


Abstract
NEK2 is a serine/threonine kinase that promotes centrosome splitting and ensures correct chromosome segregation during the G2/M phase of the cell cycle, through phosphorylation of specific substrates. Aberrant expression and activity of NEK2 in cancer cells lead to dysregulation of the centrosome cycle and aneuploidy. Thus, a tight regulation of NEK2 function is needed during cell cycle progression. In this study, we found that NEK2 localizes in the nucleus of cancer cells derived from several tissues. In particular, NEK2 co-localizes in splicing speckles with SRSF1 and SRSF2. Moreover, NEK2 interacts with several splicing factors and phosphorylates some of them, including the oncogenic SRSF1 protein. Overexpression of NEK2 induces phosphorylation of endogenous SR proteins and affects the splicing activity of SRSF1 toward reporter minigenes and endogenous targets, independently of SRPK1. Conversely, knockdown of NEK2, like that of SRSF1, induces expression of pro-apoptotic variants from SRSF1-target genes and sensitizes cells to apoptosis. Our results identify NEK2 as a novel splicing factor kinase and suggest that part of its oncogenic activity may be ascribed to its ability to modulate alternative splicing, a key step in gene expression regulation that is frequently altered in cancer cells.
INTRODUCTION
NEK2 is a member of the NIMA-related family of serine/threonine protein kinases, which share structural relationships and the involvement in cell cycle regulation (1). NEK2 displays constitutive catalytic activity and phosphorylates proteins involved in centrosome duplication and cell cycle regulation (2). Consistently, NEK2 binds to microtubules and is enriched in the centrosome, where it contributes to centrosome splitting during the G2/M phase of the cell cycle (2). Upregulation of NEK2 in human cells causes premature splitting of this organelle (3), whereas overexpression of a NEK2 kinase-dead mutant induces centrosome abnormalities and aneuploidy (4). Hence, a tight regulation of NEK2 abundance and activity is essential to ensure correct centrosome duplication and timely progression of the cell cycle.
Similar to other kinases involved in spindle assembly or duplication (5), overexpression of NEK2 was reported in several neoplastic diseases, such as preinvasive and invasive breast carcinomas (6), lung adenocarcinomas (7), testicular seminomas (8) and diffuse large B cell lymphomas (9). More recently, NEK2 expression has been proposed as a strong predictor for drug resistance and poor prognosis in human cancer, suggesting that it might represent a key therapeutic target (10). In line with this hypothesis, pharmacologic or genetic interference with NEK2 activity strongly reduced proliferation and invasiveness of cancer cells (10–12). Mechanistically, the oncogenic activity of NEK2 has been mainly linked to its ability to regulate centrosome duplication (3,6,13), whose aberrant amplification frequently leads to aneuploidy and neoplastic transformation (6). Overexpression of NEK2 in non-transformed breast epithelial cells was shown to induce centrosome overduplication (6), and increased expression of endogenous NEK2 caused centrosome amplification in breast cancer lesions expressing the oncogenic K-RAS(G12D) mutant protein (13). Furthermore, NEK2-dependent phosphorylation was required for proper localization at the kinetochores of HEC1, a protein involved in faithful chromosome segregation (14). These observations strongly suggest that NEK2-dependent centrosome amplification and aneuploidy can favour neoplastic transformation.
We previously reported that increased expression of NEK2 in human testicular seminomas correlated with its accumulation in the nucleus (8). This observation suggested that nuclear functions of NEK2 might also contribute to its role in cancer cells. Herein, we have studied in further detail the nuclear localization and function of this kinase. We found that nuclear localization of NEK2 occurs in cancer cells derived from several tissues. NEK2 localizes to splicing speckles and phosphorylates the oncogenic splicing factor SRSF1. Moreover, we found that NEK2 regulates SRSF1 activity and alternative splicing (AS) of SRSF1 target genes similarly to the SR protein kinase SRPK1. In particular, NEK2 promotes anti-apoptotic splice variants and knockdown of its expression enhanced apoptosis. Our results uncover a novel function for NEK2 in splicing regulation and suggest that phosphorylation of splicing factors and modulation of AS might contribute to its oncogenic activity.
MATERIALS AND METHODS
Immunohistochemistry and immunofluorescence analysis
Cancer patient’s tissues (14 cases of cryopreserved tissue from seminoma, breast, lung, prostate, cervix and colon cancer) were obtained from the National Cancer Institute ‘G. Pascale’ Ethical Committee approval was given in all instances. Five-micrometer sections were processed for immunohistochemistry with antibodies against NEK2 (Abgent) as described (8). Immunofluorescence was performed as described (8,15) using the following primary antibodies (1:500): rabbit anti-NEK2 (Abgent), mouse anti-SRSF1, anti-SRSF2 (Santa Cruz Biotechnology) and rabbit anti-cleaved CASPASE 3 (Sigma Aldrich). Confocal analyses were performed using a Leica confocal microscope as described (16). Images in Figure 6D, S2 and S5 were taken using a Leica inverted microscope as described (8). Images were saved as TIFF files and Photoshop (Adobe) was used for composing panels.
Figure 6.

NEK2 silencing affects splicing of SRSF1 target genes and sensitize cells to apoptosis. (A) Schematic representation of the SRSF1-regulated of BCL-X, MKNK2 and BIN1 AS events (left panel). Black arrows indicate primers used for the qRT-PCR analysis performed in HeLa cells transfected with scramble (si-SCR), NEK2 (si-NEK2), SRPK1 (si-SRPK1) or SRSF1 (si-SRSF1) siRNAs. Ratio of the AS variants are represented in the bar graph (mean ± SD, n = 3, *P < 0.05, **P < 0.01) (right panel). (B) Western blot analysis assessing NEK2, SRPK1 and SRSF1 silencing efficiency. (C and D) Western blot analysis of PARP1 cleavage (C) and quantitative analysis of cleaved-CASPASE 3 immunofluorescence (bar graph represents mean ± SD, n = 3, *P < 0.05) (D) in HeLa cells transfected with either scramble (si-SCR) or NEK2 (si-NEK2) siRNAs and treated for 24 h with increasing doses of cisplatin (CPT). Western blot analysis assessing NEK2 silencing efficiency was performed (C).
Cell culture, transfections and treatment
TCam-2 cells were grown in RPMI 1640 (Lonza), HEK293T, HeLa, MCF7, PC-3 cells were grown in Dulbecco's modified Eagle's medium (Sigma Aldrich), all supplemented with 10% FBS, gentamycin, penicillin and streptomycin. Transfection with the indicated expression vectors was performed using Lipofectamine 2000 (Invitrogen) according to manufacturer’s instructions. After 24 h, cells were harvested for protein and RNA analyses. For RNA interference, cells were transfected with siRNAs (Sigma-Aldrich) using Lipofectamine RNAiMAX (Invitrogen) according to manufacturer’s instructions and harvested 48 h later for protein and RNA analyses. Sequences for SRPK1 siRNAs were previously described (17). NEK2, SRSF1 siRNAs and scrambled siRNA sequences are listed in Supplementary Table S1. For apoptosis, HeLa cells were treated for 24 h with the indicated doses of cisplatin or starved for 8 h in Earle’s balanced salt solution (Sigma).
Protein extracts and western blot analysis
Total cellular extracts and cellular fractionations were processed and analysed by western blot as described (15,18) using the following primary antibodies (1:1000): rabbit anti-NEK2 R31 (generously provided by Prof. A.M. Fry); mouse anti-GFP, anti-MYC, anti-SRSF1, anti-SRSF3, rabbit anti-SAM68 and goat anti-lamin B (Santa Cruz Biotechnology); rabbit anti-ACTIN, mouse anti-FLAG, anti-hnRNPA1, anti-hnRNPC1/C2 and anti-TUBULIN (Sigma-Aldrich); mouse anti-hnRNPF/H (Abcam); mouse anti-SR proteins (1H4) (Invitrogen), mouse anti-SRPK1 (BD Pharmingen); rabbit anti-AKT (Novus Biologicals); rabbit anti-pAKT Ser 473 and anti-PARP1 (Cell Signaling).
In vitro kinase assay
Glutathione S-transferase (GST)-fusion proteins were expressed in Escherichia coli cells (strain BL21-DE3) and purified as previously described (19). (His)6-tagged proteins were expressed in Sf9 cells using a baculovirus system and purified on a TALON affinity resin (CLONETECH), as described (20). In vitro kinase assays were performed as described (21), using purified NEK2 active protein (Millipore).
GST–pull-down and co-immunoprecipitation assays
Nuclear extracts were incubated with GST or GST-NEK2A(271–445) adsorbed on glutathione-agarose beads; bound proteins were eluted and analyzed as described (19). For co-immunoprecipitation, nuclear extracts or total extracts from HEK293T cells, transfected with the indicated vectors, were incubated with mouse anti-FLAG or mouse anti-IgG antibodies adsorbed on Dynabeads protein A (Invitrogen) and immunocomplexes were eluted and analysed as described (15).
Extraction of RNA, reverse transcriptase-polymerase chain reaction and real-time polymerase chain reaction analysis
RNA was extracted using TRIzol reagent (Invitrogen) according to the manufacturer’s instructions. After digestion with RNase-free DNase (Roche), 1 μg of total RNA was retrotranscribed using M-MLV reverse transcriptase (Promega), used as template for polymerase chain reaction (PCR; GoTaq, Promega) and reactions were analysed on agarose or acrylamide gels. Real-time quantitative PCRs (qPCR) were performed using LightCycler 480 SYBR Green I Master and the LightCycler 480 System (Roche), according to the manufacturer’s instructions. Control reactions omitting M-MLV reverse transcriptase were also carried out. All primers used are listed in the Supplementary Table S1.
Statistical analysis
Statistical analysis was performed by the Student t-test as described in the figure legends.
RESULTS
NEK2 is enriched in the nucleus of cancer cells
NEK2 is enriched in the nucleus of human testicular seminoma cells (8). Nuclear localization of this kinase was also recently observed in myeloma cells and shown to correlate with poor prognosis (10). To investigate whether other cancer cells that overexpress NEK2 share this nuclear localization, we performed immunohistochemistry analysis of tissue specimens derived from cancer patients. Using a previously validated antibody (8), we observed that NEK2 staining was concentrated in the nucleus of breast and lung cancer cells (Figure 1A). In colon, prostate and cervix cancer cells, although it was also detected in the cytoplasm, NEK2 staining was enriched in nucleus (Figure 1A). Confocal immunofluorescence analyses of NEK2 localization in cell lines derived from breast cancer (MCF7; Figure 1B), seminoma (TCam-2), prostate cancer (PC-3), colon carcinoma (Caco-2) and cervix cancer (HeLa) (Supplementary Figure S1) suggest that nuclear localization of NEK2 is a common feature of human cancer cells.
Figure 1.

NEK2 localizes in the nucleus of cancer cells. (A) Immunohistochemistry of NEK2 in testicular seminomas, breast, prostate, lung and cervix cancer specimens (scale bar = 25 μm). (B and C) Confocal immunofluorescence analysis of MCF7 cells stained with anti-NEK2 (red) antibody and Hoechst (blue) (B, scale bar = 10 μm) or with anti-NEK2 (green), anti-SRSF1 (red, upper panel) or anti-SRSF2 (red, lower panel) (C, scale bar = 5 μm). White arrows indicate co-localization in speckles.
NEK2 is expressed as three alternative splice variants, named NEK2A, B and C (Supplementary Figure S2A) (22). NEK2A and B differ in the C-termini because an alternative polyadenylation signal in intron 7 is used in NEK2B, thus preventing inclusion of the last exon 8 (2). Notably, exon 8 encodes for protein degradation motifs in NEK2A, which mediate its degradation in mitosis (23). NEK2C is identical to NEK2A with the exception of a small internal deletion of 8 amino acids (missing residues 371–378), due to usage of a downstream splice acceptor site in exon 8 (22). Although the biochemical features of NEK2C are undistinguishable from those of NEK2A, deletion of this sequence creates a nuclear localization signal that promotes NEK2C accumulation in the nucleus (22). To investigate which splice variant was prevalently expressed in cancer cell lines, we performed reverse transcriptase-polymerase chain reaction (RT-PCR) analyses using variant-specific primers (Supplementary Figure S2A). NEK2A and B were readily detected in all cell lines analysed, whereas NEK2C was barely detectable (Supplementary Figure S2B), suggesting that its expression is unlikely to account for the nuclear localization of NEK2 in cancer cells.
Previous results indicated that a substantial fraction of NEK2A localizes in the nucleus when the protein is overexpressed (22). In line with this report, we observed that overexpression of NEK2A was sufficient to allow its accumulation in the nucleus of HeLa cells, with a localization pattern that closely resembled that of NEK2C (Supplementary Figure S2C), whereas NEK2B remained mainly cytoplasmic (Supplementary Figure S2C). These results suggest that upregulation of NEK2A is likely responsible for the nuclear localization of NEK2 in cancer cells.
NEK2 localizes in nuclear splicing speckles and co-fractionates with splicing factors
We found that NEK2 accumulated in nuclear granules of variable size and irregular shape (Figure 1B, Supplementary Figure S1), which resembled the splicing speckles, the interchromatin regions enriched in splicing factors (24). Confocal immunofluorescence analysis in MCF7 cells confirmed that NEK2 co-localizes in the nuclear speckles with SRSF1 and SRSF2 (Figure 1C), two serine/arginine-rich (SR) proteins commonly used as markers of these structures (24). We next used subcellular fractionation experiments to confirm the association of NEK2 with splicing factors. By using this technique, it was documented that splicing factors accumulate in the nuclear matrix-attached insoluble fraction (18). Analyses of cytosolic (S), nuclear soluble (NS), and nuclear matrix-attached insoluble (NI) fractions confirmed that splicing factors were enriched in the NI fraction isolated from MCF7, PC-3 and TCam-2 cells (Figure 2A). In addition, we observed that NEK2 was also enriched in both the NS and the NI fraction in all cell lines tested (Figure 2A). The molecular weight of this band corresponded to that of NEK2A and C. A faster migrating band corresponding to the molecular weight of NEK2B was instead detected in the cytosolic fraction.
Figure 2.
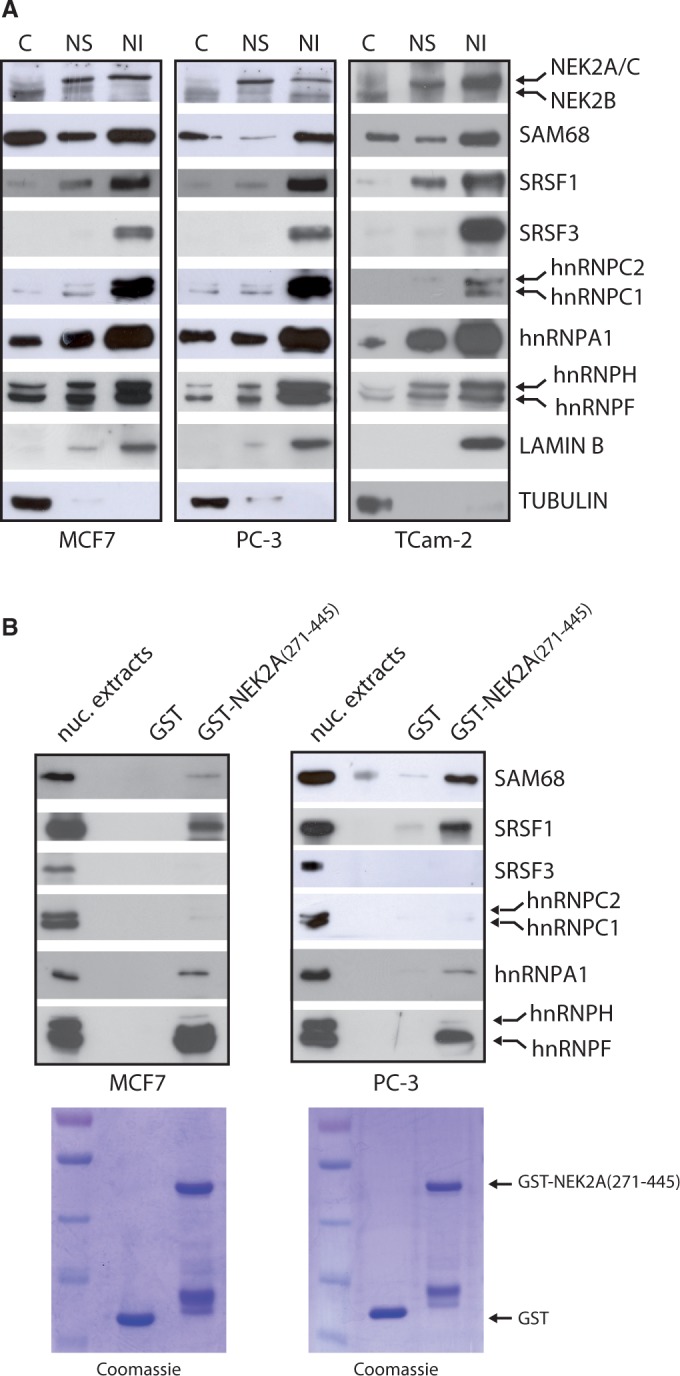
NEK2 associates with splicing factors. (A) Cytosolic (C), nuclear soluble (NS) and nuclear insoluble matrix-associated (NI) fractions of MCF7, PC-3, TCam-2 cells were analysed by western blot using antibodies for NEK2 and indicated splicing factors. LAMIN B and TUBULIN were evaluated as nuclear matrix and cytosolic markers. (B) Western blot analysis for the indicated splicing factors in pull-down assays of MCF7 and PC-3 nuclear extracts with GST-NEK2A(271–445) fusion protein and GST (as negative control). Coomassie staining shows the purified GST and GST-NEK2A(271–445) fusion protein (lower panels).
NEK2 interacts with substrates and activators through the carboxyl terminal regulatory region (residues 273–445, Supplementary Figure S2A) (2). Thus, we used purified GST-NEK2A(271–445) fusion protein as bait in affinity chromatography of nuclear extracts isolated from MCF7 and PC-3 cells. GST-NEK2A(271–445) selectively associated with some splicing regulators, as SRSF1, hnRNPA1, hnRNPF and SAM68, but not others, as SRSF3 and hnRNPC1/C2 (Figure 2B). These results suggest that NEK2 interacts with specific splicing factors in the cell nucleus.
NEK2 is a splicing factor kinase
Next, we set out to determine whether splicing factors were substrates for NEK2. We focused on SR proteins because their splicing activity is finely tuned by phosphorylation (25,26). As first step, we determined whether SRSF1 and SRSF7 were directly phosphorylated by NEK2. Kinase assays using purified full-length HIS- or GST-fusion proteins of these splicing factors showed that purified NEK2 efficiently phosphorylates SRSF1 and SRSF7 in vitro (Figure 3A). We focused the rest of our study on SRSF1 because it was more efficiently phosphorylated by NEK2. Furthermore, this splicing factor is a bona fide oncogene (27) and it is upregulated in several human cancers, including breast and prostate carcinomas, where it modulates cancer-relevant AS events (28,29).
Figure 3.
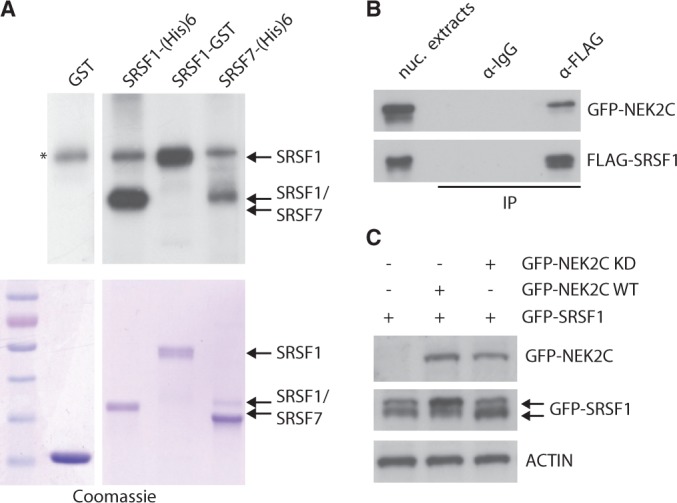
NEK2 phosphorylates splicing factors. (A) Purified NEK2 was incubated with purified GST (as negative control), SRSF1-(His)6, SRSF1-GST or SRSF7-(His)6 proteins and γ-[32P]ATP. Phosphorylation levels were detected by autoradiography (asterisk indicates autophosphorylated NEK2). Coomassie staining shows the purified proteins (lower panel). (B) Western blot analysis with anti-GFP and anti-FLAG antibodies of the immunoprecipitation assay performed with control mouse IgG or anti-FLAG antibody of nuclear extracts of HEK293T expressing FLAG-SRSF1 and GFP-NEK2C. (C) Western blot analysis using anti-GFP antibody of HEK293T cells transfected with GFP-SRSF1, wild-type NEK2C (WT) or kinase-dead NEK2C (KD). ACTIN was used as loading control.
First, we investigated whether the interaction between NEK2 and SRSF1 also occurred in live cells. We found that GFP-NEK2C (Figure 3B) and GFP-NEK2A (Supplementary Figure S3A) were efficiently co-immunoprecipitated with FLAG-SRSF1 in HEK293T cells, whereas the cytosolic GFP-NEK2B was not (Supplementary Figure S2C and S3B). Next, to verify the ability of NEK2 to phosphorylate SRSF1 in live cells, we co-expressed wild-type or kinase-dead GFP-NEK2C with GFP-SRSF1. When expressed alone, GFP-SRSF1 yielded two bands, with the faster migrating band being more abundant, indicating that the bulk of the protein is hypophosphorylated (Figure 3C). Expression of wild-type NEK2C increased the amount of the higher molecular weight band of SRSF1, suggesting its hyperphosphorylation, whereas kinase-dead NEK2C was ineffective (Figure 3C). A similar result was obtained with NEK2A (Supplementary Figure S3C), indicating that both NEK2 splice variants can phosphorylate SRSF1 in live cells.
NEK2 behaves as an SR protein kinase in live cells
Having established that NEK2 phosphorylates SRSF1, we asked whether it behaves as an SR protein kinase (SRPK). For these studies, we focused on NEK2C, which was chosen for its almost exclusive localization in the nucleus (22). Overexpression of NEK2C induced a pattern of SR protein phosphorylation similar to that elicited by SRPK1, a prototypical SRPK (26), leading to phosphorylation of SRSF6 (SRp55), SRSF5 (SRp40) and SRSF2/SRSF9 (SRp30) (Figure 4A).
Figure 4.

NEK2 phosphorylates SR proteins and modulates E1A splicing. (A) Western blot analysis of SR protein phosphorylation in HeLa cells transfected with FLAG-SRPK1 or GFP-NEK2C using the anti-SR proteins 1H4 antibody. TUBULIN was detected as loading control. (B) Schematic representation of the alternative splice variants of the E1A minigene. (C) RT-PCR analysis (upper panel) of the in vivo splicing assay in HeLa cells transfected with the E1A minigene and increasing doses of FLAG-SRPK1, GFP-NEK2C WT or GFP-NEK2C KD. Overexpression efficiency was assessed through anti-FLAG and anti-GFP western blot. TUBULIN was evaluated as loading control. Bar graph (lower panel) represents densitometric analysis for the major E1A splice variants (13S, 12S, 9S) evaluated in presence of the highest dose of vectors (mean ± SD, n = 3).
Next, we tested whether NEK2 functionally regulates the activity of SR proteins. Previous reports demonstrated that overexpression of SRPK1 modulates splicing of the E1A reporter minigene (17), a commonly used splicing target that contains several 5′ and 3′ alternative splice sites (ss) (Figure 4B). As expected (17), we observed that increasing the expression of SRPK1 in HeLa cells caused a dose-dependent switch from the 13S to the 9S 5′ splice site of the E1A minigene (Figure 4C). Remarkably, upregulation of NEK2C caused a similar switch in E1A splicing. This effect required the kinase activity of NEK2, as the kinase-dead mutant had no effect (Figure 4C). Thus, NEK2 displays features of an SRPK in live cells.
NEK2 expression modulates SRSF1-dependent BCL-X splicing
Phosphorylation of SR proteins regulates their splicing activity (25,26). Thus, we asked whether NEK2 could modulate the AS of an endogenous target of SRSF1, such as the BCL-X gene (15,30). Selection of two alternative 5′ ss in exon 2 of BCL-X leads to the production of two splice variants: the anti-apoptotic BCL-XL and the pro-apoptotic BCL-XS (31). SRSF1 promotes selection of the proximal 5′ ss leading to expression of BCL-XL (15,30). By performing real-time qPCR using exon junction-specific primers for BCL-XL and BCL-XS (Figure 5A), we found that overexpression of NEK2C in HeLa cells increased the BCL-XL/BCL-XS ratio to a similar extent as overexpression of SRSF1 (Figure 5B). Importantly, this effect was not due to activation of SRPK1. While knockdown of SRPK1 promoted the pro-apoptotic BCL-XS variant, indicating that SRPK1 also modulates this AS event, NEK2C was still capable to enhance splicing of the anti-apoptotic BCL-XL variant in SRPK1-depleted cells as observed in control cells (Figure 5C).
Figure 5.

NEK2 modulates BCL-X AS affecting SRSF1 activity. (A) Schematic representation of the BCL-X AS. Black arrows indicate exon-junction primers used for qRT-PCR analysis. (B and C) qRT-PCR analysis of endogenous BCL-X splice-variants. Bar graphs represent BCL-XL/BCL-XS ratio (mean ± SD, n = 3, *P < 0.05). HeLa cells were transfected with mock, GFP-NEK2C or GFP-SRSF1 (B) or with either scramble (si-SCR) or SRPK1 (si-SRPK1) siRNAs and then with or without GFP-NEK2C (C). Silencing and overexpression efficiency was assessed by western blot analysis. (D) RT-PCR analysis of the in vivo splicing assay of BCL-X minigene in HEK293T cells transfected with scramble (si-SCR), NEK2 (si-NEK2), SRPK1 (si-SRPK1) or both NEK2 and SRPK1 (si-NEK2/si-SRPK1) siRNAs and with or without FLAG-SRSF1. Bar graph represents the densitometric analysis of the BCL-XL/BCL-XS ratio, normalized for the value obtained in cells transfected with scramble siRNA and empty vector, set to 1 (mean ± SD, n = 3, **P < 0.01). Silencing and overexpression efficiency was assessed by western blot analysis (right panel).
To determine whether NEK2 expression affected the ability of SRSF1 to modulate BCL-X AS, we used a minigene that recapitulates the splicing of the endogenous gene (32). We found that knockdown of NEK2 in HEK293T cells slightly enhanced splicing of the pro-apoptotic BCL-XS variant (Figure 5D). Moreover, while transfection of suboptimal amounts of SRSF1 efficiently promoted splicing of the anti-apoptotic BCL-XL variant in control cells (si-SCR), this effect was partially impaired when NEK2 was silenced (Figure 5D). Importantly, similar effects were also observed when SRPK1 was knocked down, even though silencing of both NEK2 and SRPK1 did not exert additive effect on SRSF1-induced BCL-X splicing (Figure 5D).
NEK2 is involved in the regulation of apoptosis
Next, we sought out to determine whether AS of known endogenous targets of SRSF1 was affected by the knockdown of NEK2 in HeLa cells. We examined the splicing pattern of SRSF1 target transcripts from three genes with roles in cancer and for which AS variants have been characterized: BCL-X, MKNK2 and BIN1 (15,27,30,33). For comparison, we also knocked down SRSF1 and SRPK1 in parallel experiments (Figure 6A and B). Transient knockdown of NEK2 in HeLa cells resulted in decreased ratio of BCL-XL/BCL-XS and MNK2b/MNK2a, and induces skipping of exon 12A in BIN1 mRNA variants, without affecting SRSF1 expression. All the splicing changes exerted by NEK2 depletion favoured pro-apoptotic splice variants and were recapitulated by knockdown of either SRSF1 or SRPK1, although to different extent for the three genes (Figure 6A and B). Moreover, as observed for BCL-XL, overexpression of either NEK2C or SRSF1 promoted splicing of anti-apoptotic MKNK2 and BIN1 variants (Supplementary Figure 4A). These results indicate that NEK2 contributes to the regulation of SRSF1 splicing activity.
Because NEK2 knockdown induced expression of pro-apoptotic splice variants, we asked whether it also plays a role in cell viability. In line with its effect on AS, depletion of the endogenous NEK2 in HeLa cells significantly increased the basal level of apoptosis, as monitored by cleavage of PARP1 (Figure 6C) and CASPASE 3 (Figure 6D), and enhanced the apoptotic response of cells to stress, such as treatment with cisplatin (Figure 6C and D) or starvation (Supplementary Figure 4B and C). These results suggest that the effect of NEK2 on AS events regulated by SRSF1 is physiologically relevant.
DISCUSSION
NEK2 is a centrosomal kinase involved in centrosome duplication in mitosis that is frequently upregulated in human cancers (2). Recent evidence suggests that nuclear localization of NEK2 is a predictor for drug resistance and a marker of poor prognosis in patients (10). Nevertheless, the specific nuclear functions of NEK2 are still completely obscure. The present study indicates that NEK2 acts as a regulator of AS events by modulating SRSF1 activity, thus uncovering a previously unknown nuclear function for this oncogenic kinase.
NEK2 overexpression has been extensively described in several types of tumours (2,6–10). Its oncogenic activity has been primarily ascribed to the ability to induce aneuploidy by perturbing centrosome duplication and its segregation dynamics (2). However, it was recently demonstrated that in testicular seminomas (8), myelomas and other types of cancer (10), NEK2 is primarily localized in the nucleus of neoplastic cells. We now document that the nuclear localization of NEK2 is also observed in several carcinomas and cancer cell lines in which the kinase is upregulated. Thus, although our analysis is too limited to draw conclusions, these results suggest that nuclear localization of NEK2 is a common feature of neoplastic cells.
The NEK2C splice variant was reported to localize prevalently in the nucleus (22), suggesting that its selective upregulation in cancer cells might account for the observed localization of the kinase. However, our study indicates that NEK2C is expressed at low levels in all cancer cells analysed, raising doubts on its contribution to the localization of NEK2 in primary tumours and in cell lines. Conversely, upregulation of NEK2A, but not NEK2B, is sufficient to induce its nuclear localization, suggesting that NEK2A is the prevalent isoform in the nucleus of cancer cells.
Characterization of the subcellular distribution of NEK2 pointed out its co-fractionation with several splicing factors in the nuclear-insoluble material of cancer cells. Moreover, NEK2 co-localized with two SR proteins in nuclear splicing speckles. These inter-chromatin granules are particularly enriched in SR proteins and are supposed to function as nuclear storage sites for pre-mRNA processing regulators (24). Assembly of splicing speckles and active recruitment of SR proteins from these sites to the newly synthesized pre-mRNA is strictly regulated by reversible phosphorylation (34). Phosphorylation represents one of the main mechanisms by which subtle regulation of the splicing process, and especially of AS, is achieved (35,36). These observations led us to hypothesize the existence of a functional interaction between NEK2 and splicing. Several results of our study support this hypothesis. First, NEK2 interacts with and phosphorylates SRSF1. Second, we found that the splicing activity of SRSF1 is modulated by NEK2. Lastly, silencing of NEK2 negatively affects AS events that are target of SRSF1 in live cells. Collectively, these results point to NEK2 as a novel direct regulator of SRSF1 phosphorylation and activity.
The cellular localization and splicing activity of SRSF1 are regulated by reversible phosphorylation (26,37–39). We observed that NEK2 did not influence the nuclear localization of SRSF1 in HeLa and HEK293T cells (Supplementary Figure S5). By contrast, our study suggests that NEK2 can modulate SRSF1 splicing activity similarly to SRPK1, the prototype member of the SRPK family of kinases that mediate phosphorylation of SR proteins (26). Overexpression of either NEK2 or SRPK1 induced a similar pattern of SR proteins phosphorylation and caused a similar modulation of E1A AS. Likewise, knockdown of NEK2 or SRPK1 similarly reduced the splicing activity of SRSF1 toward the BCL-X minigene. Although the effects of NEK2 might be indirect (i.e. mediated by another kinase), three lines of evidence support a direct action. First, our in vitro kinase assays were performed using highly purified proteins, strongly indicating that NEK2 can directly phosphorylate SRSF1 and SRSF7. Second, although NEK2 was recently reported to induce activation of AKT in myeloma cells (10), a signalling kinase known to directly and indirectly modulate SR proteins phosphorylation (40,41), overexpression or knockdown of NEK2 did not alter the activity of AKT in HeLa cells (Supplementary Figure S6). Third, the effect of NEK2 on BCL-X splicing was not affected by knockdown of SRPK1. Thus, these experiments suggest that NEK2 behaves as a bona fide splicing factor kinase in live cells.
Our study implicates NEK2 in AS regulation of several SRSF1 target genes involved in cell viability. We found that knockdown of NEK2 mimicked that of SRSF1, or SRPK1, and induced expression of pro-apoptotic BCL-X, BIN1 and MKNK2 splice variants. Consistently, NEK2 depletion sensitized HeLa cells to spontaneous and stress-induced apoptosis, suggesting a pro-survival function for this kinase. Although protection from cell death may also involve other splicing-unrelated functions of NEK2 (10), it is likely that enhanced splicing of the anti-apoptotic variants of BCL-X, BIN1 and MKNK2 contributes to this pro-survival effect.
As NEK2, SRPK1 is also overexpressed in human cancers (42), suggesting that these kinases may act in concert to modulate SRSF1 activity. In line with this hypothesis, we found that NEK2 affected BCL-X splicing independently of SRPK1. Thus, even if apparently redundant, SRPK1 and NEK2 could regulate SR proteins phosphorylation and subsequent AS events in a coordinate manner. In fact, the subcellular localization of SRPK1, mainly cytosolic (17), and NEK2, predominantly nuclear (this study), suggest a possible coordinated activity of these kinases in different cellular compartments, as previously reported for SRPKs and CLKs (Cdc2-like kinases) (43). Notably, activation of SRPK1 in response to AKT-mediated signalling was reported to modulate gene expression by regulating AS programs (41). Because activation of NEK2 by the ERK1/2 pathway was observed in germ cells (44), and this pathway is often activated in cancer cells, it is possible that NEK2 also participates to AS regulation in response to environmental cues. Finally, as NEK2 expression and activity peak during the late S-G2 phase (45), we cannot exclude the possibility that it could affect SR protein function in a cell-cycle–dependent manner, thus contributing to coordinate gene expression regulation with cell cycle progression. Noteworthy, another centrosomal kinase, AURKA, was shown to modulate the apoptotic response to mitotic arrest of the cell cycle by regulating the stability of SRSF1 (30).
In conclusion, our study identifies NEK2 as a novel regulator of AS, which promotes SRSF1-dependent splicing of anti-apoptotic variants, thus contributing to cell survival.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Funding for open access charge: Association for International Cancer Research (AICR) [12-0150]; the Associazione Italiana Ricerca sul Cancro (AIRC); the Fondazione Santa Lucia ‘Ricerca Corrente’.
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGMENTS
We wish to thank Professors J. Stevenin for the purification of SR proteins, insightful suggestions and critical reading of the manuscript, A. Fry, F. Cecconi and G. Biamonti for the generous gift of reagents, Drs R. Busà and R. Franco for help with confocal microscopy and immunohistochemistry, P. Bielli for helpful suggestions and critical reading of the manuscript.
REFERENCES
- 1.Fry AM, O'Regan L, Sabir SR, Bayliss R. Cell cycle regulation by the NEK family of protein kinases. J. Cell Sci. 2012;125:4423–4433. doi: 10.1242/jcs.111195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hayward DG, Fry AM. Nek2 kinase in chromosome instability and cancer. Cancer Lett. 2006;237:155–166. doi: 10.1016/j.canlet.2005.06.017. [DOI] [PubMed] [Google Scholar]
- 3.Fry AM, Meraldi P, Nigg EA. A centrosomal function for the human Nek2 protein kinase, a member of the NIMA family of cell cycle regulators. EMBO J. 1998;17:470–481. doi: 10.1093/emboj/17.2.470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Faragher AJ, Fry AM. Nek2A kinase stimulates centrosome disjunction and is required for formation of bipolar mitotic spindles. Mol. Biol. Cell. 2003;14:2876–2889. doi: 10.1091/mbc.E03-02-0108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fukasawa K. Oncogenes and tumour suppressors take on centrosomes. Nat. Rev. Cancer. 2007;7:911–924. doi: 10.1038/nrc2249. [DOI] [PubMed] [Google Scholar]
- 6.Hayward DG, Clarke RB, Faragher AJ, Pillai MR, Hagan IM, Fry AM. The centrosomal kinase Nek2 displays elevated levels of protein expression in human breast cancer. Cancer Res. 2004;64:7370–7376. doi: 10.1158/0008-5472.CAN-04-0960. [DOI] [PubMed] [Google Scholar]
- 7.Landi MT, Dracheva T, Rotunno M, Figueroa JD, Liu H, Dasgupta A, Mann FE, Fukuoka J, Hames M, Bergen AW, et al. Gene expression signature of cigarette smoking and its role in lung adenocarcinoma development and survival. PLoS One. 2008;3:e1651. doi: 10.1371/journal.pone.0001651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Barbagallo F, Paronetto MP, Franco R, Chieffi P, Dolci S, Fry AM, Geremia R, Sette C. Increased expression and nuclear localization of the centrosomal kinase Nek2 in human testicular seminomas. J. Pathol. 2009;217:431–441. doi: 10.1002/path.2471. [DOI] [PubMed] [Google Scholar]
- 9.Andréasson U, Dictor M, Jerkeman M, Berglund M, Sundström C, Linderoth J, Rosenquist R, Borrebaeck CA, Ek S. Identification of molecular targets associated with transformed diffuse large B cell lymphoma using highly purified tumor cells. Am. J. Hematol. 2009;84:803–808. doi: 10.1002/ajh.21549. [DOI] [PubMed] [Google Scholar]
- 10.Zhou W, Yang Y, Xia J, Wang H, Salama ME, Xiong W, Xu H, Shetty S, Chen T, Zeng Z, et al. NEK2 induces drug resistance mainly through activation of efflux drug pumps and is associated with poor prognosis in myeloma and other cancers. Cancer Cell. 2013;23:48–62. doi: 10.1016/j.ccr.2012.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wu G, Qiu XL, Zhou L, Zhu J, Chamberlin R, Lau J, Chen PL, Lee WH. Small molecule targeting the Hec1/Nek2 mitotic pathway suppresses tumor cell growth in culture and in animal. Cancer Res. 2008;68:8393–8399. doi: 10.1158/0008-5472.CAN-08-1915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tsunoda N, Kokuryo T, Oda K, Senga T, Yokoyama Y, Nagino M, Nimura Y, Hamaguchi M. Nek2 as a novel molecular target for the treatment of breast carcinoma. Cancer Sci. 2009;100:111–116. doi: 10.1111/j.1349-7006.2008.01007.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zeng X, Shaikh FY, Harrison MK, Adon AM, Trimboli AJ, Carroll KA, Sharma N, Timmers C, Chodosh LA, Leone G, et al. The Ras oncogene signals centrosome amplification in mammary epithelial cells through cyclin D1/Cdk4 and Nek2. Oncogene. 2010;29:5103–5112. doi: 10.1038/onc.2010.253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen Y, Riley DJ, Zheng L, Chen PL, Lee WH. Phosphorylation of the mitotic regulator protein Hec1 by Nek2 kinase is essential for faithful chromosome segregation. J. Biol. Chem. 2002;277:49408–49416. doi: 10.1074/jbc.M207069200. [DOI] [PubMed] [Google Scholar]
- 15.Paronetto MP, Achsel T, Massiello A, Chalfant CE, Sette C. The RNA-binding protein Sam68 modulates the alternative splicing of Bcl-x. J. Cell Biol. 2007;176:929–939. doi: 10.1083/jcb.200701005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Busà R, Geremia R, Sette C. Genotoxic stress causes the accumulation of the splicing regulator Sam68 in nuclear foci of transcriptionally active chromatin. Nucleic Acids Res. 2010;38:3005–3018. doi: 10.1093/nar/gkq004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhong XY, Ding JH, Adams JA, Ghosh G, Fu XD. Regulation of SR protein phosphorylation and alternative splicing by modulating kinetic interactions of SRPK1 with molecular chaperones. Genes Dev. 2009;23:482–495. doi: 10.1101/gad.1752109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lin S, Xiao R, Sun P, Xu X, Fu XD. Dephosphorylation-dependent sorting of SR splicing factors during mRNP maturation. Mol. Cell. 2005;20:413–425. doi: 10.1016/j.molcel.2005.09.015. [DOI] [PubMed] [Google Scholar]
- 19.Sette C, Bevilacqua A, Geremia R, Rossi P. Involvement of phospholipase Cgamma1 in mouse egg activation induced by a truncated form of the C-kit tyrosine kinase present in spermatozoa. J. Cell Biol. 1998;142:1063–1074. doi: 10.1083/jcb.142.4.1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dreumont N, Hardy S, Behm-Ansmant I, Kister L, Branlant C, Stévenin J, Bourgeois CF. Antagonistic factors control the unproductive splicing of SC35 terminal intron. Nucleic Acids Res. 2010;38:1353–1366. doi: 10.1093/nar/gkp1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Di Agostino S, Fedele M, Chieffi P, Fusco A, Rossi P, Geremia R, Sette C. Phosphorylation of high-mobility group protein A2 by Nek2 kinase during the first meiotic division in mouse spermatocytes. Mol. Biol. Cell. 2004;15:1224–1232. doi: 10.1091/mbc.E03-09-0638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wu W, Baxter JE, Wattam SL, Hayward DG, Fardilha M, Knebel A, Ford EM, da Cruz e Silva EF, Fry AM. Alternative splicing controls nuclear translocation of the cell cycle-regulated Nek2 kinase. J. Biol. Chem. 2007;282:26431–26440. doi: 10.1074/jbc.M704969200. [DOI] [PubMed] [Google Scholar]
- 23.Hayes MJ, Kimata Y, Wattam SL, Lindon C, Mao G, Yamano H, Fry AM. Early mitotic degradation of Nek2A depends on Cdc20-independent interaction with the APC/C. Nat. Cell Biol. 2006;8:607–614. doi: 10.1038/ncb1410. [DOI] [PubMed] [Google Scholar]
- 24.Spector DL, Lamond AI. Nuclear speckles. Cold Spring Harb. Perspect. Biol. 2011;3 doi: 10.1101/cshperspect.a000646. pii: a000646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Long JC, Caceres JF. The SR protein family of splicing factors: master regulators of gene expression. Biochem. J. 2009;417:15–27. doi: 10.1042/BJ20081501. [DOI] [PubMed] [Google Scholar]
- 26.Zhou Z, Fu XD. Regulation of splicing by SR proteins and SR protein-specific kinases. Chromosoma. 2013;122:191–207. doi: 10.1007/s00412-013-0407-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Karni R, de Stanchina E, Lowe SW, Sinha R, Mu D, Krainer AR. The gene encoding the splicing factor SF2/ASF is a proto-oncogene. Nat. Struct. Mol. Biol. 2007;14:185–193. doi: 10.1038/nsmb1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Anczuków O, Rosenberg AZ, Akerman M, Das S, Zhan L, Karni R, Muthuswamy SK, Krainer AR. The splicing factor SRSF1 regulates apoptosis and proliferation to promote mammary epithelial cell transformation. Nat. Struct. Mol. Biol. 2012;19:220–228. doi: 10.1038/nsmb.2207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Olshavsky NA, Comstock CE, Schiewer MJ, Augello MA, Hyslop T, Sette C, Zhang J, Parysek LM, Knudsen KE. Identification of ASF/SF2 as a critical, allele-specific effector of the cyclin D1b oncogene. Cancer Res. 2010;70:3975–3984. doi: 10.1158/0008-5472.CAN-09-3468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Moore MJ, Wang Q, Kennedy CJ, Silver PA. An alternative splicing network links cell-cycle control to apoptosis. Cell. 2010;142:625–636. doi: 10.1016/j.cell.2010.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Boise LH, González-García M, Postema CE, Ding L, Lindsten T, Turka LA, Mao X, Nuñez G, Thompson CB. Bcl-x, a bcl-2-related gene that functions as a dominant regulator of apoptotic cell death. Cell. 1993;74:597–608. doi: 10.1016/0092-8674(93)90508-n. [DOI] [PubMed] [Google Scholar]
- 32.Massiello A, Salas A, Pinkerman RL, Roddy P, Roesser JR, Chalfant CE. Identification of two RNA cis-elements that function to regulate the 5' splice site selection of Bcl-x pre-mRNA in response to ceramide. J. Biol. Chem. 2004;279:15799–15804. doi: 10.1074/jbc.M313950200. [DOI] [PubMed] [Google Scholar]
- 33.Adesso L, Calabretta S, Barbagallo F, Capurso G, Pilozzi E, Geremia R, Delle Fave G, Sette C. Gemcitabine triggers a pro-survival response in pancreatic cancer cells through activation of the MNK2/eIF4E pathway. Oncogene. 2013;32:2848–2857. doi: 10.1038/onc.2012.306. [DOI] [PubMed] [Google Scholar]
- 34.Misteli T, Cáceres JF, Clement JQ, Krainer AR, Wilkinson MF, Spector DL. Serine phosphorylation of SR proteins is required for their recruitment to sites of transcription in vivo. J. Cell Biol. 1998;143:297–307. doi: 10.1083/jcb.143.2.297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Stamm S. Regulation of alternative splicing by reversible protein phosphorylation. J. Biol. Chem. 2008;283:1223–1227. doi: 10.1074/jbc.R700034200. [DOI] [PubMed] [Google Scholar]
- 36.Naro C, Sette C. Phosphorylation-mediated regulation of alternative splicing in cancer. Int. J. Cell Biol. 2013;2013:151839. doi: 10.1155/2013/151839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cao W, Jamison SF, Garcia-Blanco MA. Both phosphorylation and dephosphorylation of ASF/SF2 are required for pre-mRNA splicing in vitro. RNA. 1997;3:1456–1467. [PMC free article] [PubMed] [Google Scholar]
- 38.Sanford JR, Ellis JD, Cazalla D, Cáceres JF. Reversible phosphorylation differentially affects nuclear and cytoplasmic functions of splicing factor 2/alternative splicing factor. Proc. Natl Acad. Sci. USA. 2005;102:15042–15047. doi: 10.1073/pnas.0507827102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Prasad J, Colwill K, Pawson T, Manley JL. The protein kinase Clk/Sty directly modulates SR protein activity: both hyper- and hypophosphorylation inhibit splicing. Mol. Cell. Biol. 1999;19:6991–7000. doi: 10.1128/mcb.19.10.6991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Patel NA, Kaneko S, Apostolatos HS, Bae SS, Watson JE, Davidowitz K, Chappell DS, Birnbaum MJ, Cheng JQ, Cooper DR. Molecular and genetic studies imply Akt-mediated signaling promotes protein kinase CbetaII alternative splicing via phosphorylation of serine/arginine-rich splicing factor SRp40. J. Biol. Chem. 2005;280:14302–14309. doi: 10.1074/jbc.M411485200. [DOI] [PubMed] [Google Scholar]
- 41.Zhou Z, Qiu J, Liu W, Zhou Y, Plocinik RM, Li H, Hu Q, Ghosh G, Adams JA, Rosenfeld MG, et al. The Akt-SRPK-SR axis constitutes a major pathway in transducing EGF signaling to regulate alternative splicing in the nucleus. Mol. Cell. 2012;47:422–433. doi: 10.1016/j.molcel.2012.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hayes GM, Carrigan PE, Miller LJ. Serine-arginine protein kinase 1 overexpression is associated with tumorigenic imbalance in mitogen-activated protein kinase pathways in breast, colonic, and pancreatic carcinomas. Cancer Res. 2007;67:2072–2080. doi: 10.1158/0008-5472.CAN-06-2969. [DOI] [PubMed] [Google Scholar]
- 43.Ngo JC, Chakrabarti S, Ding JH, Velazquez-Dones A, Nolen B, Aubol BE, Adams JA, Fu XD, Ghosh G. Interplay between SRPK and Clk/Sty kinases in phosphorylation of the splicing factor ASF/SF2 is regulated by a docking motif in ASF/SF2. Mol. Cell. 2005;20:77–89. doi: 10.1016/j.molcel.2005.08.025. [DOI] [PubMed] [Google Scholar]
- 44.Di Agostino S, Rossi P, Geremia R, Sette C. The MAPK pathway triggers activation of Nek2 during chromosome condensation in mouse spermatocytes. Development. 2002;129:1715–1727. doi: 10.1242/dev.129.7.1715. [DOI] [PubMed] [Google Scholar]
- 45.Fry AM, Schultz SJ, Bartek J, Nigg EA. Substrate specificity and cell cycle regulation of the Nek2 protein kinase, a potential human homolog of the mitotic regulator NIMA of Aspergillus nidulans. J. Biol. Chem. 1995;270:12899–12905. doi: 10.1074/jbc.270.21.12899. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.