

Abstract
Objective:
To evaluate the mutational spectrum of NLRP7 and KHDC3L (C6orf221) in women with sporadic and recurrent androgenetic complete hydatidiform moles (AnCHM) and biparental hydatidiform moles (BiHM) to address the hypothesis that autosomal recessive mutations in these genes are only or primarily associated with biparental hydatidiform moles.
Method:
We recruited 16 women with suspected recurrent and sporadic AnCHM and 5 women with suspected BiHM in addition to their reproductive partners into our study. We then sequenced the coding exons of NLRP7 and KHDC3L from DNA isolated from either blood or saliva from the study subjects.
Results:
Sequence analysis of NLRP7 and KHDC3L revealed previously described SNPs in patients with AnCHM. However in patients with BiHM, we identified a novel homozygous mutation and a previously described intragenic duplication of exon 2-5 in NLRP7, both of which are likely to be disease causing. We did not identify mutations in KHDC3L in patients with the either forms of HM.
Conclusions:
The absence of mutations in the women with AnCHM supports a role of NLRP7 or KHDC3L in BiHM only. The absence of mutations in KHDC3L in women with BiHM is consistent with its minor role in this disease compared to NLRP7, the major BiHM gene.
Introduction
Gestational trophoblastic diseases (GTD) are a group of disorders which originate from the trophoblast with a wide phenotypic range. Among them are hydatidiform moles (HM), which are characterized by hyperproliferative trophoblast. Patients with HM can present with vaginal bleeding and increased serum β-HCG levels. HM are often first detected by their characteristic appearance on first-trimester ultrasonography. Pathologically and karyotypically, HMs can be classified into complete hydatidiform moles (CHM) and partial hydatidiform moles (PHM). CHM are typically not accompanied by a live fetus and are androgenetic diploid (AnCHM) with both genomes being paternally derived. They result from either monospermic fertilization of an oocyte without a functional maternal pronucleus and duplication of the paternal pronucleus, or from dispermic fertilization of such an oocyte. Partial HM are more focal, are usually associated with fetal development, and are diandric triploid with two paternally inherited and one maternally inherited haploid set of chromosomes. The unequal distribution of parental genomes in CHM and PHM suggests that abnormal expression of imprinted genes plays a role in their phenotype.
A third and extremely rare subset of HM are biparentally inherited (BiHM), usually recurrent and often familial (OMIM 231090). The phenotypic similarity of these BiHM to AnCHM indicates that imbalanced regulation of imprinted genes contributes to their pathogenesis. This was confirmed when loss of DNA methylation was found at imprinting control regions of maternally imprinted loci such as PEG3, SNRPN, KCNQ1OT1, together with abnormal expression patterns of p57KIP2 (1-3).
Genome wide linkage first established that chromosome 19q13.42 harbored a gene responsible for BiHM (4, 5). Subsequently, the NACHT, LRR and PYD domains-containing protein 7 gene (NLRP7) (OMIM 609661) became the first identified gene mutated in women with BiHM (6), establishing it as a true human maternal effect gene. NLRP7 belongs to the NLR family of proteins, defined by their roles in the innate immune system as intracellular sensors for Pathogen Associated Molecular Patterns (PAMPs). Several members of the NLR family are expressed in the germline and early embryos, suggesting their involvement in early developmental process (7, 8). Recently, C6orf221, also known as KH domain containing 3-like, subcortical maternal complex member (KHDC3L); an oocyte enriched gene was identified as a cause of BiHM in a few of the families that did not have mutations in NLRP7(9, 10). Whether mutations in NLRP7 and KHDC3L are the only causes of BiHM remains to be determined.
Since the recognition of mutations in NLRP7 as a cause of BiHM, studies have been conducted in diverse ethnic groups to examine if mutations in NLRP7 may also be responsible for other forms of hydatidiform moles (2, 6, 11-14) and reproductive loss. Although a few recent reports have proposed that sequence variants in NLRP7 are also responsible for some cases of recurrent androgenetic HM as well as other adverse reproductive outcomes(11, 12, 15), this data is controversial and has not been confirmed by others. Hence, the motivation for this study was to further explore this observation, because it is difficult to reconcile with the characteristic loss of methylation phenotype in BiHM tissues, which is uniquely observed at imprinted differentially methylated regions, and not in other tested methylated sequences, such as at endogenous repeats and at genes undergoing X-chromosome inactivation.
We studied the mutational spectrum of NLRP7 and KHDC3L in twenty-one women with suspected recurrent and sporadic AnCHM and BiHM. We found mutations in NLRP7 only in women with BiHM and did not find mutations in KHDC3L in any tested samples in this cohort. However, the majority of analyzed subjects carry benign sequence variations in both genes at frequencies comparable to those in the normal population. Although our series is small, our data are not consistent with a proposed role for these genes in androgenetic complete hydatidiform molar pregnancy.
Material and Methods
Study participants and samples
All patients and controls in this study were recruited under a protocol approved by the Baylor College of Medicine Institutional Review Board. Twenty-one women with recurrent and sporadic molar pregnancies were consented and recruited into this study between January 2008 and July 2012. Of these 21 women, 9 had a history of recurrent HM and 12 had a history of sporadic CHM. Of the 9 women with recurrent HM, 4 were suspected to have AnCHM and 5 were suspected to have BiHM. A description of patients with recurrent CHM is provided in Table I. Eleven reproductive partners were also recruited and sequenced for completeness of study and to rule out any incidental findings. Peripheral blood or saliva was collected from the women and from their spouses whenever available. Genomic DNA was isolated from peripheral blood using the QiagenGentraPuregene Blood Kit (Valencia, CA, USA) and from saliva using the DNA GenotekOragene Saliva Collection kit (Ontario, Canada).
Table 1.
Description of study subjects with recurrent HM.
| ID | Ethnicity | Consanguinity | Family history | Obstetric history | Molecular testing | ||
|---|---|---|---|---|---|---|---|
| NLRP7 | KHDC3L |
NLRPL
Duplication |
|||||
| 1 | Hispanic | Unknown | Unknown | RHM | + | + | − |
| 2 | Hispanic | Negative | Uterine cancer in maternal aunt | 3 CHM | + | + | − |
| 3 * | Hispanic | Yes | Yes, affected sister (4 CHM) and Maternal (3 CHM) |
3 CHM | + | − | − |
| 4* | Hispanic | Negative | Negative | 3 CHM, 1 live born |
+ | − | − |
| 14 | Est African | Yes | Yes, extensive history of CHM and NLRP7 duplication2 |
4 CHM | − | − | + |
| 16 | South East Asian | Unknown | Unknown | 1 CHM, 2 PHM, 1 SAB |
+ | − | − |
| 12 | East African | Yes | Negative | 2 CHM | + | − | + |
| 5 | Caucasian | Unknown | Unknown | RHM | + | + | − |
| 6 | Hispanic | Unknown | Unknown | 4 CHM | + | + | − |
Unknown: Information unavailable; Negative: None reported; RHM: Recurrent Hydatidiform Mole; CHM: Complete Hydatidiform Mole; (+): Sequencing done; (−): Sequencing not done.
Patients with the p.L750V change.
Kou et al, 2008; Study in which NLRP7 duplication was first described.
Control subjects
To validate a previously undescribed mutation in an Asian patient, de-identified control specimens were obtained from our institutional obstetrical biobank (Peribank) following full and informed subject consent under a protocol approved by the Baylor College of Medicine Institutional Review Board for human subject research. We searched the database for samples from women with at least one live birth, no prior obstetric complications, no history of intrauterine growth restriction or of congenital anomalies in previous pregnancies. With these criteria, we were able to extract DNA samples from Peribank of eight controls who were ethnically matched with the patient in whom the novel mutation was found and of ninety-two controls of mixed ethnic background. Genomic DNA was isolated from whole blood using the QiagenGentraPuregene Blood Kit (Valencia, CA, USA).
NLRP7 and KHDC3L Sequencing
The genomic sequence of NLRP7 was obtained from the National Center for Biotechnology Information (NCBI) Gene ID 199713. Primers for sequencing of NLRP7 were as previously described (2). Due to family history of the NLRP7 intragenic duplication, patient #14 and her spouse were first screened for presence of this duplication and therefore did not have NLRP7 or KHDC3L sequenced. A patient with sporadic HM had been sequenced for NLRP7 previously and therefore only had KHDC3L sequenced. Therefore, of the 32 study participants (21 subjects and 11 partners), sequencing of NLRP7 was carried out on 29 participants (nineteen women and ten partners). The genomic sequence of KHDC3L was obtained from the National Center for Biotechnology Information (NCBI) Gene ID 154288. This gene contains 3 coding exons which were amplified using the following primers – Exon 1 Forward 5’ CTCTAAGAGCAGCCCAGGAA 3’, Reverse 5’ GTGAGGATCGCCCTGGAAC 3’; Exon 2 Forward 5’ ACCAGTAGCCAATGCCCTCT 3’, Reverse 5’ GACTGGGAGGGCGAGACT 3’; Exon 3 Forward 5’ GATCCAGAAGGCCAAATTGA 3’, Reverse 5’ GCGCGGTTAAGGAGTACAAG 3’. KHDC3L was sequenced in fifteen women and seven partners who did not carry sequence variations in NLRP7. PCR was performed on 50 ng of genomic DNA using the amfiSure PCR Premix from GenDEPOT (Barker, TX, USA) under standard conditions. Sequencing of the purified PCR products was carried out in the forward orientation at Beckman Coulter Genomics (Danver, MA, USA). Confirmation of mutations was carried out in both orientations on repeated PCR reactions. Sequence electropherograms were analyzed using Sequencher v5.0 analysis software. Sequence variants were compared with those in the 1000 Genomes, dbSNP and HapMap databases. The predicted functional effects of novel non-synonymous SNPs were evaluated using PolyPhen-2 and SIFT.
Identification of the tandem intragenic duplication of exons 2-5 of NLRP7 was carried out using primers designed to span the junction fragment across the duplicated exons. These primers have been used by us previously and serve as an alternative to analysis by Southern Blotting and qPCR (2).
Results
Sequencing the coding exons and exon-intron boundaries of NLRP7 and KHDC3L revealed several SNPs at frequencies comparable with the minor allele frequencies (MAFs) reported in the 1000 Genomes Project as outlined in Table 2. We did not identify mutations in NLRP7 and KHDC3L in women with recurrent or sporadic CHM that are likely to be AnCHM based on history
Table 2.
Summary of NLRP7 and KHDC3L sequencing results.
| Gene | Exon | Nucleotide change |
Protein change |
dbsNP ID | MAF Score | Present study (n=29; 58 chromosomes) |
||
|---|---|---|---|---|---|---|---|---|
| Hmz | Htz | MAF | ||||||
| NLRRP7 | 2 | c.251 G>A | p. Cys84Tyr | rs104895509 | Unavailable | 0 | 1 | A=0.017/1 |
| 4 | c. 390G>A | p. Gln130Gln | rs775883 | G=0.285/344 | 6 | 6 | A=0.31/18 | |
| 4 | c.955G>A | p.Val319Ile | rs775882 | A=0.273/344 | 4 | 7 | A=0.258/15 | |
| 4 | c.1137G>C | p.Lys379Asn | rs10418277 | T=0.168/211 | 3 | 7 | C=0.224/13 | |
| 4 | c.1441G>A | p.Ala481Thr | rs61747414 | T=0.101/220 | 0 | 3 | A=0.051/3 | |
| 4 | c.1460G>A | p.Gly487Glu | rs775881 | A=0.114/250 | 2 | 0 | A=0.068/4 | |
| 4 | c.1532A>G | p.Lys511Arg | rs61743949 | C=0.021/45 | 0 | 1 | G=0.017/1 | |
| 5 | c.1981 C>T | p.Lys661Phe | Unavailable | Unavailable | 1 | 0 | T=0.034/2 | |
| 5 | c.2095G>A | p.Val699Ile | rs77072552 | T=0.005/5 | 0 | 1 | A=0.017/1 | |
| 6 | c.2148T>C | p.Pro716Pro | Unavailable | Unavailable | 0 | 1 | C=0.017/1 | |
| 6 | c.2157G>A | p. Ala719Ala | Unavailable | Unavailable | 0 | 1 | C=0.017/1 | |
| 6 | c.2248C>G | p.Leu750Val | rs104895512 | Unavailable | 2 | 1 | G=0.086/5 | |
| 9 | c.2682T>C | p. Tyr894Y | rs269951 | A=0.434/546 | 12 | 12 | A=0.62/36 | |
| 9 | c.2775A>G | p.Ala925Ala | rs269950 | A=0.434/546 | 12 | 12 | A=0.62/36 | |
| Gene | Exon |
Nucleotide
change |
Protein
change |
dbSNP | MAF Score |
Present study (n=22; 44
chromosomes) |
||
| Hmz | HTz | MAF | ||||||
| KHDC3L | 2 | c.289G>C | p.Glu97Gln | rs564533 | C=0.071/155 | 0 | 3 | C=0.068/3 |
| 3 | c.602C>G | p.Ala201Gly | rs561930 | G=0.49/1072 | 4 | 7 | G=0.341/15 | |
Three patients with recurrent HM for whom products of conception were unavailable were suspected to have biparental HM due to their ethnicity and a history of consanguinity in the family, suggesting autosomal recessive inheritance of their disease. Two of these patients, #14 and #12 were of East African ancestry. Both these patients carried a tandem intragenic duplication of exons 2-5 in NLRP7 which we previously extensively characterized and reported in patients with similar ethnic origins. Patient #14 belongs to the family in which the duplication was first described (2) and has had four HM pregnancies. Briefly, a 32bp region of homology between intron 1 and intron 5 mediates a recombination event resulting in the tandem duplication (Figure 1A). PCR primers placed in intron 4 (forward) and intron 2 (reverse) amplify a 1,221bp product from genomic DNA of patients #14 and #12 and a previously characterized positive control (#8) with this duplication (2), but not from control female DNA (Figure 1B). The presence of this duplication in these patients, together with our prior data, suggests an East African founder effect for this mutation.
Figure 1. Tandem intragenic duplication of exons 2-5 of NLRP7 in women with recurrent BiHM.

(A) A 32bp region of homology between intron 5 and intron 1 mediates recombination resulting in tandem duplication. (B) A PCR based strategy to detect the presence of the tandem duplication. A forward primer placed in intron 4 and reverse primer in intron 2 amplified a 1,221bp product in #8 (Lane 1), #14 (Lane 2) and #12 (Lane 3) but not in a control female (Lane 4).
The third subject, #16, of Southeast Asian origin had a reproductive history of one complete HM, two partial HMs and two early spontaneous uncharacterized abortions. Whether these uncharacterized abortions also presented with molar degeneration is not known. Sequencing revealed that this patient carried a novel, homozygous mutation, c.1981C>T in exon 5 of NLRP7 (Figure 2A and 2B). This transition results in an amino acid change of leucine to phenylalanine (p.L661F in NCBI Reference Sequence NM_001127255.13) which is in the linker region between the NAD and LRR domains of NLRP7 (Figure 2E). A phylogenetic comparison of NLRP7 across various primates and its ancestral gene NLRP2 gene reveals that the leucine at this position is 100% conserved (Figure 2F). This change is not documented in dbSNP build 135 and has not been seen in the Phase 1 release of the 1000 Genomes Project of 1094 worldwide individuals. Polyphen-2 predicted that this change is “probably damaging” with a score of 1.00, while SIFT predicted it is “damaging’ with a score of 0.04. Furthermore, we sequenced 100 control DNA samples from subjects with no prior obstetric complications, of whom 8% were matched for ethnicity, and did not identify this mutation in the sequenced 200 control chromosomes (Figure 2C and 2D).
Figure 2. A novel, damaging mutation in patient #14.
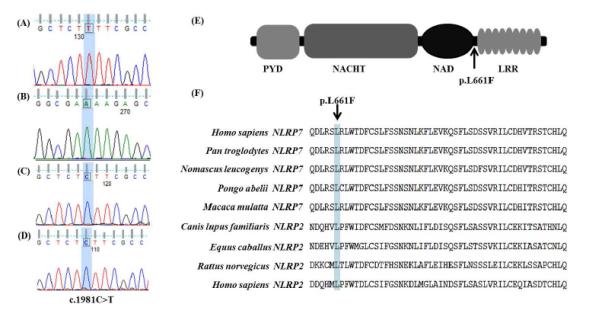
A homozygous mutation in exon 5 of a patient (A, forward orientation) and (B, reverse orientation) with recurrent HM resulting in a C>T transition. This change was not seen in 200 control chromosomes (C and D). The c.1981C>T mutation changes a leucine to phenylalanine at amino acid position 661 in the NLRP7 transcript variant 3 which links the NAD and LRR domains (E). The leucine at this position in NLRP7 is conserved across primates and in the ancestral NLRP2 as well (F).
An additional two patients, #3 and #4 of Hispanic origin with recurrent unspecified CHM carried a homozygous p.Leu750Val mutation previously shown by us to be associated with BiHM (2). While we were not able to confirm, we suspect that those were also BiHM.
Discussion
We studied women with sporadic CHM and recurrent CHM and in this cohort found that mutations in NLRP7 are causative of only the biparentally inherited, recurrent forms of HM (BiHM). We did not find mutations in KHDC3L, which is known to be a minor gene for BiHM.
It has recently been suggested that mutations in NLRP7 and KHDC3L are causative of androgenetic and triploid forms of HM in addition to recurrent BiHM and may be present in women with recurrent early spontaneous miscarriages without molar degeneration (11). However, we propose that it is more likely that androgenetic and diandric triploid HM have a different origin because the mechanisms by which pregnancies may acquire these abnormal chromosomal complements, with uniquely or excess paternally inherited chromosomes are difficult to explain by maternal mutations in NLRP7 or KHDC3L. Furthermore, loss of function of these maternal effect genes has been shown to specifically affect methylation of maternally imprinted DMRs in BiHM tissues, which is a specialized developmental defect that is not observed in either androgenetic diploid or diandric triploid moles (1, 2).
NLRP7 has evolved from the primate-specific NLRP7/NLRP2 cluster (7). Interestingly, mutations in NLRP2 have been associated with trans-imprinting defects as reported in a familial case of Beckwith-Wiedemann Syndrome (BWS), wherein the mother carried a homozygous loss of function mutation in NLRP2 which resulted in an epimutation at the BWS locus in her affected children (16). One of the two affected children also exhibited a loss of methylation at the PEG1 DMR. In addition, recent literature showing that maternal mutations in KHDC3L also impact methylation at maternally imprinted loci (9), together with the enriched expression of NLRP2, NLRP7 and KHDC3L in the female germline and early embryos, supports that they may function in the same or similar processes related to imprinting.
The search for evidence that NLRP7 and KHDC3L are associated with other forms of pregnancy loss likely originates from a proposed hypothesis for the pathogenesis of BiHM derived from known roles of NLRP proteins in the immune system. According to this hypothesis, maternal loss of function of NLRP7 results in a failure to reject defective embryos or early embryonic cleavage defects themselves in the uterine tract. This would imply that the defects in DNA methylation seen in BiHM tissues are secondary to a more generalized immune deficit caused by loss of NLRP7. In contrast, the discovery that BiHM concepti specifically lack DNA methylation marks at maternally imprinted differentially methylated regions DMRs together with the absence of DNA methylation defects at other (non-imprinted) regions (1, 2) supports an alternate hypothesis that loss of NLRP7, and perhaps KHDC3L, in the maternal germline has a more direct negative impact on the acquisition of imprinting marks in the maternal germline or their maintenance in the developing conceptus. This is supported by a recent report of a woman with BiHM who successfully reproduced after in vitro fertilization with donor oocytes (17), while to our knowledge attempts to overcome the recurrent pregnancy losses in women with hydatidiform moles with assisted reproductive measures using their own oocytes were unsuccessful (18-20). While the immune-driven hypothesis and the imprinting hypothesis are not mutually exclusive, the former cannot easily explain the specialized imprinting defects observed in BiHM. Consequently, we speculate that in addition to its participation in regulating the innate immune response, NLRP7 also participates directly or indirectly in the process of germline imprint acquisition and/or maintenance, consistent with its expression in both the female germ line and early embryos. Delineating this function is critical not only from the perspective of furthering our understanding of the biological mechanisms underlying BiHM but also to gain insight into the enigmatic processes by which mammalian germ cells reset imprints and establish parent-of-origin specific functions.
To date, KHDC3L has no documented roles in the immune system but is responsible for the same phenotype as that caused by a loss of NLRP7, further strengthening the evidence in favor of the imprinting hypothesis. KHDC3L belongs to a rapidly evolving gene family comprised of the following 4 members: KHDC1, DPPA5, KHDC3L and OOEP. These genes are all highly expressed in oocytes and embryos. Given that the maturing oocyte is the major site for imprint acquisition, a direct association of KHDC3L with this process is plausible.
Other studies have also not been able to confirm the previously suggested association between mutations in NLRP7 and androgenetic or diandric triploid forms of HM, providing additional indication that BiHM have different origins than AnCHM and diandric triploid PHM (14, 21). The evolving lack of support for the involvement of these genes in adverse reproductive outcomes other than BiHM has important implications for clinical diagnostic testing for mutations in these genes, which may only be indicated in the presence of proven BiHM or highly recurrent HM (more than two) of unknown inheritance and genotype. Since most of these studies were carried out in relatively small cohorts, larger multicenter collaborative studies may be required to conclusively demonstrate that mutations in NLRP7 or KHDC3L in women are uniquely responsible for recurrent rare BiHM.
In conclusion, we have studied the various forms of HM for mutations in NLRP7 and KHDC3L and have found no evidence that mutations in these genes are causative androgenetic forms of HM.
Acknowledgments
A statement of all funding sources that supported the work
This work was supported in part by NIH grants R21HD058081, R01HD045970 and by grant 6250-51000-055 from the USDA/ARS to I.B.V. The project was also supported by the BCM IDDRC Grant Number 5P30HD024064 from the Eunice Kennedy Shriver National Institute Of Child Health & Human Development and Grant Number C06RR029965 from the National Center For Research Resources. The content is solely the responsibility of the authors and does not necessarily represent the official views of the Eunice Kennedy Shriver National Institute Of Child Health & Human Development or the National Institutes of Health.
Footnotes
The authors declare no conflicts of interests.
- Mutations in NLRP7 and KHDC3L are causative of biparental hydatidiform moles (BiHM).
- This suggests that NLRP7 and KHDC3L may play critical roles in imprint acquisition and maintenance of DNA methylation.
- Some current literature suggests a role for NLRP7 and KHDC3L in the etiology of androgenetic HM and other adverse reproductive outcomes.
- We challenged these observations and in the present study demonstrate a lack of evidence for mutations in NLRP7 and KHDC3L in androgenetic HM and only show association of mutations in these genes with BiHM.
References
- 1.Judson H, Hayward BE, Sheridan E, et al. A global disorder of imprinting in the human female germ line. Nature. 2002 Apr 4;416(6880):539–42. doi: 10.1038/416539a. PubMed PMID: 11932746. [DOI] [PubMed] [Google Scholar]
- 2.Kou YC, Shao L, Peng HH, et al. A recurrent intragenic genomic duplication, other novel mutations in NLRP7 and imprinting defects in recurrent biparental hydatidiform moles. Mol Hum Reprod. 2008 Jan;14(1):33–40. doi: 10.1093/molehr/gam079. PubMed PMID: 18039680. [DOI] [PubMed] [Google Scholar]
- 3.Fisher RA, Hodges MD, Rees HC, et al. The maternally transcribed gene p57(KIP2) (CDNK1C) is abnormally expressed in both androgenetic and biparental complete hydatidiform moles. Hum Mol Genet. 2002 Dec 15;11(26):3267–72. doi: 10.1093/hmg/11.26.3267. PubMed PMID: 12471053. [DOI] [PubMed] [Google Scholar]
- 4.Moglabey YB, Kircheisen R, Seoud M, et al. Genetic mapping of a maternal locus responsible for familial hydatidiform moles. Hum Mol Genet. 1999 Apr;8(4):667–71. doi: 10.1093/hmg/8.4.667. PubMed PMID: 10072436. [DOI] [PubMed] [Google Scholar]
- 5.Panichkul PC, Al-Hussaini TK, Sierra R, et al. Recurrent biparental hydatidiform mole: additional evidence for a 1.1-Mb locus in 19q13.4 and candidate gene analysis. J Soc Gynecol Investig. 2005 Jul;12(5):376–83. doi: 10.1016/j.jsgi.2005.02.011. PubMed PMID: 15979551. [DOI] [PubMed] [Google Scholar]
- 6.Murdoch S, Djuric U, Mazhar B, et al. Mutations in NALP7 cause recurrent hydatidiform moles and reproductive wastage in humans. Nat Genet. 2006 Mar;38(3):300–2. doi: 10.1038/ng1740. PubMed PMID: 16462743. [DOI] [PubMed] [Google Scholar]
- 7.Tian X, Pascal G, Monget P. Evolution and functional divergence of NLRP genes in mammalian reproductive systems. BMC Evol Biol. 2009;9:202. doi: 10.1186/1471-2148-9-202. PubMed PMID: 19682372. Pubmed Central PMCID: 2735741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang P, Dixon M, Zucchelli M, et al. Expression analysis of the NLRP gene family suggests a role in human preimplantation development. PLoS One. 2008;3(7):e2755. doi: 10.1371/journal.pone.0002755. PubMed PMID: 18648497. Pubmed Central PMCID: 2447171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Parry DA, Logan CV, Hayward BE, et al. Mutations causing familial biparental hydatidiform mole implicate c6orf221 as a possible regulator of genomic imprinting in the human oocyte. Am J Hum Genet. 2011 Sep 9;89(3):451–8. doi: 10.1016/j.ajhg.2011.08.002. PubMed PMID: 21885028. Pubmed Central PMCID: 3169823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Reddy R, Akoury E, Phuong Nguyen NM, et al. Report of four new patients with protein-truncating mutations in C6orf221/KHDC3L and colocalization with NLRP7. Eur J Hum Genet. 2012 Dec 12; doi: 10.1038/ejhg.2012.274. PubMed PMID: 23232697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Messaed C, Chebaro W, Di Roberto RB, et al. NLRP7 in the spectrum of reproductive wastage: rare non-synonymous variants confer genetic susceptibility to recurrent reproductive wastage. J Med Genet. 2011 Aug;48(8):540–8. doi: 10.1136/jmg.2011.089144. PubMed PMID: 21659348. [DOI] [PubMed] [Google Scholar]
- 12.Qian J, Cheng Q, Murdoch S, et al. The genetics of recurrent hydatidiform moles in China: correlations between NLRP7 mutations, molar genotypes and reproductive outcomes. Mol Hum Reprod. 2011 Oct;17(10):612–9. doi: 10.1093/molehr/gar027. PubMed PMID: 21507883. [DOI] [PubMed] [Google Scholar]
- 13.Slim R, Coullin P, Diatta AL, et al. NLRP7 and the genetics of post-molar choriocarcinomas in Senegal. Mol Hum Reprod. 2012 Jan;18(1):52–6. doi: 10.1093/molehr/gar060. PubMed PMID: 21948117. [DOI] [PubMed] [Google Scholar]
- 14.Dixon PH, Trongwongsa P, Abu-Hayyah S, et al. Mutations in NLRP7 are associated with diploid biparental hydatidiform moles, but not androgenetic complete moles. J Med Genet. 2012 Mar;49(3):206–11. doi: 10.1136/jmedgenet-2011-100602. PubMed PMID: 22315435. [DOI] [PubMed] [Google Scholar]
- 15.Slim R, Ao A, Surti U, et al. Recurrent triploid and dispermic conceptions in patients with NLRP7 mutations. Placenta. 2011 May;32(5):409–12. doi: 10.1016/j.placenta.2011.02.001. PubMed PMID: 21421271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Meyer E, Lim D, Pasha S, et al. Germline mutation in NLRP2 (NALP2) in a familial imprinting disorder (Beckwith-Wiedemann Syndrome) PLoS Genet. 2009 Mar;5(3):e1000423. doi: 10.1371/journal.pgen.1000423. PubMed PMID: 19300480. Pubmed Central PMCID: 2650258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fisher RA, Lavery SA, Carby A, et al. What a difference an egg makes. Lancet. 2011 Dec 3;378(9807):1974. doi: 10.1016/S0140-6736(11)61751-0. PubMed PMID: 22130487. [DOI] [PubMed] [Google Scholar]
- 18.Edwards RG, Crow J, Dale S, et al. Pronuclear, cleavage and blastocyst histories in the attempted preimplantation diagnosis of the human hydatidiform mole. Hum Reprod. 1992 Aug;7(7):994–8. doi: 10.1093/oxfordjournals.humrep.a137787. PubMed PMID: 1430144. [DOI] [PubMed] [Google Scholar]
- 19.Reubinoff BE, Lewin A, Verner M, et al. Intracytoplasmic sperm injection combined with preimplantation genetic diagnosis for the prevention of recurrent gestational trophoblastic disease. Hum Reprod. 1997 Apr;12(4):805–8. doi: 10.1093/humrep/12.4.805. PubMed PMID: 9159446. [DOI] [PubMed] [Google Scholar]
- 20.Pal L, Toth TL, Leykin L, et al. High incidence of triploidy in in-vitro fertilized oocytes from a patient with a previous history of recurrent gestational trophoblastic disease. Hum Reprod. 1996 Jul;11(7):1529–32. doi: 10.1093/oxfordjournals.humrep.a019432. PubMed PMID: 8671499. [DOI] [PubMed] [Google Scholar]
- 21.Andreasen L, Bolund L, Niemann I, et al. Mosaic moles and non-familial biparental moles are not caused by mutations in NLRP7, NLRP2 or C6orf221. Mol Hum Reprod. 2012 Dec;18(12):593–8. doi: 10.1093/molehr/gas036. PubMed PMID: 22909446. [DOI] [PubMed] [Google Scholar]