

Abstract
Background:
Proximal spinal muscular atrophy (SMA) is an autosomal recessive neuromuscular disease characterized by symmetrical proximal muscle weakness and atrophy. According to the severity of the disease and the age of onset, SMA can be divided into three groups. The survival motor neuron (SMN) gene that is located on 5q13 is identified as the disease determining gene. Another gene in this region is neuronal apoptosis inhibitory protein (NAIP), and its functional role in the pathogenesis of SMA has not been fully elucidated. Here, we investigated the correlation between deletions in SMN and NAIP genes with clinical features of SMA patients.
Materials and Methods:
In the current study, 71 unrelated Iranian patients were investigated for the detection of deletions in SMN1 and NAIP genes. Polymerase chain reaction (PCR) was used to detect the deletions of exon 4 and 5 of the NAIP gene. Deletions in exon 7 and 8 of SMN1 gene were detected by RFLP-PCR with DraI and DdeI, respectively.
Results:
Our results showed that 51 patients have homozygous deletions in SMN1 and/or NAIP genes. Among these 51 patients, deletion in NAIP gene were found in 35 patients (65.7% of type I, 22.5% type II and 11.42% type III).
Conclusion:
Defect in SMN1 gene plays a major role in manifesting of the disease and NAIP (4 and 5) gene acts as a modifying factor in severity of symptoms. Correlation between NAIP gene defect and severity of the disease is confirmed. However, the exact role of NAIP gene in SMA has yet to be fully clarified.
Keywords: Deletion, neuronal apoptosis inhibitory protein gene, severity, spinal muscular atrophy, survival motor neuron gene
INTRODUCTION
Proximal spinal muscular atrophy (SMA) is an autosomal recessive neuromuscular disease characterized by symmetrical proximal muscle weakness and atrophy due to degeneration of the anterior horn cells of the spinal cord. This disease is the second most common neuromuscular disease in children after duchene muscular dystrophy.[1] The rate of its incidence is one in 10,000 newborns.[2] The patients are subdivided into three groups based on age of onset and severity of clinical features. Type I (Werding Hoffman) is the most severe form that manifests before 6 months of life and spans rarely longer than 2 years. This type of SMA is characterized by muscle weakness, hypotonia and inability to sit and walk in babies. Type II (Dubowitz) is an intermediate form with the onset after 6 months. The patients with this type of SMA are not able to walk without support. The patients with Type III have symptoms after 18 months and they are able to stand and walk, but fall frequently. This type of SMA is the mildest form.[2,3,4,5,6]
Because of the differences in severity of the disease in SMA patients, investigators prompt to find the genomic variations that contribute to these phenotypes. In this study, we were going to determine the correlation between deletion patterns in survival motor neuron (SMN) and neuronal apoptosis inhibitory protein (NAIP) genes, and the clinical phenotype of SMA among Iranian patients.
There are two SMN genes that are typically present on 5q11.2-13.3, SMN1 (telomeric copy) and SMN2 (centromeric copy).[7,8,9,10,11] Telomeric copy is the functional copy of gene but centromic gene produces truncated protein.[7,8,10] These genes are homologous and only differ by five nucleotides. Two of these differences occur in exon 7 and 8.[9] The difference in sequence is used to distinguish telomeric copy from the centromeric one. 2 of these differences occur in exon 7 and 8[9]. The PCR products from exon 7 and 8 of the 2 SMN gene copies can readily be distinguished since the centromeric (SMN2) gene contains a recognition site for the restriction enzyme DraI and DdeI, respectively. These recognition sites are absent in telomeric SMN gene. This methodology can detect homozygous absence of SMN1, but it cannot differentiate the presence of one copy from two or more copies of SMN1. SMN1 is deleted in about 90% of SMA patients in different populations.[12,13,14]
The functional role of NAIP, another gene in this region, in the pathogenesis of SMA has not been fully elucidated. However, some reports have demonstrated a correlation between deletion of the NAIP gene and severity of SMA.[15,16,17] This gene was also found to be frequently deleted in 45% of the patients who has the type I of SMA and 18% in other types of the disease.[16,18,19] The centromeric truncated NAIP gene, NAIP Ψ (pseudo gene), lacks the two coding exons, 4 and 5. In addition to deletions in NAIP gene, deletions in other genes such as p44, a subunit of transcription factor TFIIH, was found to be deleted in 45% of type I patients.[9,16,20,21] Recently, H4F5 gene has been reported as a new candidate gene with a deletion in about 90.5% of type I SMA patients together with NIAP.[21,22,23]
MATERIALS AND METHODS
We collected blood samples from 71 Iranian SMA suspected patients who were referred to the Genetics Lab of Alzahra University Hospital in Isfahan. The patients fulfilled the diagnostic criteria for SMA defined by the International SMA Consortium. The patients or their parents were informed of the aims of the study and gave their informed consent for the genetics analysis.
Genomic DNA was extracted from peripheral blood in EDTA tubes by the standard salting out method Primer sequences to amplify exon 7 and 8 were as follows, respectively: Exon 7 forward primer (7F): 5′-CTA TCA ACT TAA TTT CTG ATC A-3′, and reverse (7R), 5′-CCT TCC TTC TTT TTG ATT TTG TTT-3′ exon 8 forward primer (8F), 5′-GTA ATA ACC AAA TGC AAT GTG AA-3′ and reverse primer (8R), 5′-CTA CAA CAC CCT TCT CAC AG-3′.
Amplification of SMN exons was performed using the following conditions: 94°C for 5 min followed by 95°C for 1 min, 55°C for 1 min and 72°C for 1 min for 35 cycles and final extension at 72°C for 7 min. Polymerase chain reaction (PCR) products of centromeric and telomeric exon 7 and 8 of SMN gene were distinguished by digestion with 1 U restriction enzymes DraI and DdeI (Fermentas, Germany), respectively. The results were analyzed with 3% agarose gel.
Deletion analysis of NAIP gene was performed by primer oligonucleotide sequences for PCR amplification of exon 4: 4F, 5′-AAA GCC TCT GAC GAG AGG ATC-3′ and 4R, 5′-CTC TCA GCC TGC TCT TCA GAT-3′, and exon 5: 5F, 5′-TGC CAC TCC CAG GCA ATC TAA-3′ and 5R, 5′-CAT TTG GCA TGT TCC TTC CAA G-3′. Deletions in exons 4 and 5 of the NAIP gene were detected by amplification of these exons along with actin gene as internal control. The PCR conditions for NAIP were as follows: primary denaturation at 94°C for 6 min and 35 cycles of 95°C for 1 min, 59°C for 1 min and 72°C for 1 min and at last 7 min at 72°C.
Electrophoresis of PCR products on 2.5% agarose gel was used to confirm the presence or absence of exon 4 and 5 of NAIP gene.[26]
In this study all PCR was performed in a 25 μl reaction containing 1.5 mM MgCl2, 200mM of each dNTPs, I U of Taq DNA polymerase (Fermentase, Germany) 150 ng DNA and 25 pmol of each primers for NAIP and SMN genes in separate tubes. We used primers for actin gene PCR along with NAIP genes in same tubes. Actin is an internal control for confirmation of the PCR.
RESULTS
After digestion with DraI, the SMN1 exon 7 product will be undigested, whereas the SMN2 exon 7 product will be digested into two fragments of 164 and 24 base pairs. The 24 base pair fragment migrates off the gel so we saw two bands with 188 and 164 bp in normal samples on agarose gel. Restriction fragments of exon 8 of SMN2 gene are 122 and 78 base pairs and the SMN1 exon 8 products will be undigested. So, in normal samples three bands of 187, 123 and 65 bp were observed. On patients’ samples the undigested SMN1 alleles (188 bp in exon 7 and 187bp in exon 8) are absent and only the SMN2 gene is digested [Figure 1].
Figure 1.
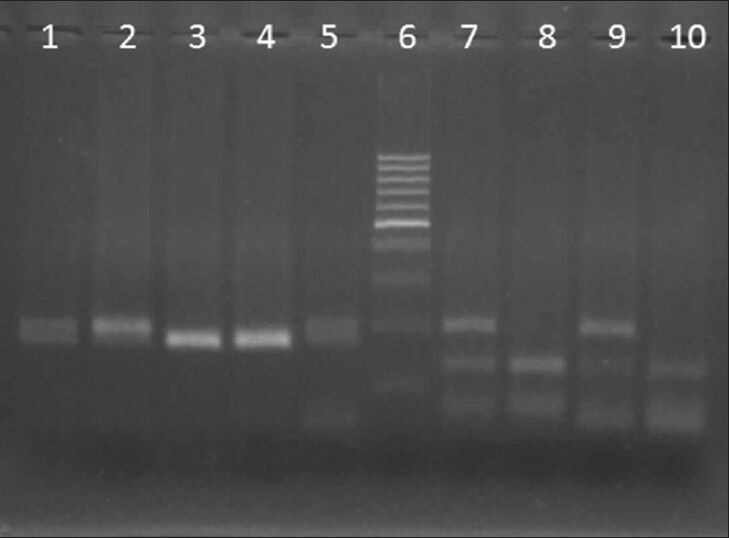
Deletion detection of survival motor neuron 1 (Exon 7, 8) performed by digestion of polymerase chain reaction product with DraI and DdeI, respectively. Digestion results were analyzed on 3% agarose gel. Lane 1 and 2: Different patients without deletion in exon 7 of survival motor neuron1 (SMN1), lane 3: Patient with deletion in exon 7, lane 4: Positive control and lane 5: Normal control, Lane 6: 100 bp marker; Lane 7: Patient without deletion in exon 8 of SMN, lane 8: Patient with deletion in exon 8, lane 9: Normal control, lane 10: Positive control
Amplification of the NAIP gene in normal samples resulted in two bands with 436 and 123 bp for exon 4 and 5, respectively, in addition to 283 bp for actin gene as an internal control. However, in affected and positive control samples, only a 283 bp of internal control was produced [Figure 2].
Figure 2.
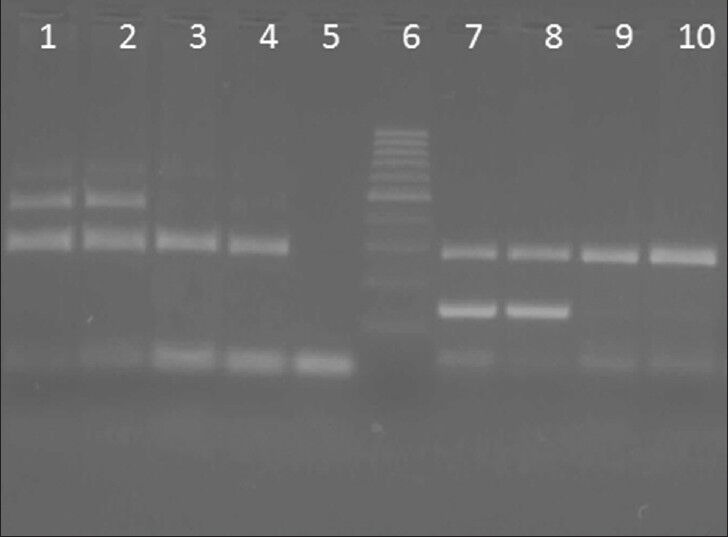
Deletion analysis of neuronal apoptosis inhibitory protein (NAIP) gene (Exon 4, 5) was done on 2.5% agrose gel lane 1 and 2: Different patients without deletion in exon 4 of NAIP, Lane 3: Patient with deletion in exon 4, lane 4: Positive control. Lane 5: Negative control without DNA template; Lane 6: 100 bp marker, Lane 7 and 8: Patients without deletion in exon 5 of NAIP, 9: Patient with deletion in exon 5, lane 10: Positive control
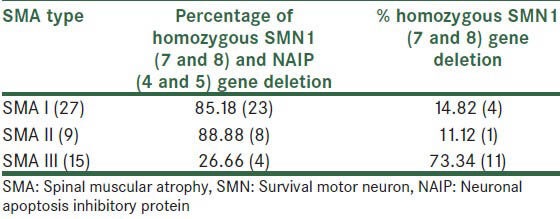
Fifty-one of 71 SMA patients analyzed in this study revealed deletion in SMN1 gene. Among these 51 patients, 29 were classified in Type I, nine had clinical features of Type II and 13 had Type III SMA. In 27 children of Type I (85%), 18 had homozygous deletions in both SMN1 (exon 7 and 8) and NAIP (exon 4 and 5) genes and 14.82% had deletions only in SMN1 gene (exon 7 and 8). Among nine patients with Type II, 88.88% had deletions in all exons that were analyzed in this study, whereas the remaining (11.12%) had deletions only in SMN1 gene, and finally all of the patients with Type III[14] had deletions in both exon 7 and 8, but only 26.66% showed deletions in the NAIP gene (exon 4 and 5) in addition to SMN gene. The summary of results is shown in Table 1.
Table 1.
Genotype-phenotype correlation of 51 Iranian spinal muscular atrophy patients who had deletion in survival motor neuron and/or neuronal apoptosis inhibitory protein gene
DISCUSSION
In fact, the molecular basis of SMA is complex. In this study, we focus on the most common proximal autosomal recessive form of SMA that is caused by deletions in SMN1 gene located on 5q chromosome. Twenty of 71 of the present SMA patients (10 with Type I, six with Type II and four with Type III) did not show deletion in SMN1 gene. Because these patients were diagnosed on the basis of the International SMA Consortium criteria, they may have point mutations in SMN1 gene or have mutated alleles of other genes either in this locus or in other loci. Patients who did not show homozygous deletion of the SMN1 and/or NAIP genes should be further analyzed by quantitative assay and linkage study.[24]
Our results showed that among proximal SMA patients, with homozygous deletions in exon 7 and 8 of SMN1 gene, only 68.6% indicated deletions in NAIP gene. The incidence of deletions in both SMN1 and NAIP genes were more frequent in type I patients (65.7%) than in Type II and III patients, which showed deletions in 22.5% and 11.42% of patients, respectively. Most of Type III showed deletions only in SMN1 gene. The data show that NAIP gene may not have a direct role in SMA manifesting but acts as a modifying factor in severity of pathologies. There is no correlation between SMN1 gene defects and severity of the disease because SMN1 gene defect was noted in all of our patients, regardless of the type of the SMA.[16,25]
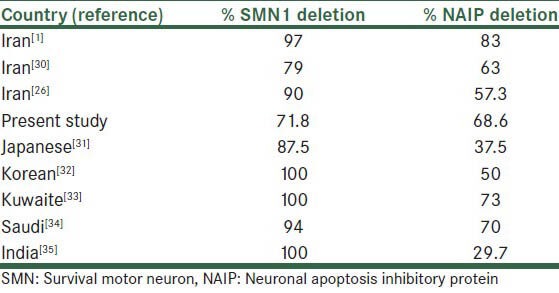
Salahshurifar et al. found 80% NAIP gene deletion in Iranian SMA patients (91% of Type I and 50% of Type II and III).[1] In another study in Iran, Omrani et al. showed the rate of SMN1 and NAIP gene deletion to be 90% and 57.3%, respectively.[26] Hasanzade et al. in 2009 analyzed deletions in exon 7 of the SMA gene only in 243 families and showed that 94% patients had deletion.[27] A comparison of deletion frequency in survival motor neuron and neuronal apoptosis inhibitory protein genes in Iranian patients and other populations are shown in Table 2.[1,26,30,31,32,33,34,35]
Table 2.
A comparison of deletion frequency in survival motor neuron and neuronal apoptosis inhibitory protein genes in Iranian patients and other populations
In our results, deletions in exon 7 of the SMN1 gene are always along with deletions in exon 8 of the gene, but sometimes one of them can be deleted alone.[9,28,29]
Our results confirm the correlation of SMA with high-frequency deletions in NAIP (4 and 5) gene, but we assay only deletions of SMN1 (7 and 8) and NAIP (4 and 5) genes in this study, and our methods do not allow analysis of other mutations or investigation of SMA carriers.
ACKNOWLEDGMENT
The authors are grateful to patients with SMA for their participating in this study.
Footnotes
Source of Support: Nil
Conflict of Interest: None declared.
REFERENCES
- 1.Salahshourifar I, Shafeghati Y, Golkar Z, Najmabadi H. Molecular analysis of the neuronal apoptosis inhibitory protein gene in families with spinal muscular atrophy. Arch Iran Med. 2007;10:509–13. [PubMed] [Google Scholar]
- 2.Pearn J. Classification of spinal muscular atrophies. Lancet. 1980;1:919–22. doi: 10.1016/s0140-6736(80)90847-8. [DOI] [PubMed] [Google Scholar]
- 3.Rodrigues NR, Owen N, Talbot K, Patel S, Muntoni F, Ignatius J, et al. Gene deletions in spinal muscular atrophy. J Med Genet. 1996;33:93–6. doi: 10.1136/jmg.33.2.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Munsat TL, Davies KE. International SMA consortium meeting. (26-28 June 1992, Bonn, Germany) Neuromuscul Disord. 1992;2:423–8. doi: 10.1016/s0960-8966(06)80015-5. [DOI] [PubMed] [Google Scholar]
- 5.Zerres K, Rudnik-Schöneborn S, Forkert R, Wirth B. Genetic basis of adult-onset spinal muscular atrophy. Lancet. 1995;346:1162. doi: 10.1016/s0140-6736(95)91835-3. [DOI] [PubMed] [Google Scholar]
- 6.Zerres K, Rudnik-Schöneborn S. Natural history in proximal spinal muscular atrophy. Clinical analysis of 445 patients and suggestions for a modification of existing classifications. Arch Neurol. 1995;52:518–23. doi: 10.1001/archneur.1995.00540290108025. [DOI] [PubMed] [Google Scholar]
- 7.Brzustowicz LM, Lehner T, Castilla LH, Penchaszadeh GK, Wilhelmsen KC, Daniels R, et al. Genetic mapping of chronic childhood-onset spinal muscular atrophy to chromosome 5q11.2-13.3. Nature. 1990;344:540–1. doi: 10.1038/344540a0. [DOI] [PubMed] [Google Scholar]
- 8.Gilliam TC, Brzustowicz LM, Castilla LH, Lehner T, Penchaszadeh GK, Daniels RJ, et al. Genetic homogeneity between acute and chronic forms of spinal muscular atrophy. Nature. 1990;345:823–5. doi: 10.1038/345823a0. [DOI] [PubMed] [Google Scholar]
- 9.Lefebvre S, Bürglen L, Reboullet S, Clermont O, Burlet P, Viollet L, et al. Identification and characterization of a spinal muscular atrophy-determining gene. Cell. 1995;80:155–65. doi: 10.1016/0092-8674(95)90460-3. [DOI] [PubMed] [Google Scholar]
- 10.Melki J, Abdelhak S, Sheth P, Bachelot MF, Burlet P, Marcadet A, et al. Gene for chronic proximal spinal muscular atrophies maps to chromosome 5q. Nature. 1990;344:767–8. doi: 10.1038/344767a0. [DOI] [PubMed] [Google Scholar]
- 11.Melki J, Sheth P, Abdelhak S, Burlet P, Bachelot MF, Lathrop MG, et al. Mapping of acute (type I) spinal muscular atrophy to chromosome 5q12-q14. The french spinal muscular atrophy investigators. Lancet. 1990;336:271–3. doi: 10.1016/0140-6736(90)91803-i. [DOI] [PubMed] [Google Scholar]
- 12.Burnett BG, Sumner CJ. Targeting splicing in spinal muscular atrophy. Ann Neurol. 2008;63:3–6. doi: 10.1002/ana.21305. [DOI] [PubMed] [Google Scholar]
- 13.Girardet A, Fernandez C, Claustres M. Efficient strategies for preimplantation genetic diagnosis of spinal muscular atrophy. Fertil Steril. 2008;90:443.e7–12. doi: 10.1016/j.fertnstert.2007.07.1305. [DOI] [PubMed] [Google Scholar]
- 14.Spinal Muscular Atrophy. ACOG committee opinion, Americam college of obstetricians and gynecologist. Obstet Gynecol. 2009;113:1194–6. doi: 10.1097/AOG.0b013e3181a6d03a. [DOI] [PubMed] [Google Scholar]
- 15.Anderson K, Talbot K. Spinal muscular atrophies reveal motor neuron vulnerability to defects in ribonucleoprotein handling. Curr Opin Neurol. 2003;16:595–9. doi: 10.1097/01.wco.0000093102.34793.13. [DOI] [PubMed] [Google Scholar]
- 16.Roy N, Mahadevan MS, McLean M, Shutler G, Yaraghi Z, Farahani R, et al. The gene for neuronal apoptosis inhibitory protein is partially deleted in individuals with spinal muscular atrophy. Cell. 1995;80:167–78. doi: 10.1016/0092-8674(95)90461-1. [DOI] [PubMed] [Google Scholar]
- 17.Velasco E, Valero C, Valero A, Moreno F, Hernández-Chico C. Molecular analysis of the SMN and NAIP genes in Spanish spinal muscular atrophy families and correlation between number of copies of cBCD541 and SMA phenotype. Hum Mol Genet. 1996;5:257–63. doi: 10.1093/hmg/5.2.257. [DOI] [PubMed] [Google Scholar]
- 18.Watihayati MS, Zabidi-Hussin AM, Tang TH, Matsuo M, Nishio H, Zilfalil BA. Deletion analyses of SMN1 and NAIP genes in Malaysian spinal muscular atrophy patients. Pediatr Int. 2007;49:11–4. doi: 10.1111/j.1442-200X.2007.02302.x. [DOI] [PubMed] [Google Scholar]
- 19.Mrad R, Dorboz I, Ben Jemaa L, Maazoul F, Trabelsi M, Chaabouni M, et al. Molecular analysis of the SMN1 and NAIP genes in 60 tunisian spinal muscular atrophy patients. Tunis Med. 2006;84:465–9. [PubMed] [Google Scholar]
- 20.Thompson TG, DiDonato CJ, Simard LR, Ingraham SE, Burghes AH, Crawford TO, et al. A novel cDNA detects homozygous microdeletions in greater than 50% of type I spinal muscular atrophy patients. Nat Genet. 1995;9:56–62. doi: 10.1038/ng0195-56. [DOI] [PubMed] [Google Scholar]
- 21.Bürglen L, Seroz T, Miniou P, Lefebvre S, Burlet P, Munnich A, et al. The gene encoding p44, a subunit of the transcription factor TFIIH, is involved in large-scale deletions associated with Werdnig-Hoffmann disease. Am J Hum Genet. 1997;60:72–9. [PMC free article] [PubMed] [Google Scholar]
- 22.Savas S, Gokgoz N, Kayserili H, Ozkinay F, Yuksel-Apak M, Kirdar B. Screening of deletions in SMN, NAIP and BTF 2p44 genes in Turkish spinal muscular atrophy patients. Hum Hered. 2000;50:162–5. doi: 10.1159/000022907. [DOI] [PubMed] [Google Scholar]
- 23.Akutsu T, Nishio H, Sumino K, Takeshima Y, Tsuneishi S, Wada H, et al. Molecular genetics of spinal muscular atrophy: Contribution of the NAIP gene to clinical severity. Kobe J Med Sci. 2002;48:25–31. [PubMed] [Google Scholar]
- 24.Simic G. Pathogenesis of proximal autosomal recessive spinal muscular atrophy. Acta Neuropathol. 2008;116:223–34. doi: 10.1007/s00401-008-0411-1. [DOI] [PubMed] [Google Scholar]
- 25.Ma H, Wang Y, Mi Z, Wu Y, Zhao P, Zhao S, Jiang M, Li Y. Study of NAIP gene in spinal muscular atrophy. Chinese journal of medical genetics. 1999;16:97–8. [PubMed] [Google Scholar]
- 26.Omrani O, Bonyadi M, Barzgar M. Molecular analysis of the SMN and NAIP genes in Iranian spinal muscular atrophy patients. Pediatr Int. 2009;51:193–6. doi: 10.1111/j.1442-200X.2008.02665.x. [DOI] [PubMed] [Google Scholar]
- 27.Hasanzad M, Golkar Z, Kariminejad R, Hadavi V, Almadani N, Afroozan F, et al. Deletions in the survival motor neuron gene in Iranian patients with spinal muscular atrophy. Ann Acad Med Singapore. 2009;38:139–41. [PubMed] [Google Scholar]
- 28.Bussaglia E, Clermont O, Tizzano E, Lefebvre S, Bürglen L, Cruaud C, et al. A frame-shift deletion in the survival motor neuron gene in Spanish spinal muscular atrophy patients. Nat Genet. 1995;11:335–7. doi: 10.1038/ng1195-335. [DOI] [PubMed] [Google Scholar]
- 29.Chang JG, Jong YJ, Huang JM, Wang WS, Yang TY, Chang CP, et al. Molecular basis of spinal muscular atrophy in Chinese. Am J Hum Genet. 1995;57:1503–5. [PMC free article] [PubMed] [Google Scholar]
- 30.Derakhshandeh-Peykar P, Esmaili M, Ousati-Ashtiani Z, Rahmani M, Babrzadeh F, Farshidi S, et al. Molecular analysis of the SMN1 and NAIP genes in Iranian patients with spinal muscular atrophy. Ann Acad Med Singapore. 2007;36:937–41. [PubMed] [Google Scholar]
- 31.Saitoh M, Sakakihara Y, Kobayashi S, Hayashi Y, Yanagisawa M. Correlation between deletion patterns of SMN and NAIP genes and the clinical features of spinal muscular atrophy in Japanese patients. Acta Paediatr Jpn. 1997;39:584–9. doi: 10.1111/j.1442-200x.1997.tb03645.x. [DOI] [PubMed] [Google Scholar]
- 32.Cho K, Ryu K, Lee E, Won S, Kim J, Yoo OJ, et al. Correlation between genotype and phenotype in Korean patients with spinal muscular atrophy. Mol Cells. 2001;11:21–7. [PubMed] [Google Scholar]
- 33.Samilchuk E, D’Souza B, Bastaki L, al-Awadi S. Deletion analysis of the SMN and NAIP genes in Kuwaiti patients with spinal muscular atrophy. Hum Genet. 1996;98:524–7. doi: 10.1007/s004390050253. [DOI] [PubMed] [Google Scholar]
- 34.Al Rajeh S, Majumdar R, Awada A, Adeyokunnu A, Al Jumah M, Al Bunyan M, et al. Molecular analysis of the SMN and NAIP genes in Saudi spinal muscular atrophy patients. J Neurol Sci. 1998;158:43–6. doi: 10.1016/s0022-510x(98)00053-7. [DOI] [PubMed] [Google Scholar]
- 35.Dastur RS, Gaitonde PS, Khadilkar SV, Udani VP, Nadkarni JJ. Correlation between deletion patterns of SMN and NAIP genes and the clinical features of spinal muscular atrophy in Indian patients. Neurol India. 2006;54:255–9. doi: 10.4103/0028-3886.27147. [DOI] [PubMed] [Google Scholar]