

Abstract
The microcolony assay developed by Withers and Elkind has been a gold standard to assess the surviving fraction of small intestinal stem cells after exposure to high (≥8 Gy) doses of ionizing radiation (IR), but is not applicable in cases of exposure to lower doses. Here, we developed a novel in vitro assay that enables assessment of the surviving fraction of small intestinal stem cells after exposure to lower IR doses. The assay includes in vitro culture of small intestinal stem cells, which allows the stem cells to develop into epithelial organoids containing all four differentiated cell types of the small intestine. We used Lgr5-EGFP-IRES-CreERT2/ROSA26-tdTomato mice to identify Lgr5+ stem cells and their progeny. Enzymatically dissociated single crypt cells from the duodenum and jejunum of mice were irradiated with 7.25, 29, 101, 304, 1000, 2000 and 4000 mGy of X-rays immediately after plating, and the number of organoids was counted on Day 12. Organoid-forming efficiency of irradiated cells relative to that of unirradiated controls was defined as the surviving fraction of stem cells. We observed a significant decrease in the surviving fraction of stem cells at ≥1000 mGy. Moreover, fluorescence-activated cell sorting analyses and passage of the organoids revealed that proliferation of stem cells surviving IR is significantly potentiated. Together, the present study demonstrates that the in vitro assay is useful for quantitatively assessing the surviving fraction of small intestinal stem cells after exposure to lower doses of IR as compared with previous examinations using the microcolony assay.
Keywords: small intestinal stem cells, ionizing radiation, survival assay, Lgr5, in vitro culture, organoid
INTRODUCTION
Adult tissue stem cells can be defined by two essential features: first, the self-renewing capacity, which enables the maintenance of stem cell populations over long periods of time, and second, the capacity to produce differentiated cell types of tissues [1]. The small intestine is among the most rapidly self-renewing tissues in adult mammals [2]. The small intestinal epithelium is composed of crypts and villi [2]. The crypts contain stem cells, transit-amplifying cells, and Paneth cells [2]. In the villi, there are differentiated, specialized cells, including absorptive enterocytes, mucous-secreting goblet cells, and hormone-secreting enteroendocrine cells [2]. The cells are newly generated from stem cells in the crypts, migrate upward along the crypt–villus axis, and are eliminated by apoptosis at the tip of the villi, with a turnover time of 4–5 days in mice [2]. Paneth cells are exceptional in that they settle at the crypt bottoms and represent the only differentiated cells that escape upward migration [2].
Unique markers for small intestinal stem cells have not been identified until recently, though stem cell characteristics have long been extensively studied using ionizing radiation (IR) [3]. Potten et al. proposed that stem cells reside at position +4 (immediately above Paneth cells) relative to the crypt bottom, on the basis of the fact that long-term DNA label-retaining cells are enriched at around position +4 during crypt regeneration after exposure to high doses of IR [4]. In contrast, Cheng and Leblond reported the presence of cycling cells between Paneth cells and proposed that cells called ‘crypt base columnar cells’ may harbor stem-cell activity [5]. In 2007, Barker et al. reported the first marker for small intestinal stem cells, leucine-rich-repeat-containing G-protein-coupled receptor 5 (Lgr5) [6]. They showed that Lgr5 is exclusively expressed in cycling crypt base columnar cells, and Lgr5+ crypt base columnar cells can generate all types of differentiated cells of the small intestinal epithelium over a 60-day period [6]. Subsequently, Bmi1 and mTert were identified as marker genes for proximal small intestinal stem cells present at position +4 [7, 8]. Lineage-tracing experiments revealed that similar to Lgr5+ stem cells, Bmi1+ or mTert+ stem cells can produce all types of differentiated cells of the small intestinal epithelium, and moreover cells positive for Bmi1 or mTert can generate Lgr5+ stem cells [8, 9]. These lines of evidence indicate that small intestinal crypts contain multiple types of stem cells, and there is hierarchy or plasticity among them.
Niches are well accepted as microenvironments that surround stem cells and support maintenance of stem cell properties [10]. Mesenchymal cells neighbouring crypts, e.g. subepithelial myofibroblasts, are well known to function as niche cells for small intestinal stem cells [11, 12]. Recently, Sato et al. reported that Paneth cells constitute the niche for Lgr5+ stem cells [13]. Taken together, it is indicated that multiple types of cells work as niche cells to support small intestinal stem cells [14].
Following genotoxic or cytotoxic insults, e.g. IR, stem cells play a critical role in the regeneration of the injured epithelium [3]. The microcolony assay developed by Withers and Elkind has been commonly used to assess the surviving fraction of stem cells after IR [3, 15]. In the assay, regenerated small crypts were directly counted by visual observation under a microscope 3–4 d after IR [3, 15]. However, the assay requires high (≥8 Gy) doses of IR to detect a numerical change in the regenerated crypts, because a crypt contains multiple (4–6) stem cells and can be regenerated from a single stem cell that survives IR [3, 15, 16]. Therefore, a more sensitive assay is needed to assess the surviving fraction of stem cells after exposure to lower doses of IR.
Recently, a long-term in vitro culture condition of Lgr5+ stem cells was established [17]. Under the culture condition, a single sorted Lgr5+ stem cell can generate a crypt–villus-like structure called an ‘organoid’. The organoid reflects the small intestinal epithelium in vivo, because it contains all types of differentiated cells of the epithelium [17].
In the present study, we developed a novel survival assay of small intestinal stem cells after IR using in vitro culture. The assay enabled us to detect a significant decrease in the surviving fraction of stem cells at ≥1000 mGy, i.e. at much lower doses than those previously examined using the microcolony assay.
MATERIALS AND METHODS
Mice
Lgr5-EGFP-IRES-CreERT2 knock-in (B6.129P2-Lgr5tm1(cre/ERT2)Cle/J, JAX mice #008875) and ROSA26-tdTomato (B6.Cg-Gt(ROSA)26Sortml4(CAG-tdTomato)Hze/J, JAX mice #007914) mice were purchased from the Jackson Laboratory (Bar Harbor, ME, USA). The mice were bred in a conventional clean room facility of the Central Research Institute of Electric Power Industry (CRIEPI) at a controlled temperature (24 ± 2°C) and humidity (45 ± 5%) and kept in a 12-h light–dark cycle, with ad libitum access to γ-sterilized food (CLEA, Tokyo, Japan) and filter-sterilized deionized water. All animal experiments were approved by the Animal Research and Ethics Committee of CRIEPI and performed following the guidelines for animal care in Japan.
Fluorescent labeling of progeny of Lgr5+ stem cells
In the Lgr5-EGFP-IRES-CreERT2 knock-in mice, Lgr5-EGFP-IRES-CreERT2 was knocked in the first exon of one of the two endogenous Lgr5 alleles [6]. The knock-in mice express enhanced green fluorescent protein (EGFP) and the Cre recombinase fused to a mutated ligand-binding domain of the human estrogen receptor (CreERT2). Another Cre reporter strain, ROSA26-tdTomato mice, harbors loxP-STOP-loxP sequences immediately before the tdTomato gene, thereby blocking the expression of the tdTomato protein. Lgr5-EGFP-IRES-CreERT2 mice were crossed with ROSA26-tdTomato mice to produce Lgr5-EGFP-IRES-CreERT2/ROSA26-tdTomato mice (hereafter called LRT mice). Administration of 4-hydroxytamoxifen (4-OHT) to LRT mice allows nuclear localization of the Cre protein and subsequent Cre-loxP recombination to remove the STOP sequence in Lgr5+ cells. Therefore, 4-OHT administration allows Lgr5+ stem cells and their progeny to be labeled with tdTomato, and the labeling persists even after 4-OHT is removed from the body. The 4-OHT was intraperitoneally administered to mice at 10–20 days of age (single administration between 10–20 days of age).
Isolation of single crypt cells
LRT mice were killed at 3–5 weeks of age, and duodenum and jejunum (10 cm from the stomach) were harvested and rinsed three times by using a jet of cold 1 × phosphate-buffered saline (PBS−). Then, the intestinal tubes were opened longitudinally and the villi were scraped using a coverslip, followed by washing three times in cold 1× PBS−. The intestinal tubes were then cut into 2–3 mm pieces and suspended in 1× PBS− + 2% fetal bovine serum (FBS). The 2–3-mm pieces were extensively washed (10–15 times) with 1× PBS− + 2% FBS until the supernatant became clear. Then, the 2–3-mm pieces were treated with 50 mM EDTA/1× PBS− for 30 min at 4°C on a rocking platform to dissociate the crypts from the intestinal tubes. The dissociated crypts were passed through a 70-µm cell strainer, washed once with 1× PBS−, and treated with TrypLE Express (Life Technologies, Carlsbad, CA, USA) for 30 min at 37°C to dissociate cell-to-cell attachment. Then, the dissociated cells were passed through a 40-µm strainer and subsequently, through a 20-µm strainer. The strained cells were pelleted, resuspended in 1× PBS− + 2% FBS, and used as single crypt cells.
Fluorescence-activated cell sorting analysis
All fluorescence-activated cell-sorting (FACS) analyses were performed using FACS Aria III (BD, Franklin Lakes, NJ, USA). For the experiment shown in Fig. 1B, dissociated single crypt cells from LRT mice were analyzed. For the experiment shown in Fig. 1C, single crypt cells were fixed with 4% paraformaldehyde for 10 min at 4°C, followed by permeabilization with IC permeabilization buffer (Life Technologies, Carlsbad, CA, USA) for 5 min at 4°C. Then, the cells were treated with a rabbit anti-lysozyme antibody (Dako, Glostrup, Denmark, EC 3.2.1.17, 1:400 dilution) for 30 min at 4°C, followed by treatment with an Alexa Fluor 405-conjugated anti-rabbit IgG (Life Technologies, Carlsbad, CA, USA, 1:1000 dilution) for 30 min at 4°C. For the experiment shown in Fig. 1D, single crypt cells were stained with phycoerythrin-conjugated rat anti-mouse antibody against cluster of differentiation 24 (CD24) (eBioscience, San Diego, CA, USA, clone M1/69, 1:667 dilution) and allophycocyanin-conjugated rat anti-mouse antibody against epithelial cell adhesion molecule (EpCAM) (eBioscience, San Diego, CA, USA, clone G8.8, 1:333 dilution) for 15 min at 4°C. All antibodies were diluted in 100 µl of 1× PBS− + 2% FBS. For the experiment shown in Fig. 4, the organoids were collected and pelleted by centrifugation (300 × g, 2 min), followed by removal of the supernatant. Then, the organoids were resuspended with 500 µl dispase (BD, Franklin Lakes, NJ, USA) and dissociated for 2 h in a humidified CO2 incubator at 37°C. After confirming dissociation with a microscope, the dissociated cells were washed once with 1× PBS−, followed by fixation with 4% paraformaldehyde. After fixation, cells were washed once with 1× PBS− and resuspended with 1× PBS− + 2% FBS. Then, the percentage of EGFPhigh cells (Lgr5high stem cells) and tdTomato+ cells (progeny of Lgr5+ stem cells) was analyzed. We confirmed that fixation by 4% paraformaldehyde did not alter the percentages of EGFPhigh- and tdTomato+ cells compared with those of cells without fixation. The positive population was gated by comparing positive and negative samples so that there were no (or very few) positive cells in the negative sample. Crypt cells harvested from wild-type mice were used as a negative sample to define Lgr5+ and tdTomato+ populations. Crypt cells treated with isotype control antibodies for anti-CD24 and EpCAM antibodies were used as a negative sample to define CD24+ and EpCAM+ populations. Crypt cells treated only with Alexa Fluor-conjugated secondary antibody were used as a negative sample to define lysozyme+ population. Side scatterhigh cells were defined as cells with upper half of side scatter value, because there are no other definitions.
Fig. 1.

Isolation of crypts and single crypt cells from the duodenum and jejunum of Lgr5-EGFP-IRES-CreERT2/ROSA26-tdTomato (LRT) mice. (A) Typical images of crypts and enzymatically dissociated single crypt cells. EGFP fluorescence in crypts or dissociated cells indicates expression of the Lgr5 gene. DIC is an abbreviation for differential interference contrast. Bars, 100 µm. (B) Lgr5high cells in dissociated single crypt cells analyzed by FACS. (C) Side-scatterhigh/lysozyme+ cells in dissociated single crypt cells analyzed by FACS. (D) CD24+/EpCAM+ cells in dissociated single crypt cells analyzed by FACS.
Fig. 4.

FACS analysis of cells in the organoids formed after exposure to various doses of X-rays. The organoids formed in the experiment shown in Fig. 3A were enzymatically dissociated into single cells, and the cells were subjected to FACS analysis. The x- and y-axes indicate the fluorescence intensity of EGFP and tdTomato, respectively. The values in parentheses represent the percentages of cells with high EGFP fluorescence (= Lgr5high stem cells). The whole population is gated in quadrants: the upper left, the upper right, the lower left, and the lower right populations represent Lgr5low/−/tdTomato+, Lgr5high/tdTomato+, Lgr5low/−/tdTomato−, and Lgr5high/tdTomato− cells, respectively. Note that the Lgr5high/tdTomato+ cells are self-renewed Lgr5high stem cells. Data for cells derived from wild-type organoids are also presented to show the validity of gating of the Lgr5high cells. The FACS analysis was performed only once.
In vitro culture of single crypt cells
The isolated single crypt cells were cultured in organoid medium established by Sato et al. [17], i.e. advanced DMEM/F12 supplemented with 1 × GlutaMAX, 10 mM HEPES, 1 × penicillin/streptomycin, 1 × N2, 1 × B27 (all obtained from Life Technologies, Carlsbad, CA, USA), N-acetylcysteine (Sigma-Aldrich, St Louis, MO, USA, 1 mM), murine epidermal growth factor (Life Technologies, Carlsbad, CA, USA, 50 ng/ml), murine Noggin (Peprotech, Rocky Hill, NJ, USA, 100 ng/ml), and murine R-Spondin I (R&D Systems, Minneapolis, MN, USA, 1 µg/ml). The Rho kinase inhibitor Y-27632 (Sigma-Aldrich, St Louis, MO, USA, 10 µM) was added on the first 2 d to avoid anoikis. First, the above organoid medium (350 µl) was added into wells of 48-well plates, and then single crypt cells were plated to the wells (1 × 105 cells/10 µl 1 × PBS− + 2% FBS per well). Subsequently, 40 µl of Matrigel (BD, Franklin Lakes, NJ, USA) was added and mixed briefly so that the final concentration of Matrigel became 10%. Freshly prepared organoid medium was added first 3 d and every 2–3 d thereafter. Cells were cultured in a humidified CO2 incubator at 37°C. For the experiments shown in Fig. 3, three wells were used in the first experiment and two wells were used in the second experiment for each dose or control. At 12 d after plating, the number of organoids was counted by visual observation under a phase-contrast microscope with a × 4 objective. The budding structure as in Fig. 2 (Day 12) was regarded as an organoid and counted.
Fig. 3.
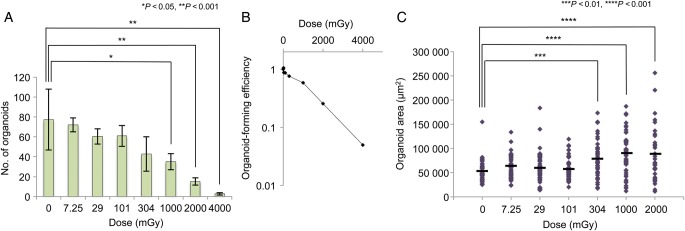
Organoid formation and organoid size after IR. (A) The number of organoids formed from 1 × 105 single crypt cells. Data represent the mean ± SEM of two independent experiments. Statistical analysis was performed by Dunnett's multiple comparison test. (B) Organoid-forming efficiency as a function of dose. Organoid-forming efficiency after exposure to the respective doses of IR relative to the unirradiated control is shown. The numbers of organoids shown in Fig. 3A were used to calculate the efficiency. Data represent the mean of two independent experiments. (C) Area of each organoid formed in the experiments shown in Fig. 3A. The organoid area was measured using Image J. A total of 50 organoids in two independent experiments were subjected to the analysis. Black bars represent the mean area of the organoids formed after exposure to the indicated doses of IR. Statistical analysis was performed using Dunnett's multiple comparison test.
Fig. 2.
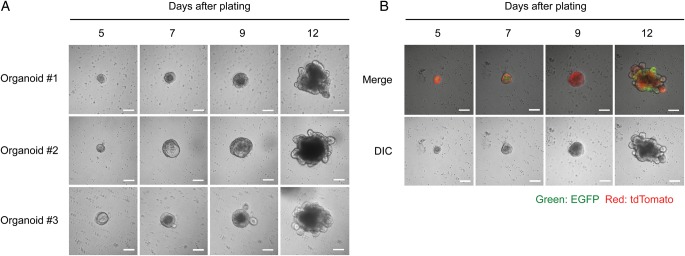
Organoid formation from single crypt cells. (A) Typical images of cells that developed into organoids. Lumen-like cystic structures started to form on Days 5–7, budding events started on Days 7–9, and robust budding was observed on Day 12. Bars, 100 µm. (B) Organoid formation from single crypt cells isolated from 4-OHT-treated LRT mice. EGFP fluorescence represents Lgr5+ stem cells, and tdTomato fluorescence indicates the progeny of Lgr5+ stem cells. Note that EGFP+ cells expand on Day 12, compared with on Days 5–9. Bars, 100 µm.
Irradiation
Immediately after plating, single crypt cells were irradiated with X-rays from an X-ray generator (Hitachi Medico, Tokyo, Japan, MBR-1505R2) with a 1-mm aluminium/0.2-mm copper filter at a dose rate of 0.10 Gy/min (7.25–304 mGy) or 0.47 Gy/min (1000–4000 mGy).
Time-lapse analysis of cell growth and measurement of organoid area
Time-lapse analysis of cell growth (shown in Fig. 2) was performed manually using a confocal laser microscope (C1si, Nikon, Tokyo, Japan). At Day 5 after plating, images of cells were captured and the coordinates of the cells were determined. Then, images of organoids located on the same coordinates as on Day 5 were captured at Days 7, 9 and 12. The captured images were analyzed by ImageJ software (National Institutes of Health, Bethesda, MD, USA, http://rsbweb.nih.gov/ij/). The organoid area was measured by encircling the periphery of each organoid. We estimated that 36 pixels in ImageJ are equivalent to 49 µm2 and calculated the area of the organoids accordingly.
Passage of organoids
The organoids formed after exposure to various doses of X-rays were mechanistically dissociated by passing the organoids 10 times through a 26G needle in the well. We microscopically confirmed dissociation of the organoids and absence of mature organoids. Then, freshly prepared medium and Matrigel (final concentration, 10%) were added for secondary organoid formation. For the experiments shown in Fig. 5A, three wells were used in the first experiment and two wells were used in the second experiment for each dose or control. Seven days after plating, the number of organoids was counted by visual observation under a phase-contrast microscope with a 4× objective. The budding structure as shown in Fig. 2 (Day 12) was regarded as an organoid and counted.
Fig. 5.
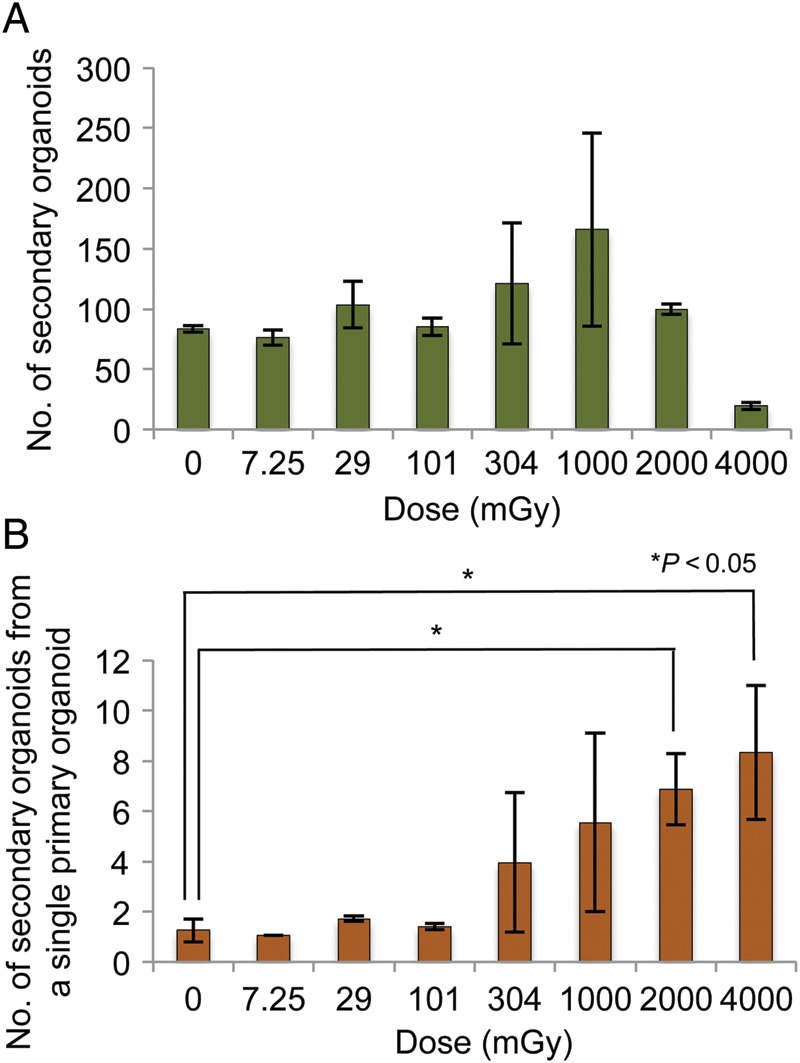
Effect of IR on the organoid-regenerating capacity of primary organoids. (A) The number of secondary organoids formed after exposure to the respective doses of IR. The primary organoids formed in the experiments shown in Fig. 3 were mechanistically dissociated and all dissociated organoids were passaged to allow secondary organoid formation. On Day 7 after passaging, the number of organoids was counted. Data represent the mean ± SEM of two independent experiments. (B) The number of secondary organoids produced from a single primary organoid. The number of secondary organoids was divided by the number of primary organoids. Data represent the mean ± SEM of two independent experiments. Statistical analysis was performed using Dunnett's multiple comparison test.
Statistical analysis
Statistical comparison was made by a Dunnett's multiple comparison test, and a P-value < 0.05 was considered significant. Data are presented as mean ± standard error of the mean (SEM).
RESULTS
Isolation of single crypt cells containing Lgr5+ stem cells and Paneth cells
Crypts harvested from duodenum and jejunum of LRT mice are shown in Fig. 1A. The crypts were dissociated from intestinal tubes by using a high concentration (50 mM) of EDTA. In our hands, this concentration did not affect the efficiency of organoid formation, while it greatly enhanced the yield of crypt cells compared with lower concentrations (Supplementary Figs. 1A and 1B). The harvested crypts were enzymatically dissociated into single cells (Fig. 1A, right panels). In crypts and single crypt cells isolated from LRT mice, EGFP fluorescence indicates expression of the Lgr5 gene, a stem cell marker [6]. We microscopically confirmed the presence of Lgr5+ stem cells in crypts and dissociated cells (Fig. 1A). Moreover, FACS analysis showed that the dissociated cells contain 3.11% Lgr5high cells (cells with high EGFP fluorescence), which are capable of initiating organoid formation (Fig. 1B) [17]. Next, we checked whether our dissociated crypt cells contain Paneth cells, which were shown to function as niche cells for Lgr5+ stem cells [13]. In the small intestine, lysozyme is specifically expressed in Paneth cells [18]. Moreover, side scatter, which indicates the complexity of the cellular content, is reportedly high in Paneth cells, because the cells contain granules [13]. Therefore, we first examined whether single crypt cells contain side scatterhigh/lysozyme+ cells. FACS analysis showed that there were 4.47% side scatterhigh/lysozyme+ cells among the crypt cells (Fig. 1C). In addition, we assessed the presence of cells positive for both CD24 and EpCAM, which were reportedly shown to be Paneth cells [13]. FACS analysis indicated that 4.36% of crypt cells were CD24+/EpCAM+ (Fig. 1D). Taken together, we confirmed that our isolated single crypt cells contain multipotent Lgr5high stem cells and their niche cells, i.e. Paneth cells.
Growth of single crypt cells into organoids
Next, we tested whether and how organoids are formed from single crypt cells in our experimental setting. Isolated crypt cells were immediately plated in organoid medium, whose composition had been established by Sato et al. [17]. Then, Matrigel was added at a final concentration of 10%. In the medium containing 10% Matrigel, cells moved around on the first 4 d after plating, but they became immobile on Day 5 and thereafter, probably due to the increase in cell mass by cell growth. Therefore, we performed a time-lapse analysis of cell growth starting from Day 5 after plating. Typical images of the cell growth are shown in Fig. 2A. On Day 5, crypt cells formed a small cystic structure, which became larger thereafter. Budding events started at Days 7–9, and the organoids robustly budded at Day 12 (Fig. 2A). In LRT mice, progeny of Lgr5+ stem cells, including both self-renewed stem cells and differentiated cells, can be labeled with tdTomato by administration of 4-OHT [19]. Therefore, we next monitored crypt cells isolated from 4-OHT-administered LRT mice (Fig. 2B). The time-lapse analysis revealed that the organoids were labeled with tdTomato. Of note, EGFP+ cells appeared to expand with the growth of the organoid, indicating that Lgr5+ stem cells robustly undergo self-renewal during organoid formation (Fig. 2B, cf. Day 12 and Days 5–9). The observations in Fig. 2 let us consider that the organoid culture may be applicable for the evaluation of survivability of small intestinal stem cells after IR.
Organoid-forming efficiency and organoid size after IR
We next examined organoid formation after exposure to various doses of X-irradiation. Immediately after plating (1 × 105 cells/well), single crypt cells were irradiated with 7.25, 29, 101, 304, 1000, 2000 or 4000 mGy of X-rays, and the number of budding organoids was counted microscopically on Day 12. Figure 3A shows the number of organoids formed after exposure to the indicated doses of X-rays. Compared with the unirradiated control, the number of organoids was unchanged at ≤304 mGy, but was significantly decreased at ≥1000 mGy. Figure 3B shows the organoid-forming efficiency as a function of dose. The organoid-forming efficiency was reduced to 45, 20 and 4% of that of the control at 1000, 2000 and 4000 mGy, respectively (Fig. 3B). The organoid-forming efficiency reflects the surviving fraction of stem cells among crypt cells because organoids are produced from stem cells [17]. Intriguingly, we noticed that the size of the organoids appeared to become larger in the irradiated population than in the unirradiated population. Therefore, we measured the area of each organoid using image-analyzing software to quantitatively estimate the size of organoids formed after exposure to 7.25–2000 mGy. We failed to measure the area of organoids formed after exposure to 4000 mGy because the number of analyzable organoids was too small. We found that the organoid size became significantly greater at ≥304 mGy compared with that of the control (Fig. 3C). The larger size of the organoids formed after IR indicates that the stem cells surviving IR proliferate more robustly than unirradiated stem cells. Thus, we next examined the percentage of stem cells in the organoids formed after IR. For this, we enzymatically dissociated the organoids into single cells and performed FACS analysis to determine the percentage of Lgr5high stem cells. Immediately before plating, 96.4% of Lgr5+ stem cells were labeled with tdTomato, indicating that there was a enough time interval between 4-OHT injection and cell harvesting for stem cell labeling to be maximized (Fig. 4, contour plot of ‘Immediately before plating’). However, both the whole Lgr5+ population and Lgr5+/tdTomato+ population decreased in the unirradiated control organoids, compared with the dissociated crypt cells immediately before plating. (Figure 4, control, cf. ‘Immediately before plating’.) Instead, the Lgr5+/tdTomato− population was increased in the unirradiated organoid, in comparison with the dissociated cells before plating (discussed later). By comparing irradiated and unirradiated organoids, we found that more Lgr5high cells were present in organoids formed after exposure to 304–4000 mGy than in unirradiated organoids, and that the percentage of Lgr5high cells increased dose-dependently between 304–4000 mGy (Fig. 4). Moreover, the percentages of Lgr5high/tdTomato+ cells also increased after exposure to 304–2000 mGy, indicating that the self-renewal capacity of the Lgr5high stem cells was enhanced (304 mGy, 1.56%; 1000 mGy, 2.00%; 2000 mGy, 2.30%; cf. control, 1.28%; Fig. 4). Although the number of Lgr5high/tdTomato+ cells did not markedly increase after exposure to 4000 mGy, we observed an increase in the Lgr5high/tdTomato− population (3.60%; cf. control, 2.00%; Fig. 4). Moreover, the Lgr5low/−/tdTomato+ population also expanded, indicating that differentiation of Lgr5high stem cells was accelerated at 4000 mGy (35.9%; cf. control, 10.6%; Fig. 4).
Organoid-regenerating capacity of the primary organoids after IR
The results presented in Figs. 3C and 4 imply that the stem cells surviving ≥304 mGy of IR became hyper-proliferative in the organoids. To substantiate the hyper-proliferative activity of the surviving stem cells in the irradiated organoids, we mechanistically dissociated the organoids used in the experiments shown in Fig. 3 (hereafter called primary organoids) by passing them through a 26G needle, then, passaged the dissociated organoids. Seven days after passaging, the number of regenerated organoids (hereafter called secondary organoids) was counted. The number of secondary organoids is shown in Fig. 5A. Unlike the case of primary organoids, we did not observe a significant decrease in the number of secondary organoids at ≥1000 mGy, indicating that more secondary organoids were produced from primary organoids after exposure to ≥1000 mGy compared with the control. To quantify the capacity of a single primary organoid to produce secondary organoids, the number of secondary organoids (Fig. 5A) was divided by the number of primary organoids (Fig. 3A), and the numbers of secondary organoids derived from a single primary organoid are shown in Fig. 5B. The analysis revealed that the organoid-regenerating capacity of a single primary organoid was potentiated 5.5-fold after exposure to 2000 mGy and 6.6-fold after exposure to 4000 mGy, compared with the control (Fig. 5B).
DISCUSSION
Small intestinal crypts exhibit high regenerative capacity upon tissue damage [3]. The regenerative capacity of crypts after IR has been extensively studied using the microcolony assay developed by Withers and Elkind [15]. The microcolony assay provided extensive information about the characteristics of small intestinal stem cells, including the surviving fraction of stem cells after exposure to high (≥8 Gy) doses of IR [3, 15]. However, in the assay, the IR dose must be large enough to ensure that the proportion of surviving stem cells is small, so that local areas of confluence of regenerated nodules are minimal enough to be countable [15]. Thus, a more sensitive assay is required to assess the surviving fraction of stem cells after exposure to lower IR doses. In the present study, we employed in vitro culture of stem cells to estimate the surviving fraction of stem cells after IR. On one hand, the in vitro assay enabled us to detect a reduction in the organoid-forming efficiency at ≥1000 mGy, showing that our assay system is more sensitive than the microcolony assay. However, on the other, we identified several points concerning our assay that need to be discussed.
In the present study, we plated 1 × 105 single crypt cells per well in a 48-well plate and could count 50–100 organoids in the unirradiated population. From the FACS analysis results shown in Fig. 1B (3.11% Lgr5high cells among crypt cells), it can be estimated that ∼ 3000 Lgr5high stem cells were present among the 1 × 105 crypt cells. Therefore, only 1.7–3.3% of the Lgr5high stem cells could initiate organoid formation in our experiments, assuming that the organoids originate solely from Lgr5high stem cells. The low efficiency of organoid formation may be due to cell damage caused by enzymatic dissociation or culture stress, both of which are unavoidable in the assay. The comparison between dissociated cells immediately before plating and cells in unirradiated control organoids revealed that, while the whole Lgr5+ population and the Lgr5+/tdTomato+ population were decreased, the Lgr5+/tdTomato− population was increased in the control organoids compared with the dissociated crypt cells immediately before plating. We consider that a substantial fraction of Lgr5+ stem cells die during cell culture, probably due to culture stress, and that the increase in the Lgr5+/tdTomato− population indicates de novo production of Lgr5+ stem cells by some mechanism, such as conversion of other stem cells to Lgr5+ stem cells. Despite the low organoid-forming efficiency and the decrease in Lgr5+ stem cells during cell culture, we could reproducibly observe a significant difference in the organoid number between irradiated (1000–4000 mGy) and unirradiated populations, indicating that a dose-dependent reduction in the surviving fraction of stem cells could be detected by our assay.
Importantly, we noticed the survival curve presented here does not have an obvious shoulder component. Lack of shoulder means that the extrapolation number is nearly 1. It indicates that (i) a single organoid is derived from a single stem cell, and (ii) the stem cells tend to choose cell death rather than DNA repair. Strictly, single-cell sorting is required to demonstrate that a single organoid is derived from a single stem cell, however, it has already been shown that a single-sorted Lgr5+ small intestinal stem cell has the ability to generate an organoid [17]. Thus, we consider that most of the organoids we counted were derived from single stem cells. As for (ii), we recently showed that Lgr5+ small intestinal stem cells undergo apoptosis within 24 h after exposure to 1 Gy of X-rays [19]. We observed caspase cleavage at 6 h after 1 Gy, at a time when DNA repair should not have been completed. Therefore, we consider that Lgr5+ stem cells have a tendency to choose cell death rather than DNA repair to protect genome integrity after exposure to IR. It is well known that the classical survival curve of small intestinal stem cells after irradiation has a large shoulder component [3, 15]. However, the survival curves were depicted using the microcolony assay, in which crypts regenerated after high doses of radiation were directly counted by visual observation under a microscope [15]. It is estimated that a crypt has 4–6 stem cells, and can be regenerated if a single stem cell survives radiation damage [3, 15, 16]. Therefore, the large shoulder of the classical survival curve of small intestinal stem cells is attributable to the above characteristics of crypts and the microcolony assay.
We noticed several advantages of the in vitro assay. First, as described above, the efficiency of primary organoid formation after IR reflects the surviving fraction of stem cells. Second, organoid size may represent the proliferative activity of stem cells in each organoid. Third, passage of the organoids allows a quantitative estimation of the proliferative activity of stem cells in primary organoids. By passaging the primary organoids, we were able to quantitatively show that the organoid-regenerating capacity of a single primary organoid was increased more than 5-fold after exposure to ≥2000 mGy. From the results, we conclude that small intestinal stem cells surviving ≥2000 mGy of IR acquire hyper-proliferative activity. We consider that the hyper-proliferative activity of surviving stem cells is of biological significance for the regeneration of an epithelium damaged by IR.
Recently, van Es et al. reported that secretory progenitor cells positive for the expression of Delta-like 1 can revert to Lgr5+ stem cells and regenerate the epithelium upon exposure to 6 Gy of γ-rays [20]. On the basis of this report, it can be considered that the expansion of Lgr5high cells at ≥304 mGy in our study may be, in part, attributable to the reversion of Delta-like 1+ progenitor cells to stem cells. Moreover, Bmi1+ or mTert+ stem cells can produce Lgr5+ stem cells [8, 9]. Therefore, multiple types of stem or progenitor cells may contribute to the enhanced secondary organoid formation after exposure to ≥2000 mGy by de novo production of Lgr5+ stem cells, in addition to hyper-proliferation of Lgr5+ stem cells themselves. In the FACS analysis, we observed a dose-dependent increase in the percentage of Lgr5+ stem cells upon exposure to ≥304 mGy; however, at 4000 mGy, the increase in the stem cell population is mainly attributable to the increase in Lgr5+/tdTomato− stem cells (3.6%, cf. 2.0% in the control population), not to the increase in Lgr5+/tdTomato+ stem cells (1.48%, cf. 1.28% in the control population). In our experimental setting, the increase in Lgr5+/tdTomato− stem cells indicates de novo generation of Lgr5+ stem cells, because the culture medium did not contain 4-OHT, which is necessary for Cre-LoxP recombination to allow tdTomato expression. We recently showed that the de novo production of Lgr5+ stem cells is accelerated in the colon, but not in the duodenum, after exposure of mice to 1 Gy of X-rays [19]. The present study indicates the possibility that de novo production of Lgr5+ stem cells is also facilitated in the duodenum after exposure to higher doses, e.g. 4 Gy.
Reportedly, a single sorted Lgr5+ stem cell can generate an organoid without niche cells under the same culture conditions as those used in the present study [17]. However, in our assay, niche cells, e.g. Paneth cells, should contribute to organoid formation, because all crypt cells were plated. In addition, our isolated cells contained non-epithelial cells, which are indicated as EpCAM-negative cells in Fig. 1D. Therefore, non-epithelial cells, e.g. subepithelial myofibroblasts may also support in vitro organoid formation.
The in vitro culture of intestinal stem cells can be used for many other applications, e.g. examination of the effect of other genotoxic or cytotoxic agents on stem cell survival and proliferation. Sato et al. reported optimal culture conditions of organoids from human small intestine, colon, and colon cancer [21]. The in vitro organoid culture using human colon cancer cells may greatly contribute to the development of cancer therapies, e.g. screening of chemical compounds with anti-tumor activities.
SUPPLEMENTARY DATA
Supplementary data is available at the Journal of Radiation Research online.
ACKNOWLEDGEMENTS
We are grateful to Dr. Toshiro Sato (Keio Univ., Japan) for technical instruction of crypt isolation and crypt cell culture, and for a number of invaluable advices. We also thank Mr. Takeshi Oda and Ms. Naomi Iizawa for animal care, and Ms. Mikie Okada for genotyping of the mice. We greatly appreciate helpful comments from Drs. Otsura Niwa (Fukushima Medical Univ., Japan) and Keiji Suzuki (Nagasaki Univ., Japan).
REFERENCES
- 1.Keller R. Stem cells on the way to restorative medicine. Immunol Lett. 2002;83:1–12. doi: 10.1016/s0165-2478(02)00065-2. [DOI] [PubMed] [Google Scholar]
- 2.Van der Flier LG, Clevers H. Stem cells, self-renewal, and differentiation in the intestinal epithelium. Annu Rev Physiol. 2009;71:241–60. doi: 10.1146/annurev.physiol.010908.163145. [DOI] [PubMed] [Google Scholar]
- 3.Potten CS. Radiation, the ideal cytotoxic agent for studying the cell biology of tissues such as the small intestine. Radiat Res. 2004;161:123–36. doi: 10.1667/rr3104. [DOI] [PubMed] [Google Scholar]
- 4.Potten CS, Owen G, Booth D. Intestinal stem cells protect their genome by selective segregation of template DNA strands. J Cell Sci. 2002;115:2381–8. doi: 10.1242/jcs.115.11.2381. [DOI] [PubMed] [Google Scholar]
- 5.Cheng H, Leblond CP. Origin, differentiation and renewal of the four main epithelial cell types in the mouse small intestine. V. Unitarian Theory of the origin of the four epithelial cell types. Am J Anat. 1974;141:537–61. doi: 10.1002/aja.1001410407. [DOI] [PubMed] [Google Scholar]
- 6.Barker N, van Es JH, Kuipers J, et al. Identification of stem cells in small intestine and colon by marker gene Lgr5. Nature. 2007;449:1003–7. doi: 10.1038/nature06196. [DOI] [PubMed] [Google Scholar]
- 7.Sangiorgi E, Capecchi MR. Bmi1 is expressed in vivo in intestinal stem cells. Nat Genet. 2008;40:915–20. doi: 10.1038/ng.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Montgomery RK, Carlone DL, Richmond CA, et al. Mouse telomerase reverse transcriptase (mTert) expression marks slowly cycling intestinal stem cells. Proc Natl Acad Sci U S A. 2011;108:179–84. doi: 10.1073/pnas.1013004108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yan KS, Chia LA, Li X, et al. The intestinal stem cell markers Bmi1 and Lgr5 identify two functionally distinct populations. Proc Natl Acad Sci U S A. 2012;109:466–71. doi: 10.1073/pnas.1118857109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Morrison SJ, Spradling AC. Stem cells and niches: mechanisms that promote stem cell maintenance throughout life. Cell. 2008;132:598–611. doi: 10.1016/j.cell.2008.01.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yen TH, Wright NA. The gastrointestinal tract stem cell niche. Stem Cell Rev. 2006;2:203–12. doi: 10.1007/s12015-006-0048-1. [DOI] [PubMed] [Google Scholar]
- 12.Shaker A, Rubin DC. Intestinal stem cells and epithelial–mesenchymal interactions in the crypt and stem cell niche. Transl Res. 2010;156:180–7. doi: 10.1016/j.trsl.2010.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sato T, van Es JH, Snippert HJ, et al. Paneth cells constitute the niche for Lgr5 stem cells in intestinal crypts. Nature. 2011;469:415–8. doi: 10.1038/nature09637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yeung TM, Chia LA, Kosinski CM, et al. Regulation of self-renewal and differentiation by the intestinal stem cell niche. Cell Mol Life Sci. 2011;68:2513–23. doi: 10.1007/s00018-011-0687-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Withers HR, Elkind MM. Microcolony survival assay for cells of mouse intestinal mucosa exposed to radiation. Int J Radiat Biol. 1970;17:261–7. doi: 10.1080/09553007014550291. [DOI] [PubMed] [Google Scholar]
- 16.Bjerknes M, Cheng H. Clonal analysis of mouse intestinal epithelial progenitors. Gastroenterology. 1999;116:7–14. doi: 10.1016/s0016-5085(99)70222-2. [DOI] [PubMed] [Google Scholar]
- 17.Sato T, Vries RG, Snippert HJ, et al. Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature. 2009;459:262–5. doi: 10.1038/nature07935. [DOI] [PubMed] [Google Scholar]
- 18.Ouellette AJ. Paneth cells and innate immunity in the crypt microenvironment. Gastroenterology. 1997;113:1779–84. doi: 10.1053/gast.1997.v113.pm9352884. [DOI] [PubMed] [Google Scholar]
- 19.Otsuka K, Hamada N, Magae J, et al. Ionizing radiation leads to the replacement and de novo production of colonic Lgr5(+) stem cells. Radiat Res. 2013;179:637–46. doi: 10.1667/RR3253.1. [DOI] [PubMed] [Google Scholar]
- 20.van Es JH, Sato T, van de Wetering M, et al. Dll1+ secretory progenitor cells revert to stem cells upon crypt damage. Nat Cell Biol. 2012;14:1099–104. doi: 10.1038/ncb2581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sato T, Stange DE, Ferrante M, et al. Long-term expansion of epithelial organoids from human colon, adenoma, adenocarcinoma, and Barrett's epithelium. Gastroenterology. 2011;141:1762–72. doi: 10.1053/j.gastro.2011.07.050. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.