

Abstract
Phospholamban (PLN) inhibits the sarco(endo)plasmic reticulum Ca2+-ATPase (SERCA), thereby regulating cardiac diastole. In membranes, PLN assembles into homopentamers that in both the phosphorylated and non-phosphorylated states have been proposed to form ion-selective channels. Here, we determined the structure of the phosphorylated pentamer using a combination of solution and solid-state nuclear magnetic resonance methods. We found that the pinwheel architecture of the homopentamer is preserved upon phosphorylation, with each monomer having an L-shaped conformation of each monomer. The TM domains form a hydrophobic pore of approximately 24 Å long, and 2 Å in diameter, which is inconsistent with canonical Ca2+ selective channels. Phosphorylation, however, enhances the conformational dynamics of the cytoplasmic region of PLN, causing the partial unwinding of the amphipathic helix. We propose that PLN oligomers act as storage for active monomers, keeping SERCA function within a physiological window.
Keywords: Hybrid NMR, cardiac proteins, heart failure, calcium transport
INTRODUCTION
Phospholamban (PLN), a 52-residue membrane protein, is involved in the Ca2+ regulation pathway in cardiac myocytes (MacLennan and Kranias, 2003; Simmerman and Jones, 1998; Tada et al., 1975). To initiate muscle contraction, Ca2+ ions enter the sarcolemmal membrane and stimulate the release of Ca2+ from the sarcoplasmic reticulum (SR) by the ryanodine receptors (Calcium-induced-Calcium-release mechanism). Ca2+ ions then bind the troponin complex, signaling muscle contraction. The relaxation phase takes place when Ca2+ is transferred back into the SR by the sarco(endo)plasmic reticulum Ca2+-ATPase SERCA2a. A small membrane protein, PLN, binds and down-regulates SERCA function by reducing its apparent Ca2+ binding affinity. PLN’s secondary structure comprises two helical segments joined by a short dynamic loop; the transmembrane (TM) helix, spanning residues 25–51, constitutes the inhibitory domain (Kimura et al., 1996), and the cytoplasmic helix bearing the phosphorylation sites has a regulatory role, reversing PLN inhibitory function upon phosphorylation at Ser16 (Li et al., 1998). Both in vitro and in vivo studies demonstrated that PLN forms stable homopentamer (Li et al., 1998). However, the role of this homopentamer is at the center of an active debate.
One hypothesis is that PLN forms Ca2+ (Kovacs et al., 1988) or Cl− (Oxenoid and Chou, 2005) selective channels, based on initial patch-clamp measurements and electrochemical measurements with PLN reconstituted in artificial membranes, which indicated that several different ions were likely transported by PLN. In contrast, other groups hypothesize that the PLN homopentameric assembly may act as a buffer or as storage, regulating the concentration of active monomers available to bind SERCA and keeping its function (Ca2+ concentration) within a physiological window. The latter is supported by biochemical and molecular biology data showing that the presence of SERCA shifts the homopentamer/monomers equilibrium toward PLN monomers, forming 1:1 SERCA/PLN complexes (Kimura et al., 1997). Additionally, it has been shown that mutants promoting PLN oligomerization upregulate SERCA, whereas mutants that enhance deoligomerization down-regulate the ATPase, reducing Ca2+ flux and resulting in heart failure progression in mice models (Chu et al., 1997).
The recent structure of the non-phosphorylated state of PLN in lipid membranes revealed a pinwheel topology of the homopentamer, with a long hydrophobic TM pore (~24 Å) of relatively small diameter (~2 Å), which cannot accommodate passive transport of hydrated Ca2+ or Cl− ions (Becucci et al., 2009; Verardi et al., 2011). However, recent physiological studies reinstated the Ca2+ channel hypothesis, proposing that phosphorylated PLN pentamer may form ion channels that increase Ca2+ leak (Smeazzetto et al., 2011).
In the present work, we seek to clarify the role of phosphorylation on pentameric PLN. Using a combination of solution and solid-state nuclear magnetic resonance (NMR) spectroscopy, we determined the structure, topology, and conformational dynamics of the phosphorylated PLN pentamer. We found that upon phosphorylation, pentameric PLN retained its overall architecture within the lipid membranes, with an average structure resembling the pinwheel conformation found for the non-phosphorylated PLN. Taken with earlier electrochemical measurements (Becucci et al., 2013), the structural topology of phosphorylated PLN is not consistent with the formation of an ion-selective channel, and instead supports an active role of oligomerization in maintaining the population of active monomer within a physiological window of SERCA inhibition.
Experimental Procedures
Protein Preparation
Wild-type PLN (PLNWT) was cloned in a pMal expression vector (New England Biolabs; Ipswich, MA) and expressed in Escherichia coli cells grown in M9 media, supplemented with 15NH4Cl as the sole source of 15N (uniform labeling), or in combination with the non-labeled amino acids (residue type reverse labeling) (Vuister et al., 1994). Isotope labeled compounds were purchased from either Isotec or Cambridge Isotope Laboratories. Maltose binding protein – PLN fusion protein was purified on amylose resin, cleaved with tobacco etch virus protease and purified by high performance liquid chromatography (HPLC), utilizing a Vydac® C8 column (Grace; Deerfield, IL) with a linear water-isopropanol gradient at 1.5% per minute at a 2 ml/min flow rate. (Buck et al., 2003; Veglia et al., 2010). The phosphorylation of S16 was accomplished by incubating PLNWT in β-D-octylglucoside (1/100 w/w) with the recombinant catalytic subunit of protein kinase A (PKA-C) at pH 7.0 and 30 °C for 12–16 hrs. The completion of the phosphorylation reaction and sample purity was assessed by gel shift assays and ESI-MS (Figure S1).
Selectively labeled PLNWT was prepared using a Liberty microwave assisted peptide synthesizer (CEM, Matthews, NC). The synthesis was performed on a 0.05 mmol scale, using preloaded PEG-polysterene resin. Activators used were HBTU (regular coupling, 10-fold amino acid excess) and PyClock (isotope coupling, 2-fold amino acid excess) (Subiros-Funosas et al., 2008). Each residue was double coupled. Methionine and cysteine residues were coupled in the conventional (non-microwave) fashion to reduce the risk of racemization. Side chain deprotection and cleavage were accomplished using modified Reagent K (room temperature, 3 hours), with 1,2-ethanedithiol substituted with 3,6-dioxa-1,8-octanedithiol (King et al., 1990; Teixeira et al., 2002). Synthetic PLNWT was purified using reverse phase HPLC utilizing the conditions described above for the recombinant protein.
Solution NMR
Solution NMR experiments were performed on a 600 MHz Varian and a 750 MHz Bruker Avance III DMX equipped with cryogenic probe. PLN was reconstituted in DPC-d38 micelles (1:70 w/w) by mixing the appropriate amounts of protein and detergent powder and dissolving in 50 mM phosphate buffer with 100 mM NaCl, pH 6.0. For PLN/detergent NOE, U-2H,15N-PLNWT and fully protonated DPC were employed. Relaxation experiments were carried out at 37 °C at the two magnetic field strengths, utilizing the pulse sequences described by Farrow et al. (Farrow et al., 1994). Experimental uncertainties were estimated from duplicate delays (R1 and R2) or duplicate experiments (1H-15N NOE). Errors in the products or ratios of R1 and R2 were calculated according to:
Solid-state NMR
Separated local field (SLF) PISEMA (Wu et al., 1994) data were acquired on PLNWT incorporated in DOPC/DOPE (4/1 mol/mol) (Avanti Polar Lipids, Alabaster, AL). Total amount of lipid was 60 μmol with a pentamer/lipid ratio of 1/100 mol/mol (Traaseth et al., 2009; Verardi et al., 2011). Protein in trifluoroethanol and lipids in chloroform were co-mixed, organic solvent was removed under a stream of nitrogen gas and residual organic solvent was removed under high vacuum. Protein-lipid mixture was resuspended in water, deposited evenly between 36 glass slides (Marienfeld, Lauda-Konigshofen, Germany), partially dehydrated at 33% relative humidity (saturated solution of MgCl2) and re-hydrated by adding water up to 45% w/w (Cornell et al., 1988; van der Wel et al., 2002). Lipid alignment was verified using 31P (Figure S2). Typically 300–500 μs cross-polarization contact time was employed, the dipolar evolution was 400–600 μs and 1500–3000 scans per increment were acquired. Data were acquired at 600 MHz (Bruker DRX) and 700 MHz (Varian), utilizing a low-E 15N-1H probe (Gor’kov et al., 2006). Bicelle samples were prepared using the long chain lipid DMPC/POPC (4/1 mol/mol) and the short chain lipid DHPC. Importantly, the presence of POPC lipid reduces the optimal temperature necessary to form an aligned bicellar phase. To induce parallel orientation of lipid bilayers relative to B0, YbCl3 was added to the sample to a final concentration of 10 mM. The q value employed was 4.5, as the lower values (namely q=3.2) resulted in incomplete alignment and smaller dipolar couplings (data not shown). The SLF spectra for bicelle preparations were acquired on a Varian spectrometer operating at 700 MHz.
Dipolar assisted rotational resonance (DARR) experiments (Takegoshi et al., 2001) were performed on a phosphorylated PLN sample consisting of an asymmetric isotopic mixture of 15N/13C-Leu PLNWT and 15N/13C-Ile PLNWT reconstituted in DOPC/DOPE 4/1 (mol/mol) multilamellar vesicles (MLV), 20 μmol total lipid. The magic angle spinning (MAS) rate was 8 kHz and DARR mixing time was 400 ms. Data were acquired at −15 °C.
Structure calculations were performed with Xplor-NIH 2.33 (Schwieters et al., 2003). Structural restraints (Table 1) were obtained through HSQC-NOESY at 600 MHz field (Varian), utilizing 200 ms mixing time and binned as described previously (Verardi et al., 2011). Chemical shift data were converted to dihedral restraints with Talos+ (Shen et al., 2009). SLF data were modeled using the Xplor-NIH routines (Straus et al., 2011), using 15N chemical shift tensor components of σ11=64, σ22=77, σ33=216 ppm (Shi et al., 2009). The Da component of the dipolar coupling alignment tensor was set to 9.5 kHz, corresponding to the value of the generalized order parameter of 0.93 (assuming 1.06 Å NH bond length, the maximum dipolar coupling therefore being 10.22 kHz) and the chemical shift alignment tensor was scaled accordingly. As noted previously, minor variations in the order parameter or chemical shift tensor components do not have a profound effect on the quality of the structure, but do lead to higher energies of the corresponding terms resulting from poor data fit (Shi et al., 2009; Vostrikov et al., 2011a). DARR data were modeled as ambiguous restraints between L37, L44, L51 and I33, I40, I47 based on the structure of the non-phosphorylated form as well as mutagenesis data, indicating that all these residues are essential for the stability of the pentamer (Simmerman et al., 1996). The distance range for the observed DARR cross peaks was set between 1.8 and 6.5 Å irrespective of the peak intensity. Ez potential energy function (Senes et al., 2007; Shi et al., 2009) was employed at the monomer calculation step for the depth of insertion, and then switched to planar restraints during the pentamer refinement. In addition to the standard Xplor-NIH potentials (Table S1), knowledge based hydrogen bonding, Ramachandran, and residue affinity potentials were employed. Structures were visualized with PyMOL (DeLano, 2008).
Table 1.
Experimental structural restraints employed during the structure calculation (per protomer).
| NOE | |
| Strong | 42 |
| Medium | 111 |
| Weak | 259 |
| Intra residue (i = j) | 175 |
| Sequential (|i − j| = 1) | 140 |
| Medium range (|i − j| = 2) | 47 |
| Long range (|i − j| > 2) | 62 |
| Dihedral restraints | |
| Phi | 43 |
| Psi | 43 |
| DARR restraints | 9 |
| Planar restraints | 3 |
| SLF restraints, unique/ambiguous | |
| Chemical shifts | 10/4 |
| Dipolar couplings | 10/4 |
ATPase activity assays
Consumption of ATP by SERCA has been measured using enzyme coupled assay in DOPC:DOPE 4:1 multilamellar vesicles as described previously (Ha et al., 2007; Madden et al., 1979). Temperature was maintained at 37 °C, PLN : SERCA : lipid ratio was 6 : 1 : 700. Enzyme activity was monitored at 340 nm continuously for 8 minutes with 9 second dwell time and fitted to a linear curve. The curve slopes at different Ca2+ concentrations were fitted to Hill equation, the midpoint of the sigmoidal curve being the pKCa value.
Results
Hybrid Solution/Solid-state NMR Structure of Phosphorylated PLN pentamer
On the SDS-PAGE gel, we observed the pentameric assembly of PLN at an apparent molecular weight of ~25 kDa, which is lower than its formula weight (~30 kDa) (Wegener and Jones, 1984). Despite this difference in mobility, we readily identified the phosphorylated species by the gel shift relative to the non-phosphorylated form of the protein (Figure S1). The correct molecular weight was confirmed by electrospray ionization mass spectra (ESI-MS). Under the ESI-MS conditions, deoligomerization of the pentamer occurred and the protomers were detectable in the 700–1300 m/z window, corresponding to the PLN forms bearing five to nine charges. In all cases, PLN incubated with PKA-C exhibited a mass shift corresponding to one phosphate group per monomer.
The [1H,15N]-heteronuclear single-quantum correlation spectroscopy (1H, 15N-HSQC) spectra of the non-phosphorylated and phosphorylated forms of wild type PLN (PLNWT) reconstituted in dodecylphosphocholine (DPC) micelles are very similar (Figure S3). A single resonance is observed for each residue, indicative of the symmetric nature of the pentamer. The hydrophobic segments of the proteins (clusters of peaks centered at ~8.5 ppm in 1H dimension) are essentially superimposable. The residues mostly affected by phosphorylation are located close to Ser16, with other chemical shift perturbations detected for residues in the flexible loop and domain Ib. These structural features closely resemble those observed for the phosphorylated monomeric PLN, where the three cysteine residues in the TM domain are mutated into Ala and Phe to prevent pentamerization (Metcalfe et al., 2004). The plot of the chemical shift perturbations of the phosphorylated proteins (monomer and pentamer) relative to the non-phosphorylated form are very similar, showing that phosphorylation perturbs both forms and these changes are independent of the oligomeric state of PLN. The chemical shift data have been deposited to the Biological Magnetic Resonance Bank with accession code 18952.
The [1H, 15N]-NOESY-HSQC spectra of PLNWT, phosphorylated at S16 (pS16-PLNwt) show the typical signature of regular α-helices, including (α, α+3), (α, α+4), and (β, β+3) NOE connectivities. This pattern is present in the TM domain and the N-terminal segment of the cytoplasmic domain, but is interrupted for the residues spanning 16–26, indicating a disruption of the helical secondary structure.
To determine the architecture of the phosphorylated pentamer in lipid bilayers, we reconstituted pS16-PLNWT in mechanically aligned 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC)/1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE) lipid bilayers and carried out [1H, 15N] polarization inversion spin exchange at the magic angle ([1H, 15N]-PISEMA) experiments. The PISEMA spectrum displays three main features: a cluster of resonances corresponding to the TM segment, a region where the amide groups of the in-plane cytoplasmic helix resonate, and high intensity signals centered around the isotropic chemical shift values that correspond to highly dynamic residues as well as the side chain resonances. The TM region of PLN comprises 25 hydrophobic residues arranged in an α-helical conformation, giving rise to a complex spectrum with only a few resolved resonances (Traaseth et al., 2007). To assign the individual signals, we employed a combination of residue-type labeling schemes using recombinant proteins as well as synthetic samples (Figure 1). Synthetic samples with four sequentially labeled residues yielded well-resolved spectra in which we were able to identify the individual residues. The α-helix geometry dictates the unique assignments of the labeled four amino acid stretches that constitute part of the hydrophobic zipper motif (Landschulz et al., 1988; Simmerman et al., 1996). The PISEMA spectral signature is similar to that observed for the non-phosphorylated form (Traaseth et al., 2007). This spectrum enabled us to define the assignment of all the resonances.
Figure 1.

PISEMA spectra of the selectively labeled pS16-PLNWT in mechanically aligned DOPC:DOPE lipid bilayers. Samples were prepared with synthetic (A) or recombinant (B–C) proteins. Two spectra obtained from different synthetic samples are color coded in panel A. Definition of topological angles τ (tilt), ρ (rotation), θ (angle between helices) (D) and their calculation from the structural data (E). Structural model of pS16-PLNWT pentamer, illustrating the tilt angles of the transmembrane (τTM) and cytoplasmic (τCYT) helices defined as the angles between the applied magnetic field (B0) and the corresponding α-helix vectors (h→). The inter-domain angle θ is defined between the vectors h→TM and h→CYT. Performing successive right-handed rotations of an α-helix by negative τ and negative ρ aligns the reference atom (blue) with the X-axis (see Table 1). Helical axes were calculated using the average direction of carbonyl bonds, employing script from PyMOL repository.
The spectrum of the 15N-Leu labeled sample confirmed the tilt and azimuthal rotation angle adopted by the TM domain. The dispersion of the chemical shifts and dipolar couplings as well as the intensities of the resonances are indicative of residues clustered primarily on one face of the helix (Figure 1, C). The 15N-Leu spectrum obtained using synthetic samples provided additional assignments for the remaining Leu residues; these were then used as restraints in the structure calculations and classified as ambiguous (Nilges, 1997) (Figure S4). The resonances at the C-terminus show scaled dipolar and chemical shift values due to their dynamic nature; therefore, these values were not used as constraints for the structure calculation. To assess the orientation of the juxtamembrane domain Ib and the loop, we labeled PLN with 15N-Gln and 15N-Glu. Whereas the values of the dipolar couplings and chemical shifts of those residues located in the loop are scaled by conformational dynamics, residues in the juxtamembrane domain Ib are more rigid, giving valuable constraints to determine PLN topology and assisting in the determination of the orientation of the cytoplasmic domain Ia (Figure 1).
To compare the tilt angles between the non-phosphorylated and phosphorylated forms of PLNWT, we recorded PISEMA spectra of U-15N labeled proteins in dimyristoylphosphatidylcholine (DMPC)/1-pamlitoyl,2-oleoyl-sn-glycero-3-phosphocholine (POPC)/dihexaneoylphosphatidylcholine (DHPC) bicelles. The samples were prepared in D2O to eliminate those resonances that are in contact with the bulk solvent (Figure S5, B–C). From the analysis of the two spectra, we concluded that the DC/CSA pattern for the TM domains overlaps with that of the non-phosphorylated form, indicating that tilt and rotation angles of the pS16-PLNWT species are similar to the non-phosphorylated protein. Since the hydrophobic length of the DMPC/POPC is ~4 Å shorter than the DOPC/DOPE membrane (de Planque and Killian, 2003), the tilt angle of PLN is affected by the protein-lipid interactions. In fact, the size of the PISA wheel (DC/CSA pattern) (Marassi and Opella, 2000; Mascioni and Veglia, 2003; Wang et al., 2000) in bicelles is larger than that measured in mechanically aligned lipid bilayers. The dipolar couplings measured for the TM domain in mechanically aligned samples range from 4 to 9 kHz; in contrast, those measured in bicelles range from 2 to 8 kHz. In principle, the data from both bicelles and mechanically aligned samples can be used for determining the topology of PLN; however, we used only those determined in the DOPC/DOPE mixture that closely mimics SR membrane composition for the structural refinement (Gustavsson et al., 2011b).
To determine inter-monomer distances in the pentameric assembly in the lipid bilayer membrane, we reconstituted the pentameric phosphorylated PLN in lipid vesicles and performed magic angle spinning (MAS) experiments. We employed an asymmetric labeling scheme previously utilized for both solution and solid-state NMR experiments, labeling 13C Leu of one protomer and 13C Ile of the other protomer (Verardi et al., 2012). The isotopically labeled proteins were expressed individually and mixed together in a 1:1 molar ratio (Traaseth et al., 2008b). Using this isotopic scheme in concert with dipolar-assisted rotational resonance (DARR) experiments (Takegoshi et al.), we were able to detect the dipolar contacts between adjacent protomers. The DARR spectrum of pS16-PLNWT reported in Figure 2 exhibits several cross-peaks between Leu and Ile of the adjacent protomers. Remarkably, the interactions are not limited to the methyl-methyl contacts alone; several inter-protomer cross peaks between Cβ and Cα resonances are also detected. The DARR cross-peaks show multiple dipolar interactions between one or more Leu (nine residues in the domain II) and one or more Ile (six residues), the latter indicating the tight packing of the protomers’ interface. Although this labeling scheme does not provide resonance specific assignments, the dipolar contacts can be used in the structure calculations as ambiguous long-range distances, and can be easily assigned when combined with the orientational restrains from SLF experiments (Figure 2, C). The network of dipolar interactions coincides with that observed for PLNWT, indicating a similar arrangement of the TM domains. The rotation angles calculated from the SLF data restrict the interfacial orientations between the protomers, defining the Leu-Ile zipper motif between residues 33, 40, 47 (Ile) and 37, 44, 51 (Leu). Furthermore, the rotation angle of the TM helices positions the helices such that Ile residues of a given protomer interlock with Leu of an adjacent protomer located clockwise, resulting in a left-handed coiled-coil arrangement (Figure 3) (Crick, 1953; Walters and DeGrado, 2006).
Figure 2.

Arrangement of Leu (red) and Ile (green) side chains on the structure of pS16 form (one protomer is removed for clarity) (A). Strips from DARR spectrum of the mixed Ile and Leu pS16-PLNWT (each protomer labeled with only one amino acid type) (B). Ambiguous restraints modeled from the DARR data (C).
Figure 3.

Structural ensembles of PLNWT in the non-phosphorylated (left) and pS16 (right) forms as viewed along the membrane normal (A, B) or perpendicular to it (E, F). Structures are aligned on all backbone atoms. The residues 1–17 are shown in ribbon projection, illustrating disorder near the phosphorylation site when the alignment is performed on the heavy atoms of only these residues (C, D). Side chains of the cytoplasmic domain are shown as sticks. Carbon atoms of the side chains are colored green for hydrophobic residues and cyan for hydrophilic ones. Structural statistics are reported in the Supplemental Information (Tables S1–S3, Figures S7–S8).
Summaries of the experimental restraints and the common metrics used to assess the structure quality are provided in Tables 1 and 2, respectively. Since the azimuthal orientation of the cytoplasmic domains relative to the transmembrane domains is restrained only by steric interactions between them, the cytoplasmic helix for an individual protomer can cover an azimuthal range of about 180° (Figure 3), leading to the large root-mean-square deviation (RMSD) for the whole pentameric assembly in both the non-phosphorylated and phosphorylated forms. Nevertheless, if the structures are analyzed in terms of the individual domains, the RMSD values drop sharply to the acceptable values. Orientation of the TM domain is still governed by the Leu-Ile side chain packing, which tolerates little variation in topology of the TM helix (Table 3). The ordered segment of the cytoplasmic domain is not altered by phosphorylation either, as the negatively charged phosphate group does not shift the domain’s amphiphilic character. The structural ensemble has been deposited to the Protein Data Bank with accession code 2M3B.
Table 2.
Structural statistics of apo (PDB entry 2KYV) and pS16 forms of WT PLN (ensemble of 20 structures). Data was evaluated using Protein Structure Validation Suite (Bhattacharya et al., 2007), Molprobity (Davis et al., 2007), Pymol (DeLano, 2008) and in-house scripts.
| Criterion | S16 | pS16 |
|---|---|---|
| Ramachandran angles (Procheck), per cent | ||
| Most favored | 93.9 | 93.0 |
| Additionally allowed | 5.8 | 6.4 |
| Generously allowed | 0.3 | 0.3 |
| Disallowed | 0.0 | 0.3 |
| Helical residues (DSSP) | 3–17 and 25–51 | 2–14 and 24–51 |
| Clashscore (Molprobity) | 3.1 ± 1.6 | 6.0 ± 1.7 |
| Molprobity score | 1.8 ± 0.3 | 2.4 ± 0.2 |
| RMSD of helical domains, Å1 | ||
| Transmembrane domain: | ||
| Backbone (pentamer) | 0.6 ± 0.1 | 1.6 ± 0.3 |
| Heavy atom (pentamer) | 1.3 ± 0.1 | 2.1 ± 0.3 |
| Backbone (protomer) | 0.4 ± 0.1 | 1.3 ± 0.3 |
| Heavy atom (protomer) | 1.1 ± 0.1 | 1.8 ± 0.3 |
| Cytoplasmic domain: | ||
| Backbone (pentamer) | 4.8 ± 0.8 | 5.6 ± 1.5 |
| Heavy atom (pentamer) | 5.4 ± 0.8 | 5.8 ± 1.5 |
| Backbone (protomer) | 1.0 ± 0.2 | 0.2 ± 0.1 |
| Heavy atom (protomer) | 1.6 ± 0.2 | 0.5 ± 0.2 |
Root mean squared deviation was calculated using the helical domains of each of the ensemble members as a reference structure. The reported values correspond to the average ± standard deviation of the individual RMSDs. The calculation was performed in Pymol, using in-house scripts built around “intra_fit” function.
Table 3.
Topological parameters of α-helices in the structural ensemble of pS16-PLNWT.
| Span, residues1 | Tilt τ, degrees | Rotation ρ, degrees2 | |
|---|---|---|---|
| pS16-PLNWT | |||
| Cytoplasmic helix | 2–14 | 100 ± 5 | 314 ± 10 |
| Transmembrane helix | 24–51 | 11 ± 3 | 302 ± 18 |
| S16-PLNWT, 3 | |||
| Cytoplasmic helix | 3–17 | 86 ± 3 | 308 ± 18 |
| Transmembrane helix | 25–51 | 10 ± 1 | 274 ± 13 |
Calculated using DSSP.
Reference atoms are amide nitrogens of T8 (cytoplasmic helix) and L37 (transmembrane helix). For definition of angles see Figure 1.
PDB entry 2KYV (Verardi et al., 2011).
Angle θ between the helices was calculated to be 100±8° (pS16-PLNWT) and 94±6° (S16-PLNWT).
As shown previously, the monomeric mutant of PLN is capable of inhibiting SERCA, and the monomer is likely to be the biologically active species, although recent works have proposed the pentamer to be the inhibitory species (Cornea et al., 2000; Glaves et al., 2011). To assess the inhibitory activity of PLNWT in the non-phosphorylated and pS16 states we have performed coupled enzyme assays to establish the shift of the calcium concentration required for SERCA in the presence of PLN to regain its initial activity (ΔpKCa). As seen in Figure 4 the addition of PLNWT shifts the activity curve toward the higher calcium concentrations, causing a pKCa shift of 0.3, which is nearly 50% of the monomeric PLNAFA ΔpKCa (Gustavsson et al., 2011b). For the phosphorylated species of PLNWT we observe the complete relief of inhibition, in lines with the pS16-PLNAFA. If the pentamer was indeed the inhibitory form of PLN, one would expect it to increase SERCA inhibition in comparison with the monomeric PLNAFA; nevertheless we observe the opposite trend. As noted earlier, pentameric PLNWT exists at an equilibrium with the monomeric species, the equilibrium being shifted toward the former. The decrease of the available monomeric units for PLNWT and the decrease in SERCA inhibition caused by PLNWT in comparison with PLNAFA are in line with the explanation that monomeric PLN is the active inhibitory form.
Figure 4.

Coupled enzyme assays for SERCA alone (black), SERCA in the presence of PLNWT (red) or pS16-PLNWT (blue).
Conformational Dynamics of pentameric phosphorylated PLN
To probe the motions of PLN, we measured the longitudinal (T1) and transverse (T2) nuclear relaxation times and converted them into R1 and R2, the longitudinal and transverse relaxation rates, respectively. As a proxy for conformational dynamics, we also measured 1H-15N heteronuclear steady-state NOE (HX NOE, Figure S6). These relaxation parameters were measured at two magnetic field strengths. The relaxation parameters for the transmembrane regions of both phosphorylated and non-phosphorylated forms of PLN are very similar. Domains Ia and II exhibited different spin-lattice relaxation rates, and the mean values of the cytoplasmic domain were approximately 20–50% higher for the non-phosphorylated and pS16 forms (Figure S6). Both non-phosphorylated and phosphorylated PLNWT have very similar R1 values, which are mostly sensitive to the global tumbling of the proteins. At higher fields, there was a uniform increase in the longitudinal relaxation rates, suggesting that the global correlation time is not influenced by phosphorylation.
The relaxation parameters for pentameric wild-type and monomeric (AFA) PLN are compared in Figures 5 and S6. To estimate the local correlation times, we plotted the R2/R1 ratios along the sequence of PLN (Jarymowycz and Stone, 2006). We note substantial differences between resonances located in the cytoplasmic domains and those belonging to the TM domains. However, the dimensionless values of the R2/R1 ratios of the pentamers are similar to those of the monomeric species, indicating that the dominant factor contributing to the rotational correlation times is the size of the DPC micelle coating the proteins (de Medeiros et al., 2010; Vostrikov et al., 2011b). Similar to the case with monomeric PLNAFA (Metcalfe et al., 2005; Metcalfe et al., 2004), phosphorylation of PLNWT did not affect the motions of TM helix. Nevertheless, the differences in the R2 values for the non-phosphorylated and phosphorylated proteins led to a decreased R2/R1 ratio for pS16-PLN. These changes are apparent for the PLNWT, as the R2/R1 ratios are relatively constant throughout the sequence, with the exception of residues located near Ser16.
Figure 5.

Model independent relaxation parameters for non-phosphorylated and pS16 forms of pentameric PLNWT and monomeric PLNAFA at 600 MHz (A, C). Model independent relaxation parameters derived from the 600 MHz data plotted as a color map on the structures of PLN in the non-phosphorylated and pS16 forms (B, D). Color scales for a given parameter are indicated and ramped in increment of one unit, absent data is in white. See also Figure S6.
The difference between the two major helical domains of PLN is also apparent in the R1 × R2 products (Kneller et al., 2002) (Figure S6). While the values for the TM helices are similar within the experimental error, the values for the cytoplasmic domain of the pS16-PLN are consistently lower than those of the non-phosphorylated form. Moreover, the changes near Ser16 are more pronounced and also present in the flexible loop. Other variations are also observed in the N-terminal residues of domain Ib from residue 22 to approximately residue 25 – a crucial region for SERCA regulation (Kimura et al., 1998). Interestingly, this region shows opposite effects upon phosphorylation, exhibiting the highest R1 × R2 values throughout the entire sequence. These elevated R1 × R2 values are indicative of the presence of motions in the μs-ms timescales. The pentameric and monomeric forms of pS16-PLN differ in the length of this region, i.e., while PLNWT has higher R1 × R2 values immediately after P21, the monomer PLN has a longer stretch of residues with higher values, extending to the loop.
Finally, we calculated R1 × (1 − NOE) values that are used to characterize fast (ps to ns) timescale dynamics (Jarymowycz and Stone, 2006). Figure 5 Shows that phosphorylation at S16 leads to an increase of the local motions manifested by the elevated R1 × (1 − NOE) values. Considerable differences are observed between the pS16 forms of wild-type and monomeric proteins. For pS16-PLNWT, the N-terminal residues are largely unaffected, while those located in the middle of the cytoplasmic domain up to the beginning of domain II display increased motions. Conversely, pS16-PLNAFA displays changes in the fast motion regime throughout the entire cytoplasmic region, including domain Ia, loop, and domain Ib.
The increased conformational dynamics of the phosphorylated cytoplasmic helix were confirmed by solvent accessibility data. To determine the exposure of each residue to the solvent or its depth of insertion into the aliphatic core of the micelle, we performed [1H, 15N]-NOESY experiments on pS16-PLNWT in protonated DPC and quantified the NOE cross peaks between the amide resonances and the water peak (for solvent-exposed residues), or between amides and the aliphatic resonances of the DPC molecules (for residues buried in the membrane) (Figure S5) (Fernandez et al., 2002). While the non-phosphorylated PLN exhibited an oscillatory NOE pattern demonstrating a strong association between the amphipathic domain Ia and the micelle (Verardi et al., 2011), the majority of the pS16 cytoplasmic domain exhibits NOE cross peaks between amide groups and the water resonance. The only exceptions are Y6 and L7, which show cross peaks with both the bulk solvent and the micelles, showing that these residues populate two major states: one in contact with the micelle and the other solvent exposed. Residues 26–27 in domain Ib located at the membrane interface show several NOE cross peaks with the aliphatic chains of the DPC micelle. Nonetheless, these residues are still partially accessible to water, while the rest of the sequence makes contact only with the micelle.
Taken with the spin relaxation data, the NOE patterns indicate that phosphorylation increases the dynamics in this domain, and shifts the population of PLN from the micelle absorbed T state toward the micelle desorbed R state (Gustavsson et al., 2011a).
Discussion
The physiological response to PLN phosphorylation is a net increase of the ATPase’s apparent affinity for Ca2+, with a concomitant augmentation of muscle contractility. The molecular mechanisms for this process are unknown. One possibility is that PLN forms ion channels that increase Ca2+ flux into the SR membrane, thereby increasing diastole. Patch-clamp experiments, electrochemical measurements with synthetic membranes, and PLN pentamer structure determined in detergent micelles support this hypothesis (Oxenoid and Chou, 2005; Smeazzetto et al., 2013). However, the structural topology of the PLN pentamer in lipid bilayers does not reveal the canonical architecture of an ion channel (Traaseth et al., 2008a; Traaseth et al., 2007; Verardi et al., 2011). The pentameric assembly with a pinwheel topology displays a narrow and hydrophobic pore ~25 Å in length and ~2 Å in diameter on average (Traaseth et al., 2008a; Traaseth et al., 2007; Verardi et al., 2011). Free energy calculations show that the energy barrier for Ca2+ ion transport is ~40 kcal/mol, which is prohibitive in the absence of an energy source (Becucci et al., 2009). A recent paper opened up the possibility that phosphorylation at Ser16 might be responsible for ion transport or Ca2+ leakage (Aschar-Sobbi et al., 2012), motivating our structural studies.
Several previous biophysical studies were devoted to pS16-PLN including circular dichroism (Terzi et al., 1992), fluorescence (Li et al., 1998), EPR (Cornea et al., 1997), solution NMR in organic/aqueous mixture (Pollesello and Annila, 2002), and site-specific solid-state NMR (Chu et al., 2010); these studies exclude major conformational changes in the TM domain of PLN, which would justify the formation of a channel. Cornea et al. showed that upon phosphorylation the thermostability of the pentamer increases possibly affecting the number of regulatory monomeric protomers (Cornea et al., 1997), suggesting that electrostatic interactions due to phosphorylation at Ser16 cause a complete rearrangement of the cytoplasmic amphipathic helix. However, the structure and dynamics of the Ser16 phosphorylated monomeric PLN in DPC micelles revealed only a local unwinding of the cytoplasmic helix in the proximity of the phosphorylation site, without substantial long-range structural effects in the TM domain (Metcalfe et al., 2005). The latter was supported by a study carried out by Lorigan and coworkers, who used site-specific ssNMR measurements to demonstrate that the TM domains of PLN are essentially intact upon phosphorylation, with minor structural and dynamical effects localized to the cytoplasmic domain (Abu-Baker and Lorigan, 2006). On the other hand, fluorescence resonance energy transfer (FRET) experiments with probes engineered both in the cytoplasmic and TM domains of PLN suggested an increase in helical content (Li et al., 2003). A comparison between PLNWT and its pseudophosphorylated analog, in which negatively charged glutamic acid is substituted for serine, did not reveal significant differences between the two states (Oxenoid et al., 2007).
The present study, which was carried out with phosphorylated PLN in lipid bilayers and micelles, establishes that the overall architecture of the pS16-PLNWT is similar to that of the non-phosphorylated form, with a reduced helicity of the cytoplasmic domain in proximity to the phosphorylation site. The calculated cavity and its maximum diameter as a function of Z-coordinate are represented in Figure 6. As for the non-phosphorylated pentamer, the pore of the phosphorylated pentameric assembly does not exceed 3 Å. The pore is coated exclusively by side chains; none of the polar backbone atoms are accessible from the inside. Three of the side chains located in the juxtamembrane region are moderately hydrophilic (Q23, Q26, Q29), and form inter-protomer (Q29) and possibly intra-protomer (Q23 to Q26 or Q26 to Q29) polar contacts. The rest of the residues in the pore are hydrophobic methyl groups of the zipper motif (Leu 37, 44, 51 and Ile 33, 40, 47). Although this organization is reminiscent of ion channel architecture (hydrophilic residues that provide selectivity and hydrophobic ones which strip the water shell), there is a restriction of the pore around residues 26, 29, 33 and 37 with the average diameter ranging from 1.4 to 1.8 Å, which would prevent Ca2+ ion transport (Tsien et al., 1987). In a recently solved structure of the Orai Ca2+ channel, the average pore diameter is 3 Å and can reach 1.5 Å (Hou et al., 2012). In this case, however, the charges present in the side chains coating the inner pore and their rearrangement would facilitate ion translocation.
Figure 6.
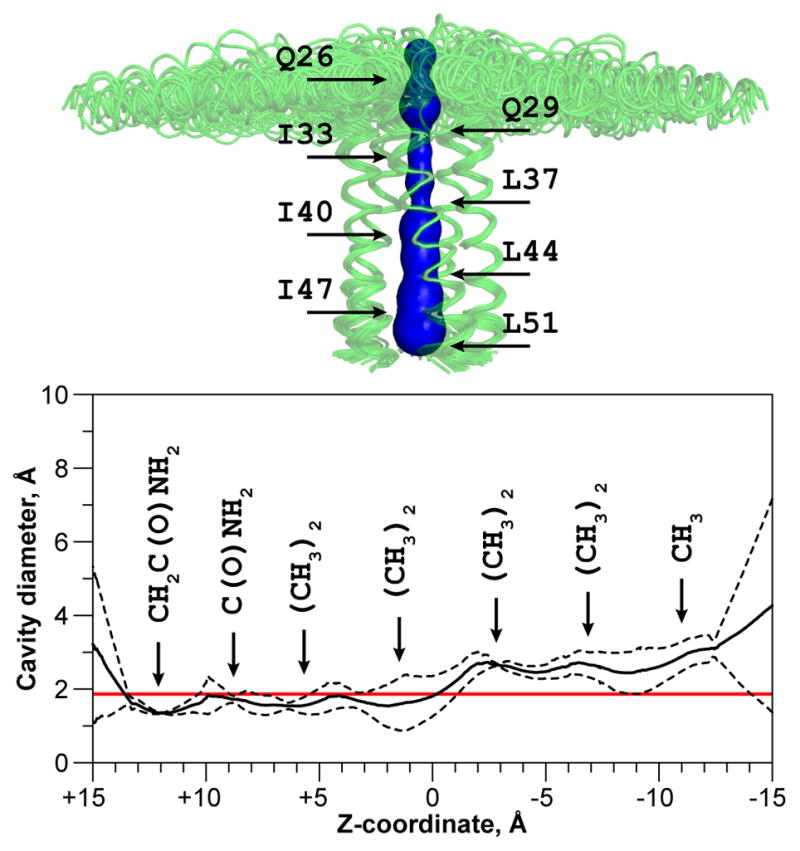
Inner pore of the pentameric assembly of pS16-PLNWT. The structural features were analyzed with the program MOLE (Petrek et al., 2007). The surface of the pore is shown in blue and is overlaid with the structural ensemble, aligned at the transmembrane domains. The maximum pore diameter has been calculated for the five lowest energy structures and is presented in the mean ± standard deviation form. Red line indicates the diameter of non-hydrated Ca2+ ion (1.98 Å, (Tsien et al., 1987)).
For the pS16-PLNWT pentamer, the juxtamembrane region does not possess negatively charged residues that could constitute a selectivity filter, which in Orai closely matches the diameter of the ion. The pore constrictions at glutamines 26 and 29 are mainly defined by the amide group, a less efficient ligand for Ca2+ (Forsen and Kordel, 1994). Importantly, the hydrophobic residues span five helical turns, constituting a high energy barrier to ion transport. Also, the preferred coordination geometry for Ca2+ ions is pentagonal bipyramid (akin to EF-hand motif, (Yang et al., 2002)), which requires an additional ligand positioned deeper in the pore. The pore of PLN is devoid of such a ligand, and it is unlikely to be water (Becucci et al., 2009). Recent electrochemical impedance spectroscopy measurements on pS16-PLNWT also revealed its inability to conduct ions at physiologically relevant transmembrane potentials (Becucci et al., 2013).
The most significant changes that occur upon phosphorylation are located in the cytoplasmic domains. The amphipathic cytoplasmic domain Ia of PLN populates both folded, membrane associated (T-state) and unfolded, membrane detached (R-state) conformations (Gustavsson et al., 2011a; Karim et al., 2004). This equilibrium, critical for SERCA regulation, is present for the phosphorylated protein with the population of the unfolded state increasing upon phosphorylation (Gustavsson et al., 2011a; Gustavsson et al., 2012). The phosphate group in position 16 increases conformational dynamics in the pico- to nanosecond timescale, encompassing residues in the loop up to the distal sites of the N-terminus. These conformational dynamics facilitate the folded-to-unfolded (order-to-disorder) transition of the cytoplasmic domain, which can bind and regulate SERCA function.
Previously, we demonstrated that there is an inverse correlation between enhanced conformational dynamics of domain Ia and PLN inhibitory potency (Ha et al., 2012; Ha et al., 2007). Site-specific mutations that increase PLN internal dynamics, promoting detachment of domain Ia from the membrane and concomitant unfolding, also upregulate SERCA. Thus the non-phosphorylated form of monomeric PLNAFA bearing the P21G mutation shows enhanced dynamics in this region and behaves functionally as the pseudophosphorylated S16E mutation, while two glycine mutations fully mimic the effects of phosphorylation (Chien, 2006; Ha et al., 2007). Nonetheless the coupling between the two domains remains essential, and an overly flexible loop has been shown not to follow the linear trend (Ha et al., 2012). The phosphorylation event described here introduces disorder in the C-terminal portion of the cytoplasmic domain, leading to its partial unfolding and detachment from the membrane, the conformation of the cytoplasmic domain being akin to the T′ state (Gustavsson et al., 2011a), along the energy landscape of this polymorphic domain.
Although they dismiss the formation of a classical ion channel, our results on the pentameric pS16-PLNWT support our regulatory model, where oligomerization has buffer effects on SERCA regulation, fine-tuning the concentration of phosphorylated monomers that can bind and upregulate SERCA. Both phosphorylated and non-phosphorylated PLN keep SERCA function within a well-defined physiological window, and exceeding this window either by superinhibition or loss of function causes the progression of heart disease (Kranias and Hajjar, 2012). Keeping PLN regulation of SERCA within this physiological window requires maintaining a correct equilibrium between its monomeric and oligomeric states (Chu et al., 1997; Ha et al., 2011), its ability to be effectively phosphorylated (Ceholski et al., 2012; Said et al., 2003), net charge of the regulatory domain (Hou et al., 2008; Medeiros et al., 2011), and structural communication between TM and regulatory domains for effective signal transduction (Ha et al., 2012).
Since SERCA and PLN are emerging as targets for gene therapy by adeno-associated gene transfer, knowledge about all the functional states of these proteins is central to the design of appropriate strategies for intervention (Anderson et al., 2011; Hadri et al., 2010; Marks, 2013; Naim et al., 2013).
Supplementary Material
Acknowledgments
This work was supported by the National Institute of Health (GM 64742)
This work is supported by a grant from National Institute of Health (GM64742 to G.V.) and postdoctoral fellowship from American Heart Association (13POST14670054 to V.V.). The authors would like to thank Dr. Martin Gustavsson for helpful discussions, and Drs. Lucia Becucci and Rolando Guidelli for sharing electrochemistry data prior to publication. Solid-state NMR data were acquired at the Minnesota NMR Center and at the NHMFL. Relaxation data at 750 MHz were acquired at NMRFAM. Structure calculation utilized computing resources at the University of Minnesota Supercomputing Institute. V.V, K.R.M., R.V. and G.V. designed the experiments and participated in manuscript preparation, V.V., K.R.M. and R.V. performed the experiments and analyzed the data.
Abbreviations
- DARR
dipolar assisted rotational resonance
- DPC
dodecylphosphocholine
- PKA
protein kinase A
- PLNAFA
phospholamban monomer bearing mutations C36A C41F C46A
- PLNWT
wild type phospholamban pentamer
- pS
phosphoserine
- SERCA
sarco(endo)plasmic reticulum Ca2+ ATPase
- SLF
separated local field
- SR
sarcoplasmic reticulum
- TM
transmembrane
Footnotes
Supplemental Information includes 8 figures and 1 table and can be found with this article online at ***
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abu-Baker S, Lorigan GA. Phospholamban and its phosphorylated form interact differently with lipid bilayers: a 31P, 2H, and 13C solid-state NMR spectroscopic study. Biochemistry. 2006;45:13312–13322. doi: 10.1021/bi0614028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson ME, Brown JH, Bers DM. CaMKII in myocardial hypertrophy and heart failure. J Mol Cell Cardiol. 2011;51:468–473. doi: 10.1016/j.yjmcc.2011.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aschar-Sobbi R, Emmett TL, Kargacin GJ, Kargacin ME. Phospholamban phosphorylation increases the passive calcium leak from cardiac sarcoplasmic reticulum. Pflugers Arch. 2012;464:295–305. doi: 10.1007/s00424-012-1124-9. [DOI] [PubMed] [Google Scholar]
- Becucci L, Cembran A, Karim CB, Thomas DD, Guidelli R, Gao J, Veglia G. On the function of pentameric phospholamban: ion channel or storage form? Biophys J. 2009;96:L60–L62. doi: 10.1016/j.bpj.2009.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becucci L, Foresti ML, Schwan A, Guidelli R. Can proton pumping by SERCA enhance the regulatory role of phospholamban and sarcolipin? Biochim Biophys Acta. 2013;1828:2682–2690. doi: 10.1016/j.bbamem.2013.07.012. [DOI] [PubMed] [Google Scholar]
- Bhattacharya A, Tejero R, Montelione GT. Evaluating protein structures determined by structural genomics consortia. Proteins. 2007;66:778–795. doi: 10.1002/prot.21165. [DOI] [PubMed] [Google Scholar]
- Buck B, Zamoon J, Kirby TL, DeSilva TM, Karim C, Thomas D, Veglia G. Overexpression, purification, and characterization of recombinant Ca-ATPase regulators for high-resolution solution and solid-state NMR studies. Protein Expr Purif. 2003;30:253–261. doi: 10.1016/s1046-5928(03)00127-x. [DOI] [PubMed] [Google Scholar]
- Ceholski DK, Trieber CA, Holmes CF, Young HS. Lethal, hereditary mutants of phospholamban elude phosphorylation by protein kinase A. J Biol Chem. 2012;287:26596–26605. doi: 10.1074/jbc.M112.382713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chien KR. Beyond small molecule drugs for heart failure: prospects for gene therapy. Novartis Found Symp. 2006;274:244–256. [PubMed] [Google Scholar]
- Chu G, Dorn GW, 2nd, Luo W, Harrer JM, Kadambi VJ, Walsh RA, Kranias EG. Monomeric phospholamban overexpression in transgenic mouse hearts. Circ Res. 1997;81:485–492. doi: 10.1161/01.res.81.4.485. [DOI] [PubMed] [Google Scholar]
- Chu S, Abu-Baker S, Lu J, Lorigan GA. 15N Solid-state NMR spectroscopic studies on phospholamban at its phosphorylated form at Ser-16 in aligned phospholipid bilayers. Biochim Biophys Acta. 2010;1798:312–317. doi: 10.1016/j.bbamem.2009.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornea RL, Autry JM, Chen Z, Jones LR. Reexamination of the role of the leucine/isoleucine zipper residues of phospholamban in inhibition of the Ca2+ pump of cardiac sarcoplasmic reticulum. J Biol Chem. 2000;275:41487–41494. doi: 10.1074/jbc.M008195200. [DOI] [PubMed] [Google Scholar]
- Cornea RL, Jones LR, Autry JM, Thomas DD. Mutation and phosphorylation change the oligomeric structure of phospholamban in lipid bilayers. Biochemistry. 1997;36:2960–2967. doi: 10.1021/bi961955q. [DOI] [PubMed] [Google Scholar]
- Cornell BA, Separovic F, Baldassi AJ, Smith R. Conformation and orientation of gramicidin A in oriented phospholipid bilayers measured by solid state carbon-13 NMR. Biophys J. 1988;53:67–76. doi: 10.1016/S0006-3495(88)83066-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crick FHC. The Packing of α-Helices: Simple Coiled-Coils. Acta Crystallogr. 1953;6:689–697. [Google Scholar]
- Davis IW, Leaver-Fay A, Chen VB, Block JN, Kapral GJ, Wang X, Murray LW, Arendall WB, 3rd, Snoeyink J, Richardson JS, Richardson DC. MolProbity: all-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res. 2007;35:W375–W383. doi: 10.1093/nar/gkm216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Medeiros LN, Angeli R, Sarzedas CG, Barreto-Bergter E, Valente AP, Kurtenbach E, Almeida FC. Backbone dynamics of the antifungal Psd1 pea defensin and its correlation with membrane interaction by NMR spectroscopy. Biochim Biophys Acta. 2010;1798:105–113. doi: 10.1016/j.bbamem.2009.07.013. [DOI] [PubMed] [Google Scholar]
- de Planque MR, Killian JA. Protein-lipid interactions studied with designed transmembrane peptides: role of hydrophobic matching and interfacial anchoring. Mol Membr Biol. 2003;20:271–284. doi: 10.1080/09687680310001605352. [DOI] [PubMed] [Google Scholar]
- DeLano WL. The PyMOL Molecular Graphics System 2008 [Google Scholar]
- Farrow NA, Muhandiram R, Singer AU, Pascal SM, Kay CM, Gish G, Shoelson SE, Pawson T, Forman-Kay JD, Kay LE. Backbone dynamics of a free and phosphopeptide-complexed Src homology 2 domain studied by 15N NMR relaxation. Biochemistry. 1994;33:5984–6003. doi: 10.1021/bi00185a040. [DOI] [PubMed] [Google Scholar]
- Fernandez C, Hilty C, Wider G, Wuthrich K. Lipid-protein interactions in DHPC micelles containing the integral membrane protein OmpX investigated by NMR spectroscopy. Proc Natl Acad Sci U S A. 2002;99:13533–13537. doi: 10.1073/pnas.212515099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forsen S, Kordel J. Calcium in Biological Systems. In: Bertini I, Gray HB, Lippard SJ, Valentine JS, editors. Bioinorganic Chemistry. Mill Valley, CA: University Science Books; 1994. pp. 107–166. [Google Scholar]
- Glaves JP, Trieber CA, Ceholski DK, Stokes DL, Young HS. Phosphorylation and mutation of phospholamban alter physical interactions with the sarcoplasmic reticulum calcium pump. J Mol Biol. 2011;405:707–723. doi: 10.1016/j.jmb.2010.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gor’kov PL, Chekmenev EY, Fu R, Hu J, Cross TA, Cotten M, Brey WW. A large volume flat coil probe for oriented membrane proteins. J Magn Reson. 2006;181:9–20. doi: 10.1016/j.jmr.2006.03.008. [DOI] [PubMed] [Google Scholar]
- Gustavsson M, Traaseth NJ, Karim CB, Lockamy EL, Thomas DD, Veglia G. Lipid-mediated folding/unfolding of phospholamban as a regulatory mechanism for the sarcoplasmic reticulum Ca2+-ATPase. J Mol Biol. 2011a;408:755–765. doi: 10.1016/j.jmb.2011.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gustavsson M, Traaseth NJ, Veglia G. Activating and deactivating roles of lipid bilayers on the Ca2+-ATPase/phospholamban complex. Biochemistry. 2011b;50:10367–10374. doi: 10.1021/bi200759y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gustavsson M, Traaseth NJ, Veglia G. Probing ground and excited states of phospholamban in model and native lipid membranes by magic angle spinning NMR spectroscopy. Biochim Biophys Acta. 2012;1818:146–153. doi: 10.1016/j.bbamem.2011.07.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ha KN, Gustavsson M, Veglia G. Tuning the structural coupling between the transmembrane and cytoplasmic domains of phospholamban to control sarcoplasmic reticulum Ca2+-ATPase (SERCA) function. J Muscle Res Cell Motil. 2012;33:485–492. doi: 10.1007/s10974-012-9319-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ha KN, Masterson LR, Hou Z, Verardi R, Walsh N, Veglia G, Robia SL. Lethal Arg9Cys phospholamban mutation hinders Ca2+-ATPase regulation and phosphorylation by protein kinase A. Proc. Natl Acad Sci U S A. 2011;108:2735–2740. doi: 10.1073/pnas.1013987108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ha KN, Traaseth NJ, Verardi R, Zamoon J, Cembran A, Karim CB, Thomas DD, Veglia G. Controlling the inhibition of the sarcoplasmic Ca2+-ATPase by tuning phospholamban structural dynamics. J Biol Chem. 2007;282:37205–37214. doi: 10.1074/jbc.M704056200. [DOI] [PubMed] [Google Scholar]
- Hadri L, Bobe R, Kawase Y, Ladage D, Ishikawa K, Atassi F, Lebeche D, Kranias EG, Leopold JA, Lompre AM, et al. SERCA2a gene transfer enhances eNOS expression and activity in endothelial cells. Mol Ther. 2010;18:1284–1292. doi: 10.1038/mt.2010.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou X, Pedi L, Diver MM, Long SB. Crystal structure of the calcium release-activated calcium channel Orai. Science. 2012;338:1308–1313. doi: 10.1126/science.1228757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou Z, Kelly EM, Robia SL. Phosphomimetic mutations increase phospholamban oligomerization and alter the structure of its regulatory complex. J Biol Chem. 2008;283:28996–29003. doi: 10.1074/jbc.M804782200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarymowycz VA, Stone MJ. Fast time scale dynamics of protein backbones: NMR relaxation methods, applications, and functional consequences. Chem Rev. 2006;106:1624–1671. doi: 10.1021/cr040421p. [DOI] [PubMed] [Google Scholar]
- Karim CB, Kirby TL, Zhang Z, Nesmelov Y, Thomas DD. Phospholamban structural dynamics in lipid bilayers probed by a spin label rigidly coupled to the peptide backbone. Proc Natl Acad Sci U S A. 2004;101:14437–14442. doi: 10.1073/pnas.0402801101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura Y, Asahi M, Kurzydlowski K, Tada M, MacLennan DH. Phospholamban domain Ib mutations influence functional interactions with the Ca2+-ATPase isoform of cardiac sarcoplasmic reticulum. J Biol Chem. 1998;273:14238–14241. doi: 10.1074/jbc.273.23.14238. [DOI] [PubMed] [Google Scholar]
- Kimura Y, Kurzydlowski K, Tada M, MacLennan DH. Phospholamban regulates the Ca2+-ATPase through intramembrane interactions. J Biol Chem. 1996;271:21726–21731. doi: 10.1074/jbc.271.36.21726. [DOI] [PubMed] [Google Scholar]
- Kimura Y, Kurzydlowski K, Tada M, MacLennan DH. Phospholamban inhibitory function is activated by depolymerization. J Biol Chem. 1997;272:15061–15064. doi: 10.1074/jbc.272.24.15061. [DOI] [PubMed] [Google Scholar]
- King DS, Fields CG, Fields GB. A cleavage method which minimizes side reactions following Fmoc solid phase peptide synthesis. Int J Pept Protein Res. 1990;36:255–266. doi: 10.1111/j.1399-3011.1990.tb00976.x. [DOI] [PubMed] [Google Scholar]
- Kneller JM, Lu M, Bracken C. An effective method for the discrimination of motional anisotropy and chemical exchange. J Am Chem Soc. 2002;124:1852–1853. doi: 10.1021/ja017461k. [DOI] [PubMed] [Google Scholar]
- Kovacs RJ, Nelson MT, Simmerman HK, Jones LR. Phospholamban forms Ca2+-selective channels in lipid bilayers. J Biol Chem. 1988;263:18364–18368. [PubMed] [Google Scholar]
- Kranias EG, Hajjar RJ. Modulation of cardiac contractility by the phospholamban/SERCA2a regulatome. Circ Res. 2012;110:1646–1660. doi: 10.1161/CIRCRESAHA.111.259754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landschulz WH, Johnson PF, McKnight SL. The leucine zipper: a hypothetical structure common to a new class of DNA binding proteins. Science. 1988;240:1759–1764. doi: 10.1126/science.3289117. [DOI] [PubMed] [Google Scholar]
- Li J, Bigelow DJ, Squier TC. Phosphorylation by cAMP-dependent protein kinase modulates the structural coupling between the transmembrane and cytosolic domains of phospholamban. Biochemistry. 2003;42:10674–10682. doi: 10.1021/bi034708c. [DOI] [PubMed] [Google Scholar]
- Li M, Cornea RL, Autry JM, Jones LR, Thomas DD. Phosphorylation-induced structural change in phospholamban and its mutants, detected by intrinsic fluorescence. Biochemistry. 1998;37:7869–7877. doi: 10.1021/bi9801053. [DOI] [PubMed] [Google Scholar]
- MacLennan DH, Kranias EG. Phospholamban: a crucial regulator of cardiac contractility. Nat Rev Mol Cell Biol. 2003;4:566–577. doi: 10.1038/nrm1151. [DOI] [PubMed] [Google Scholar]
- Madden TD, Chapman D, Quinn PJ. Cholesterol modulates activity of calcium-dependent ATPase of the sarcoplasmic reticulum. Nature. 1979;279:538–541. doi: 10.1038/279538a0. [DOI] [PubMed] [Google Scholar]
- Marassi FM, Opella SJ. A solid-state NMR index of helical membrane protein structure and topology. J Magn Reson. 2000;144:150–155. doi: 10.1006/jmre.2000.2035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marks AR. Calcium cycling proteins and heart failure: mechanisms and therapeutics. J Clin Invest. 2013;123:46–52. doi: 10.1172/JCI62834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mascioni A, Veglia G. Theoretical analysis of residual dipolar coupling patterns in regular secondary structures of proteins. J Am Chem Soc. 2003;125:12520–12526. doi: 10.1021/ja0354824. [DOI] [PubMed] [Google Scholar]
- Medeiros A, Biagi DG, Sobreira TJ, de Oliveira PS, Negrao CE, Mansur AJ, Krieger JE, Brum PC, Pereira AC. Mutations in the human phospholamban gene in patients with heart failure. Am Heart J. 2011;162:1088–1095. doi: 10.1016/j.ahj.2011.07.028. [DOI] [PubMed] [Google Scholar]
- Metcalfe EE, Traaseth NJ, Veglia G. Serine 16 phosphorylation induces an order-to-disorder transition in monomeric phospholamban. Biochemistry. 2005;44:4386–4396. doi: 10.1021/bi047571e. [DOI] [PubMed] [Google Scholar]
- Metcalfe EE, Zamoon J, Thomas DD, Veglia G. 1H/15N heteronuclear NMR spectroscopy shows four dynamic domains for phospholamban reconstituted in dodecylphosphocholine micelles. Biophys J. 2004;87:1205–1214. doi: 10.1529/biophysj.103.038844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naim C, Yerevanian A, Hajjar RJ. Gene therapy for heart failure: where do we stand? Curr Cardiol Rep. 2013;15:333. doi: 10.1007/s11886-012-0333-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilges M. Ambiguous distance data in the calculation of NMR structures. Fold Des. 1997;2:S53–S57. doi: 10.1016/s1359-0278(97)00064-3. [DOI] [PubMed] [Google Scholar]
- Oxenoid K, Chou JJ. The structure of phospholamban pentamer reveals a channel-like architecture in membranes. Proc Natl Acad Sci U S A. 2005;102:10870–10875. doi: 10.1073/pnas.0504920102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oxenoid K, Rice AJ, Chou JJ. Comparing the structure and dynamics of phospholamban pentamer in its unphosphorylated and pseudo-phosphorylated states. Protein Sci. 2007;16:1977–1983. doi: 10.1110/ps.072975107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrek M, Kosinova P, Koca J, Otyepka M. MOLE: a Voronoi diagram-based explorer of molecular channels, pores, and tunnels. Structure. 2007;15:1357–1363. doi: 10.1016/j.str.2007.10.007. [DOI] [PubMed] [Google Scholar]
- Pollesello P, Annila A. Structure of the 1–36 N-terminal fragment of human phospholamban phosphorylated at Ser-16 and Thr-17. Biophys J. 2002;83:484–490. doi: 10.1016/S0006-3495(02)75184-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Said M, Vittone L, Mundina-Weilenmann C, Ferrero P, Kranias EG, Mattiazzi A. Role of dual-site phospholamban phosphorylation in the stunned heart: insights from phospholamban site-specific mutants. Am J Physiol Heart Circ Physiol. 2003;285:H1198–H1205. doi: 10.1152/ajpheart.00209.2003. [DOI] [PubMed] [Google Scholar]
- Schwieters CD, Kuszewski JJ, Tjandra N, Clore GM. The Xplor-NIH NMR molecular structure determination package. J Magn Reson. 2003;160:65–73. doi: 10.1016/s1090-7807(02)00014-9. [DOI] [PubMed] [Google Scholar]
- Senes A, Chadi DC, Law PB, Walters RF, Nanda V, DeGrado WF. Ez, a depth-dependent potential for assessing the energies of insertion of amino acid side-chains into membranes: derivation and applications to determining the orientation of transmembrane and interfacial helices. J Mol Biol. 2007;366:436–448. doi: 10.1016/j.jmb.2006.09.020. [DOI] [PubMed] [Google Scholar]
- Shen Y, Delaglio F, Cornilescu G, Bax A. TALOS+: a hybrid method for predicting protein backbone torsion angles from NMR chemical shifts. J Biomol NMR. 2009;44:213–223. doi: 10.1007/s10858-009-9333-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi L, Traaseth NJ, Verardi R, Cembran A, Gao J, Veglia G. A refinement protocol to determine structure, topology, and depth of insertion of membrane proteins using hybrid solution and solid-state NMR restraints. J Biomol NMR. 2009;44:195–205. doi: 10.1007/s10858-009-9328-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simmerman HK, Jones LR. Phospholamban: protein structure, mechanism of action, and role in cardiac function. Physiol Rev. 1998;78:921–947. doi: 10.1152/physrev.1998.78.4.921. [DOI] [PubMed] [Google Scholar]
- Simmerman HK, Kobayashi YM, Autry JM, Jones LR. A leucine zipper stabilizes the pentameric membrane domain of phospholamban and forms a coiled-coil pore structure. J Biol Chem. 1996;271:5941–5946. doi: 10.1074/jbc.271.10.5941. [DOI] [PubMed] [Google Scholar]
- Smeazzetto S, Saponaro A, Young HS, Moncelli MR, Thiel G. Structure-function relation of phospholamban: modulation of channel activity as a potential regulator of SERCA activity. PLoS One. 2013;8:e52744. doi: 10.1371/journal.pone.0052744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smeazzetto S, Schroder I, Thiel G, Moncelli MR. Phospholamban generates cation selective ion channels. Phys Chem Chem Phys. 2011;13:12935–12939. doi: 10.1039/c1cp20460b. [DOI] [PubMed] [Google Scholar]
- Straus SK, Scott WR, Schwieters CD, Marvin DA. Consensus structure of Pf1 filamentous bacteriophage from X-ray fibre diffraction and solid-state NMR. Eur Biophys J. 2011;40:221–234. doi: 10.1007/s00249-010-0640-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subiros-Funosas R, Moreno JA, Bayo-Puxan N, Abu-Rabeah K, Ewenson A, Atias D, Marks RS, Albericio F. PyClock, the phosphonium salt derived from 6-Cl-HOBt. Chimica Oggi - Chemistry Today. 2008;26(supplement):10–12. [Google Scholar]
- Tada M, Kirchberger MA, Katz AM. Phosphorylation of a 22,000-dalton component of the cardiac sarcoplasmic reticulum by adenosine 3′:5′-monophosphate-dependent protein kinase. J Biol Chem. 1975;250:2640–2647. [PubMed] [Google Scholar]
- Takegoshi K, Nakamura S, Terao T. 13C-1H dipolar-assisted rotational resonance in magic-angle spinning NMR. Chem Phys Lett. 2001;344:631–637. [Google Scholar]
- Teixeira A, Benckhuijsen WE, de Koning PE, Valentijn AR, Drijfhout JW. The use of DODT as a non-malodorous scavenger in Fmoc-based peptide synthesis. Protein Pept Lett. 2002;9:379–385. doi: 10.2174/0929866023408481. [DOI] [PubMed] [Google Scholar]
- Terzi E, Poteur L, Trifilieff E. Evidence for a phosphorylation-induced conformational change in phospholamban cytoplasmic domain by CD analysis. FEBS Lett. 1992;309:413–416. doi: 10.1016/0014-5793(92)80819-3. [DOI] [PubMed] [Google Scholar]
- Traaseth NJ, Ha KN, Verardi R, Shi L, Buffy JJ, Masterson LR, Veglia G. Structural and dynamic basis of phospholamban and sarcolipin inhibition of Ca2+-ATPase. Biochemistry. 2008a;47:3–13. doi: 10.1021/bi701668v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traaseth NJ, Shi L, Verardi R, Mullen DG, Barany G, Veglia G. Structure and topology of monomeric phospholamban in lipid membranes determined by a hybrid solution and solid-state NMR approach. Proc Natl Acad Sci U S A. 2009;106:10165–10170. doi: 10.1073/pnas.0904290106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traaseth NJ, Verardi R, Torgersen KD, Karim CB, Thomas DD, Veglia G. Spectroscopic validation of the pentameric structure of phospholamban. Proc Natl Acad Sci U S A. 2007;104:14676–14681. doi: 10.1073/pnas.0701016104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traaseth NJ, Verardi R, Veglia G. Asymmetric methyl group labeling as a probe of membrane protein homo-oligomers by NMR spectroscopy. J Am Chem Soc. 2008b;130:2400–2401. doi: 10.1021/ja711499r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsien RW, Hess P, McCleskey EW, Rosenberg RL. Calcium channels: mechanisms of selectivity, permeation, and block. Annu Rev Biophys Biophys Chem. 1987;16:265–290. doi: 10.1146/annurev.bb.16.060187.001405. [DOI] [PubMed] [Google Scholar]
- van der Wel PC, Strandberg E, Killian JA, Koeppe RE., 2nd Geometry and intrinsic tilt of a tryptophan-anchored transmembrane α-helix determined by 2H NMR. Biophys J. 2002;83:1479–1488. doi: 10.1016/S0006-3495(02)73918-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veglia G, Ha KN, Shi L, Verardi R, Traaseth NJ. What can we learn from a small regulatory membrane protein? Methods Mol Biol. 2010;654:303–319. doi: 10.1007/978-1-60761-762-4_16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verardi R, Shi L, Traaseth NJ, Walsh N, Veglia G. Structural topology of phospholamban pentamer in lipid bilayers by a hybrid solution and solid-state NMR method. Proc Natl Acad Sci U S A. 2011;108:9101–9106. doi: 10.1073/pnas.1016535108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verardi R, Traaseth NJ, Masterson LR, Vostrikov VV, Veglia G. Isotope labeling for solution and solid-state NMR spectroscopy of membrane proteins. Adv Exp Med Biol. 2012;992:35–62. doi: 10.1007/978-94-007-4954-2_3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vostrikov VV, Grant CV, Opella SJ, Koeppe RE., 2nd On the combined analysis of 2H and 15N/1H solid-state NMR data for determination of transmembrane peptide orientation and dynamics. Biophys J. 2011a;101:2939–2947. doi: 10.1016/j.bpj.2011.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vostrikov VV, Gu H, Ingolfsson HI, Hinton JF, Andersen OS, Roux B, Koeppe RE., 2nd Gramicidin A backbone and side chain dynamics evaluated by molecular dynamics simulations and nuclear magnetic resonance experiments. II: nuclear magnetic resonance experiments. J Phys Chem B. 2011b;115:7427–7432. doi: 10.1021/jp200906y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vuister WV, Kim SJ, Wu C, Bax A. 2D and 3D NMR Study of Phenylalanine Residues in Proteins by Reverse Isotopic Labeling. J Am Chem Soc. 1994;116:9206–9210. [Google Scholar]
- Walters RF, DeGrado WF. Helix-packing motifs in membrane proteins. Proc Natl Acad Sci U S A. 2006;103:13658–13663. doi: 10.1073/pnas.0605878103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Denny J, Tian C, Kim S, Mo Y, Kovacs F, Song Z, Nishimura K, Gan Z, Fu R, et al. Imaging membrane protein helical wheels. J Magn Reson. 2000;144:162–167. doi: 10.1006/jmre.2000.2037. [DOI] [PubMed] [Google Scholar]
- Wegener AD, Jones LR. Phosphorylation-induced mobility shift in phospholamban in sodium dodecyl sulfate-polyacrylamide gels. Evidence for a protein structure consisting of multiple identical phosphorylatable subunits. J Biol Chem. 1984;259:1834–1841. [PubMed] [Google Scholar]
- Wu CH, Ramamoorthy A, Opella SJ. High-resolution heteronuclear dipolar solid-state NMR spectroscopy. J Magn Reson Ser A. 1994;109:270–272. [Google Scholar]
- Yang W, Lee HW, Hellinga H, Yang JJ. Structural analysis, identification, and design of calcium-binding sites in proteins. Proteins. 2002;47:344–356. doi: 10.1002/prot.10093. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.