


Abstract
Natural transformation (NT) in bacteria is a complex process, including binding, uptake, transport and recombination of exogenous DNA into the chromosome, consequently generating genetic diversity and driving evolution. DNA processing protein A (DprA), which is distributed among virtually all bacterial species, is involved in binding to the internalized single-stranded DNA (ssDNA) and promoting the loading of RecA on ssDNA during NTs. Here we present the structures of DNA_processg_A (DprA) domain of the Helicobacter pylori DprA (HpDprA) and its complex with an ssDNA at 2.20 and 1.80 Å resolutions, respectively. The complex structure revealed for the first time how the conserved DprA domain binds to ssDNA. Based on structural comparisons and binding assays, a unique ssDNA-binding mode is proposed: the dimer of HpDprA binds to ssDNA through two small, positively charged binding pockets of the DprA domains with classical Rossmann folds and the key residue Arg52 is re-oriented to ‘open’ the pocket in order to accommodate one of the bases of ssDNA, thus enabling HpDprA to grasp substrate with high affinity. This mode is consistent with the oligomeric composition of the complex as shown by electrophoretic mobility-shift assays and static light scattering measurements, but differs from the direct polymeric complex of Streptococcus pneumoniae DprA–ssDNA.
INTRODUCTION
Natural transformation (NT) is one of conserved ways of acquiring genetic diversity to drive bacterial evolution (1). Except for a few species such as those from the genera Neisseria, most of the naturally transformable bacteria become competent only for short period of time and the regulation of competence development is tightly controlled by a complex organism-specific process (2). Some genes encoding NT-related proteins are widely distributed in non-NT competent species such as Escherichia coli. Therefore, there probably exist more species that are transformation competent, but the conditions to trigger their competence are yet to be discovered (3). Among the more than 60 NT-competent bacteria, Bacillus subtilis and Streptococcus pneumoniae are used as prototypes for NT in Gram-positive microorganisms, whereas Neisseria gonorrhoeae and Haemophilus influenza for Gram-negative microorganisms. Researches on these organisms revealed that NT of bacterial cells generally involves three steps: (i) exogenous DNA binding to the bacterial surface; (ii) uptake and translocation of the exogenous DNA across the bacterial membrane(s); and (iii) homologous recombination between donor DNA and the recipient chromosome or plasmid (4,5). Some similar competence devices have been utilized. For instance, multi-protein machines analogous to type IV pili (T4P) or type II secretion systems (T2SS) are in charge of importing DNA, and the recombination systems are universally RecA dependent (2,3,6–8).
As a representative of epsilon proteobacteria, Helicobacter pylori is a good model for NT research. NT contributes to the genetic plasticity of H. pylori and yields better-adapted pathogen variants to colonize the stomach, a very hostile environment, for many decades and to resist eradication attempts by various methods (9–11). Helicobacter pylori exhibits a comparatively long-time competence state during both logarithmic and stationary phases and DNA damage can activate NT-related genes (12,13). A two-step uptake mechanism is proposed for the transport of external DNA from the cell surface to the cytosol in H. pylori. dsDNA uptake across the outer membrane is mediated by the type IV secretion system (T4SS) known as ComB, whereas ComEC (HP1361), an inner membrane channel, is implicated in DNA transport across the inner membrane (14–16). Some proteins related to NT, such as ComL (HP1378), ComH (HP1527) and NucT (HP0323), are located in periplasm (16–18). Two intracellular proteins, DprA (HP0333) and RecA (HP0153), are essential for NT of this bacterium, but the mechanism of how internalized ssDNA is transferred to RecA remains unclear (19–21). In most organisms, homologous recombination based on RecA is usually initiated by the RecBCD pathway or the RecFOR pathway. Although AddAB (RecBCD complex homolog) and RecOR exist in H. pylori, they are unrelated to NT (4,22,23). Hence, NT-mediated homologous recombination of H. pylori is believed to be closely linked with DprA.
The biological function of the conserved DprA protein was extensively studied in S. pneumoniae and B. subtilis. SpDprA can bind to ssDNA cooperatively through self-interaction and protects ssDNA from nuclease digestions. In addition to free ssDNA, SpDprA interacts with heterologous E. coli Ssb-coated ssDNA, alleviates the EcSsb barrier and facilitates the loading of EcRecA onto ssDNA (24). The results that BsDprA facilitates homologous BsRecA assembly onto ssDNA coated by BsSsbB further confirmed DprA’s mediator role (25).The latest study indicated that residues involved in SpRecA interaction might overlap partially with SpDprA dimerization and the authors proposed a model in which the SpDprA dimer was rearranged or disrupted by SpRecA interaction during RecA-dependent recombination (26). As of now, DprA has been demonstrated to be a novel recombination-mediator protein (RMP) that plays a crucial role in NT. Hence, the (Ssb)–DprA–RecA pathway is considered as the third recombination pathway and the studies of which contribute to more comprehensive understandings of homologous recombination. Although the function of SpDprA or BsDprA as a representative of the highly conserved protein family has been described very well, no 3D structure of DprA protein in complex with ssDNA has been reported so far, and the mechanism of how DprA binds to ssDNA remains elusive. Here we present the high-resolution crystal structures of conserved DNA_processg_A (DprA) domain of H. pylori DprA (HpDprA) and its complex with an ssDNA. The complex structure shows the first classical Rossmann fold (RF) protein to be structurally characterized in complex with ssDNA and a unique ssDNA-binding mode. Unlike SpDprA–ssDNA polymeric complex reported previously (24), HpDprA dimers firstly tend to form an oligomeric complex at 1:1 molar ratio with ssDNA. In association with a series of structure-based mutagenesis analyses and binding property assays, one possible structural mechanism for HpDprA–ssDNA interaction was proposed, in which HpDprA functions as a dimer resembling a barbell and relies on two binding pockets to grasp ssDNA effectively with a switch mediated by a critical residue Arg52.
MATERIALS AND METHODS
Protein expression and purification
The ORF fragment encoding DNA processing chain A protein (hp0333, Gene ID 900099) from H. pylori strain 26 695 was cloned into an expression vector pET-22 b (Novagen) and a 6-histidine tag was constructed at the C-terminus of the recombinant protein. Escherichia coli BL21 (DE3) strain transformed with the pET-22 b plasmid containing the HpDprA gene were grown in the presence of ampicillin and were induced overnight with 0.2 mM IPTG at 16°C. The cells were harvested by centrifugation (4000 g, 4°C, 30 min). The cell pellets were suspended in lysis buffer containing 50 mM Na2HPO4/NaH2PO4 (pH 7.5), 300 mM NaCl, 10 mM imidazole and 5% (v/v) glycerol, and then 1% (v/v) PMSF (10 mg/ml, dissolved in isopropanol) was added to inhibit proteases prior to lysis using ultra-sonication. After removal of the insoluble debris by centrifugation (16 000 g, 4°C, 30 min), the supernatant was applied to a Ni-NTA (Novagen) column at 4°C. The fraction eluted with 250 mM imidazole added in the lysis buffer contained target protein and was concentrated by ultrafiltration using Amicon Ultra-15 concentrators (Millipore). The protein was then loaded onto a HiLoad 16/60 Superdex 200 column (GE Healthcare) equilibrated in 50 mM Tris-HCl (pH 7.2), 300 mM NaCl, 1 mM DTT and 5% (v/v) glycerol. The eluted peak corresponding to HpDprA dimer was collected and the protein was concentrated to 10 mg/ml by ultrafiltration. The HpDprA mutants were purified in the same way as the wild-type (WT) protein.
The C-terminal truncated genes of HpDprA(5-225) and HpDprA(5-217) were also cloned into the expression vector pET-22 b (Novagen). These truncated mutants are more stable than the full-length HpDprA, and cation-exchange chromatography (HiTrap SP HP column, GE Healthcare) was added before gel-filtration chromatography to avoid chromosome DNA pollution. The truncated proteins were concentrated to 20 mg/ml in the buffer containing 50 mM Tris-HCl (pH7.2), 150 mM NaCl, 1 mM DTT. Additional 5 mM DTT and 0.2 mM EDTA were added for the Se-Met substituted HpDprA(5-225). The gene of HpDprA(5-225) was also cloned into the vector pGEX-6 p-2 (GE Healthcare) with an N-terminal GST tag. After first GST-affinity chromatography, the GST tag was truncated with PreScission Protease and removed by a second GST-affinity chromatography to obtain the tag-free HpDprA(5-225) sample. The sample was further purified by cation-exchange and gel-filtration chromatography in tandem. The gel-filtration chromatography analysis was performed on a HR 10/300 Superdex 75 column (GE Healthcare). DprA homolog from S. pneumonia strain tigr4 (SpDprA) was cloned into pET-22 b vector and purified as described (26). Detailed information of all constructed proteins was listed in Supplementary Table S1. The concentrations of all proteins were measured with NanoDrop 2000 UV-Vis Spectrophotometer (Thermo).
Crystallization, data collection and structure determination
Crystals of native HpDprA(5-225) were obtained using hanging drop method under the condition of 20–25% PEG3350 and 100 mM Bis-Tris (pH 5.5–6.5) or 100 mM HEPES (pH 6.8–7.5). One dataset was collected on beamline BL17U of the Shanghai Synchrotron Radiation Facility (Shanghai, People’s Republic of China). Crystals of the Se-Met substituted protein were obtained under the condition of 25% PEG3350, 100 mM HEPES (pH 6.8) and 100 mM potassium thiocyanate. The dataset for the Se-Met substituted HpDprA(5-225) was collected at beamline BL17A of Photon Factory, KEK (Tsukuba, Japan) at the peak wavelength of 0.9789 Å. For the crystallization of HpDprA–ssDNA complex, HpDprA(5-217)–dT35 mixtures were incubated at 25°C for 30 min with a protein to nucleic acid molar ratio of 1:5. The complex crystal was obtained under the condition of 8% PEG3350 and 100 mM NaAc (pH 5.2). One x-ray diffraction dataset for the complex was collected at the same beamline of KEK. Prior to data collection, all of the crystals were transferred to their corresponding well solutions supplemented with 10% (v/v) ethylene glycol as a cryoprotectant.
All the datasets were processed with the program MOSFLM and scaled with SCALA from the CCP4 program suite (27). Phase determination of HpDprA(5-225) was performed by SAD method and automatic model building was carried out with software package PHENIX (28). The rest of the model was manually built with COOT (29). The refinement was carried out with PHENIX. The structure of HpDprA(5-217)–dT35 complex was solved by molecular replacement with PHASER of the CCP4 program suite using monomer of HpDprA(5-225) structure as a search model. Model building and structural refinement were performed with COOT and PHENIX, respectively. The statistics of data collection and refinement are summarized in Table 2. The quality of these final models was checked with MolProbity (30). All protein information is from the database of National Center for Biotechnology Information (NCBI). Sequence alignments were performed with ClustalW2 (31). Sequence alignment figures were produced by ESPript (32). All the structure figures were rendered in PyMOL (http://www.pymol.org).
Table 2.
Data collection and refinement statistics
| Se-HpDprA(5-225) | HpDprA(5-225) | HpDprA(5-217)–ssDNA | |
|---|---|---|---|
| Data collection | |||
| Space group | P21 | P21212 | P21 |
| Cell dimensions | |||
| a, b, c (Å) | 117.88, 42.43, 181.33 | 171.09, 107.86, 37.47 | 37.40, 40.51, 154.68 |
| α, β, γ (°) | 90, 108.65, 90 | 90, 90, 90 | 90, 92.18, 90 |
| Peak | |||
| Wavelength (Å) | 0.9789 | 0.9792 | 1.0000 |
| Resolution (Å) | 59.23-2.35 (2.48-2.35) | 45.62-2.20 (2.32-2.20) | 40.51-1.80 (1.90-1.80) |
| Unique reflections | |||
| Rmergea (%) | 7.4 (24.8) | 8.6 (22.7) | 6.3 (31.1) |
| I/σ | 14.7 (6.0) | 17.9 (7.8) | 9.8 (2.9) |
| Completeness (%) | 99.1 (97.3) | 95.9 (80.9) | 83.6 (77.0) |
| Redundancy | 6.7 (6.2) | 11.1 (8.4) | 3.3 (3.5) |
| Refinement | |||
| Resolution (Å) | 39.77-2.20 | 33.16-1.80 | |
| No. of reflections | 34 008 | 34 988 | |
| Rwork/Rfreeb (%) | 16.96/21.30 | 19.40/23.71 | |
| No. of atoms | |||
| Protein | 3440 | 3318 | |
| ssDNA | 142 | ||
| Water | 321 | 385 | |
| B-factors (Å2) | |||
| Protein | 36.525 | 30.502 | |
| ssDNA | 51.915 | ||
| Water | 42.673 | 40.080 | |
| R.m.s.d. | |||
| Bond lengths (Å) | 0.0060 | 0.0040 | |
| Bond angles (°) | 0.976 | 0.870 |
Values in parentheses indicate the specific values in the highest resolution shell.
aRmerge =  , where
, where  is the intensity of the measured reflection, and
is the intensity of the measured reflection, and 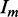 is the mean intensity of all the symmetry-related reflections.
is the mean intensity of all the symmetry-related reflections.
bRwork =  , where
, where 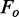 denotes the observed structure factor amplitude and
denotes the observed structure factor amplitude and 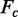 denotes the structure factor amplitude calculated from the model. Rfree is as for Rwork but calculated with 5.0% of randomly chosen reflections omitted from the refinement.
denotes the structure factor amplitude calculated from the model. Rfree is as for Rwork but calculated with 5.0% of randomly chosen reflections omitted from the refinement.
Electrophoretic mobility-shift binding assays
Protein–DNA binding interactions were assayed by electrophoretic mobility-shift assay (EMSA). HPLC grade oligomeric ssDNAs were synthesized with 5′ biotin label. Protein and DNA samples were dialysed overnight at 4°C into the buffer E containing 25 mM HEPES (pH 7.0), 150 mM NaCl, and 1 mM DTT. Protein and DNA were mixed in the binding buffer with 25 mM HEPES (pH 7.0), 150 mM NaCl, 10% glycerol, 1 mM DTT, and 0.05% IGEPAL (v/v) (Sigma-Aldrich). After 20 min incubation at room temperature, samples were applied to 8% non-denaturing PAGE electrophoresis and then transferred to Hybond-N+ membrane (Amersham Biosciences) at 4°C in 0.5X TBE buffer. Fluorescence detection was performed with streptavidin-conjugated alkaline phosphatase and CDP-Star (Roche) as described by product specifications.
MST assays
The microscale thermophoresis (MST) method to assay binding interactions between proteins and DNA has been described in detail elsewhere (33). Proteins were labeled with fluorescence according to the manufacturer’s protocol. The synthesized oligomeric ssDNAs were purified by Mono Q column chromatography (GE Healthcare) and were dialysed overnight at 277 K into the buffer E. These samples were quantified with NanoDrop 2000 UV-Vis Spectrophotometer (Thermo). A series of 16 ssDNA solutions with different concentrations were prepared by consecutive 2-fold dilutions from the highest concentration. ssDNA with different concentrations and protein at 200 nM concentration were mixed at a volume ratio of 1:1. The samples were loaded into silica capillaries (Polymicro Technologies) after incubation at room temperature for 20 min. Measurements were performed at 25°C in 25 mM HEPES buffer (pH 7.0) with 150 mM NaCl, 1 mM DTT and 0.05% (v/v) Tween 20, by using 40–90% LED power and 40% IR-laser power. Measurements were also carried out on 40–90% LED power and 80% IR-Laser power for comparison. Data analyses were performed using Nanotemper Analysis software, v.1.2.229.
SLS measurements
Static light scattering (SLS) measurements were performed at 25°C on a DAWN HELEOS II instrument (Wyatt Technology, Santa Barbara, CA). HpDprA(5-217) and HpDprA(5-217)–dT35 complex were diluted to 1.0 mg/ml in the buffer S containing 20 mM HEPES (pH 7.5) and 150 mM NaCl for SLS analyses. Calibration of the light scattering detector was verified with BSA monomer standard before the assays. The data were analysed with ASTRA software (Wyatt Technology, version 5.3.4.11).
RESULTS
The conserved DprA domain is sufficient for HpDprA to bind to ssDNA
The full-length HpDprA consists of two domains (Figure 1A). The N-terminal domain (residues Ser12–Asp217) belongs to Pfam02481 (DNA_processg_A domain, referred to as DprA domain hereafter), which is characterized by the DprA/Smf protein family. Two DprA homologous structures have been reported as of now, including SpDprA (PDB code 3UQZ) from S. pneumoniae and RpDprA (PDB code 3MAJ) from Rhodopseudomonas palustris (26). Structural comparisons showed that the DprA domains are conserved in all three structures. SpDprA and RpDprA possess a N-terminal five-helix fold SAM-like domain, respectively, which probably participates in various protein–protein interactions (26). In addition, RpDprA has a C-terminal DML1-like domain, which was proposed to be involved in Z-DNA binding (Figure 1A) (26,34). Sequence alignments demonstrated that DprA domains are remarkably conserved among these three proteins (sequence identities between HpDprA and SpDprA/RpDprA = 31/33%, positives = 51/52%) (Figure 1C). The C-terminal domain of HpDprA (residues Met226–Ala270) was predicted to be of the DML1-like fold, which was also present in RpDprA, by I-TASSER server (Supplementary Figure S1C) (35).
Figure 1.

Sequence alignment of three DprA homologous proteins from R. palustris, S. pneumonia and H. pylori. (A) Linear representation of the three DprA proteins with multi-domains, including SAM domain (orange), DNA_processg_A domain (pale green) and DML1 domain (salmon). (B) EMSA results of full-length HpDprA–dT35 (left panel) and HpDprA(5-225)–dT35 (right panel). Different concentrations of HpDprA (0.77 nM to 5 μM) with 5 nM biotin-labeled ssDNA are incubated for 20 min at 25°C before loading on the gel. Samples were electrophoresed on 8% PAGE and detected by fluorography. (C) Sequence alignment of three DprAs (color code as in (A)). Important residues are indicated by symbols in blue: residues involved in hydrophobic interaction of dimerization interface (stars), conserved residues near the binding pocket (dots) and residues directly contacted by ssDNA (triangles).
The ssDNA-binding activity of HpDprA was determined by EMSA, with oligomeric ssDNA dT35 as substrate. To confirm whether the additional C-terminal domain is involved in ssDNA binding, binding assays with full-length HpDprA or truncated HpDprA(5-225) were performed (Figure 1B). In low protein to dT35 ratios, an initial oligomeric complex (A1 band, the ratio of HpDprA dimer to ssDNA is 1:1, proved below) formed as demonstrated by gel mobility retardation compared to free DNA (FD band). In the same protein concentration gradient, the ssDNA-binding modes of the two proteins were almost identical except for the molecular weight (MW) of A1 band. However, in the cases of higher protein to dT35 ratios, the quantity of the oligomeric complex gradually reduced, accompanied by the increase of polymeric complexes (A2, A3 and A4 bands), some of which (A4 band) was retained in the wells in full-length HpDprA-binding assay. It is similar to full-length SpDprA with high MW complexes retained in loading wells but no oligomeric complexes formed (24). In contrast, polymeric complexes did not appear in the truncated HpDprA experiment. To gain insight into the precise affinity of HpDprA–ssDNA complex, the MST method was used to assay the dissociation constants (Kd) of full-length HpDprA and HpDprA(5-225) toward dT35 (Supplementary Figure S5A). Full-length HpDprA bound to dT35 with a Kd of 2.26 ± 0.167 nM, and HpDprA(5-225) bound to it with a Kd of 30.6 ± 0.998 nM (Table 1). Both EMSA and MST results indicated that the ssDNA-binding ability of full-length HpDprA is higher than that of HpDprA(5-225), probably because of additional C-terminal domain. The DML1-like domain might facilitate the formation of polymeric complex by mediating protein–protein interactions or by forming additional protein–ssDNA interactions (see Discussion section). Nevertheless, the binding mode of the truncated HpDprA to ssDNA is in accordance with the full-length HpDprA at low protein to dT35 ratios. Another truncated HpDprA(5-217) behaved as HpDprA(5-225) in ssDNA-binding assay (Supplementary Figure S3G). Hence, truncated HpDprA(5-225) and HpDprA(5-217) that contain the complete DprA domain can be used to study the ssDNA-binding activity in place of the full-length HpDprA to a large extent.
Table 1.
The dissociation constants (Kd) of different constructions of HpDprA toward different types of ssDNA determined by MST
| Protein | ssDNA | Dissociation constant (Kd)nM |
|---|---|---|
| Full-length HpDprA | dT35 | 2.26 ± 0.167 |
| HpDprA(5-225) | dT35 | 30.6 ± 0.998 |
| HpDprA(5-217)WT | dT35 | 31.5 ± 0.806 |
| HpDprA(5-217)H8E | dT35 | 66.3 ± 0.721 |
| HpDprA(5-217)Y11E | dT35 | 55.9 ± 1.82 |
| HpDprA(5-217)R52E | dT35 | 9950 ± 367 |
| HpDprA(5-217)Y108E | dT35 | 351 ± 6.88 |
| HpDprA(5-217)K137E | dT35 | 7220 ± 216 |
| HpDprA(5-217)F140E | dT35 | 556 ± 28 |
| HpDprA(5-217)R143E | dT35 | 5470 ± 246 |
| HpDprA(5-217)R52E/K137E | dT35 | nd |
| HpDprA(5-217)K137E/R143E | dT35 | nd |
| HpDprA(5-217)R52E/R143E | dT35 | nd |
| HpDprA(5-217) | dT6 | nd |
| HpDprA(5-217) | dT10 | nd |
| HpDprA(5-217) | dT15 | 18 400 ± 574 |
| HpDprA(5-217) | dT20 | 2230 ± 52.3 |
| HpDprA(5-217) | dA35 | 1200 ± 35.3 |
| HpDprA(5-217) | dR30 | 370 ± 8.82 |
| HpDprA(5-217) | dsDNA30 | 831 ± 8.48 |
nd, not detected.
Other ssDNA-binding properties of HpDprA were also assayed using EMSA techniques. The results showed that neither the presence or absence of 5 mM Mg2+, nor the different ionic strengths of 150 mM NaCl (protein precipitation occurred when the salt concentration is <150 mM) or 300 mM NaCl (data not shown), nor the presence of six-histidine tag at the C-terminus of HpDprA(5-225) had any noticeable effects on the binding of dT35 with the truncated mutant protein (Supplementary Figure S3F).
Overall structures of HpDprA(5-225) and HpDprA(5-217)–dT35 complex
Despite many attempts to obtain the crystals of the full-length HpDprA, natural degradation always occurs spontaneously during the crystallization process. The crystalline sample was found through sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) to be a degraded fragment instead of the full-length protein. The degraded fragment is confirmed to be HpDprA(5-225) by protein N-terminal sequencing and mass spectrometry analysis (data not shown). We first solved the crystal structure of HpDprA(5-225), by selenium single-wavelength anomalous diffraction. It has a dimer per asymmetric unit. The final model includes residues 5-223 for both monomers. The details of the crystallographic analysis are summarized in Table 2. There is one obvious dimerization interface (Figure 2A), which is consistent with the fact that both full-length HpDprA and HpDprA(5-225) exist as dimers in solution. The monomer of the HpDprA(5-225) adopts a classical RF that consists of nine α-helices and nine β-strands. The nine strands constitute an extended sheet in the center of the structure, and the helices flank on both sides of the β-strands to form a sandwiched structure (Figure 2B). Structural comparisons between HpDprA(5-225) and DprA domain of SpDprA or RpDprA yield a root mean square deviations (r.m.s.d.) of 1.9 or 2.0 Å for 205 or 209 aligned Cα atoms, indicative of notable structural similarity (Supplementary Figure S1E). The dimerization is mediated mainly through hydrophobic inter-molecular interactions consisting of residues Pro183, Leu196 and Phe205. On the reverse side of the hydrophobic core, four inter-molecular hydrogen bonds between residues Arg185 and Glu188 from the loop (β8–α8) enhance the HpDprA dimer. In addition, the Tyr57 side-chain hydroxyl donates a hydrogen bond to the main-chain carbonyl group of Leu186 (Figure 2C). Structural comparisons among three DprA homologous structures showed that dimer is widely adopted as a universal quaternary structure (Supplementary Figure S1). Sequence alignments showed lower similarities among residues on the loop (β8–α8), but the fact that the dimerization mode of one hydrophobic interface is conserved among DprA domains demonstrated that dimers are essential for the biochemical functions of these proteins (Figures 1C and 2D).
Figure 2.

Structures of apo-HpDprA(5-225). (A) The dimer of HpDprA(5-225) colored in pale green. (B) The secondary structure elements of the monomer are labeled in different colors. (C) The dimerization interface of HpDprA(5-225). Residues involved in dimerization are shown in stick models. (D) The superimpositions of the hydrophobic cores. The hydrophobic residues are shown as stick models with those of RpDprA in orange, of SpDprA in white and of HpDprA in cyan. These residues are also marked as blue stars in Figure 1C.
The structure of truncated HpDprA(5-217) in complex with dT35 contained one protein dimer per asymmetric unit, while majority of ssDNA were flexible and not observed. Only seven deoxythymidine nucleotides (dT) were traced in the electron density maps, including six consecutive dTs bound to molecule B and a single dT interacting with molecule A (Figure 3A). HpDprA(5-217) exists as a dimer both in crystal and in solution. The structures of the two HpDprA(5-217) monomers in ssDNA-binding complex are generally very similar to the structures of apo-HpDprA(5-225) (r.m.s.d. of 0.4 and 0.5 Å over 212 Cα atoms for monomers A and B, respectively). Moreover, the conformation of the dimeric interface is not affected by ssDNA binding.
Figure 3.

Models of the HpDprA(5-217)–ssDNA complex. (A) Ribbon representation of the HpDprA(5-217)–ssDNA complex. There are HpDprA(5-217) dimer (cartoon in palegreen), six consecutive dTs (sticks in cyan) bound by molecule B and a single dT (dT5*) bound by molecule A. (B) The electrostatic surface potential (from blue = 74 kT/e to red = –74 kT/e) in molecule B of HpDprA(5-217)–ssDNA. dT4–dT6 are displayed as cartoons and the binding pocket is in ‘open’ state. Two protruding helices (α6 and α7) and the loop (β3–α3) divide the positive-charged region into three separate entrances (E1–E3). The dots show the possible trend of ssDNA between the two binding pockets of dimer. (C) The electrostatic surface potential shows the ‘closed’ pocket in molecule B of apo-HpDprA(5-225). (D) The ssDNA involved in binding shown in cyan stick representation covered by a σA-weighted 2Fo-Fc omit map contoured at 1.0 σ. (E) Stereo view of binding pockets in comparison showing conformational changes of active residues in ssDNA binding. The residues in apo-HpDprA(5-225) and HpDprA(5-217)–ssDNA complex are colored in red and green, respectively. The key residue, Arg52, rotates ∼80° owing to binding to ssDNA.
ssDNA docking onto specific surface pockets of HpDprA dimer
Surface potential calculations showed that there were no obvious grooves on the HpDprA dimer, but only two positively charged pockets covering small areas at the opposite ends (Figure 3B). The pocket consists of three helices α4, α6, α7 and a flexible loop (β3–α3), as a deep hydrophobic hole with a diameter of ∼6.0 Å. Two protruding helices (α6 and α7) and the loop (β3–α3) (located at the top of α4) surrounding the pocket form three independent entrances (E1–E3) (Figure 3B and Supplementary Figure S1D). Among them, E1, an extensive positively charged region below the pocket, and E2, a hydrophobic region, can reasonably accommodate ssDNA. Similar binding pockets were also observed in RpDprA and SpDprA structures based on surface potential calculations (Supplementary Figure S1A and B). Sequence alignments of DprA family members from both NT-competent and non-NT-competent bacteria showed that most of the highly conserved amino acid residues are concentrated near the pocket (Supplementary Figure S6). The results indicated that the highly conserved, positively charged area is most likely to be the DNA-binding sites for all DprA proteins.
In HpDprA(5-217)–dT35 complex, the electron density of the six dTs was visible at the binding pocket of molecule B, but only one dT could be traced at the binding pocket of molecule A (Figure 3D). dT1 and dT4–dT6 of the six consecutive dTs were bound by molecule B and dT2–dT3 were in contact with molecule B of a neighboring, crystallographic equivalent dimer (referred to as molecule B′ hereafter) (Figure 4A and Supplementary Figure S2B). The single dT (dT5*) binding to the molecule A is at the position corresponding to that of dT5 grasped by the molecule B. Oligonucleotide–HpDprA(5-217) interaction is maintained through hydrophobic interactions and hydrogen bonds, which are contributed by 12 residues (Supplementary Figure S2A). Interestingly, several residues, such as His8′, Phe9′, Gln10′ and Tyr11′, from molecule B′ of another crystallographic symmetric dimer are also involved in the interaction with dT2 and dT3 near the binding site of molecule B, including a three-member stack contributed by dT2, dT3 and Tyr11′ (Supplementary Figure S2C). However, there is no such interactions between molecule A and its partner molecule A′ from the symmetric dimer, which should be the reason that the molecule A of the dimer could bind to only one nucleotide dT5*. In fact, the ssDNA-binding experiments showed that the mutations of His8 and Tyr11 had almost no effect on the formation of oligomeric complex (A1 band). This implies that the interactions from the molecule B′ are not essential for the formation of the HpDprA–dT35 complex (Table 1 and Figure 5E), but may be significant for stability of ssDNA docking onto the target site of HpDprA. Furthermore, a triple mutant HpDprAH8A-Q10A-Y11A (Phe9 is not mutated because only its main-chain is involved in ssDNA binding through two water-mediated hydrogen bonds) was assayed using EMSA techniques (Supplementary Figure S3D) and the results confirmed that these three residues are not required for the formation of stable oligomeric complex.
Figure 4.

The HpDprA(5-217)–ssDNA interface. (A) Schematic view of extensive hydrogen bonds network. Residues of molecule B and its crystallographic symmetric partner, molecule B′, are labeled in green and yellow, respectively. The red dots represent waters. (B) Stereo view of interactions between HpDprA and dT4–dT6. ssDNA-contacting residues are shown as stick representations. The water molecules are represented as sphere models. Dashed lines represent H-bond interactions within typical range (2.5–3.5 Å).
Figure 5.

ssDNA binding and chromatographic analyses of HpDprA and its mutants. (A) Comparison between size-exclusion chromatographies of WT-HpDprA and HpDprAL196E-F205E showing that WT-HpDprA behaves as a dimer, but HpDprAL196E-F205E is monomeric in solution. (B) EMSA results of HpDprAL196E-F205E–dT35 show that the monomers almost lose ssDNA-binding activity. (C) Elution profiles of apo-HpDprA(5-217) (Peak2), apo-dT35 (Peak3) and HpDprA(5-217)–dT35 complexes (Peak1) by size-exclusion chromatography. The elution volume of linear ssDNA deviated from standard globular proteins marker, but one of the complexes fits well with protein marker because of ssDNA wrapping the HpDprA(5-217). (D) SLS analyses of HpDprA(5-217)–dT35 complex and apo-protein, demonstrating that their MWs are 62.25 ± 0.6 kDa and 50.25 ± 1.0 kDa, respectively. (E) Quantitation of different mutations of HpDprA binding to ssDNA. The data points are obtained from densitometric analysis of EMSA results in Supplementary Figure S5B. The oligomeric complex (Band A1) and polymeric complexes (Bands A2, A3 and A4) are regarded as NPCs.
The ssDNA binding of DprA domain is achieved through the extensive interactions between dT4–dT6 and the binding pocket. Firstly, Tyr108, Pro135 and Phe140 constitute one hydrophobic core near the pocket. dT5 is directly contacted through an edge-on stack by Phe140, and the nucleobase group of dT4 is stacked onto the imidazole group of His8′ and the hydrophobic core (Supplementary Figure S2B). Secondly, there is an extensive hydrogen bond network contributed to ligand binding (Figure 4A). The dT4 is sandwiched in the gap of two symmetrical molecules by polar interactions from Arg52 and Pro135 (Figure 4B). dT5 is the most important nucleotide in HpDprA(5-217)–dT35 interactions because of its thymine inserting into the most conserved, positively charged pocket of the molecule B. Four residues Tyr108, Arg143, Asn144 and Gly164 further stabilize it. With its phosphate group fastened by Lys137, dT6 extends into the narrow groove formed by two helices (α6 and α7). In the binding pocket of molecule A, the pyrimidine ring of dT5* inserts into the cavity like that of dT5, but the densities of its sugar and phosphate group were so poor that it was not possible to predict the locations of other nucleobases, which might result from lacking of interactions from symmetric molecule A′. These results demonstrated the intrinsic flexible and dynamic natures of ssDNA on one hand, and the strong interactions between HpDprA and ssDNA on the other hand, since a single base was sufficient to hold ssDNA in place near molecule A.
Conformational changes of the binding pocket and critical residues for binding
Structural comparisons showed some side-chains of interactive residues were re-oriented because of substrate binding. What is more noteworthy is that Arg52, which plays a key role in the hydrogen bond network, showed remarkable conformational change. Its guanidine group pointed outside with a Cβ-Cγ-Cσ-Nσ dihedral of 80° in molecule B, and inward with Cβ-Cγ-Cσ-Nσ dihedral of 40° in molecule A (Supplementary Figure S2E and Figure 3E). In the absence of ssDNA substrate, the long side-chain of Arg52 extends to ‘cover’ the binding pocket. We designated it the closed state (Figure 3C). Once interacting with ssDNA, the side-chain of Arg52 were re-oriented to make the binding pocket open widely enough to accommodate one base of ssDNA (Figure 3B and Supplementary Figure S2F). Only in the ‘opened’ state HpDprA could bind to ssDNA effectively even though only one base was located in the pocket. In search of the effects of these residues on ssDNA binding, different mutants of HpDprA(5-217) were assayed for their Kd-values toward dT35 using MST (Table 1 and Supplementary Figure S5B). The same mutations were subsequently introduced to full-length HpDprA in EMSA experiments, whose results were mostly in accordance with those of HpDprA(5-217) mutations (Figure 5E and Supplementary Figure S4). The Kd-value of HpDprA(5-217)R52E mutant to dT35 markedly decreased to 9.950 ± 0.367 μM, a ∼315-fold reduction. Full-length HpDprAR52E showed the most loss of binding affinities, indicating that the Arg52 is essential for ssDNA binding as a switch of the binding pocket. Actually, preliminary EMSA experiments in which these active residues were mutated to alanines did not show large differences (data not shown). So we adopted a more radical mutation strategy to change the basic or hydrophobic residues to acidic ones. Mutation experiments under this strategy verified the complex formation model. Mutant R52A might lose the role as a switch but still allowed the docking of thymine of ssDNA. However, mutant R52E has drastically reduced substrate binding affinities because of the change of a positively charged side-chain to a negatively charged one repelling the phosphate groups of ssDNA.
Although other active residues lack obvious conformational change, mutation experiments indicated the important roles in ssDNA binding. Firstly, mutants HpDprA(5-217)K137E and HpDprA(5-217)R143E have ∼230-fold and ∼173-fold reductions, respectively, in their ssDNA-binding affinities. Although not a conserved residue, the mutation of positively charged Lys137 into a negatively charged one may exclude the phosphate backbone of ssDNA (dT6) to destabilize docked ssDNA. Mutation of Arg143 may impede seriously its role in guiding one ssDNA base into the binding pocket. Furthermore, any double mutations of basic residues of Arg52, Lys137 and Arg143 led to loss of ssDNA-binding ability, which meant polar interactions are essential for ssDNA-binding stability. Secondly, among four hydrophobic residues, only changes of Phe140 and Tyr108 had the influences on ssDNA-binding affinity, with ∼15 - and ∼10-fold reductions, respectively. The damage of the hydrophobic core resulted in instability of thymine of ssDNA docked onto the binding pocket.
To sum up, we proposed a molecular mechanism of ssDNA docking into the binding pocket of HpDprA as following. When ssDNA is nearby, the phosphate groups form polar interactions with the side-chain of Arg52 and force it to switch to the ‘opened’ state. The side-chain of Arg143, which is held in place by extensive polar interactions, and that of Asn144, orient dT5 into the binding pocket. Phe140 provides a hydrophobic platform for insertion of pyrimidine ring. In addition, dT4, which interacts with the E2 hydrophobic region with its thymine ring, and dT6, which is stuck in the ‘pliers’ formed by α6 and α7, stabilize ssDNA binding together (Figures 3B and 4B).
The stoichiometry and sequence preference of HpDprA dimer interaction with dT35
Quevillon-Cheruel et al. demonstrated that SpDprA dimerization is crucial for the formation of poly-nucleoprotein complexes (26). To study the importance of HpDprA dimerization for ssDNA binding, a double mutant HpDprAL196E-F205E was purified and assayed. Because of the disruption of the hydrophobic core, HpDprAL196E-F205E behaved as a monomer in solution from size-exclusion chromatography (Figure 5A). Interestingly, the same mutations in HpDprA(5-217) would result in the formation of insoluble inclusion bodies. This result indicated that the additional C-terminal domain might be indispensable in the correct folding of HpDprA. We then carried out EMSA assays to compare HpDprAL196E-F205E with the WT HpDprA on ssDNA-binding ability (Figure 5B). Similar to SpDprAH264A-L273R monomer that completely lost its ssDNA-binding activity (26), HpDprAL196E-F205E was extremely weak on ssDNA-binding activity. The result indicated that HpDprA could not rely on a single-binding pocket to bind to ssDNA stably.
Regardless of the full-length or truncated protein, the results of EMSA experiments suggest that with low protein to ssDNA ratio, they tend to form a stable oligomeric complex. The size-exclusion chromatography and SLS experiments were applied to measure the precise MW of this complex. A mixture of the HpDprA(5-217) and dT35 were prepared with excess ssDNA (the molar ratio of protein to dT35 = 1:3) and incubated for 20 min at 25°C. The size-exclusion chromatography showed the complex peak eluted prior to apo-HpDprA(5-217) dimer peak (Figure 5C). The MW of the complex between HpDprA(5-217) and dT35 was measured by SLS as 62.25 kDa (there existed partially dissociated apo-protein from the complex sample after more than 24 h at 4°C), and the control sample of apo-HpDprA(5-217) dimer was measured as 50.25 kDa (Figure 5D), whereas the theoretical MW of dT35 is 10.59 kDa. It is therefore clear that the A1 bands in EMSA experiments were from the 1:1 complex between HpDprA dimer and dT35.
In addition to dT35, dC35, dA35 and dR35 with random sequence (Supplementary Table S2) were assayed for their affinities with full-length HpDprA using EMSA techniques. The binding affinities of HpDprA toward dC35 or dR35 were equivalent to that of dT35 (Supplementary Figure S3A and B), while the affinity to dA35 was very weak (Supplementary Figure S3C). MST experiments determined the Kd of HpDprA(5-217)–dA35 at 1200 ± 35.3 nM, ∼40 times lower than dT35 (Table 1 and Supplementary Figure S5D). The differences between the results of dA35 from these two techniques could be accounted for by the different nature of MST and EMSA: MST measures dissociation constant at dynamic equilibrium, whereas EMSA requires stable nucleocomplexes in order to resist electrophoresis. We concluded that HpDprA can bind to dA35 with lower affinity than dT35, and the dA35–HpDprA complex is not stable enough to survive EMSA. These results demonstrated that there may be a certain preference for HpDprA to bind to different types of ssDNA, but it has no sequence specificity in ssDNA binding, which is also consistent with the biological function of HpDprA as a non-specific ssDNA processing protein.
Chain length requirement of ssDNA for stable interactions with DprA domain of HpDprA
In our complex crystal structure, the majority of the dT35 ssDNA is disordered. We were thus intrigued by the question that whether short six dTs, which is the length of the ssDNA with traceable electron density in the structure, is enough to interact with the DprA domain. Firstly, dT6 was used as the shortest substrate to measure the affinity of HpDprA(5-217) using MST technique. The results showed that these two molecules did not bind. The Kd-values of HpDprA(5-217) toward longer ssDNA substrates dT10, dT15 and dT20 were not detectable, 18.4 ± 0.574 μM and 2.23 ± 0.0523 μM, respectively (Table 1 and Supplementary Figure S5C). The affinities of these ssDNA toward HpDprA increase with their lengths, but still remain significantly lower than that of dT35. A simple modeling of the HpDprA–ssDNA complex structure revealed that minimal length of ssDNA required for interacting with the two binding pockets of the HpDprA dimer is ∼14–16 nt (data not shown). But considering the surface potential of the protein is not optimal for the ssDNA to tightly wrap around and the nucleotides between the two binding pockets are structurally flexible, longer ssDNA chain is probably needed. EMSA assays of HpDprA(5-217) interacting with ssDNAs of different lengths were performed (Supplementary Figure S3H). When the lengths of the ssDNA are 20–35 nt, there existed only one oligomeric complex (A1 band). But with the increase of the length of the ssDNA, two retarded bands appeared with HpDprA(5-217) and dT40 or dT50, and three such bands with DprA(5-217) and dT60. Since the truncated HpDprA, instead of the full-length protein, was used in these experiments, the appearance of the polymeric bands cannot be attributed to the existence of the C-terminal domain. We speculated that HpDprA(5-217) dimer tends to form 1:1 complex with ssDNA at low protein concentrations, while longer ssDNA (>35 nt) can bind more than one HpDprA(5-217) dimer at higher protein concentrations. Then gel-filtration chromatography and SLS analyses of HpDprA(5-217) dimer with dT60 at a molar ratio of 5:1 were performed (Supplementary Figure S3I). The SLS result confirmed that the MW of a stable complex purified using gel-filtration chromatography was 118.7 kDa, demonstrating that the A′2 band was a 2:1 complex of two HpDprA(5-217) with one dT60. Combined with MST results and crystal structure analysis, we conclude that the minimal length of ssDNA bound by DprA domain dimer is more than 17 nt, but longer ssDNA is essential in the formation of stable complexes. These results additionally confirmed that ssDNA-binding ability is stronger as the length of ssDNA increases (<40 nt) but does not change substantially when the length of ssDNA is above 40 nt. In conclusion, ssDNA must reach certain length to stride across the distance between two conserved binding sites of the dimer, and the flexibility of the bound ssDNA may have certain biological signification, such as the facilitating the interactions with RecA.
DISCUSSION
Two motifs to identify DprA domain
The DprA proteins are widely distributed in eubacteria, not only in transformable bacteria but also in bacteria whose transformability was not discovered. As of now, 4658 such sequences from 4181 species have been recorded in the Sanger database (36). A highly conserved 205 residues domain designated as pfam02481/SMF/DprA is shared among these proteins. In this study, we demonstrated that four conserved residues, Arg52, Phe140, Arg143 and Asn144, play important roles in ssDNA binding of HpDprA. Coupling the result with sequence alignments, we here propose that two motifs of DprA family proteins play essential roles in binding to ssDNA. The first motif is ‘G-S/T/A-R’ located in loop (β3–α3) corresponding to residues Gly50–Arg52 in HpDprA, and the second one is ‘F/L/Y-X-X-R-N/D’ located in helix α6 corresponding to residues Phe140–Asn144 in HpDprA (Supplementary Figure S6). Interestingly, there is another motif ‘A-M-X-R-N/D’ in place of ‘F/L/Y-X-X-R-N/D’ in many bacteria containing two homologs DprA proteins (such as E. coli O157:H7 strain Sakai). In the same CL0349 superfamily, one shorter conserved domain (∼133 residues), PF03641 (Lysine_decarbox), adopts similar RF and sequences, which is easily confused with DprA domain. PF03461 owns a discernible motif ‘P-G-G-X-G-T-X-X-E’ and is annotated as lysine decarboxylases (37). Therefore, these two motifs are signatures to identify DprA domain that binds to ssDNA.
DprA proteins may play different roles in NT in different bacterium
Sequence alignments of DNA processing protein A from different bacteria classifications showed that those consisted of three domains are the most frequently observed, but those with two domains are also widely distributed. Some DprA proteins were found to contain a single Pfam02481 domain (such as the DprA protein from Rhodobacter sphaeroides strain ATCC 17025), but there always existed another homolog with more than two domains in the same strain. Therefore, a single DprA domain may not be sufficient for this novel RMP to function in NT.
Those additional domains are highly diversified in their sequences and functions. Both S. pneumoniae and H. pylori, for example, are NT-bacteria, which suggest that their DprAs are active, but distinct mechanisms of regulations of their NT competence status were observed. NT in S. pneumoniae is regulated by a quorum-sensing system in which phosphorylated ComE (ComE-P) plays a key role in activating competence-specific genes (38). Direct interaction between SAM domain of SpDprA and ComE-P has been observed to shut off the competence, demonstrating that additional domains indeed provide DprA proteins with additional functions. In H. pylori, however, natural competence is induced by DNA damage (13) and the HpDprA protein possesses a C-terminal DML1-like domain instead of SAM domain. So the highly species-specific competence regulations correspond to the highly diversified functions of DprA proteins in addition to their DprA domains. GST pull-down and Isothermal Titration Calorimetry (ITC) experiments also revealed that HpDprA does not interact with HpRecA in vitro (unpublished data), which are significantly different from those reported for SpDprA (24,26). Thirdly, our EMSA results showed SpDprA tends to form high MW complexes directly, as reported previously (24) and verified in this study (Supplementary Figure S3E), but HpDprA tends to form oligomeric complex at first, and polymeric complexes were observed with higher protein concentrations. Besides, HpDprA was recently reported to have dsDNA-binding activity (39). We further determined the Kd-value between HpDprA(5-217) and dsDNA30 at 831 ± 8.48 nM, which is moderately weaker than 370 ± 8.82 nM of HpDprA(5-217)–dR30 (Table 1 and Supplementary Figure S5D). Taken these facts together, DprAs from different bacteria may perform species-specific multi-functions.
The oligomeric complex mode of DprA domain of HpDprA with ssDNA
The present study demonstrates several important HpDprA’s ssDNA-binding activities. (i) HpDprA tends to bind to ssDNA without sequence specificity. (ii) Only dimers, but not monomers, of HpDprA could bind ssDNA effectively. (iii) The highly flexible ssDNA must reach enough length to be effectively docked onto two specific binding pockets of DprA domain dimer. (iv) The stable oligomeric complex of DprA domain dimer with ssDNA at a 1:1 molar ratio appears first, and is the basis of polymeric complexes. Taking all results reported above into account, we propose an oligomeric model of how conserved DprA domains bind to ssDNA in which one dimer is wrapped by one ssDNA, with the ssDNA docking onto two conserved, positively charged binding pockets at two monomeric surfaces.
It is mysterious that full-length HpDprA and dT35 form polymeric complexes gradually as protein concentration increases. The property of HpDprA is similar to BsDprA which forms larger complexes spot with increasing protein concentration as revealed by atomic force microscope (25). However, truncated HpDprAs only formed oligomeric complex with dT35. There is no doubt that deletion of C-terminal DML1-like domain caused the difference. Two possible interpretations can account for the roles played by the DML1-like domain. (i) The DML1-like domain could possess a second ssDNA-binding site, but the site is much weaker than main site of DprA domain, because the mutants HpDprAL196E-F205E, which is monomeric, and HpDprAR52E, whose mutation is quite removed from the DML1-like domain, have the weakest ssDNA-binding affinity. (ii) The DML1-like domain is not involved in ssDNA binding, but instead mediates the nucleoprotein complex (NPC) formation through protein–protein interaction.
As more DprA family members are studied, a polymeric mode that multiple DprAs and ssDNAs molecules aggregate densely on a limited area was observed and considered as the universal mode (25,26). Our oligomeric mode might be the initial step of the process that eventually leads to the formation of DprA–ssDNA polymeric complex. More research is needed to illustrate how the oligomeric complexes come into being from initial polymeric complexes, but our studies reported here provide a solid starting point for future research.
The novel ssDNA-binding mode of DprA domain
Recent studies have identified several protein folds that are often involved in the non-specific binding of ssDNA. (i) The oligonucleotide/oligosaccharide binding (OB) fold. A conserved topology of a β-barrel is formed by two β-sheets arranged by five β-strands and is surrounded by extended ssDNA (Supplementary Figure S7A). A large superfamily of proteins, such as single-stranded DNA binding (Ssb) proteins and Cold shock proteins (Csp), belong to this fold (40–42). Some other proteins utilize a variation of the OB fold, in which a flat β-sheet or β-barrel is involved in ssDNA binding. These include plant Whirly protein, DNA damage response B (DdrB), Human transcription cofactor PC4 and Lactococcus lactis YdbC (43–46). (ii) RecA-like fold. Proteins with RecA-like fold are usually enzymes involved in DNA metabolism, such as RecA, Rep helicase and superfamily 1B helicase RecD2 (47–49). These proteins mainly employ several domains cooperatively to form a deep main groove to bind to the phosphate backbone of ssDNA (Supplementary Figure S7B). Apart from these two folds, some ssDNA-binding proteins consist of wholly α-helices to bind to their substrates, such like the HhH domain of Human XPF protein (Supplementary Figure S7C) (50).
The structural and biochemical characterizations on HpDprA we reported here revealed a distinctive ssDNA-binding mode with a RF in which a small binding pocket consisted of highly conserved residues is responsible for the affinities of DprA domain toward ssDNA. A highly conserved arginine residue, which corresponds to Arg52 in HpDprA, may play essential roles in ssDNA binding by regulating the access of the binding pocket with different side-chain conformations. But more interesting questions remain for HpDprA and other members of this superfamily. For example, does HpDprA bind to other ssDNA in a way different to what we observed here with dT35? What are the functions of the C-terminal domain of HpDprA? The fact that most DprA domain-containing proteins also have other domains indicates that these proteins may have very diversified functions in addition to their roles in interacting with ssDNA. This is very evident when we consider the difference among the dsDNA-binding activities of SpDprA, BsDprA and HpDprA. SpDprA and BsDprA have very weak affinities toward dsDNA (24,25), but our experiments indicated that HpDprA has distinctive affinities toward dsDNA. Are these additional functions synergetic to the ssDNA-related functions of the DprA domains, or are they independent of it? The complex structure between HpDprA and ssDNA we report here provides us with a good platform and starting point for further studies into HpDprA and other related proteins.
ACCESSION NUMBERS
PDB accession numbers: 4LJK, 4LJL and 4LJR.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Chinese Ministry of Science and Technology 973 Program [2011CB911100, 2011CB910304]; National Natural Science Foundation of China [31070658]. Funding for open access charge: Institute of Biophysics, Chinese Academy of Sciences.
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Quanming Zou (Third Military Medical University) and Deqiang Wang (Chongqing Medical University) for providing us with the genomes of H. pylori and S. pneumonia, respectively. We are grateful to Li Guo (Institute of Microbiology, Chinese Academy of Sciences) and Zhouliang Yu (Institute of Biophysics, Chinese Academy of Sciences) for advice on the electrophoretic mobility-shift assay experiments. We also thank Liang Zhou (Quantum Design China & NanoTemper Technologies GmbH) for providing data analyses of the Microscale Thermophoresis assay. We thank Xiaoxia Yu (Institute of Biophysics, Chinese Academy of Sciences) for assistance in static light scattering analysis. We thank the staff at the beamline 17 A of the Photon Factory (KEK, Tsukuba, Japan) and at the beamline 17U of the Shanghai Synchrotron Radiation Facility (Shanghai, People’s Republic of China) for technical assistance during data collection.
REFERENCES
- 1.Lorenz MG, Wackernagel W. Bacterial gene transfer by natural genetic transformation in the environment. Microbiol. Rev. 1994;58:563–602. doi: 10.1128/mr.58.3.563-602.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Johnsborg O, Eldholm V, Havarstein LS. Natural genetic transformation: prevalence, mechanisms and function. Res. Microbiol. 2007;158:767–778. doi: 10.1016/j.resmic.2007.09.004. [DOI] [PubMed] [Google Scholar]
- 3.Claverys JP, Martin B. Bacterial “competence” genes: signatures of active transformation, or only remnants? Trends Microbiol. 2003;11:161–165. doi: 10.1016/s0966-842x(03)00064-7. [DOI] [PubMed] [Google Scholar]
- 4.Gilbreath JJ, Cody WL, Merrell DS, Hendrixson DR. Change is good: variations in common biological mechanisms in the epsilonproteobacterial genera Campylobacter and Helicobacter. Microbiol. Mol. Biol. Rev. 2011;75:84–132. doi: 10.1128/MMBR.00035-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kidane D, Ayora S, Sweasy JB, Graumann PL, Alonso JC. The cell pole: the site of cross talk between the DNA uptake and genetic recombination machinery. Crit. Rev. Biochem. Mol. Biol. 2012;47:531–555. doi: 10.3109/10409238.2012.729562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen I, Christie PJ, Dubnau D. The ins and outs of DNA transfer in bacteria. Science. 2005;310:1456–1460. doi: 10.1126/science.1114021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen I, Dubnau D. DNA uptake during bacterial transformation. Nat. Rev. Microbiol. 2004;2:241–249. doi: 10.1038/nrmicro844. [DOI] [PubMed] [Google Scholar]
- 8.Wang L, Sheng D, Han W, Huang B, Zhu S, Ni J, Li J, Shen Y. Sulfolobus tokodaii RadA paralog, stRadC2, is involved in DNA recombination via interaction with RadA and Hjc. Sci. China Life Sci. 2012;55:261–267. doi: 10.1007/s11427-012-4292-0. [DOI] [PubMed] [Google Scholar]
- 9.Baltrus DA, Guillemin K, Phillips PC. Natural transformation increases the rate of adaptation in the human pathogen Helicobacter pylori. Evolution. 2008;62:39–49. doi: 10.1111/j.1558-5646.2007.00271.x. [DOI] [PubMed] [Google Scholar]
- 10.Hofreuter D, Odenbreit S, Haas R. Natural transformation competence in Helicobacter pylori is mediated by the basic components of a type IV secretion system. Mol. Microbiol. 2001;41:379–391. doi: 10.1046/j.1365-2958.2001.02502.x. [DOI] [PubMed] [Google Scholar]
- 11.Israel DA, Lou AS, Blaser MJ. Characteristics of Helicobacter pylori natural transformation. FEMS Microbiol. Lett. 2000;186:275–280. doi: 10.1111/j.1574-6968.2000.tb09117.x. [DOI] [PubMed] [Google Scholar]
- 12.Baltrus DA, Guillemin K. Multiple phases of competence occur during the Helicobacter pylori growth cycle. FEMS Microbiol. Lett. 2006;255:148–155. doi: 10.1111/j.1574-6968.2005.00066.x. [DOI] [PubMed] [Google Scholar]
- 13.Dorer MS, Fero J, Salama NR. DNA damage triggers genetic exchange in Helicobacter pylori. PLoS Pathog. 2010;6:e1001026. doi: 10.1371/journal.ppat.1001026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Smeets LC, Kusters JG. Natural transformation in Helicobacter pylori: DNA transport in an unexpected way. Trends Microbiol. 2002;10:159–162; discussion 162. doi: 10.1016/s0966-842x(02)02314-4. [DOI] [PubMed] [Google Scholar]
- 15.Stingl K, Muller S, Scheidgen-Kleyboldt G, Clausen M, Maier B. Composite system mediates two-step DNA uptake into Helicobacter pylori. Proc. Natl Acad. Sci. USA. 2010;107:1184–1189. doi: 10.1073/pnas.0909955107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yeh YC, Lin TL, Chang KC, Wang JT. Characterization of a ComE3 homologue essential for DNA transformation in Helicobacter pylori. Infect. Immun. 2003;71:5427–5431. doi: 10.1128/IAI.71.9.5427-5431.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Smeets LC, Bijlsma JJ, Boomkens SY, Vandenbroucke-Grauls CM, Kusters JG. comH, a novel gene essential for natural transformation of Helicobacter pylori. J. Bacteriol. 2000;182:3948–3954. doi: 10.1128/jb.182.14.3948-3954.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.O'Rourke EJ, Pinto AV, Petroni EA, Tolmasky ME, Ielpi L. Evidence for the active role of a novel nuclease from Helicobacter pylori in the horizontal transfer of genetic information. J. Bacteriol. 2004;186:2586–2593. doi: 10.1128/JB.186.9.2586-2593.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ando T, Israel DA, Kusugami K, Blaser MJ. HP0333, a member of the DprA family, is involved in natural transformation in Helicobacter pylori. J. Bacteriol. 1999;181:5572–5580. doi: 10.1128/jb.181.18.5572-5580.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Smeets LC, Bijlsma JJ, Kuipers EJ, Vandenbroucke-Grauls CM, Kusters JG. The DprA gene is required for natural transformation of Helicobacter pylori. FEMS Immunol. Med. Microbiol. 2000;27:99–102. doi: 10.1111/j.1574-695X.2000.tb01418.x. [DOI] [PubMed] [Google Scholar]
- 21.Fischer W, Haas R. The RecA protein of Helicobacter pylori requires a posttranslational modification for full activity. J. Bacteriol. 2004;186:777–784. doi: 10.1128/JB.186.3.777-784.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Marsin S, Mathieu A, Kortulewski T, Guerois R, Radicella JP. Unveiling novel RecO distant orthologues involved in homologous recombination. PLoS Genet. 2008;4:e1000146. doi: 10.1371/journal.pgen.1000146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Marsin S, Lopes A, Mathieu A, Dizet E, Orillard E, Guerois R, Radicella JP. Genetic dissection of Helicobacter pylori AddAB role in homologous recombination. FEMS Microbiol. Lett. 2010;311:44–50. doi: 10.1111/j.1574-6968.2010.02077.x. [DOI] [PubMed] [Google Scholar]
- 24.Mortier-Barriere I, Velten M, Dupaigne P, Mirouze N, Pietrement O, McGovern S, Fichant G, Martin B, Noirot P, Le Cam E, et al. A key presynaptic role in transformation for a widespread bacterial protein: DprA conveys incoming ssDNA to RecA. Cell. 2007;130:824–836. doi: 10.1016/j.cell.2007.07.038. [DOI] [PubMed] [Google Scholar]
- 25.Yadav T, Carrasco B, Hejna J, Suzuki Y, Takeyasu K, Alonso JC. Bacillus subtilis DprA recruits RecA onto single-stranded DNA and mediates annealing of complementary strands coated by SsbB and SsbA. J. Biol. Chem. 2013;288:22437–22450. doi: 10.1074/jbc.M113.478347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Quevillon-Cheruel S, Campo N, Mirouze N, Mortier-Barriere I, Brooks MA, Boudes M, Durand D, Soulet AL, Lisboa J, Noirot P, et al. Structure-function analysis of pneumococcal DprA protein reveals that dimerization is crucial for loading RecA recombinase onto DNA during transformation. Proc. Natl Acad. Sci. USA. 2012;109:E2466–E2475. doi: 10.1073/pnas.1205638109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Collaborative Computational Project,N. The CCP4 suite: programs for protein crystallography. Acta Crystallogr. D Biol. Crystallogr. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 28.Adams PD, Grosse-Kunstleve RW, Hung LW, Ioerger TR, McCoy AJ, Moriarty NW, Read RJ, Sacchettini JC, Sauter NK, Terwilliger TC. PHENIX: building new software for automated crystallographic structure determination. Acta Crystallogr. D Biol. Crystallogr. 2002;58:1948–1954. doi: 10.1107/s0907444902016657. [DOI] [PubMed] [Google Scholar]
- 29.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 30.Davis IW, Leaver-Fay A, Chen VB, Block JN, Kapral GJ, Wang X, Murray LW, Arendall WB, III, Snoeyink J, Richardson JS, et al. MolProbity: all-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res. 2007;35:W375–W383. doi: 10.1093/nar/gkm216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, et al. Clustal W and Clustal X version 2.0. Bioinformatics. 2007;23:2947–2948. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- 32.Gouet P, Courcelle E, Stuart DI, Metoz F. ESPript: analysis of multiple sequence alignments in PostScript. Bioinformatics. 1999;15:305–308. doi: 10.1093/bioinformatics/15.4.305. [DOI] [PubMed] [Google Scholar]
- 33.Jerabek-Willemsen M, Wienken CJ, Braun D, Baaske P, Duhr S. Molecular interaction studies using microscale thermophoresis. Assay Drug Dev. Technol. 2011;9:342–353. doi: 10.1089/adt.2011.0380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ha SC, Lokanath NK, Van Quyen D, Wu CA, Lowenhaupt K, Rich A, Kim YG, Kim KK. A poxvirus protein forms a complex with left-handed Z-DNA: crystal structure of a Yatapoxvirus Zalpha bound to DNA. Proc. Natl Acad. Sci. USA. 2004;101:14367–14372. doi: 10.1073/pnas.0405586101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Roy A, Kucukural A, Zhang Y. I-TASSER: a unified platform for automated protein structure and function prediction. Nat. Protoc. 2010;5:725–738. doi: 10.1038/nprot.2010.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Punta M, Coggill PC, Eberhardt RY, Mistry J, Tate J, Boursnell C, Pang N, Forslund K, Ceric G, Clements J, et al. The Pfam protein families database. Nucleic Acids Res. 2012;40:D290–D301. doi: 10.1093/nar/gkr1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kukimoto-Niino M, Murayama K, Kato-Murayama M, Idaka M, Bessho Y, Tatsuguchi A, Ushikoshi-Nakayama R, Terada T, Kuramitsu S, Shirouzu M, et al. Crystal structures of possible lysine decarboxylases from Thermus thermophilus HB8. Protein Sci. 2004;13:3038–3042. doi: 10.1110/ps.041012404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mirouze N, Berge MA, Soulet AL, Mortier-Barriere I, Quentin Y, Fichant G, Granadel C, Noirot-Gros MF, Noirot P, Polard P, et al. Direct involvement of DprA, the transformation-dedicated RecA loader, in the shut-off of pneumococcal competence. Proc. Natl Acad. Sci. USA. 2013;110:E1035–E1044. doi: 10.1073/pnas.1219868110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dwivedi GR, Sharma E, Rao DN. Helicobacter pylori DprA alleviates restriction barrier for incoming DNA. Nucleic Acids Res. 2013;41:3274–3288. doi: 10.1093/nar/gkt024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chan KW, Lee YJ, Wang CH, Huang H, Sun YJ. Single-stranded DNA-binding protein complex from Helicobacter pylori suggests an ssDNA-binding surface. J. Mol. Biol. 2009;388:508–519. doi: 10.1016/j.jmb.2009.03.022. [DOI] [PubMed] [Google Scholar]
- 41.Max KE, Zeeb M, Bienert R, Balbach J, Heinemann U. Common mode of DNA binding to cold shock domains. Crystal structure of hexathymidine bound to the domain-swapped form of a major cold shock protein from Bacillus caldolyticus. FEBS J. 2007;274:1265–1279. doi: 10.1111/j.1742-4658.2007.05672.x. [DOI] [PubMed] [Google Scholar]
- 42.Antony E, Weiland EA, Korolev S, Lohman TM. Plasmodium falciparum Ssb tetramer wraps single-stranded DNA with similar topology but opposite polarity to E. coli Ssb. J. Mol. Biol. 2012;420:269–283. doi: 10.1016/j.jmb.2012.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cappadocia L, Marechal A, Parent JS, Lepage E, Sygusch J, Brisson N. Crystal structures of DNA-Whirly complexes and their role in Arabidopsis organelle genome repair. Plant Cell. 2010;22:1849–1867. doi: 10.1105/tpc.109.071399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sugiman-Marangos S, Junop MS. The structure of DdrB from Deinococcus: a new fold for single-stranded DNA binding proteins. Nucleic Acids Res. 2010;38:3432–3440. doi: 10.1093/nar/gkq036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Werten S, Moras D. A global transcription cofactor bound to juxtaposed strands of unwound DNA. Nat. Struct. Mol. Biol. 2006;13:181–182. doi: 10.1038/nsmb1044. [DOI] [PubMed] [Google Scholar]
- 46.Rossi P, Barbieri CM, Aramini JM, Bini E, Lee HW, Janjua H, Xiao R, Acton TB, Montelione GT. Structures of apo- and ssDNA-bound YdbC from Lactococcus lactis uncover the function of protein domain family DUF2128 and expand the single-stranded DNA-binding domain proteome. Nucleic Acids Res. 2013;41:2756–2768. doi: 10.1093/nar/gks1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chen Z, Yang H, Pavletich NP. Mechanism of homologous recombination from the RecA-ssDNA/dsDNA structures. Nature. 2008;453:489–484. doi: 10.1038/nature06971. [DOI] [PubMed] [Google Scholar]
- 48.Korolev S, Hsieh J, Gauss GH, Lohman TM, Waksman G. Major domain swiveling revealed by the crystal structures of complexes of E. coli Rep helicase bound to single-stranded DNA and ADP. Cell. 1997;90:635–647. doi: 10.1016/s0092-8674(00)80525-5. [DOI] [PubMed] [Google Scholar]
- 49.Saikrishnan K, Powell B, Cook NJ, Webb MR, Wigley DB. Mechanistic basis of 5'-3' translocation in SF1B helicases. Cell. 2009;137:849–859. doi: 10.1016/j.cell.2009.03.036. [DOI] [PubMed] [Google Scholar]
- 50.Das D, Folkers GE, van Dijk M, Jaspers NG, Hoeijmakers JH, Kaptein R, Boelens R. The structure of the XPF-ssDNA complex underscores the distinct roles of the XPF and ERCC1 helix- hairpin-helix domains in ss/ds DNA recognition. Structure. 2012;20:667–675. doi: 10.1016/j.str.2012.02.009. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.