15 (also annotated as telo126), which has two promoters of highly similar sequence (P1 and P2). After normalizing for the different number of reads obtained with input and immunoprecipitated DNA, peaks of reads were observed in several BES regions, most of them in the 70-bp repeat regions, the VSG gene and the VSG pseudogene (Figure 6, second panel). However, these peaks were also observed in input DNA that was not enriched by IP (Figure 6, third panel), which suggests that they were formed owing to an underrepresentation of these repetitive sequences in the published genome. Two peaks in BES15, however, were absent in the input. They precisely mapped to the two BES15 promoters and, therefore, are the result of ChIP enrichment. Accordingly, when we calculated the fold enrichment of ChIP reads over input reads, 13.6- and 9.2-fold enrichments were obtained for P1 and P2, respectively. Closer inspection of the 67-bp-long P2 promoter showed that the highest number of reads and the highest folds of enrichment precisely mapped to all seven windows representing the promoter sequence (Figure 6, zoomed-in panels). The number of reads on either side of the promoter rapidly declined, suggesting that the transcription factor occupies only the promoter and does not follow the polymerase after transcription initiation. This notion was backed up by our finding that RPA1, the largest subunit of RNA pol I, did not co-immunoprecipitate with CITFA-7-PTP in a DNA-independent manner (Supplementary Figure S3) and by the fact that subunits of RNA pol I or of CITFA never cross-purified in several tandem affinity purifications (24,25,27,37,38). Hence, we concluded that, within a BES, CITFA-7 occupancy is restricted to the promoter region and that there are no additional binding sites that could explain CITFA concentration in the ESB. This result is in close agreement with our standard ChIP results, which showed that CITFA-7 occupancy was strongly decreased downstream of the BES promoter (Figure 1). Given that the single 67-bp BES promoter is unlikely to support binding of multiple CITFA complexes, and that CITFA is not found away from this promoter, our data raise the interesting possibility that CITFA is concentrated in the nucleolus and the ESB independent of DNA and RNA pol I association, thereby restricting high RNA pol I transcription initiation rates to these two compartments.
Figure 6.

CITFA-7 enrichment at BES15 (telo126). For BES15, the fold enrichment of DNA immunoprecipitated with CITFA-7 compared with input DNA (first panel) and the normalized read coverages obtained from the CITFA-7 IP (second panel) and the input material (third panel) are shown. The read coverage was normalized to account for differences in library size (see ‘Materials and Methods’ section) and calculated for a window size of 11 bp. Orange boxes represent ESAGs identified by numbers (e.g. seven stands for ESAG7) and of VSG genes (Ψ identifies pseudogenes), green boxes indicate promoters, and black boxes depict 70-bp repeats. For the zoomed-in panels, sequences for each window are specified. The two BES promoter elements and the TIS position are identified by red lettering and the remaining promoter sequence by blue lettering. The bars corresponding to the promoter sequence are depicted in blue. The green bar at the bottom represents the promoter region annotation in the genome data bank.
CITFA-7 was enriched to some extent on all BES promoters analyzed (Supplementary Figure S4 and data not shown). This likely resulted from reads of the active BES being distributed among other BESs. In support of this notion, we found more reads at RRNA promoters than on BES promoters. Within the nine annotated RRNA promoters, the peak window accumulated an average of 1233 normalized reads, which is 4.25-fold more than the BES15 promoter peak, suggesting that reads from the active BES were aligned to silent BESs owing to sequence identities. Moreover, we observed several-fold differences in the number of normalized reads assigned to the different BES promoters that were phylogenetically linked (Supplementary Figure S5). The combined reads for the seven promoter windows ranged from 154 for the single BES1 promoter to 2148 for the single BES5 promoter. Promoters whose sequence closely resembled those of the similar BES15/BES5 promoters obtained high numbers of reads, whereas more distantly related BES promoters obtained much fewer reads. These findings are in agreement with CITFA being predominantly bound to the promoter of the active BES.
DISCUSSION
VSG expression in T. brucei is an ideal system to study the mechanism of monoallelic gene expression because RNA pol I transcription of the active BES in the ESB is spatially separated from RRNA transcription in the nucleolus. We took advantage of this fact and showed that CITFA-7, an essential subunit of the CITFA complex, is concentrated in the nucleolus and the ESB, substantially co-localizing with RNA pol I in these compartments. Consistent with this observation, we found that CITFA-7 and the previously characterized, essential subunit CITFA-2 were predominantly associated with the promoter of the marked active BES versus that of a marked silent BES. In situ switching between these two BESs reversed the promoter occupancy pattern of the two CITFA subunits, which established that BES activation is coupled with high CITFA interaction. Because CITFA is a promoter-binding transcription initiation factor that is absolutely required for RNA pol I transcription in vivo and in vitro (24,25), high CITFA occupancy should lead to high rates of RNA pol I recruitment and promoter-proximal transcription rates. Accordingly, we found the RNA pol I-specific subunit RPB6z to be highly abundant in the promoter region of the active BES, and promoter-proximal RNA levels of the active BES to exceed those of the silent BES ∼70-fold. Furthermore, knockdown of CITFA-7 led to a sharp decrease of RNA pol I occupancy at the RRNA promoter and the downstream 18S RRNA coding region, as well as a high reduction of the promoter-proximal pre-rRNA. Together, these data show that monoallelic VSG expression is controlled, at least partially, at the level of transcription initiation and indicate that this control is mediated by binding of a functional CITFA complex to the active BES promoter. This is the first demonstration in any system of antigenic variation that basal transcription initiation factor occupancy differs between the active and the silent genes.
Although CITFA predominantly occupied the active BES promoter, we clearly observed above background CITFA occupancy at the silent BES promoter. One possibility is that the marked silent BES in our study was not completely silenced but remained in a ‘pre-active’ state. Previously, as a model to explain co-expression of two BESs under antibiotic selection, it was proposed that between silent and active BESs there is a pre-active state that precedes BES activation (39). The pre-active state of a BES is characterized by close spatial proximity to the active BES and thought to enable rapid back and forth switching between these two BESs under selective pressure. Because we switched repeatedly between BES1 and BES17, it is possible that the silenced BES was locked in the pre-active state giving it greater access to CITFA than a completely silenced BES. On the other hand, our detection of CITFA at silent BES promoters is consistent with previous studies demonstrating that a low level of transcription does initiate at silent BESs but does not reach the promoter-distal VSG gene (40,41). A previous expression analysis of the promoter-proximal ESAG6 gene revealed that ∼20% of all ESAG6 mRNA were derived from silent BESs, whereas 80% originated from the active site (40). Taking 15 BESs into account, this implies an ∼56-fold stronger transcription initiation rate from the active BES than from any of the silent sites. This fold difference closely reflects our pre-mRNA-based estimate of an ∼70-fold higher initiation rate at the active BES promoter. However, cloning and sequencing of RT-PCR products along the complete BES revealed a more diverse picture and promoter-proximal expression from silent BESs varied strongly between different T. brucei strains (41). Nevertheless, in all cases, promoter-proximal RNA levels derived from the active BES did exceed those from any silent BES manifold. Together, these results confirm that activation of a BES not only requires ablation of telomeric silencing but also a mechanism that leads to higher transcription initiation rates.
Knowledge about proteins that preferentially interact with the promoter regions of silenced or active BESs is limited. While we do know that silent BESs are occupied by nucleosomes that are absent from the active BES (22,23), it is not known whether this difference is a consequence of transcriptional activity or represents an actual control mechanism (42). In agreement with the latter, a recent study showed that the HMG (high mobility group) box protein TDP1, which is thought to be a general architectural chromatin protein, exhibited high occupancy of the active BES and RRNA gene units, thereby likely facilitating their transcription (43). Additionally, histone H1 (18) and NLP (17) were found to be mainly associated with silent BES promoters, which suggest that they have specific roles in reducing transcription from these promoters. However, it remains unclear whether the differential distributions of these proteins among BESs are regulatory in nature or whether they reinforce the activated or silenced status of a BES. Nevertheless, these studies support our model that there are control mechanisms that operate at BES promoters.
Our data show that the active BES promoter recruits CITFA more efficiently than a promoter of a silent BES. One possible explanation for this difference is that repressive chromatin interferes with CITFA binding to silent BES promoters. This notion is supported by experiments that showed that silencing of DAC3, ISWI and NLP led to an upregulation in promoter-proximal gene expression at silent BESs (14,16,17). However, in these cases the derepressed expression level was not compared with that of the active BES and it is likely that it remained considerably lower. The high transcription level of the active BES (44) likely impedes upregulation of all silent BESs to the same level. If BES activation is achieved by derepression only, one would expect that general BES derepression would effectively impede transcription of the active site, owing to competition for CITFA and RNA pol I. Thus far, promoter-proximal transcription was only assessed in the NLP knockdown, which led to a moderate ∼3-fold reduction of mRNA from a selectable marker gene that was introduced downstream of the active BES promoter (17). Because our results suggest that the rate of RNA pol I transcription initiation is limited by CITFA availability, it will be important to analyze whether BES derepression leads to higher CITFA occupancy.
An alternative explanation for the predominant association of CITFA with the active BES would be a mechanism that confines this basal transcription factor to the nucleolus and the ESB, limiting its availability and high RNA pol I transcription initiation rates to these compartments. Because we did not find evidence for a direct interaction between CITFA-7 and RNA pol I and CITFA appeared to be cross-linked only to the promoter region in our ChIP-seq analysis, the high concentration of CITFA-7 in these compartments seemed to be independent of RNA pol I and the capability of CITFA to bind DNA. A model of confined CITFA availability would be in agreement with previous analyses of VSG expression. The spatial proximity of co-expressed BESs observed by Chaves et al. (39) could reflect the need to acquire CITFA from the ESB for selectable marker expression. This model would also be consistent with previous findings showing that functional replacement of a BES promoter with an RRNA promoter left BES regulation intact (45) because CITFA also enables RRNA promoter transcription (24). Finally, it concurs with results obtained on silencing of RAP1, which led to a significant increase of detectable RNA pol I accumulations outside the nucleolus but did not alter the transcription efficiency of the promoter-proximal BES regions (12). These findings imply that transcription elongation of RNA pol I along BESs is mainly blocked epigenetically and that, in accordance with our results, VSG activation occurs at the BES promoter region. Lastly, these results show that RNA pol I accumulation at derepressed BESs is not sufficient to drive promoter-proximal BES transcription, suggesting that RNA pol I sequestration is not regulating BES transcription, whereas confinement of CITFA may do so.
It should be noted here that CITFA subunits are conserved among all trypanosomatids including those that do not have a multifunctional RNA pol I. Except for some structural differences between RRNA and BES promoters (46), RRNA transcription initiation in the nucleolus and BES transcription in the ESB appear to function equivalently. Nevertheless, because protein sequestration to the nucleolus is a well-established phenomenon (47), it is possible that the ESB is a nucleolus-like structure that offers interaction sites for the CITFA complex. As discussed previously, the dynein component of CITFA is a likely candidate to facilitate factor sequestration (24). However, DYNLL1 binding motifs, which are conserved from yeast to mammals, were not detected in CITFA sequences and, thus, will have to be determined experimentally.
Spatial confinement of an essential gene expression factor may have relevance beyond T. brucei’s RNA pol I system. In P. falciparum, the active var gene is transcribed by RNA pol II in a perinuclear site and activation of a silent var gene requires relocation from the heterochromatic nuclear periphery to this euchromatic site possibly to interact with a sequestered var gene-specific transcription factor [reviewed in (9)]. Similarly, DNA FISH analysis in the mouse system showed that the H enhancer element co-localized with OR genes on different chromosomes, suggesting that nuclear positioning of the active OR gene is important in this system as well (48).
In summary, our results favor a bipartite model of monoallelic VSG expression in which BES repression is achieved epigenetically and the expressed BES is not only derepressed by chromatin changes but also activated at the promoter, a process that entails recruitment of CITFA.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
This work was funded by the National Institutes of Health grant [R01 AI059377 to A.G.]; the Young Investigator Program of the Research Center of Infectious Diseases (ZINF) of the University of Würzburg, Germany, and the Human Frontier Science Program (to T.N.S.). Funding for open access charge: National Institutes of Health [R01 AI059377 to A.G.].
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
We are grateful to Drs George Cross and Luisa Figueiredo (Rockefeller University) for providing the PN13 cell line and the BES switching protocol, to Dr Ziyin Li (University of Texas, Houston) for construct pC-eYFP-NEO, to Drs Ji Yu and Dongmyung Oh (UConn Health Center) for help with fluorescence microscopy, to Ms Bao Nguyen for help with plasmid construction and to Dr Justin Radolf and Justin K. Kirkham for critical reading of the manuscript.
REFERENCES
- 1.Horn D, McCulloch R. Molecular mechanisms underlying the control of antigenic variation in African trypanosomes. Curr. Opin. Microbiol. 2010;13:700–705. doi: 10.1016/j.mib.2010.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Magklara A, Lomvardas S. Stochastic gene expression in mammals: lessons from olfaction. Trends Cell Biol. 2013;23:449–456. doi: 10.1016/j.tcb.2013.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schwede A, Jones N, Engstler M, Carrington M. The VSG C-terminal domain is inaccessible to antibodies on live trypanosomes. Mol. Biochem. Parasitol. 2011;175:201–204. doi: 10.1016/j.molbiopara.2010.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hertz–Fowler C, Figueiredo LM, Quail MA, Becker M, Jackson A, Bason N, Brooks K, Churcher C, Fahkro S, Goodhead I, et al. Telomeric expression sites are highly conserved in Trypanosoma brucei. PLoS One. 2008;3:e3527. doi: 10.1371/journal.pone.0003527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Günzl A, Bruderer T, Laufer G, Schimanski B, Tu LC, Chung HM, Lee PT, Lee MG. RNA polymerase I transcribes procyclin genes and variant surface glycoprotein gene expression sites in Trypanosoma brucei. Eukaryot. Cell. 2003;2:542–551. doi: 10.1128/EC.2.3.542-551.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chaves I, Zomerdijk J, Dirks MA, Dirks RW, Raap AK, Borst P. Subnuclear localization of the active variant surface glycoprotein gene expression site in Trypanosoma brucei. Proc. Natl Acad. Sci. USA. 1998;95:12328–12333. doi: 10.1073/pnas.95.21.12328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Navarro M, Gull K. A pol I transcriptional body associated with VSG mono-allelic expression in Trypanosoma brucei. Nature. 2001;414:759–763. doi: 10.1038/414759a. [DOI] [PubMed] [Google Scholar]
- 8.Magklara A, Yen A, Colquitt BM, Clowney EJ, Allen W, Markenscoff-Papadimitriou E, Evans ZA, Kheradpour P, Mountoufaris G, Carey C, et al. An epigenetic signature for monoallelic olfactory receptor expression. Cell. 2011;145:555–570. doi: 10.1016/j.cell.2011.03.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Scherf A, Lopez-Rubio JJ, Riviere L. Antigenic variation in Plasmodium falciparum. Annu. Rev. Microbiol. 2008;62:445–470. doi: 10.1146/annurev.micro.61.080706.093134. [DOI] [PubMed] [Google Scholar]
- 10.Duraisingh MT, Voss TS, Marty AJ, Duffy MF, Good RT, Thompson JK, Freitas-Junior LH, Scherf A, Crabb BS, Cowman AF. Heterochromatin silencing and locus repositioning linked to regulation of virulence genes in Plasmodium falciparum. Cell. 2005;121:13–24. doi: 10.1016/j.cell.2005.01.036. [DOI] [PubMed] [Google Scholar]
- 11.Tonkin CJ, Carret CK, Duraisingh MT, Voss TS, Ralph SA, Hommel M, Duffy MF, Silva LM, Scherf A, Ivens A, et al. Sir2 paralogues cooperate to regulate virulence genes and antigenic variation in Plasmodium falciparum. PLoS Biol. 2009;7:e84. doi: 10.1371/journal.pbio.1000084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yang X, Figueiredo LM, Espinal A, Okubo E, Li B. RAP1 is essential for silencing telomeric variant surface glycoprotein genes in Trypanosoma brucei. Cell. 2009;137:99–109. doi: 10.1016/j.cell.2009.01.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Figueiredo LM, Janzen CJ, Cross GA. A histone methyltransferase modulates antigenic variation in African trypanosomes. PLoS Biol. 2008;6:e161. doi: 10.1371/journal.pbio.0060161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hughes K, Wand M, Foulston L, Young R, Harley K, Terry S, Ersfeld K, Rudenko G. A novel ISWI is involved in VSG expression site downregulation in African trypanosomes. EMBO J. 2007;26:2400–2410. doi: 10.1038/sj.emboj.7601678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Denninger V, Fullbrook A, Bessat M, Ersfeld K, Rudenko G. The FACT subunit TbSpt16 is involved in cell cycle specific control of VSG expression sites in Trypanosoma brucei. Mol. Microbiol. 2010;78:459–474. doi: 10.1111/j.1365-2958.2010.07350.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang QP, Kawahara T, Horn D. Histone deacetylases play distinct roles in telomeric VSG expression site silencing in African trypanosomes. Mol. Microbiol. 2010;77:1237–1245. doi: 10.1111/j.1365-2958.2010.07284.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Narayanan MS, Kushwaha M, Ersfeld K, Fullbrook A, Stanne TM, Rudenko G. NLP is a novel transcription regulator involved in VSG expression site control in Trypanosoma brucei. Nucleic Acids Res. 2011;39:2018–2031. doi: 10.1093/nar/gkq950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Povelones ML, Gluenz E, Dembek M, Gull K, Rudenko G. Histone H1 plays a role in heterochromatin formation and VSG expression site silencing in Trypanosoma brucei. PLoS Pathog. 2012;8:e1003010. doi: 10.1371/journal.ppat.1003010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tiengwe C, Marcello L, Farr H, Dickens N, Kelly S, Swiderski M, Vaughan D, Gull K, Barry JD, Bell SD, et al. Genome-wide analysis reveals extensive functional interaction between DNA replication initiation and transcription in the genome of Trypanosoma brucei. Cell Rep. 2012;2:185–197. doi: 10.1016/j.celrep.2012.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Alsford S, Horn D. Cell-cycle-regulated control of VSG expression site silencing by histones and histone chaperones ASF1A and CAF-1b in Trypanosoma brucei. Nucleic Acids Res. 2012;40:10150–10160. doi: 10.1093/nar/gks813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kim HS, Park SH, Günzl A, Cross GA. MCM-BP is required for repression of life-cycle specific genes transcribed by RNA polymerase I in the mammalian infectious form of Trypanosoma brucei. PLoS One. 2013;8:e57001. doi: 10.1371/journal.pone.0057001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Figueiredo LM, Cross GA. Nucleosomes are depleted at the VSG expression site transcribed by RNA polymerase I in African trypanosomes. Eukaryot. Cell. 2010;9:148–154. doi: 10.1128/EC.00282-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stanne TM, Rudenko G. Active VSG expression sites in Trypanosoma brucei are depleted of nucleosomes. Eukaryot. Cell. 2010;9:136–147. doi: 10.1128/EC.00281-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brandenburg J, Schimanski B, Nogoceke E, Nguyen TN, Padovan JC, Chait BT, Cross GA, Günzl A. Multifunctional class I transcription in Trypanosoma brucei depends on a novel protein complex. EMBO J. 2007;26:4856–4866. doi: 10.1038/sj.emboj.7601905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nguyen TN, Nguyen BN, Lee JH, Panigrahi AK, Günzl A. Characterization of a novel class I transcription factor A (CITFA) subunit that is indispensable for transcription by the multifunctional RNA polymerase I of Trypanosoma brucei. Eukaryot. Cell. 2012;11:1573–1581. doi: 10.1128/EC.00250-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li Z, Umeyama T, Wang CC. The Aurora Kinase in Trypanosoma brucei plays distinctive roles in metaphase-anaphase transition and cytokinetic initiation. PLoS Pathog. 2009;5:e1000575. doi: 10.1371/journal.ppat.1000575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nguyen TN, Schimanski B, Günzl A. Active RNA polymerase I of Trypanosoma brucei harbors a novel subunit essential for transcription. Mol. Cell. Biol. 2007;27:6254–6263. doi: 10.1128/MCB.00382-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Laufer G, Schaaf G, Bollgönn S, Günzl A. In vitro analysis of alpha-amanitin-resistant transcription from the rRNA, procyclic acidic repetitive protein, and variant surface glycoprotein gene promoters in Trypanosoma brucei. Mol. Cell. Biol. 1999;19:5466–5473. doi: 10.1128/mcb.19.8.5466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Park SH, Nguyen TN, Kirkham JK, Lee JH, Günzl A. Transcription by the multifunctional RNA polymerase I in Trypanosoma brucei functions independently of RPB7. Mol. Biochem. Parasitol. 2011;180:35–42. doi: 10.1016/j.molbiopara.2011.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schimanski B, Nguyen TN, Günzl A. Highly efficient tandem affinity purification of trypanosome protein complexes based on a novel epitope combination. Eukaryotic Cell. 2005;4:1942–1950. doi: 10.1128/EC.4.11.1942-1950.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lee JH, Cai G, Panigrahi AK, Dunham-Ems S, Nguyen TN, Radolf JD, Asturias FJ, Günzl A. A TFIIH-associated mediator head is a basal factor of small nuclear spliced leader RNA gene transcription in early-diverged trypanosomes. Mol. Cell. Biol. 2010;30:5502–5513. doi: 10.1128/MCB.00966-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.French SL, Osheim YN, Cioci F, Nomura M, Beyer AL. In exponentially growing Saccharomyces cerevisiae cells, rRNA synthesis is determined by the summed RNA polymerase I loading rate rather than by the number of active genes. Mol. Cell Biol. 2003;23:1558–1568. doi: 10.1128/MCB.23.5.1558-1568.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Navarro M, Penate X, Landeira D. Nuclear architecture underlying gene expression in Trypanosoma brucei. Trends Microbiol. 2007;15:263–270. doi: 10.1016/j.tim.2007.04.004. [DOI] [PubMed] [Google Scholar]
- 35.Panov KI, Friedrich JK, Zomerdijk JC. A step subsequent to preinitiation complex assembly at the ribosomal RNA gene promoter is rate limiting for human RNA polymerase I-dependent transcription. Mol. Cell Biol. 2001;21:2641–2649. doi: 10.1128/MCB.21.8.2641-2649.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hirumi H, Hirumi K, Doyle JJ, Cross GA. In vitro cloning of animal-infective bloodstream forms of Trypanosoma brucei. Parasitology. 1980;80:371–382. doi: 10.1017/s0031182000000822. [DOI] [PubMed] [Google Scholar]
- 37.Nguyen TN, Schimanski B, Zahn A, Klumpp B, Günzl A. Purification of an eight subunit RNA polymerase I complex in Trypanosoma brucei. Mol. Biochem. Parasitol. 2006;149:27–37. doi: 10.1016/j.molbiopara.2006.02.023. [DOI] [PubMed] [Google Scholar]
- 38.Walgraffe D, Devaux S, Lecordier L, Dierick JF, Dieu M, Van Den AJ, Pays E, Vanhamme L. Characterization of subunits of the RNA polymerase I complex in Trypanosoma brucei. Mol. Biochem. Parasitol. 2005;139:249–260. doi: 10.1016/j.molbiopara.2004.11.014. [DOI] [PubMed] [Google Scholar]
- 39.Chaves I, Rudenko G, Dirks MA, Cross M, Borst P. Control of variant surface glycoprotein gene-expression sites in Trypanosoma brucei. EMBO J. 1999;18:4846–4855. doi: 10.1093/emboj/18.17.4846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ansorge I, Steverding D, Melville S, Hartmann C, Clayton C. Transcription of ‘inactive’ expression sites in African trypanosomes leads to expression of multiple transferrin receptor RNAs in bloodstream forms. Mol. Biochem. Parasitol. 1999;101:81–94. doi: 10.1016/s0166-6851(99)00060-2. [DOI] [PubMed] [Google Scholar]
- 41.Vanhamme L, Poelvoorde P, Pays A, Tebabi P, Xong HV, Pays E. Differential RNA elongation controls the variant surface glycoprotein gene expression sites of Trypanosoma brucei. Mol. Microbiol. 2000;36:328–340. doi: 10.1046/j.1365-2958.2000.01844.x. [DOI] [PubMed] [Google Scholar]
- 42.Rudenko G. Epigenetics and transcriptional control in African trypanosomes. Essays Biochem. 2010;48:201–219. doi: 10.1042/bse0480201. [DOI] [PubMed] [Google Scholar]
- 43.Narayanan MS, Rudenko G. TDP1 is an HMG chromatin protein facilitating RNA polymerase I transcription in African trypanosomes. Nucleic Acids Res. 2013;41:2981–2992. doi: 10.1093/nar/gks1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ehlers B, Czichos J, Overath P. RNA turnover in Trypanosoma brucei. Mol. Cell Biol. 1987;7:1242–1249. doi: 10.1128/mcb.7.3.1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rudenko G, Blundell PA, Dirks Mulder A, Kieft R, Borst P. A ribosomal DNA promoter replacing the promoter of a telomeric VSG gene expression site can be efficiently switched on and off in T. brucei. Cell. 1995;83:547–553. doi: 10.1016/0092-8674(95)90094-2. [DOI] [PubMed] [Google Scholar]
- 46.Günzl A, Vanhamme L, Myler PJ. Transcription in trypanosomes: a different means to the end. In: Barry JD, Mottram JC, McCulloch R, Acosta-Serrano A, editors. Trypanosomes—After the Genome. Pittsburgh: Horizon Press; 2007. pp. 177–208. [Google Scholar]
- 47.Emmott E, Hiscox JA. Nucleolar targeting: the hub of the matter. EMBO Rep. 2009;10:231–238. doi: 10.1038/embor.2009.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lomvardas S, Barnea G, Pisapia DJ, Mendelsohn M, Kirkland J, Axel R. Interchromosomal interactions and olfactory receptor choice. Cell. 2006;126:403–413. doi: 10.1016/j.cell.2006.06.035. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.