

Abstract
Antisense-mediated exon skipping is currently in clinical development for Duchenne muscular dystrophy (DMD) to amend the consequences of the underlying genetic defect and restore dystrophin expression. Due to turnover of compound, transcript, and protein, chronic treatment with effector molecules (antisense oligonucleotides) will be required. To investigate the dynamics and persistence of antisense 2′-O-methyl phosphorothioate oligonucleotides, exon skipping, and dystrophin expression after dosing was concluded, mdx mice were treated subcutaneously for 8 weeks with 100 mg/kg oligonucleotides twice weekly. Thereafter, mice were sacrificed at different time points after the final injection (36 hours–24 weeks). Oligonucleotide half-life was longer in heart (~65 days) compared with that in skeletal muscle, liver, and kidney (~35 days). Exon skipping half-lives varied between 33 and 53 days, whereas dystrophin protein showed a long half-life (>100 days). Oligonucleotide and exon-skipping levels peaked in the first week and declined thereafter. By contrast, dystrophin expression peaked after 3–8 weeks and then slowly declined, remaining detectable after 24 weeks. Concordance between levels of oligonucleotides, exon skipping, and proteins was observed, except in heart, wherein high oligonucleotide levels but low exon skipping and dystrophin expression were seen. Overall, these results enhance our understanding of the pharmacokinetics and pharmacodynamics of 2′-O-methyl phosphorothioate oligos used for the treatment of DMD.
Introduction
Duchenne muscular dystrophy (DMD) is the most prevalent form of inherited muscular dystrophies, affecting around 1 in 5,000 newborn boys.1,2 Patients display severe progressive muscular weakness due to the absence of the dystrophin protein, which functions as mechanical stabilizer during muscle contraction. In the absence of dystrophin, the muscle fibers are easily damaged and are gradually replaced by fibrotic and adipose tissues. First symptoms generally become apparent at 2–3 years of age, after which, pathology develops rapidly. Patients often die before the age of 30 years due to respiratory and/or cardiac failure.3
In humans, lack of dystrophin is generally caused by out-of-frame deletions or small mutations that introduce premature stop codons in the DMD gene.4 Several animal models are available for DMD, among which, the mdx mouse (C57Bl/10ScSn-DMDmdx/J)—which does not express dystrophin due to a premature stop codon in exon 23—is the most widely used.5 However, the lack of dystrophin is less disastrous in mdx mice than in humans. These mice have a nearly normal life expectancy, only slightly impaired muscle function and a better muscle quality compared with DMD patients.6 Nevertheless, this mouse model displays several features of the DMD pathology, i.e., muscle degeneration and leaky fibers due to the absence of dystrophin, and is very useful for preclinical studies.7
During the past several years, a lot of progress has been made toward the development of antisense oligonucleotide (AON)–mediated exon skipping as a potential therapy targeting the underlying genetic defect of DMD. This aims to restore the reading frame or bypass a small mutation by skipping one or more exons. Thereby, translation can continue and a largely functional protein can be formed, as is found in the related, but much milder, Becker muscular dystrophy. After obtaining proof of principle in vitro in cultured cells and in vivo in animal models, clinical trials for AONs with two different backbone chemistries, 2′-O-methyl phosphorothioate RNA (2OMePS) and phosphorodiamidate morpholino oligomers (PMO), are currently ongoing.8 Systemic (subcutaneous, s.c.) treatment with drisapersen (2OMePS AON, targeting exon 51) for 5 weeks induced dystrophin expression in 10 of 12 patients by up to 15.6% of levels found in healthy persons.9 Treatment with eteplirsen (PMO AON, targeting exon 51) by intravenous (i.v.) infusion resulted in dystrophin restoration in 7 of 19 patients at highly variable percentages up to 18% of control levels.10 Placebo-controlled phase 2 and 3 trials have been conducted for drisapersen. Encouraging results were reported for the phase 2 trials, suggesting improved walking distance in 6 minutes for treated patients compared with placebo-administered patients after 24 and 48 weeks of treatment. However, it was recently reported that the primary outcome measure (distance walked in 6 minutes) in the phase 3 trial,11 although slightly improved in the treated patients, did not differ significantly from the placebo-treated patients. More detailed analysis is pending. In addition to drisapersen (targeting exon 51), AONs targeting other exons, i.e., exons 44, 45, and 53, are under clinical evaluation.
Because RNA-mediated therapies are subject to clearance and turnover of AONs and the dystrophin transcripts and proteins, repeated, lifelong injections will be required to maintain therapeutic effects. Therefore, insight into the pharmacokinetic (PK) and pharmacodynamic properties of these compounds is essential to determine how long the effects persist and to assess dosing frequencies and regimens. Previous studies have revealed a plasma half-life for 2OMePS AONs of around 4 weeks in patients9 and a tissue half-life of 2–6 weeks in mdx mouse muscle.12 It has previously been observed that uptake of AONs is better in dystrophic muscle than in healthy muscle, probably due to the dystrophic “leaky” nature of the muscle fibers. Both s.c. and i.v. routes of administration resulted in muscle uptake and exon skipping, but i.v. injections led to much higher AON levels in liver and kidneys, although AON levels in muscle were comparable with those after s.c. treatment.12 Furthermore, for 2OMePS AONs, exon-skipping and dystrophin protein levels vary among different muscle tissues. Generally, exon-skipping levels in heart are lower, whereas the levels are comparable in limb muscle and diaphragm.12,13,14,15,16,17 Increase in exon skipping and accumulation of dystrophin protein were seen up to 12 weeks of treatment, and long-term treatment for 6 months has been shown to be feasible.13,15 Comparison of various maintenance regimens revealed that 8 weeks after the final dose, the decline in AON levels was faster than the decline in exon-skipping and protein levels, indicating differences in turnover.17 However, the relation between clearance and turnover of the compound, the induced skipped transcript, and restored dystrophin protein and long-term analysis of the persistence of different effects after s.c. treatment have not been studied. In order to expand our understanding of the PK/pharmacodynamic relationship of 2OMePS AONs, mdx mice, in which the mutation can be bypassed by skipping exon 23, were dosed with 100 mg/kg twice weekly for 8 weeks via the s.c. route, after which, tissues were harvested at different time points for the analysis of 23AON, exon-skipping, and protein levels.
Results
Dynamics of 23AON levels in plasma, muscle, and organs
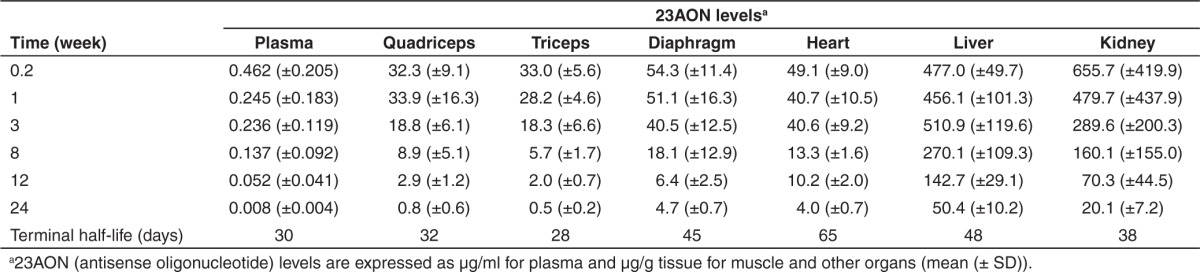
To investigate the dynamics of the effects induced by treatment with 2OMePS AONs, mice were treated subcutaneously with 100 mg 23AON/kg body weight twice weekly for 8 weeks. Six mice per group were sacrificed at different time points after the final injection, ranging from 36 hours up to 24 weeks. To assess the uptake, turnover, and clearance of AON in plasma and in different tissues, 23AON levels were determined with an AON-specific hybridization-ligation assay, and average results were calculated (Figure 1; Table 1). For all tissues, this revealed a decline in AON levels after the final injection, and 23AON was still detectable in muscle, liver, and kidney after 24 weeks (on average, around 5–10% of the 23AON levels at 36 hours). Terminal half-lives ranged from 28 days in triceps to 65 days in heart (Table 1). The half-life in heart was much longer than that in the other tissues. Although the terminal half-life in diaphragm was comparable with that in liver and kidney, it was slightly shorter in limb muscles (quadriceps and triceps) and comparable with that found in plasma. As observed previously,12 AON levels were higher in liver and kidney than in muscle. When comparing the different muscles with each other, 23AON levels were higher in diaphragm and heart than in limb muscles (P < 0.05). No differences were observed in 23AON levels when comparing liver and kidney. Oligonucleotide levels in plasma decreased rapidly after the final dose, and by 24 weeks, 23AON was barely detectable, with levels in only two out of six mice reaching the limit of detection.
Figure 1.

23AON levels in plasma, muscle, and other organs over time. A hybridization-ligation assay was used to determine 23AON levels in plasma, muscle, and organs at different time points ranging from 36 hours up to 24 weeks after the final injection. Data are represented on a logarithmic scale. Each dot represents an individual measurement, and lines are fitted through the averages for each tissue. AON, antisense oligonucleotide.
Table 1. Average AON levels at each time point per tissue and terminal half-life for each tissue.
Dynamics of exon-skipping levels in muscle
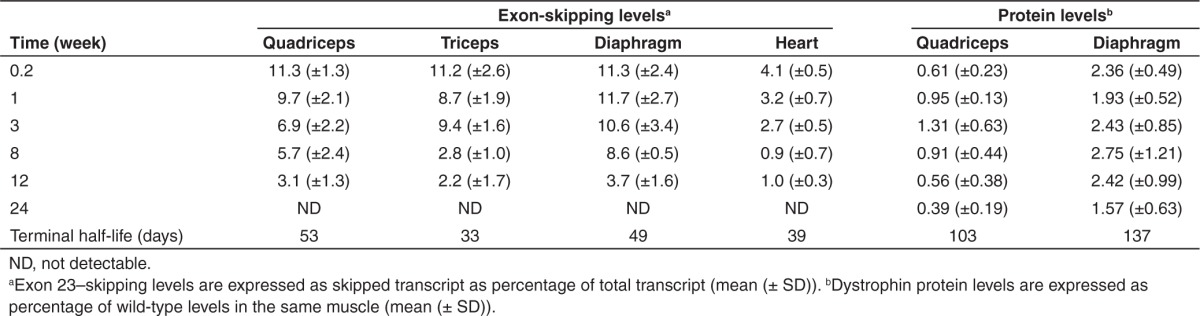
The exon 23–skipping levels were determined in several limb muscles, diaphragm, and heart (Figure 2; Table 2). Exon-skipping levels were highest at the early time points (36 hours and 1 week after final AON injection) and declined thereafter. Exon-skipping levels were significantly lower in heart compared with those in skeletal muscle (P < 0.01). After 24 weeks, skipping was undetectable in all samples. No large differences in rate of decline between individual muscles were observed, considering the interindividual variations within a tissue and the variations in half-life between 33 and 53 days.
Figure 2.

Exon-skipping levels in different muscles over time. (a) Representative example of an reverse transcription–polymerase chain reaction (RT–PCR) analysis for the quadriceps. Unskipped product consists of 334 base pairs and exon 23 skipping results in a 121-base pair product. “PCR ctrls” indicates PCR controls, M is marker. (b) Quantification of exon 23–skipping levels by RT–PCR analysis at several time points after the final 23AON injection. Data are represented as mean ± SD. AON, antisense oligonucleotide.
Table 2. Average exon-skipping and protein levels at each time point per muscle and terminal half-life for each muscle.
Dynamics of dystrophin protein expression
Restoration of dystrophin protein expression after AON treatment was detectable at each time point in the quadriceps and diaphragm (Figure 3; Table 2). No dystrophin was observed for untreated mdx mice (data not shown). In treated muscles, dystrophin levels were initially low, peaked somewhere between 3 and 8 weeks after the final injection for both diaphragm and quadriceps, and then slowly declined. Levels in diaphragm were and remained significantly (P < 0.01) higher than those in quadriceps and also were slightly more stable, with half-lives of ~100 and 130 days, respectively. In heart, no dystrophin protein levels >0.5% could be detected (data not shown).
Figure 3.

Dystrophin protein levels in muscle over time. (a) Representative examples of western blot analyses for the diaphragm showing low levels of dystrophin protein (top) at all analyzed time points. As loading control, α-actinin (bottom) was used. (b) Quantification of dystrophin protein levels, detected by western blot analysis. Data are represented as mean ± SD for each tissue.
Correlation among 23AON levels, exon skipping, and protein expression
For the majority of muscles analyzed, a correlation among 23AON levels, exon skipping, and protein expression was observed. The highest 23AON levels were observed in the diaphragm, resulting in highest exon skipping and dystrophin protein expression. Lower 23AON levels were observed in quadriceps and triceps, resulting in exon-skipping levels that were, at most time points analyzed, slightly lower than or comparable with exon-skipping levels in the diaphragm. Furthermore, the level of dystrophin protein expression in quadriceps was lower than the corresponding value observed in diaphragm. By contrast, this correlation was not observed in heart, wherein 23AON levels were relatively high (comparable with diaphragm), while exon-skipping levels were significantly lower than those in skeletal muscle; moreover, dystrophin protein expression was also very low.
When comparing patterns of the different parameters, both 23AON and exon-skipping levels peaked shortly after treatment, within the first week, and thereafter showed a similar declining pattern, whereas the profile of dystrophin protein expression was shifted, peaking a few weeks after treatment and displaying a more prolonged effect (Figure 4).
Figure 4.

Comparison of time effects at different levels. Comparison of time course of 23AON levels, exon skipping, and protein expression after the final injection for (a) quadriceps and (b) diaphragm. Data are represented as percentages of levels observed at time point t = 36 hours (set at 100%) for 23AON, exon-skipping, and protein levels separately. Error bars represent the SD. AON, antisense oligonucleotide.
Discussion
AON-mediated exon skipping is currently in advanced stages of clinical development as a potential therapy for DMD aiming to partly correct consequences of the genetic defect. However, due to clearance of AONs, skipped transcripts, and restored dystrophin protein, chronic treatment will be required. One way to try and prolong the effect is to deliver antisense sequences using (viral) vectors in order to achieve a stabler expression of the AONs. Adenoassociated virus–expressing modified small nuclear ribonucleoprotein particles, i.e., U1 and U7 small nuclear ribonucleoprotein particles, in which the small nuclear RNA is replaced by the desired AON sequence, have shown to induce long-term dystrophin rescue in the mdx mouse.18 This approach has also been proved to result in long-term cardiac expression of dystrophin in the Golden retriever muscular dystrophy dog,19 harboring a deletion of exon 7, requiring skipping of exons 6 and 8 to restore the reading frame.20 Drawbacks of using adenoassociated viruses as vectors are toxicity concerns and the possibility of an immune response against the viral vector, resulting in the loss of transduced fibers.21 A recent 5-year follow-up study in Golden retriever muscular dystrophy dogs did not show immune rejection; however, after 5 years, disappearance of dystrophin protein expression was observed, probably due to instability of the newly formed protein.22 The same was observed in a study in mdx mice, wherein it was shown also shown that adenoassociated virus vectors and transgene expression were lost quicker from dystrophic muscle than from healthy muscle.23 Therefore, repetition of treatment would still be required, which is not possible due to immunization against the adenoassociated virus after the first injection. This makes it unlikely that this approach will be clinically applicable in the near future. Thus, the AON approach is more viable in the short term, which involves repeated injections, and knowledge about the PK and pharmacodynamic effects is, therefore, valuable. The detailed longitudinal analysis of different muscles, kidney, and liver is ethically and practically challenging in humans but more straightforward in mice. In this study, the clearance/turnover of AON, transcript, and protein over time in mdx mice after treatment with 2OMePS AON, one of the two background chemistries mainly used in clinical trials, was studied.
First of all, the decline in 23AON levels over time was studied in plasma, muscle, and other organs following s.c. injections. After injection, AON levels in plasma decline rapidly due to uptake by organs and because of clearance, which occurs primarily through the kidney. For 2OMePS AONs, this is partly modulated by the plasma protein binding properties of the phosphorothioate backbone of the AONs, but when high doses are injected exceeding the binding capacity, unbound AONs will be cleared rapidly.24,25 The first time point analyzed was 36 hours after the final injection, the time point when previous studies indicated that the majority of the plasma clearance has already occurred.17 The mean half-life determined for 23AONs in mice (30 days) was comparable with that observed for 51AON in patients (29 days).9 When comparing different muscle types, higher 23AON levels in diaphragm and heart were observed, whereas the longest terminal half-life was found in heart. This was probably due to reduced AON turnover because the turnover rate of cardiac muscle cells is much slower than that of skeletal muscle fibers (especially dystrophic skeletal muscle fibers). This is in line with previous observations of a longer half-life of AONs in heart compared with that in skeletal muscle. In the previous study, a markedly shorter half-life in triceps (~10 days) was observed compared with that in quadriceps (~33 days), whereas in the current study, half-lives are comparable (~30 days). Furthermore, the currently calculated half-life in heart is longer (65 days versus 46 days).12 These discrepancies are probably due to the fact that in the previous study, only four time points up to 14 days after the final injection were measured, consisting of two mice per time point, making the estimations less accurate. Furthermore, in the previous study, i.v. injections were used, which show higher levels of AON uptake by the kidney and liver,12 which can also have contributed to the observed differences. As expected, AON levels in liver and kidney were much higher than those in muscle. It has been noticed before that for 2OMePS AONs, levels are similar in liver and kidney,14 and here it is shown that the decline pattern is comparable too. Unfortunately, when comparing the terminal half-lives in the nontarget organs (liver and kidney) with those in the targeted organ (muscle), half-lives were slightly shorter in limb muscles compared with those in liver and kidney. A longer muscle half-life would have been advantageous for planning off-treatment periods, e.g., to prevent accumulation of compounds in liver and kidney. Nevertheless, the absolute reduction of AON levels in liver and kidney is greater than that in muscles.
Second, exon-skipping levels were determined in several skeletal muscles and the heart. Here, highest levels were also observed shortly after the final injection (within the first week), whereupon they declined in a similar pattern as the 23AON levels. For diaphragm, the higher 23AON levels were also accompanied by high exon-skipping levels, whereas by contrast, this correlation was not observed in heart. wherein, although 23AON levels were relatively high (comparable with diaphragm), exon-skipping levels were significantly lower than those in skeletal muscle.
Third, dystrophin protein levels were determined. These showed a delayed pattern, peaking a few weeks later, compared with AON and exon-skipping levels. Importantly, although levels were low in general, dystrophin protein expression was observed up to 24 weeks. Wu et al. determined the half-life of the dystrophin protein in mdx mice after a single i.v. injection with peptide-conjugated PMOs (pPMOs) targeting exon 23. This resulted in a protein half-life of ~2 months for skeletal muscles, and levels had dropped to around 10% after 5 months.26 The half-lives measured in our experiments were a bit longer (3–4 months), resulting in a smaller decrease in dystrophin levels; 30–50% of initial levels were still observed after ~5 months (levels at 24 weeks compared with peak levels) in both muscles. A possible explanation is the difference in experimental setup and backbone chemistry of the AONs between both studies. In this study, 2OMePS AONs were used, whereas the study of Wu et al. used pPMOs. These two chemistries probably have a different biodistribution pattern, as was also seen between 2OMePS AONs and naked PMOs.14 Because AON PKs were not measured in the study of Wu et al., it is not known how much AON remained in the muscle to induce further exon skipping and dystrophin production, resulting in differences over time. Furthermore, Wu et al. only used a single AON injection, whereas mice were treated for a longer period in our study. A single injection is more likely to give variation in AON levels between mice, which influences the preciseness of the half-life estimations. Therefore, both results are not directly comparable. Unfortunately, dystrophin protein was barely detectable in heart. This was also seen in a study using PMO AONs, wherein dystrophin expression could be detected in all analyzed muscles, except for the heart, after 7 weeks of treatment.27 Probably, overall, higher skipping levels and/or longer treatment times are needed for protein restoration. This is underlined by the fact that after 6 months of weekly treatment with 200 mg/kg, dystrophin was readily detectable in heart.15
Overall, there was a good correlation between AON effects at different levels. In diaphragm, the higher 23AON levels were accompanied by higher exon-skipping levels and protein expression. Only in heart, the higher 23AON levels did not result in higher exon-skipping and protein levels. This can be explained by inherent differences between cardiac and skeletal muscle. As mentioned before, skeletal muscles become permeable in the absence of dystrophin, facilitating the uptake of AONs. The heart is built up of individual cardiomyocytes, which do not display this leakiness, thereby making it harder for AONs to get inside.12,28 The assay used for determination of 23AON levels does not discriminate between AON inside the heart as whole organ and AON that is actually inside the cardiomyocytes. Therefore, the presence of higher 23AON levels, but lower exon-skipping levels, suggests that in heart, the majority of AON is probably located in the interstitium and therefore ineffective. This might also be the reason why, although low 23AON levels were still detectable after 24 weeks, no exon skipping could be observed at this time point. Other possibilities are that this is due to differences in sensitivity of the assay used for determination of AON levels versus those for determining exon-skipping levels or that the low AON levels are insufficient to induce exon skipping. Expression percentages for dystrophin protein are much lower than those for exon skipping. This is usually seen for both 2OMePS15,17 and PMO/pPMO AONs,14,26,29,30 and this discrepancy can be explained by the fact that exon-skipping levels are compared with the total transcript levels in the mdx mouse, whereas dystrophin protein levels are compared with wild-type dystrophin levels. It has been reported that compared with wild-type dystrophin transcripts, a 5′-to-3′ imbalance exists for mdx dystrophin transcripts. This means that not all mdx dystrophin transcripts are complete and cannot be translated into dystrophin protein.31
In conclusion, 23AON and exon-skipping levels show a similar decline pattern over time after the final injection. Exon skipping is not detectable after 6 months, but its effects on dystrophin protein restoration remain quite stable. After treatment, AONs are taken up by muscles and other organs, partly leading to immediate excretion without inducing further effects and partly leading to exon skipping inside the muscle fibers. Thereafter, new dystrophin protein can be formed, which displays a long half-life. The differences in rates of turnover of the compound itself, RNA, and the protein influence these effects, in addition to differences in composition and amount of degeneration and regeneration between different muscle groups. Our results provided further insight into how these processes interact and are thus useful for studying the long-term effects of AON treatment.
Materials and Methods
All experiments were approved by the local ethical committee for animal experiments of the Leiden University Medical Center. Mice were housed in individually ventilated cages in the animal facility of the Leiden University Medical Center and received food and drink ad libitum. Moreover, the mdx mice for the study (C57Bl/10ScSn-DMDmdx/J) were obtained from our own breeding facility.
Treatment of mdx mice with 23AON and sample preparation. Starting at an age of 3–4 weeks, 36 mdx mice were treated subcutaneously with 100 mg of 23AON/kg body weight in 100 µl saline twice weekly for 8 weeks. The 23AON molecule, previously described as M23D(+2–18), is a 2′-O-methyl phosphorothioate RNA oligonucleotide with a full-length phosphorothioate backbone, specifically targeting exon 2332 (Prosensa Therapeutics, Leiden, the Netherlands). Mice were sacrificed by cervical dislocation at different time points after the final injection: t = 36 hours and 1, 3, 8, 12, and 24 weeks (6 mice (3 males, 3 females) per time point). Before sacrifice, blood samples were taken for plasma PK analysis (see below). Plasma was generated by centrifuging at 18,000g for 5 minutes and it was stored at −80 °C until analysis. After sacrifice, muscles (triceps, tibialis anterior, quadriceps, heart, and diaphragm) and liver, kidney, and spleen were isolated, snap-frozen in liquid nitrogen–cooled 2-methylbutane, and stored at −80 °C.
Assessment of 23AON levels with a hybridization-ligation assay. An assay based on a previously published hybridization-ligation assay was used for the determination of the 23AON level in different tissues at Prosensa Therapeutics.33 Tissues were homogenized in 100 mmol/l Tris–HCl (pH 8.5), 200 mmol/l NaCl, 0.2% sodium dodecyl sulfate, 5 mmol/l ethylenediaminetetraacetic acid, and 2 mg/ml protK using zirconium beads (1.4 mm; OPS Diagnostics, Lebanon, NJ) in a MagNA Lyser (Roche Diagnostics, Almere, the Netherlands) according to manufacturer's protocol. Plasma samples were diluted tenfold, muscle samples 500 and 1,000 times, and organs 1,000 and 5,000 times in pooled control mdx tissue in phosphate-buffered saline. A signal probe (containing the peptide for antibody recognition) and a template (complementary to 23AON and the probe) were added to homogenized samples. Only when both 23AON and probe are bound to the template, a ligation step will take place. After this step and washing away of the unbound probe, enzyme-linked antibodies were used to detect the amount of probe–23AON. Calibration curves of the analyzed 23AON prepared in 10% pooled plasma or in pooled control mouse mdx tissue in phosphate-buffered saline were used to quantify 23AON. All analyses were performed in duplicate.
RNA extraction and analysis of exon skipping by reverse transcription–polymerase chain reaction. Muscles were homogenized in TriPure isolation reagent (Roche Diagnostics) using zirconium beads (1.4 mm; OPS Diagnostics) by grinding in a MagNA Lyser (Roche Diagnostics). Total RNA was extracted, and 600 ng was used for reverse transcription–polymerase chain reactionanalysis. Complementary DNA was generated by incubating at 42 °C for 45 minutes with random hexamer primers (20 ng/µl) and Transcriptor reverse transcriptase polymerase (Roche Diagnostics) in 20 µl. Subsequently, 2 µl of complementary DNA was amplified in a 50-µl polymerase chain reaction reaction with 30 cycles of 94 °C for 30 seconds, 60 °C for 30 seconds, and 72 °C for 30 seconds, as previously described.34 PCR products were visualized on 1.5% agarose gels and quantified using a DNA high-sensitivity chip on the LabChip GX, in combination with the LabChip GX software (Caliper Life Sciences, Teralfene, Belgium).
Analysis of dystrophin protein expression by western blot. Muscles were minced in treatment buffer containing 75 mmol/l Tris–HCl (pH 6.8)–15% (w/v) sodium dodecyl sulfate using zirconium beads (1.4 mm; OPS Diagnostics) or MagNA Lyser green beads (Roche Diagnostics) in a BBY24M Bullet Blender Storm (Next Advance, Averill Park, NY). Protein concentrations were determined using a Pierce bicinchoninic acid protein assay kit (Thermo Fisher Scientific, Waltham, MA) according to manufacturer's instructions. Samples containing 30 µg of protein were made in treatment buffer with 20% (v/v) glycerol, 5% (v/v) β-Mercaptoethanol, and 0.001% (w/v) Bromophenol blue and heated for 5 minutes at 95 °C. Wild-type control samples containing 10%, 3.3%, 1.1%, and 0.4% of protein were used as reference. Samples were loaded on 1.0-mm-thick native polyacrylamide Tris-acetate gels, with a linear resolving gel gradient of 3–8% (BioRad, Veenendaal, the Netherlands) and run on the Trans-Blot Turbo system for 1 hour at 75 V (0.07 A) and for 2.5 hours at 150 V (0.12 A) in an ice container. Proteins were blotted onto a nitrocellulose membrane using the ready to use Trans-Blot Turbo transfer packs in combination with the Trans-Blot Turbo transfer system (BioRad) at 2.5 A and ~25 V for 10 minutes. Membranes were blocked in 10 mmol/l Tris–HCl (pH 8) and 0.15 mol/l NaCl (Tris-buffered saline (TBS))–5% nonfat dried milk (Elk, Campina Melkunie, Zaltbommel, the Netherlands) and washed in TBS–0.05% (v/v) Tween20 (TBST). Membranes were incubated overnight with 1:125 NCL-Dys1 (Dy4; NovoCastra, Newcastle Upon Tyne, UK) and α-actinin was used as loading control (1:7,500; AB72592; Abcam, Cambridge, UK) in TBS. The next day, membranes were washed in TBST, incubated 1 hour with the fluorescent secondary antibodies IRDye 800CW goat-α-mouse IgG (1:5,000; Li-Cor; NE) and IRDye 680LT donkey-α-rabbit IgG (1:10 000; Li-Cor) in TBS, washed in TBST and TBS, and analyzed with the Odyssey system and software (Li-Cor) (M. Hulsker et al., manuscript in preparation).
Statistical analysis. For 23AON levels, individual data are represented for each measurement, and exponential curves were fitted for each tissue. For exon-skipping and protein levels, data are represented as mean ± SD for each tissue. PK plasma parameters (terminal half-lives of the molecule in plasma and tissue) were derived by noncompartmental analysis using Phoenix WinNonLin software (version 6.2; Pharsight, Mountain View, CA). For samples in which 23AON was not detectable, values were set at 50% of limit of quantification of mouse 23AON in neat plasma (i.e., 50% of 0.011 µg/ml). For statistical testing, SPSS version 20.0 (IBM, Armonk, NY) was used. A P value ≤0.05 was considered significant.
Acknowledgments
The authors are grateful to Rick Vermue (Prosensa Therapeutics) for assistance with the hybridization-ligation assay. This work was supported by grants from the Prinses Beatrix Spierfonds and Spieren voor Spieren (the Netherlands) and the Dutch Duchenne Parent Project. This work was carried out in Leiden, the Netherlands. A.A.-R. discloses being employed by Leiden University Medical Center, which has patent applications on exon skipping that are licensed to Prosensa Therapeutics. As a coinventor on some of these patents, A.A.-R. is entitled to a share of royalties. J.A.S., S.d.K., I.G.M.K., and J.C.T.v.D. report being employed by Prosensa Therapeutics. J.C.T.v.D. discloses being coinventor on exon-skipping patents and being entitled to a share of royalties. L.L., J.E.R., and S.R.H. report being employed by GlaxoSmithKline. The other authors declare no conflict of interest.
References
- Moat SJ, Bradley DM, Salmon R, Clarke A, Hartley L. Newborn bloodspot screening for Duchenne muscular dystrophy: 21 years experience in Wales (UK). Eur J Hum Genet. 2013;21:1049–1053. doi: 10.1038/ejhg.2012.301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendell JR, Lloyd-Puryear M. Report of MDA muscle disease symposium on newborn screening for Duchenne muscular dystrophy. Muscle Nerve. 2013;48:21–26. doi: 10.1002/mus.23810. [DOI] [PubMed] [Google Scholar]
- Emery AE. The muscular dystrophies. Lancet. 2002;359:687–695. doi: 10.1016/S0140-6736(02)07815-7. [DOI] [PubMed] [Google Scholar]
- Blake DJ, Weir A, Newey SE, Davies KE. Function and genetics of dystrophin and dystrophin-related proteins in muscle. Physiol Rev. 2002;82:291–329. doi: 10.1152/physrev.00028.2001. [DOI] [PubMed] [Google Scholar]
- Sicinski P, Geng Y, Ryder-Cook AS, Barnard EA, Darlison MG, Barnard PJ. The molecular basis of muscular dystrophy in the mdx mouse: a point mutation. Science. 1989;244:1578–1580. doi: 10.1126/science.2662404. [DOI] [PubMed] [Google Scholar]
- Torres LF, Duchen LW. The mutant mdx: inherited myopathy in the mouse. Morphological studies of nerves, muscles and end-plates. Brain. 1987;110 (Pt 2):269–299. doi: 10.1093/brain/110.2.269. [DOI] [PubMed] [Google Scholar]
- Banks GB, Chamberlain JS. The value of mammalian models for duchenne muscular dystrophy in developing therapeutic strategies. Curr Top Dev Biol. 2008;84:431–453. doi: 10.1016/S0070-2153(08)00609-1. [DOI] [PubMed] [Google Scholar]
- Verhaart IE, Aartsma-Rus A. Gene therapy for Duchenne muscular dystrophy. Curr Opin Neurol. 2012;25:588–596. doi: 10.1097/WCO.0b013e328357b0be. [DOI] [PubMed] [Google Scholar]
- Goemans NM, Tulinius M, van den Akker JT, Burm BE, Ekhart PF, Heuvelmans N, et al. Systemic administration of PRO051 in Duchenne's muscular dystrophy. N Engl J Med. 2011;364:1513–1522. doi: 10.1056/NEJMoa1011367. [DOI] [PubMed] [Google Scholar]
- Cirak S, Arechavala-Gomeza V, Guglieri M, Feng L, Torelli S, Anthony K, et al. Exon skipping and dystrophin restoration in patients with Duchenne muscular dystrophy after systemic phosphorodiamidate morpholino oligomer treatment: an open-label, phase 2, dose-escalation study. Lancet. 2011;378:595–605. doi: 10.1016/S0140-6736(11)60756-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prosena GSK and Prosensa announce primary endpoint not met in phase III study of drisapersen in patients with duchenne muscular dystrophy. <http://ir.prosensa.eu/releasedetail.cfm?ReleaseID=791929> .
- Heemskerk H, de Winter C, van Kuik P, Heuvelmans N, Sabatelli P, Rimessi P, et al. Preclinical PK and PD studies on 2'-O-methyl-phosphorothioate RNA antisense oligonucleotides in the mdx mouse model. Mol Ther. 2010;18:1210–1217. doi: 10.1038/mt.2010.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heemskerk H, de Winter CL, van Ommen GJ, van Deutekom JC, Aartsma-Rus A. Development of antisense-mediated exon skipping as a treatment for duchenne muscular dystrophy. Ann N Y Acad Sci. 2009;1175:71–79. doi: 10.1111/j.1749-6632.2009.04973.x. [DOI] [PubMed] [Google Scholar]
- Heemskerk HA, de Winter CL, de Kimpe SJ, van Kuik-Romeijn P, Heuvelmans N, Platenburg GJ, et al. In vivo comparison of 2'-O-methyl phosphorothioate and morpholino antisense oligonucleotides for Duchenne muscular dystrophy exon skipping. J Gene Med. 2009;11:257–266. doi: 10.1002/jgm.1288. [DOI] [PubMed] [Google Scholar]
- Tanganyika-de Winter CL, Heemskerk H, Karnaoukh TG, van Putten M, de Kimpe SJ, van Deutekom J, et al. Long-term exon skipping studies with 2'-O-methyl phosphorothioate antisense oligonucleotides in dystrophic mouse models. Mol Ther Nucleic Acids. 2012;1:e43. doi: 10.1038/mtna.2012.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verhaart IE, Heemskerk H, Karnaoukh TG, Kolfschoten IG, Vroon A, van Ommen GJ, et al. Prednisolone treatment does not interfere with 2'-O-methyl phosphorothioate antisense-mediated exon skipping in Duchenne muscular dystrophy. Hum Gene Ther. 2012;23:262–273. doi: 10.1089/hum.2011.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verhaart IE, Tanganyika-de Winter CL, Karnaoukh TG, Kolfschoten IG, de Kimpe SJ, van Deutekom JC, et al. Dose-dependent pharmacokinetic profiles of 2'-O-methyl phosphorothioate antisense oligonucleotidesin mdx mice. Nucleic Acid Ther. 2013;23:228–237. doi: 10.1089/nat.2012.0398. [DOI] [PubMed] [Google Scholar]
- Brun C, Suter D, Pauli C, Dunant P, Lochmüller H, Burgunder JM, et al. U7 snRNAs induce correction of mutated dystrophin pre-mRNA by exon skipping. Cell Mol Life Sci. 2003;60:557–566. doi: 10.1007/s000180300047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bish LT, Sleeper MM, Forbes SC, Wang B, Reynolds C, Singletary GE, et al. Long-term restoration of cardiac dystrophin expression in golden retriever muscular dystrophy following rAAV6-mediated exon skipping. Mol Ther. 2012;20:580–589. doi: 10.1038/mt.2011.264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharp NJ, Kornegay JN, Van Camp SD, Herbstreith MH, Secore SL, Kettle S, et al. An error in dystrophin mRNA processing in golden retriever muscular dystrophy, an animal homologue of Duchenne muscular dystrophy. Genomics. 1992;13:115–121. doi: 10.1016/0888-7543(92)90210-j. [DOI] [PubMed] [Google Scholar]
- Wang Z, Tapscott SJ, Chamberlain JS, Storb R. Immunity and AAV-Mediated Gene Therapy for Muscular Dystrophies in Large Animal Models and Human Trials. Front Microbiol. 2011;2:201. doi: 10.3389/fmicb.2011.00201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vulin A, Barthélémy I, Goyenvalle A, Thibaud JL, Beley C, Griffith G, et al. Muscle function recovery in golden retriever muscular dystrophy after AAV1-U7 exon skipping. Mol Ther. 2012;20:2120–2133. doi: 10.1038/mt.2012.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Hir M, Goyenvalle A, Peccate C, Précigout G, Davies KE, Voit T, et al. AAV genome loss from dystrophic mouse muscles during AAV-U7 snRNA-mediated exon-skipping therapy. Mol Ther. 2013;21:1551–1558. doi: 10.1038/mt.2013.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown DA, Kang SH, Gryaznov SM, DeDionisio L, Heidenreich O, Sullivan S, et al. Effect of phosphorothioate modification of oligodeoxynucleotides on specific protein binding. J Biol Chem. 1994;269:26801–26805. [PubMed] [Google Scholar]
- Yu RZ, Kim TW, Hong A, Watanabe TA, Gaus HJ, Geary RS. Cross-species pharmacokinetic comparison from mouse to man of a second-generation antisense oligonucleotide, ISIS 301012, targeting human apolipoprotein B-100. Drug Metab Dispos. 2007;35:460–468. doi: 10.1124/dmd.106.012401. [DOI] [PubMed] [Google Scholar]
- Wu B, Lu P, Cloer C, Shaban M, Grewal S, Milazi S, et al. Long-term rescue of dystrophin expression and improvement in muscle pathology and function in dystrophic mdx mice by peptide-conjugated morpholino. Am J Pathol. 2012;181:392–400. doi: 10.1016/j.ajpath.2012.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alter J, Lou F, Rabinowitz A, Yin H, Rosenfeld J, Wilton SD, et al. Systemic delivery of morpholino oligonucleotide restores dystrophin expression bodywide and improves dystrophic pathology. Nat Med. 2006;12:175–177. doi: 10.1038/nm1345. [DOI] [PubMed] [Google Scholar]
- Wu B, Lu P, Benrashid E, Malik S, Ashar J, Doran TJ, et al. Dose-dependent restoration of dystrophin expression in cardiac muscle of dystrophic mice by systemically delivered morpholino. Gene Ther. 2010;17:132–140. doi: 10.1038/gt.2009.120. [DOI] [PubMed] [Google Scholar]
- Betts C, Saleh AF, Arzumanov AA, Hammond SM, Godfrey C, Coursindel T, et al. Pip6-PMO, A New Generation of Peptide-oligonucleotide Conjugates With Improved Cardiac Exon Skipping Activity for DMD Treatment. Mol Ther Nucleic Acids. 2012;1:e38. doi: 10.1038/mtna.2012.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin H, Saleh AF, Betts C, Camelliti P, Seow Y, Ashraf S, et al. Pip5 transduction peptides direct high efficiency oligonucleotide-mediated dystrophin exon skipping in heart and phenotypic correction in mdx mice. Mol Ther. 2011;19:1295–1303. doi: 10.1038/mt.2011.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spitali P, van den Bergen JC, Verhaart IE, Wokke B, Janson AA, van den Eijnde R, et al. DMD transcript imbalance determines dystrophin levels. FASEB J. 2013;27:4909–4916. doi: 10.1096/fj.13-232025. [DOI] [PubMed] [Google Scholar]
- Mann CJ, Honeyman K, McClorey G, Fletcher S, Wilton SD. Improved antisense oligonucleotide induced exon skipping in the mdx mouse model of muscular dystrophy. J Gene Med. 2002;4:644–654. doi: 10.1002/jgm.295. [DOI] [PubMed] [Google Scholar]
- Yu RZ, Baker B, Chappell A, Geary RS, Cheung E, Levin AA. Development of an ultrasensitive noncompetitive hybridization-ligation enzyme-linked immunosorbent assay for the determination of phosphorothioate oligodeoxynucleotide in plasma. Anal Biochem. 2002;304:19–25. doi: 10.1006/abio.2002.5576. [DOI] [PubMed] [Google Scholar]
- Spitali P, Heemskerk H, Vossen RH, Ferlini A, den Dunnen JT, ‘t Hoen PA, et al. Accurate quantification of dystrophin mRNA and exon skipping levels in duchenne muscular dystrophy. Lab Invest. 2010;90:1396–1402. doi: 10.1038/labinvest.2010.98. [DOI] [PubMed] [Google Scholar]