

Abstract
Artificial antigen-presenting cells (aAPCs) have shown great initial promise for ex vivo activation of cytotoxic T cells. The development of aAPCs has focused mainly on the choice of proteins to use for surface presentation to T cells when conjugated to various spherical, microscale particles. We review here biomimetic nanoengineering approaches that have been applied to the development of aAPCs that move beyond initial concepts about aAPC development. This article also discusses key technologies that may be enabling for the development of nano- and micro-scale aAPCs with nanoscale features, and suggests several future directions for the field.
Keywords: anisotropy, artificial antigen-presenting cell, biomimetic, immunology, nanobiotechnology, nanoparticle, particle shape, polymer
Synthetic biomaterials and particles can be engineered to be ‘biomimetic’, where they act similarly in an organism to natural biological molecules and cells. This biological mimicry can be beneficial for wound healing and regenerative medicine, new types of advanced therapeutics, coatings on medical devices and in other applications. One particular area where biomimetic particles have significant potential to advance medicine is in the field of tumor immunotherapy.
The main goal of tumor immunotherapy is to encourage the body's own immune responses to aberrant cells. The recognition that there are tumor-infiltrating immune cells, T lymphocytes or T cells, and the identification of the genes that are targeted by T cells in melanoma led to the identification of tumor antigens for immunotherapy [1,2]. Tumor antigens are specific biological molecules that are present on the surface of cancer cells, but are not present on the surface of healthy cells. Numerous human tumor antigens recognized by T cells have now been identified, and these antigens are promising targets for tumor immunotherapy [3].
A variety of approaches for tumor immunotherapy have been investigated, with the most common approach involving natural biological antigen-presenting cells (APCs) interacting with T cells. However, there are challenges with these approaches, including efficacy, safety, cost and general flexibility. As an example, Sipuleucel-T, the only currently US FDA-approved cell-based artificial APC (aAPC) therapy, requires a complex process to isolate a patient's own white blood cells, activate them ex vivo and reinfuse them to the patient, at a total cost of nearly US$100,000 for a 4-month survival benefit in late-stage metastatic prostate cancer [4]. If there was an off-the-shelf synthetic product that was relatively easy to manufacture, biocompatible and safe, and could engender strong and specific T-cell responses against an antigen either ex vivo or, preferably, in situ, it would be a boon for the treatment of many types of cancer as well as being beneficial for the treatment of other diseases such as infectious diseases [5]. To construct such an off-the-shelf synthetic product (i.e., biomimetic acellular aAPCs) there are many important nanomedicine design parameters to consider, and this is the subject of this article.
Much of the focus to date in the development of aAPCs has centered on the identity of the surface-presented proteins used to target the T-cell population and induce a response. Spherical, cell-sized (2–10 μm), isotropic (homogeneous surface presentation) aAPCs that present some of the same protein signals on their surface as biological APCs have been developed and well characterized, and are already attractive options for ex vivo T-cell proliferation [6] and have shown in vivo efficacy in mouse models [5–13]. For the development of next-generation nanoengineered aAPCs, special attention should be paid to particle size and particle shape, and at the protein level, surface density, spatial organization and dynamics are key parameters of interest (Table 1). This article will focus both on the promise of nanosized constructs as aAPCs and the application of nanoengineering to develop nanosized features on microsized particles.
Table 1.
Key parameters of current- and future-generation artificial antigen-presenting cells and their biological rationale.
| Key parameter | Activated biological APC/DC | Current aAPC | Future aAPC |
|---|---|---|---|
| Signal 1: recognition | pMHC | Antigen specific: pMHC/pMHC multimers General T-cell targeting: anti-CD3 mAb | pMHC multimers |
| Signal 2: costimulation | B7.1 and B7.2, among others | Anti-CD28 mAb | Anti-CD28 mAb, among others |
| Signal 3: secretable, immunomodulatory signals | Various (e.g., IL-2) or chemokine (e.g., CCL3 and CCL4) [101] | IL-2 release [9] | Cytokine or chemokine release |
| Size | 20 μm in diameter | 4–5 μm in diameter; >1 μm for spherical aAPC [11] | Micro- or nano-scale |
| Shape | Long, thin, sheet-like projections/veils in many directions from the cell body; highly dynamic [102] | Spherical | Nonspherical |
| Density | Dynamic local density | 2000, >10,000 or 40,000 MHC/μm2 [10] | Preclustered, intermediate density |
| Organization | Before T-cell contact: microclustering After contact with T cell: dynamic reorganization; immune synapse formation [18,19,62,67,68,103] | Liposomes/lipid bilayers allow for preclustering and dynamic reorganization [28,69] | Preclustered with dynamic reorganization via supported lipid bilayers |
aAPC: Artificial antigen-presenting cell; APC: Antigen-presenting cell; DC: Dendritic cell; mAb: Monoclonal antibody; pMHC: Peptide in MHC.
There are potentially many advantages to a nanosized immunostimulatory platform for use in vivo. Smaller particles (20–200 nm in diameter) can transit directly to the lymphatics after subcutaneous injection without the aid of phagocytosis [14], where they could reach the T-cell population in the ideal setting for T-cell expansion. Nanosized constructs are also potentially intravenously injectable, as microsized particles would potentially be trapped in the capillary bed of the lungs, blocking capillary flow. In addition, control over surface topology on the nanoscale (shape), or surface density or organization on larger scale constructs would both more precisely mimic the biological setting, and also allow for greater understanding about the role that geometry, protein surface organization and dynamic rearrangement plays in the biological setting.
How biological APCs work
Antigens on a tumor cell are recognized by a T cell by its T-cell receptor (TCR). To accomplish this, naive T cells must be directed to productively respond to TCR binding by APCs that present specific antigens to T cells via MHC molecules. There are two classes of MHCs with different functions and that present different peptides. MHC class II molecules present peptides obtained via the endosomal–lysosomal route and serve to present peptides that come from outside the cell; thus, presentation of nonself-peptides in class II MHC is crucial to mediate the immune response to extracellular pathogens. MHC class I molecules, on the other hand, bind to peptides generated by the proteasome, and are generally used to present peptides whose source is internal to the cell; thus, presentation of peptides in class II MHC is crucial in mediating the immune response to intracellular pathogens and cancer. Class I MHC function is to activate CD8+ T cells or cytotoxic T lymphocytes (CTLs), whose primary function within the adaptive immune system is the recognition and killing of infected or cancerous cells within the body. There is a strong argument that class I MHC-restricted tumor antigens that can be recognized by CTLs make the best ‘tumor rejection antigens’, as class I MHCs are present in most nucleated cells. CTLs are the primary effector arm of the immune response against cancer and loss of HLA class I expression in cancer patients is strongly associated with disease progression [15]. Thus, control over CD8+ T-cell fate is critical to the success of tumor immunotherapy.
T-cell fate is dictated not only by the antigen recognized, but also the context in which the antigen is recognized (Figure 1). Naive T cells that recognize peptide in MHC (pMHC), without costimulation by secondary signals, are directed to become unresponsive to further stimuli (anergy) or die, which allows for T-cell tolerance to form outside of the immune organs. Professional APCs, such as dendritic cells (DCs), in addition to presenting pMHC on the surface (which is termed signal 1), provide secondary signals to the T cell in two main ways. Principally, recognition-dependent activation is modulated by expression of surface molecules, such as B7.1, which interact with other surface molecules on the T-cell surface such as CD28 (termed signal 2) [16]. Secretable immunostimulatory factors, such as cytokines, also serve to help direct T-cell fate; these are often termed signal 3 [17]. In addition to the identities of the molecules involved in the process, the interaction between a biological APC and a T cell requires close apposition of membranes over a large area of surface contact, and results in large-scale protein rearrangements and the subsequent formation of the immunological synapse (IS) (Figure 2A) [18,19].
Figure 1. A biological antigen-presenting cell interacting with a T cell and an artificial antigen-presenting cell interacting with a T cell.
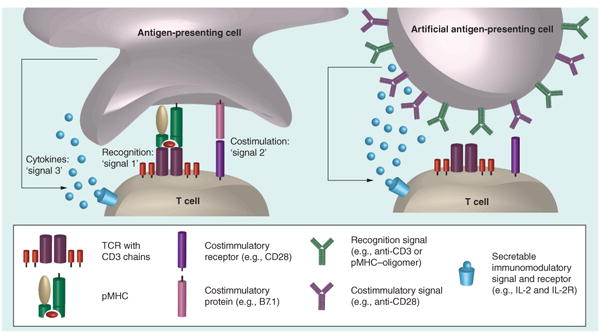
In the biological setting, the recognition signal (‘signal 1’) is provided by the interaction of pMHC with the TCR complex, composed of a TCR heterodimer and signaling CD3 chains. Costimulation (‘signal 2’) occurs through a variety of cell surface protein mediators; the B7.1–CD28 interaction is often used in artificial antigen-presenting cell constructs. T-cell fate is also determined classically by the cytokine milieu-sensed signal (‘signal 3’).
pMHC: Peptide in MHC; TCR: T-cell receptor.
Figure 2. Immune synapse formation is dependent on the shape of the substrate.

T cells with fluorescently stained TCRs (green) were added to supported lipid bilayers containing peptide in MHC (unlabeled) and ICAM (red) on the surfaces (A) without patterning, (B) with 2-μm parallel lines, (C) with 5-μm boxes and (D) with hexagonal lines (1-μm spacing).
TCR: T-cell receptor.
Reproduced with permission from [71].
aAPCs
aAPCs are a promising, but relatively early-stage concept, that relies on cell-sized constructs mostly for ex vivo expansion. Currently, the more commonly used approach for T-cell immunotherapy involves the use of ex vivo-expanded autologous tumor-specific T cells [20,21]. Other currently used approaches involve pulsing autologous, in vitro-expanded and -activated DCs with synthetic peptide epitopes from tumor-associated antigens or the tumor-associated antigen themselves for induction of tumor-specific CTLs [22]. However, these approaches are often unsuccessful and lead to induction of a T-cell response that is unable to recognize the tumor cells. Isolation of specific T-cell subsets has been greatly aided by the development of peptide-specific tetramers/multimers that consist of pMHC (or HLA) arranged into a multimeric particle by linking four (or more) HLA molecules to an avidin-derivative core [23,24]. One major advantage of the ‘streptamers’ is that the function of the T cell is preserved following selection, whereas with tetramers the selected T-cell population shows impaired function [24]. Alternative approaches involve transfecting or transducing DCs with DNA or RNA to produce the tumor-associated antigens [25], or inserting conventional or chimeric TCRs into T cells to impart the desired specificity [26,27].
As an alternative to the conventional approach of accessing native APCs and using them to direct the immune response, there has been increasing interest in developing particle systems that can substitute for the function of biological APCs. These particles as a class are termed aAPCs. Cellular aAPC approaches are beyond the scope of this current article, but are reviewed by Turtle and Riddell [13].
An ideal aAPC system would provide the same signals (antigen recognition, surface costimulation and secretable factors) to the T-cell target as the biological APCs (Figure 1). aAPCs have been generated by coupling proteins that deliver signal 1 and 2 to the surface of particles made from a range of materials, including liposomes [10,28], magnetic particles [5,29–31], polystyrene (PS) [11] and degradable polymeric particles [8,9,32]. Each material has its own advantages and disadvantages for their use as aAPCs. Liposomes have fluid membranes that closely mimic biological membranes and can be used for delivery of drugs, but are substantially less stable than hard particles. Magnetic particles are of particular interest for ex vivo T-cell expansion because they can be readily removed from the expanded T-cell population before reinfusion. Biodegradable particles can be useful for their release properties, and are very biocompatible. Therefore, they may be excellent for in vivo applications, even though from a materials perspective, biodegradable aAPC may have difficulty with extended surface presentation. We will review here the key parameters for consideration in the design of aAPCs and the progress made with each. We will also suggest future directions to this work, with a focus on nanoengineering approaches.
aAPCs have been mostly investigated for their immunostimulatory properties towards CD4+ or CD8+ T cells. We will focus here mainly on efforts directed at activation of CD8+ T cells. However, it is important to note that the same bioengineering approaches can be used to target CD4+ T cells [31,33], to synthesize killer aAPCs that kill targeted T-cell subsets to eliminate autoreactive clones that are responsible for autoimmune disease [34,35], to generate aAPCs that stimulate and expand natural killer T cells [36,37], or stimulate and expand CD1–lipid–antigen-restricted T cells [38].
One of the challenges associated with aAPCs for ex vivo T-cell expansion is that T-cell quality may become compromised with sequential rounds of expansion and long-term ex vivo culture. Sauer et al. showed that antiacute myelogenous leukemia CTLs that were generated by initially coculturing splenocytes with acute myelogenous leukemia-lysate pulsed DCs, then cultured ex vivo for 9 days with magnetic anti-CD3/anti-CD28 aAPCs, were superior to CTLs cultured ex vivo for 16 days. Short-term expanded CTLs increased persistence in lymphoid organs and conferred a survival advantage at high CTL doses. Short-term expanded CTLs also had higher levels of CTL l-selectin expression as compared with long-term expanded CTLs [39]. Attempting to overcome this tradeoff between increased expansion time (which allows for greater numbers of CTLs to be generated) and decreased T-cell quality should be one of the goals for developers of next-generation aAPCs to overcome.
Signals 1 & 2: antigen presentation & costimulation
One of the advantages with a synthetic approach is the ability to pattern the surface with defined surface molecules and specified ratios. This allows the precise study of the effects of those particular protein sets in isolation or in combination. The biological recognition signal consists strictly of pMHC and TCR, so TCR-subset-specific aAPC systems have utilized surface pMHC or pMHC multimers (dimers [40] or tetramers [41]) for the purpose of targeting. The vast majority of aAPC systems tested, however, instead use anti-CD3 monoclonal antibody (mAb) as their targeting ligand. CD3 itself is a coreceptor that is part of the TCR complex regardless of TCR antigen specificity, and when used in aAPC systems, is used to stimulate the TCR of any T cell in an antigen independent fashion.
The first study detailing the use of HLA molecules in a completely artificial system to activate T cells was published in 1978 by Engelhard et al., who showed that CTL induction was possible using 100-nm phospholipid vesicles presenting purified HLAs on their surface [10]. The critical finding of this study revealed that CTL activation was very sensitive to the density of antigen presented. The highest level of CTL function was elicited by liposomes containing 20 molecules per vesicle (2000 molecules/μm2), which was the lowest density tested in this initial study. Higher density vesicles (100 and 400 molecules per vesicle, or 10,000 and 40,000 molecules/μm2) showed lower maximal levels of function. For optimal function, however, this system required a high ratio of liposomes to target cells (100 liposomes/target cell).
Multiple biological molecules can function as signal 2 and can induce positive or negative costimulation (repression) depending on their identity. These surface molecules interact dynamically to modulate the response to TCR triggering. Studies with PS microparticles have helped to identify the importance of costimulation to effective aAPC function. PS microparticles functionalized with anti-TCR antibody and B7-1 (a positive costimulatory signal 2) showed that costimulation by B7-1 is sufficient for the induction of effector function in CD4+ and CD8+ T cells [42], but that CD8+ responses were transient in nature and required higher density of costimulatory molecules on the surface than that required by CD4+ cells. In addition, ICAM-1 (critical for cell–cell adhesion and the major component of the pSMAC) was also shown to be able to provide costimulation for CD8+ T-cell activation and synergize with B7-1 for induction of IL-2 production, but failed to do so for CD4+ T cells [43].
One major difference between classic APCs and aAPCs is that following activation, biological APCs upregulate inhibitory recognition molecules (iRLs), such as PD-1, which help to tune down subsequent responses and avoid over-exuberant inflammatory responses. As an important example, PD-L1 has been shown to bind to B7-1 and they interact specifically to inhibit T-cell activation [44]. Multifunctionality (the production of multiple cytokines by a single CD8+ T cell in response to a specific antigen challenge) is an important predictor of T-cell-mediated immune protection and memory development. Interestingly, Ndhlovu et al. showed that aAPC-expanded antigen specific CD8+ T cells were multifunctional (produced multiple cytokines at higher levels) compared with DC-expanded CTLs, which were mostly monofunctional [45]. However, aAPC expanded cells, which provided continuous positive stimulation, also expressed higher PD-1 levels than DC-expanded cells. This increased PD-1 expression may be due to the lack of any iRLs on aAPCs.
As a result, some groups have added anti-PD-L1–Fc to aAPCs and have found differential responses. Recently, however, Fuertes Marraco et al. showed that some of these results may be due to competition of ligands for surface presentation on aAPCs when incubated simultaneously [46]. They found that simultaneous incubation of anti-CD3 mAbs with either PDL1–Fc or HLA–DR1 reduced expansion due to a reduction in the amount of anti-CD3 presented on the particles. When the anti-CD3 mAbs and either iRL were added in a stepwise fashion (1 h incubation with anti-CD3 mAbs, followed by a wash step, then 1 h incubation with an iRL) they found consistent anti-CD3 presentation and no subsequent effect due to the addition of these iRLs, suggesting that protein competition may lead to erroneous conclusions, and highlighting the importance of checking protein coating efficiency for aAPC quality control [46].
Signal 3: cytokine release
One of the advantages of a biodegradable aAPC system compared with a nondegradable aAPC system is that the biodegradable system can be engineered to release soluble factors from within the aAPC in addition to presenting factors that are attached to the aAPC surface. In addition, for in vivo administration, biodegradable aAPCs would offer the promise of avoiding in vivo accumulation and increasing biocompatability, as the eventual dissolution of the particle would allow for complete elimination of the system from the body.
One major difficulty with constructing biodegradable aAPCs is that degradation of the particle tends to lead to loss of surface function. Initial work by Shalaby et al. showed that poly(glycolic acid) microparticles could be used to release immunomodulatory compounds, and could also be made into aAPCs by irreversible adsorption of anti-CD3 and anti-CD28 onto the surface [47]. Subsequently, using a novel method that relies on the incorporation of avidin–palmitic acid conjugates into the surface of the poly(lactic-co-glycolic acid) (PLGA) particles [48], Steenblock and Fahmy were able to develop a PLGA-based biodegradable aAPC that could stably present ligands on its surface in solution for over 20 days [8]. This method relies on the hydrophobic chain of the palmitic acid partitioning into the hydrophobic PLGA core, while the avidin partitions to the surface of the particles and is available to bind biotinylated anti-CD3 and anti-CD28 antibodies.
In addition, because PLGA is biodegradable, IL-2 could be released from the particle core, allowing the aAPC to incorporate cytokine signaling (signal 3) in addition to antigen recognition and surface costimulation (Figure 1). Importantly, paracrine release of IL-2 from the particles resulted in superior T-cell expansion compared with exogenous administration of equivalent amounts of cytokines [8]. Paracrine delivery of IL-2 upon T-cell contact resulted in increased IL-2 in the contact region, and increased proliferation of CD8+ T cells in vitro, tenfold compared with bulk IL-2 administration. In addition, these responses appear to require sustained release of low levels of IL-2 and depend on close contact between the aAPC and T cell [9]. These results indicating the importance of paracine delivery of cytokines were established using microscale PLGA platforms, but probably would be useful for nanoscale aAPC platforms.
Nanoscale approaches to mimicking the organization of the IS
Nanoscale surface patterning
Most aAPC systems utilize uniform presentation of ligands on the surface, due to ease of fabrication and simplicity of design. However, the interaction of a biological APC with a T cell results in the formation of an organized, anisotropic arrangement of surface proteins termed the IS [18,19]. When mature, the IS consists of two main distinct concentric rings of organized proteins. The cSMAC consists primarily of TCRs in contact with pMHC molecules on the APC surface and other costimulatory molecules (e.g., as the B7–CD28 interaction). This is surrounded by a pSMAC, formed by integrin–adhesion molecule interactions (principally LFA-1 on the T-cell surface binding to ICAM-1 on the APC surface). Taking advantage of these dynamic rearrangements and anisotropic protein arrangements could be of great advantage in future aAPC systems.
One potential way of mimicking the bio-molecular organization of the IS on aAPC systems would be to utilize recent advances in the synthesis and design of patchy particles [49]. Through the formation of particles with at least two distinct surface subdomains, one could synthesize a particle that had the components of the cSMAC (pMHC and various costimulatory molecules) in one subdomain, with the components of the pSMAC (principally ICAM-1) in the other subdomain. As these designs get more advanced, more precise biomimicry could be achieved with such systems.
Janus particles, named after the Roman god Janus who had two faces, are particles that have two distinct faces. The various synthetic approaches to Janus particles are helpfully reviewed by Walther and Muller [50], and these approaches to double-sided particles could prove useful for the development of aAPCs.
Lithography has proved to be an incredibly useful tool in a number of fields, from computers to microfluidics and biology. In the 2D setting, researchers have developed IS arrays, which are stimulatory patches surrounded by integrin fields in a flat lithographically patterned substrate [51], and used them to study how the disruption of these 2D ISs might affect the response of T cells to these substrates. Complex particle lithography, on the micro- and, particularly, nano-scale, has proven to be challenging. However, some work has been done in this area to develop particles that could be suitable for nanoscale aAPC design. Snyder et al. used polyelectrolytes to cover the exposed surface of amine-functionalized PS spheres adhered to negatively charged cover slips as a mask [52]. Using micro-contact printing techniques and polydimethylsiloxane (PDMS) molds, Cayre et al. were able to synthesize dipolar particles [53]. In addition, they extended their method to allow printing of one colloidal layer onto another, which enables the formation of ‘raspberry’ particles if the particles are of very dissimilar sizes [54].
Another approach to the formation of patchy particles with multiple patches involves the use of glancing angle deposition [55]. The glancing angle deposition technique involves the deposition of gold or silver vapor onto a close-packed colloidal monolayer at low pressure. The technique is referred to as ‘glancing angle’ because the sample is angled with respect to the vapor deposition. Changing the angle allows alterations in the geometry of, and location of, the vapor deposition. To produce multiple patches of different functionalities, the vapor deposition is performed in two steps. To position the second group differently, the angle of deposition is changed. This can allow for two separate patches on the same side of the particle that also interconnect. Patches can be produced on opposite poles of the particles by using a PDMS stamp to flip the particles and then allow vapor deposition to proceed on the opposite side [56]. These techniques have also been applied to colloidal crystals. By using upper single or double layers in colloidal crystals as masks during the vapor deposition, Zhang et al. were able to develop patterns on the third layer of nanoparticles with nanoscale feature resolution [57].
Recently, Kamalasanan et al. reported a novel technique to produce multiple circular patches on the surface of microspheres. The technique involves applying liquid PDMS to a 3D colloidal crystal or various 2D arrays of spherical PS microparticles. During this process, selective solidification of the PDMS happens at particle interfaces, allowing for those sites to be selectively blocked by the PDMS masks (Figure 3) [58]. This process allows ligands to be conjugated at the unblocked sites, followed by exposure of the PDMS patches, and addition of a second ligand or set of ligands to the newly exposed sites. The number of PDMS patches is determined by the coordination number of the particle in the colloidal crystal/2D array [58]. This technique could enable the fabrication of more biomimetic anisotropic aAPCs with patches of pMHC and costimulatory molecules (cSMAC components) surrounded by areas covered by pSMAC components.
Figure 3. Colloidal crystal-templated synthesis of patchy particles.
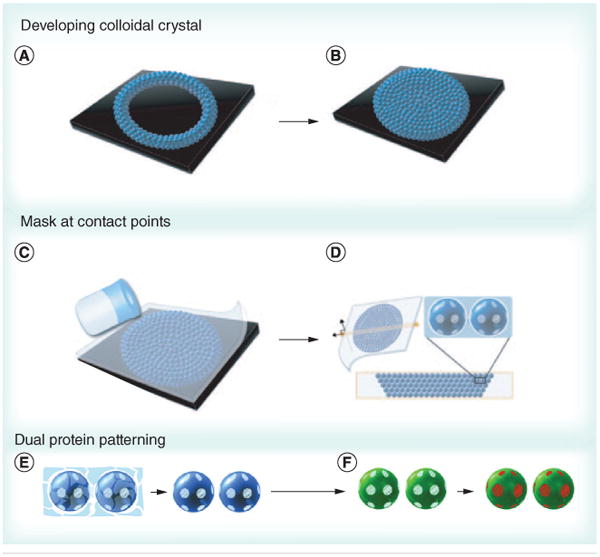
(A & B) Patchy particles can be formed by filling a well with a colloidal crystal iteratively, (C) then forming a polydimethylsiloxane (PDMS) mask by adding PDMS solution. (D) As dewetting occurs at the interface between particles, PDMS patches form at the contact points. The particles can be (E) separated from the scaffold and (F) two different proteins can be added by adding the first protein to the exposed region (green) followed by removal of the mask and addition of a second protein (red).
Reproduced with permission from [58].
Microfluidics has also been used to address the problem of synthesizing particles with multiple functionalities. Particles are synthesized by flowing multiple polymers or monomers into a single stream [59]. Janus and ternary particles can be synthesized by mixing two or three monomers with a photoinitiator in a microfluidic device. Two surfactant-containing streams and a monomer-containing stream between them are forced through a narrow opening that causes the fluid to break up into droplets. The droplets are then polymerized by UV irradiation [60]. This approach does not, however, generate particles with nanoscale features, as the particle sizes generated are approximately 100 μm. Biphasic Janus particles were generated with nanoscale features by the use of simultaneous electrohydrodynamic jetting [61]. In this case, instead of the particles being generated by being forced through a narrow opening, an electric field is applied and the nano-Janus particles are collected on the collecting plate, which houses the counter electrode (Figure 4) [61]. This method can be performed using two of the same polymers while loading different drugs in each half, and/or can allow selective chemical modification of one portion of the particle, making this setup amenable to the generation of biphasic nanoscale aAPCs. This technique does not allow the precise patterning afforded by some of the printing strategies, but future advances in the technique may make this a viable option. In addition, one could envision using this dual approach to provide cytokine release from one portion of the nanoparticle and stable ligand presentation from another portion of the nanoparticle, successfully avoiding some of the compromises often required for controlled release and surface presentation of ligands. Critically, this type of approach allows generation of a ‘top’ and ‘bottom’, or ‘front’ and ‘back’, which can each contain appropriate surface molecules, and in this manner better mimic biological cells.
Figure 4. Nanoscale biphasic Janus particles.
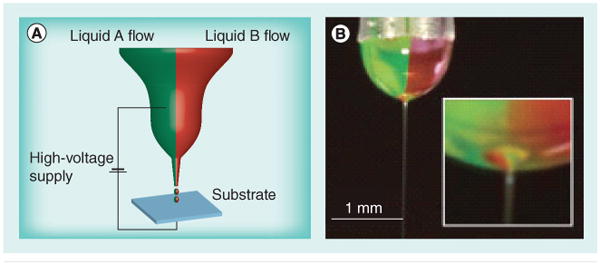
(A) The experimental setup used for generation of nanoscale biphasic Janus particles. The bipolar jetting fluid is exposed to applied electrical potential and the particles are collected on the counter electrode. (B) Digital image of the biphasic taylor cone with jet.
Reproduced with permission from [61].
Dynamic surface rearrangement: liposomes & protocells
More recent advances in the use of liposomes as aAPCs help to demonstrate additional parameters of likely importance for nanoengineered approaches to aAPC design. In particular, the signal 1–signal 2–signal 3 model that emphasizes the identity of the molecular signals involved in the directed signaling by an APC to a T cell may be supplemented by developing approaches to mimic other critical aspects of the biological system.
Liposomes afford two advantages to solid particle systems that mimic the biological system more directly. First of all, liposomal membrane fluidity enables the aAPC to dynamically rearrange the proteins on its surface, in a similar fashion as the dramatic protein rearrangements that occur during APC/T-cell interaction and generate the IS. In addition to the ability of liposomes to allow for biomimetic nanoscale reorganization of the proteins expressed on the surface, further advances in the ability to control initial organization of the expressed proteins might also be enabling. With respect to IS formation, an innovative study using 2D supported lipid bilayers (SLBs) showed that the imposition of patterned lines disrupted the geometry of the IS [62]. These data indicate that initial TCR engagement is followed by formation of TCR microdomains, which are then followed by directed transport of microclusters to form the final cSMAC. Lipid rafts, which are small, 10–200 nm domains that are highly enriched in sterols (e.g., cholesterol) or sphingolipids [63], have been shown to be critical organizing features of biological membranes [64,65], and molecular simulations have indicated the importance of these nanodomains to improve protein–protein interactions [66]. In fact, biological APCs have been shown to precluster antigen even in the absence of T cells [67], and the concentration of MHC molecules into lipid rafts has been shown to improve antigen presentation in biological APCs [68]. Perhaps partitioning cSMAC components into small lipid rafts for initial TCR signaling, and allowing flow in the membrane to rearrange into larger clusters, could be a particularly effective method to mimic the biology and achieve optimal stimulation from aAPCs.
Giannoni et al. showed that preclustering of pMHC and costimulatory signals in a nanoscale liposomal system resulted in higher T-cell stimulation than with soluble tetramers or liposomal aAPCs without preclustering [28]. The β-subunit of cholera toxin interacts strongly with cholesterol, a major component of lipid rafts. Preclustering was accomplished by linking the surface proteins to cholera toxin by biotin–neutravidin interactions (with biotin on the antibodies and neutravidin on the β-subunit of the cholera toxin). Importantly, the distribution alone (with no alteration in the quantity of any ligand) critically modulated the strength of the stimulation by the aAPCs. A subsequent study where the same group added anti-LFA-1 (the major adhesion molecule involved) to anti-CD3 (for general T-cell activation) and anti-CD28 (for costimulation), all preclustered in microdomains as before, resulted in increased expansion of polyclonal T cells and MART-1-specific CD8+ T cells compared with commercially available systems [69]. Interestingly, this second approach of preclustering LFA-1 with cSMAC components does not allow for subsequent segregation of the LFA-1 into the surrounding pSMAC. Perhaps future experiments might investigate whether preclustering of signal 1 and signal 2 separate from adhesion molecules, such as LFA-1, could more directly mimic the biological situation and further enhance activation.
A major difficulty that has hindered the use of liposomes as aAPC surrogates is their relative instability when compared with solid particles. One potential novel solution that has been used for drug delivery, but has yet to be applied to the design of aAPCs, is the concept of particle–SLBs.
Particle systems that incorporate lipid monolayers or lipid bilayers on their surfaces have been developed, which offer improved stability and drug delivery particles to standard liposomal formulations [70]. To synthesize next-generation aAPCs, the focus should be on SLBs as opposed to the systems that utilize only lipid monolayers at the particle surface, because SLBs enable replication of the fluidity of biological membranes in artificial systems. In particular, for the design of future aAPCs, mimicking the biological situation that allows for large-scale protein rearrangement subsequent to TCR triggering is likely to be of significant benefit.
SLBs have been used extensively as a model system for the study of the molecular dynamics of ISs, as they provide a flexible platform that is compatible with modern imaging techniques and have most of the characteristics of a real APC membrane [71]. This technique, extensively studied in 2D, has been recently applied to the synthesis of nanoparticles with SLBs on a hydrogel [72], silica particle [73,74] or polymeric (PLGA) particle core [75]. These particle–SLBs can be anchored (by covalently attaching the inner layer to the surface) or unanchored, with a lipid bilayer sitting on the surface of the silica or polymeric particle core.
Ashley et al. developed a protocell that consists of a nanoporous silica core and a SLB, which can be modified with targeting ligands, fusogenic peptides and PEG, which enables increased stability and drug delivery capacity (Figure 5) [76]. Interestingly, the nanoporous silica particles showed increased membrane fluidity when compared with protocells formed from nonporous solid silica nanoparticles or unsupported liposomes. These protocells can be loaded with a variety of cargoes, such as small molecule drugs, siRNA, toxins or quantum dots, and show vastly improved (106-fold) anti-cancer activity when compared with comparable liposomes. Critically, this indicates the potential to use this system to release immunomodulatory cytokines from the construct, potentially allowing the synthesis of an aAPC that can provide signal 1, 2 and 3, while replicating the fluidity of biological membranes in a stable fashion.
Figure 5. A particle-supported lipid bilayer with the range of cargoes and surface ligands that can be encapsulated or presented on the surface of the particle.
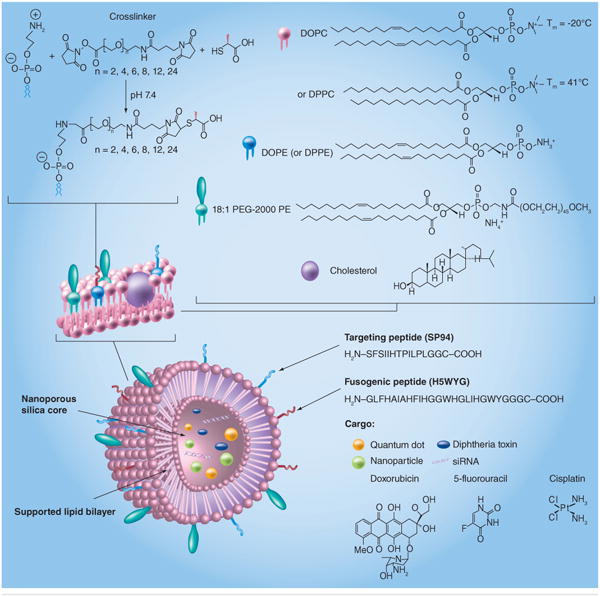
DOPC: 1,2-dioleoyl-sn-glycero-3-phosphocholine; DOPE: 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine; DPPC: 1,2-dipalmitoyl-sn- glycero-3-phosphorylcholine; DPPE: 1,2-dipalmitoyl-sn-glycero-3-phosphorylethanolamine; PE: Polyethylene.
Reproduced with permission from [76].
Porotto et al. have used larger (3 μm) protocells designed to inactivate enveloped viruses by presenting viral entry receptors on the surface of the protocells. This enables the protocells to act as cellular decoys, inactivating viruses that would otherwise infect healthy cells by triggering premature fusion of the viruses. Interestingly, at low temperatures (4°C), the protocells were unable to activate the virus, whereas at 37°C, considerable inactivation was demonstrated. This indicates that membrane fluidity is required for protocell inactivation of the virus. Importantly, the protocells did not accumulate the virus, but rather were renewable, as binding led directly to premature fusion and permanent inactivation of the viruses [77].
In addition to fully chemically synthesized SLBs, natural erythrocyte membranes have been used to coat biodegradable polymeric microparticles [75], and leukocyte membranes have been fused to silica cores [78]. These approaches present an alternative to the reductionist systems typically used in acellular aAPC systems. They offer the potential to generate particles that are coated with real DC membranes and perhaps subsequently modified to generate an off-the-shelf acellular aAPC, which has much of the benefits of an acellular system while retaining all the critical components of a real DC membrane.
Importance of the area of contact between a cell & an artificial surface
Particle size as a critical parameter
PS particles have been extensively studied as platforms for the development of aAPCs and studies in this system have enhanced our understanding of critical parameters in designing acellular aAPCs. Perhaps most critically, the effect of particle size on aAPC function was first studied using PS particles [11]. Testing various sized spherical PS particles with class I MHC immobilized on the surface, Mesher found that 4–5 μm particles provided optimal stimulus. Smaller particles showed decreased stimulation, and this decreased stimulation could not be overcome by increasing the dose of the smaller particles. These results indicated that receptor occupancy over a large surface area of contact is a critical determinant for activation [11]. While efficient and effective nanoscale aAPCs might have better properties for in vivo applications, such as improved draining to lymph nodes, and reduced likelihood of safety concerns (e.g., as aAPC becoming trapped in a capillary bed), these data point to the potential limitations of nanoscale spherical aAPCs.
Altered particle shape
The use of nonspherical particles has generated increasing interest in recent years for biomedical applications. From a biomimetic perspective, the various morphologies of bacteria and viruses are suspected to play a role in their ability to efficiently invade and colonize cells. Generally, the cytoskeleton, its organization and the physical cues that it can transmit can result in dramatic effects on cell fate [79]. This is seen during the interaction of a T cell with an APC, which is a critical determinant of T-cell fate and effector function. With activation, APCs, such as DCs, have major changes in their cell morphology resulting in significant increases in their overall cell surface area, facilitating interaction with naive T cells to direct T-cell fate. In addition to surface area consideration, the geometry at the interface of an APC and a T cell is dramatically dissimilar from two spheres interacting. Nonspherical microscale aAPCs would allow greater surface area for a given volume and flatter interfacial geometries can allow for increased interaction with T cells compared with spherical alternatives. In addition, for nanoscale aAPCs, nonspherical particles could allow for interfacial geometry similar to successful microparticulate systems, while nanoscale size could facilitate improved in vivo performance due to easy access to draining lymph nodes and suitability for intravenous injection.
A wide variety of shapes have been generated by top-down and bottom-up approaches [80,81]. Particles have been made that mimic the mechanobiology of red blood cells, resulting in increased circulation times and dramatically improving their biodistribution profiles [82]. Recent advances in the ability to generate particles with diverse shapes has enabled the study of the effect of shape on cellular internalization, phagocytosis, particle attachment and circulation half-life [83]. Local particle curvature appears to dictate whether or not the particle will be phagocytosed [84,85]. In particular, high aspect ratio (AR) ellipsoidal microparticles (in particular, those with AR >20) have reduced phagocytosis compared with spherical particles, because when the low curvature sides of the particle encounter a phagocytic cell, the low curvature prevents the cell from engulfing the particle. In addition, the shape of the particle also modulates the degree of particle attachment to macrophages independently of the rate it internalizes the particle. In particular, prolate ellipsoids (one axis longer than the other two; i.e., a > b = c) showed the lowest internalization rates, but most efficient particle attachment, when compared with oblate ellipsoids (one axis shorter than the other two; i.e., a = b > c) or spherical particles [86]. Particle shape has also been implicated in increasing circulation time for particles injected into the bloodstream of mice, by aligning with blood flow in a superior fashion to spherical particles and reducing phagocytosis [87,88].
Particle shape may also play a role in aAPC function. Barua et al. recently developed spherical, rod- and disk-shaped PS micro- and nano-particles with anti-Her2 antibody on their surface [89]. Anti-Her2 mAb conjugated rods showed reduced nonspecific uptake and enhanced specific uptake into cancer cell lines, and greater inhibition of cancer cell growth in vitro as compared with the spheres; this was posited to be due to interplay between shape and binding/unbinding events. These same physical principles are also key for the interaction of an APC and a T cell, and so strongly support the idea that particle shape may play a key role in improving aAPC function.
To date, nearly all acellular aAPC systems have utilized spherical particles in their constructs. However, both the dramatic morphological changes that come with activation of DCs, and the potential improvements in terms of decreased particle internalization and increased particle attachment indicates that particle shape may play a key role in future aAPC systems.
Fadel et al. have developed single-walled carbon nanotube (SWNT) bundles for immune stimulation by nonspecific absorption of antibodies to the surface of chemically treated carbon nanotubes [90,91]. In their initial study, they adsorbed anti-CD3 antibody to their SWNT bundles, and found that functionalized SWNT bundles activated T cells more efficiently than CD3 mAb alone, by over an order of magnitude. This was superior to activated PS nanoparticles and buckeyballs. Interestingly, the nanotubes appear to cluster only once functionalized with an antibody, forming large-scale aggregates (>5 μm in size). One advantage of this system is that the nanoscale features of the SWNT bundles provide a very high surface area.
SWNT bundles with absorbed anti-CD3 and anti-CD28 mAb showed random regions of higher density that mimic the biological situation, as activated aAPCs before contact with T cells have preclustered pMHC and costimulatory signals such as B7.1. This clustering occurred principally in the chemically treated SWNT bundles and these showed substantial improvements in activity. Thus, this ‘preclustering’ of smaller areas of high density appeared to be beneficial for activity.
Methods to engineer polymeric aAPC shape
Perhaps the simplest and most accessible method developed for generating nonspherical particles uses a film-stretching technique originally developed by Ho et al. [92], and more recently adapted to generate polymeric micro- and nano-particles of varied shape (Figure 6) [93]. Polymeric particles are suspended in a solution containing high quantities of poly(vinyl)alcohol and some glycerol as a plasticizer. The particle solution is then cast into a film by pouring it onto a leveling table and allowing the water to evaporate over time. This film can then be cut into pieces, stretched under heating on a stretching device, cooled and then the particles are removed by dissolving the film. The advantages of this approach are that it does not require complex technology and generation of nanoparticles with a complex shape is possible. This process lends itself less efficiently to large-scale batch synthesis and batch-to-batch variability due to inhomogenous stretching can be a moderate issue. This approach has also been used to generate biodegradable PLGA micro-and nano-particles with altered shape using the film-stretching approach [94]. Our laboratory has recently constructed functional ellipsoidal aAPCs using this technique that showed improvements in in vitro T-cell activation and in vivo tumor prevention as compared with their spherical counterparts [95].
Figure 6. Potential particle shapes of interest for artificial antigen-presenting cell development.

Diverse shapes can be made using a film-stretching method using (A–H) microparticles and (I) nanoparticles. (J) Polymeric particles can be synthesized with a single metallic flat patch using a hybrid etching and deposition process based on colloidal lithography.
(A–I) Adapted with permission from [93].
(J) Reproduced with permission from [100] © American Chemical Society (2012).
There are several alternative methods to the film-stretching approach to construct non-spherical particles. One of the most versatile approaches to generating a vast array of potential shapes is particle replication in nonwetting templates (PRINT) technology [96]. By using photocurable perfluoropolyether molds and fluorinated surfaces, PRINT is able to produce isolated, harvestable individual particles. It can be used to generate monodisperse micro- and nano-particles (<100 nm) of various polymers, such as PLGA, poly(pyrrole) and PEG, and can be used to incorporate proteins, DNA and small molecules into the shaped particles. In addition, particles can be printed that are 100% protein [97].
High AR particles, as noted above, are of considerable interest owing to favorable reduction in phagocytosis and increase in cellular binding properties. High AR particles can be generated using PRINT by using a mechanical elongation strategy. This involves fabricating an initial PDMS mold, deforming that mold and using the deformed mold to generate a new mold [98]. Due to limitations in deformation extent, this process can be repeated in cycles to generate very high AR particles.
Continuous flow lithography has also been developed for synthesis of particles with diverse shapes [99]. In this process, an acrylate oligomer stream containing a photoinitiator flowing through a PDMS microfluidic device is exposed to controlled pulses of UV light shone through a transparency mask that is patterned by lithography. The limitation here is that continuous flow lithography cannot readily fabricate <3-μm shapes, as the process is limited by polymerization times and the feature sizes that are printable on a transparency mask (polymerization time is inversely related to the size of the transparency mask). A modified stop–polymerize–flow method was suggested to enable synthesis of 1-μm particles.
Yu et al. have developed binary and ternary hybrid particles, which consist of spherical PS particles with a flat spherical metallic stamp (Figure 6J), using a combination of etching and deposition processes [100]. This method is of particular interest, as it might enable dual functionalization of a putative aAPC (with one set of proteins conjugated to the metallic surface, and another set of proteins conjugated to the rest of the polymeric particle) while presenting a much different, flat shape at the APC–T-cell interface.
Conclusion & future perspective
Acellular aAPCs, in particular, have shown great initial promise for ex vivo activation of CTLs, and have also been investigated for in vivo applications. The development of aAPCs has focused mainly on the choice of proteins to use for surface presentation to T cells when conjugated to various spherical, microscale particles. Key recent advances have allowed for the development of acellular aAPCs that incorporate more biological cues than antigen recognition (signal 1) and costimulation (signal 2). Some aAPCs have been developed that incorporate secretable cues (cytokines; ‘signal 3’) and surface geometric cues that operate from the nanoscale to the microscale, such as interfacial geometry, surface protein organization and segregation, and dynamic protein rearrangement. Early work has demonstrated a critical role for particle size, showing that the surface area available for contact is crucial in these systems. In addition, preclustering of protein signals into nanodomains (lipid rafts) has been shown to be of substantial benefit for aAPC-based stimulation.
New synthetic particle technology has been developed to synthesize patchy particles with varying geometry and nanoscale features. These technologies may allow for the development of nanoscale anisotropic aAPCs that mimic the physical segregation evident in the IS.
Liposome-based aAPCs, and lipid rafts, enable the generation of nanoclustered surface functionality, and the fluidity of the membranes allows for more biomimetic dynamic rearrangement of surface proteins upon contact with target cells. However, liposomes have suffered from relative instability compared with solid particles. Recent advances in the development of SLBs could enable the generation of SLB-based aAPCs that offer the advantages of liposomal systems with superior stability and improved drug release.
Biodegradable particles offer strong biocompatablility, and are useful for the release of secretable cues or other immunomodulatory factors. Recent advances in the development of methods for the generation of nonspherical biodegradable particles may enable next-generation aAPCs with interfacial geometry that more closely mimics the biological situation. In addition, nonspherical aAPCs offer the potential of developing nanoparticles with interfacial geometry similar to successful microparticulate systems, with improved in vivo performance due to easy access to draining lymph nodes and suitability for intravenous injection.
When designing future aAPCs, a biomaterials/nanomedicine scientist should carefully consider not just the choice of surface protein (signal 1 and signal 2) and soluble (signal 3) signals, but also consider the spatial organization of ligands on the nanoscale, ligand orientation, ligand anisotropy (either fixed or dynamic) and the optimal geometry at the T-cell–aAPC interface. The end goal of such efforts should be directed at not only accomplishing efficient T-cell expansion, but also engendering memory formation, optimizing T-cell functionality and enabling effective immune responses in clinically relevant treatment models.
Executive summary.
Acellular artificial antigen-presenting cells
Acellular artificial antigen-presenting cells (aAPCs) are micro- or nano-particle-based reductionist systems that mimic the biological cues given by activated biological antigen-presenting cells to T cells to direct T-cell fate and induce cellular immunity.
aAPCs can be equipped with signals for antigen recognition (‘signal 1’; peptide in MHC), costimulation (‘signal 2’) and cytokine release (‘signal 3’).
Nanoengineering approaches for next-generation aAPCs
Most current-generation aAPCs employ static isotropic presentation of protein signals, but advances in nanoengineering approaches may allow for better biomimicry through the addition of nanoscale features to microscale aAPCs, and may improve the potential of nanoparticle-based aAPCs.
Engineering surface protein organization for increased biomimicry
New techniques for the synthesis of patchy particles allows for fixed ligand organization that may be able to mimic the geometry and organization of the mature immunological synapse.
Liposomes and particle-supported lipid bilayers can allow for surface presentation of organized nanoclusters prior to T-cell contact, and allow for the dynamic surface rearrangements seen in the biological setting.
Modulation of particle shape to increase surface contact area
Experiments with aAPC particle size have indicated that receptor occupancy over a large surface area of contact is a critical determinant for activation.
New techniques enable the generation of defined, nonspherical micro- and nano-particles.
Alterations in particle shape enable increased surface area for cell contact, allowing for nanoparticles with interfacial geometry similar to successful microparticulate systems, with improved in vivo performance due to easy access to draining lymph nodes, and suitability for intravenous injection.
Acknowledgments
The authors thank the NIH (R01-EB016721) for support and JC Sunshine thanks the Medical Scientist Training Program (MSTP) program for support. JJ Green is a consultant for NexImmune.
Footnotes
Financial & competing interests disclosure: Conflicts of interest are managed by the Johns Hopkins Medical Institutions Committee on Outside Interests.
The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
References
Papers of special note have been highlighted as:
▪ of interest
▪ of considerable interest
- 1.Kawakami Y, Eliyahu S, Delgado CH, et al. Cloning of the gene coding for a shared human melanoma antigen recognized by autologous T cells infiltrating into tumor. Proc Natl Acad Sci USA. 1994;91(9):3515–3519. doi: 10.1073/pnas.91.9.3515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.van der Bruggen P, Traversari C, Chomez P, et al. A gene encoding an antigen recognized by cytolytic T lymphocytes on a human melanoma. Science. 1991;254(5038):1643–1647. doi: 10.1126/science.1840703. [DOI] [PubMed] [Google Scholar]
- 3.Novellino L, Castelli C, Parmiani G. A listing of human tumor antigens recognized by T cells: March 2004 update. Cancer Immunol Immunother. 2005;54(3):187–207. doi: 10.1007/s00262-004-0560-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kantoff PW, Higano CS, Shore ND, et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N Engl J Med. 2010;363(5):411–422. doi: 10.1056/NEJMoa1001294. [DOI] [PubMed] [Google Scholar]
- 5.Oelke M, Maus MV, Didiano D, June CH, Mackensen A, Schneck JP. Ex vivo induction and expansion of antigen-specific cytotoxic T cells by HLA-Ig-coated artificial antigen-presenting cells. Nat Med. 2003;9(5):619–624. doi: 10.1038/nm869. [DOI] [PubMed] [Google Scholar]
- 6.Maus MV, Thomas AK, Leonard DG, et al. Ex vivo expansion of polyclonal and antigen-specific cytotoxic T lymphocytes by artificial APCs expressing ligands for the T-cell receptor, CD28 and 4–1BB. Nat Biotechnol. 2002;20(2):143–148. doi: 10.1038/nbt0202-143. [DOI] [PubMed] [Google Scholar]
- 7.Durai M, Krueger C, Ye Z, et al. In vivo functional efficacy of tumor-specific T cells expanded using HLA-Ig based artificial antigen presenting cells (aAPC) Cancer Immunol Immunother. 2009;58(2):209–220. doi: 10.1007/s00262-008-0542-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Steenblock ER, Fahmy TM. A comprehensive platform for ex vivo T-cell expansion based on biodegradable polymeric artificial antigen-presenting cells. Mol Ther. 2008;16(4):765–772. doi: 10.1038/mt.2008.11. [DOI] [PubMed] [Google Scholar]
- 9▪.Steenblock ER, Fadel T, Labowsky M, Pober JS, Fahmy TM. An artificial antigen-presenting cell with paracrine delivery of IL-2 impacts the magnitude and direction of the T cell response. J Biol Chem. 2011;286(40):34883–34892. doi: 10.1074/jbc.M111.276329. Demonstrated paracrine cytokine release from biodegradable artificial antigen-presenting cells with stable presentation of surface ligands. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Engelhard VH, Strominger JL, Mescher M, Burakoff S. Induction of secondary cytotoxic T lymphocytes by purified HLA-A and HLA-B antigens reconstituted into phospholipid vesicles. Proc Natl Acad Sci USA. 1978;75(11):5688–5691. doi: 10.1073/pnas.75.11.5688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mescher MF. Surface contact requirements for activation of cytotoxic T lymphocytes. J Immunol. 1992;149(7):2402–2405. [PubMed] [Google Scholar]
- 12.Curtsinger J, Deeths MJ, Pease P, Mescher MF. Artificial cell surface constructs for studying receptor-ligand contributions to lymphocyte activation. J Immunol Methods. 1997;209(1):47–57. doi: 10.1016/s0022-1759(97)00146-4. [DOI] [PubMed] [Google Scholar]
- 13.Turtle CJ, Riddell SR. Artificial antigen-presenting cells for use in adoptive immunotherapy. Cancer J. 2010;16(4):374–381. doi: 10.1097/PPO.0b013e3181eb33a6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Manolova V, Flace A, Bauer M, Schwarz K, Saudan P, Bachmann MF. Nanoparticles target distinct dendritic cell populations according to their size. Eur J Immunol. 2008;38(5):1404–1413. doi: 10.1002/eji.200737984. [DOI] [PubMed] [Google Scholar]
- 15.Gilboa E. The makings of a tumor rejection antigen. Immunity. 1999;11(3):263–270. doi: 10.1016/s1074-7613(00)80101-6. [DOI] [PubMed] [Google Scholar]
- 16.Jenkins MK, Taylor PS, Norton SD, Urdahl KB. CD28 delivers a costimulatory signal involved in antigen-specific IL-2 production by human T cells. J Immunol. 1991;147(8):2461–2466. [PubMed] [Google Scholar]
- 17.Curtsinger JM, Schmidt CS, Mondino A, et al. Inflammatory cytokines provide a third signal for activation of naive CD4+ and CD8+ T cells. J Immunol. 1999;162(6):3256–3262. [PubMed] [Google Scholar]
- 18.Grakoui A, Bromley SK, Sumen C, et al. The immunological synapse: a molecular machine controlling T cell activation. Science. 1999;285(5425):221–227. doi: 10.1126/science.285.5425.221. [DOI] [PubMed] [Google Scholar]
- 19.Lee KH, Holdorf AD, Dustin ML, Chan AC, Allen PM, Shaw AS. T cell receptor signaling precedes immunological synapse formation. Science. 2002;295(5559):1539–1542. doi: 10.1126/science.1067710. [DOI] [PubMed] [Google Scholar]
- 20.Rosenberg SA, Packard BS, Aebersold PM, et al. Use of tumor-infiltrating lymphocytes and interleukin-2 in the immunotherapy of patients with metastatic melanoma A preliminary report. N Engl J Med. 1988;319(25):1676–1680. doi: 10.1056/NEJM198812223192527. [DOI] [PubMed] [Google Scholar]
- 21.Dudley ME, Wunderlich JR, Robbins PF, et al. Cancer regression and autoimmunity in patients after clonal repopulation with antitumor lymphocytes. Science. 2002;298(5594):850–854. doi: 10.1126/science.1076514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kurokawa T, Oelke M, Mackensen A. Induction and clonal expansion of tumor-specific cytotoxic T lymphocytes from renal cell carcinoma patients after stimulation with autologous dendritic cells loaded with tumor cells. Int J Cancer. 2001;91(6):749–756. doi: 10.1002/1097-0215(200002)9999:9999<::aid-ijc1141>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- 23.Altman JD, Moss PA, Goulder PJ, et al. Phenotypic analysis of antigen-specific T lymphocytes. Science. 1996;274(5284):94–96. doi: 10.1126/science.274.5284.94. [DOI] [PubMed] [Google Scholar]
- 24.Neudorfer J, Schmidt B, Huster KM, et al. Reversible HLA multimers (streptamers) for the isolation of human cytotoxic T lymphocytes functionally active against tumor- and virus-derived antigens. J Immunol Methods. 2007;320(1–2):119–131. doi: 10.1016/j.jim.2007.01.001. [DOI] [PubMed] [Google Scholar]
- 25.Nguyen DN, Green JJ, Chan JM, Longer R, Anderson DG. Polymeric materials for gene delivery and dna vaccination. Adv Mater. 2009;21(8):847–867. doi: 10.1002/adma.200801478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rosenberg SA. Raising the bar: the curative potential of human cancer immunotherapy. Sci Transl Med. 2012;4(127):127ps8. doi: 10.1126/scitranslmed.3003634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Morgan RA, Dudley ME, Wunderlich JR, et al. Cancer regression in patients after transfer of genetically engineered lymphocytes. Science. 2006;314(5796):126–129. doi: 10.1126/science.1129003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28▪▪.Giannoni F, Barnett J, Bi K, et al. Clustering of T cell ligands on artificial APC membranes influences T cell activation and protein kinase C theta translocation to the T cell plasma membrane. J Immunol. 2005;174(6):3204–3211. doi: 10.4049/jimmunol.174.6.3204. Demonstrates the utility and importance of protein signal preclustering in liposomal acellular artificial antigen-presenting cells. [DOI] [PubMed] [Google Scholar]
- 29.Ugel S, Zoso A, De Santo C, et al. In vivo administration of artificial antigen-presenting cells activates low-avidity T cells for treatment of cancer. Cancer Res. 2009;69(24):9376–9384. doi: 10.1158/0008-5472.CAN-09-0400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Levine BL, Bernstein WB, Connors M, et al. Effects of CD28 costimulation on long-term proliferation of CD4+ T cells in the absence of exogenous feeder cells. J Immunol. 1997;159(12):5921–5930. [PubMed] [Google Scholar]
- 31.Maus MV, Riley JL, Kwok WW, Nepom GT, June CH. HLA tetramer-based artificial antigen-presenting cells for stimulation of CD4+ T cells. Clin Immunol. 2003;106(1):16–22. doi: 10.1016/s1521-6616(02)00017-7. [DOI] [PubMed] [Google Scholar]
- 32.Han H, Peng JR, Chen PC, et al. A novel system of artificial antigen-presenting cells efficiently stimulates Flu peptide-specific cytotoxic T cells in vitro. Biochem Biophys Res Commun. 2011;411(3):530–535. doi: 10.1016/j.bbrc.2011.06.164. [DOI] [PubMed] [Google Scholar]
- 33.Haveman LM, Bierings M, Klein MR, et al. Selection of perforin expressing CD4+ adenovirus-specific T-cells with artificial antigen-presenting cells. Clin Immunol. 2013;146(3):228–239. doi: 10.1016/j.clim.2013.01.002. [DOI] [PubMed] [Google Scholar]
- 34.Schutz C, Fleck M, Mackensen A, et al. Killer artificial antigen-presenting cells: a novel strategy to delete specific T cells. Blood. 2008;111(7):3546–3552. doi: 10.1182/blood-2007-09-113522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schutz C, Oelke M, Schneck JP, Mackensen A, Fleck M. Killer artificial antigen-presenting cells: the synthetic embodiment of a ‘guided missile’. Immunotherapy. 2010;2(4):539–550. doi: 10.2217/imt.10.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Webb TJ, Bieler JG, Schneck JP, Oelke M. Ex vivo induction and expansion of natural killer T cells by CD1d1-Ig coated artificial antigen-presenting cells. J Immunol Methods. 2009;346(1–2):38–44. doi: 10.1016/j.jim.2009.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sun W, Subrahmanyam PB, East JE, Webb TJ. Connecting the dots: artificial antigen-presenting cell-mediated modulation of natural killer T cells. J Interferon Cytokine Res. 2012;32(11):505–516. doi: 10.1089/jir.2012.0045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shiratsuchi T, Schneck J, Kawamura A, Tsuji M. Human CD1 dimeric proteins as indispensable tools for research on CD1-binding lipids and CD1-restricted T cells. J Immunol Methods. 2009;345(1–2):49–59. doi: 10.1016/j.jim.2009.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sauer MG, Ericson ME, Weigel BJ, et al. A novel system for simultaneous in vivo tracking and biological assessment of leukemia cells and ex vivo generated leukemia-reactive cytotoxic T cells. Cancer Res. 2004;64(11):3914–3921. doi: 10.1158/0008-5472.CAN-03-3991. [DOI] [PubMed] [Google Scholar]
- 40.O'Herrin SM, Slansky JE, Tang Q, et al. Antigen-specific blockade of T cells in vivo using dimeric MHC peptide. J Immunol. 2001;167(5):2555–2560. doi: 10.4049/jimmunol.167.5.2555. [DOI] [PubMed] [Google Scholar]
- 41.Pareja E, Tobes R, Martin J, Nieto A. The tetramer model: a new view of class II MHC molecules in antigenic presentation to T cells. Tissue Antigens. 1997;50(5):421–428. doi: 10.1111/j.1399-0039.1997.tb02896.x. [DOI] [PubMed] [Google Scholar]
- 42.Deeths MJ, Mescher MF. B7-1-dependent co-stimulation results in qualitatively and quantitatively different responses by CD4+ and CD8+ T cells. Eur J Immunol. 1997;27(3):598–608. doi: 10.1002/eji.1830270305. [DOI] [PubMed] [Google Scholar]
- 43.Deeths MJ, Mescher MF. ICAM-1 and B7-1 provide similar but distinct costimulation for CD8+ T cells, while CD4+ T cells are poorly costimulated by ICAM-1. Eur J Immunol. 1999;29(1):45–53. doi: 10.1002/(SICI)1521-4141(199901)29:01<45::AID-IMMU45>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 44.Butte MJ, Keir ME, Phamduy TB, Sharpe AH, Freeman GJ. Programmed death-1 ligand 1 interacts specifically with the B7-1 costimulatory molecule to inhibit T cell responses. Immunity. 2007;27(1):111–122. doi: 10.1016/j.immuni.2007.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ndhlovu ZM, Oelke M, Schneck JP, Griffin DE. Dynamic regulation of functionally distinct virus-specific T cells. Proc Natl Acad Sci USA. 2010;107(8):3669–3674. doi: 10.1073/pnas.0915168107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fuertes Marraco SA, Baumgaertner P, Legat A, Rufer N, Speiser DE. A stepwise protocol to coat aAPC beads prevents out-competition of anti-CD3 mAb and consequent experimental artefacts. J Immunol Methods. 2012;385(1–2):90–95. doi: 10.1016/j.jim.2012.07.017. [DOI] [PubMed] [Google Scholar]
- 47.Shalaby WS, Yeh H, Woo E, et al. Absorbable microparticulate cation exchanger for immunotherapeutic delivery. J Biomed Mater Res B Appl Biomater. 2004;69(2):173–182. doi: 10.1002/jbm.b.20040. [DOI] [PubMed] [Google Scholar]
- 48.Fahmy TM, Samstein RM, Harness CC, Mark Saltzman W. Surface modification of biodegradable polyesters with fatty acid conjugates for improved drug targeting. Biomaterials. 2005;26(28):5727–5736. doi: 10.1016/j.biomaterials.2005.02.025. [DOI] [PubMed] [Google Scholar]
- 49.Pawar AB, Kretzschmar I. Fabrication, assembly, and application of patchy particles. Macromol Rapid Commun. 2010;31(2):150–168. doi: 10.1002/marc.200900614. [DOI] [PubMed] [Google Scholar]
- 50.Walther A, Muller AHE. Janus particles. Soft Matter. 2008;4(4):663–668. doi: 10.1039/b718131k. [DOI] [PubMed] [Google Scholar]
- 51.Doh J, Irvine DJ. Immunological synapse arrays: patterned protein surfaces that modulate immunological synapse structure formation in T cells. Proc Natl Acad Sci USA. 2006;103(15):5700–5705. doi: 10.1073/pnas.0509404103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Snyder CE, Yake AM, Feick JD, Velegol D. Nanoscale functionalization and site-specific assembly of colloids by particle lithography. Langmuir. 2005;21(11):4813–4815. doi: 10.1021/la050715l. [DOI] [PubMed] [Google Scholar]
- 53.Cayre O, Paunov VN, Velev OD. Fabrication of dipolar colloid particles by microcontact printing. Chem Commun (Camb) 2003;18:2296–2297. doi: 10.1039/b307296g. [DOI] [PubMed] [Google Scholar]
- 54.Cayre O, Paunov VN, Velev OD. Fabrication of asymmetrically coated colloid particles by microcontact printing techniques. J Mater Chem. 2003;13(10):2445–2450. doi: 10.1039/b307296g. [DOI] [PubMed] [Google Scholar]
- 55.Zhao YP, Ye DX, Wang GC, Lu TM. Novel nano-column and nano-flower arrays by glancing angle deposition. Nano Lett. 2002;2(4):351–354. [Google Scholar]
- 56.Pawar AB, Kretzschmar I. Multifunctional patchy particles by glancing angle deposition. Langmuir. 2009;25(16):9057–9063. doi: 10.1021/la900809b. [DOI] [PubMed] [Google Scholar]
- 57.Zhang G, Wang DY, Mohwald H. Patterning microsphere surfaces by templating colloidal crystals. Nano Lett. 2005;5(1):143–146. doi: 10.1021/nl048121a. [DOI] [PubMed] [Google Scholar]
- 58▪.Kamalasanan K, Jhunjhunwala S, Wu J, Swanson A, Gao D, Little SR. Patchy, anisotropic microspheres with soft protein islets. Angew Chem Int Ed Engl. 2011;50(37):8706–8708. doi: 10.1002/anie.201101217. Novel method to generate fixed ligand anisotropy with appropriate geometry for acellular artificial antigen-presenting cell generation. [DOI] [PubMed] [Google Scholar]
- 59.Serra CA, Chang ZQ. Microfluidic-assisted synthesis of polymer particles. Chem Eng Technol. 2008;31(8):1099–1115. [Google Scholar]
- 60.Nie ZH, Li W, Seo M, Xu SQ, Kumacheva E. Janus and ternary particles generated by microfluidic synthesis: design, synthesis, and self-assembly. J Am Chem Soc. 2006;128(29):9408–9412. doi: 10.1021/ja060882n. [DOI] [PubMed] [Google Scholar]
- 61▪▪.Roh KH, Martin DC, Lahann J. Biphasic Janus particles with nanoscale anisotropy. Nat Mater. 2005;4(10):759–763. doi: 10.1038/nmat1486. Novel method for generating Janus nanoparticles with nanoscale anisotropy. [DOI] [PubMed] [Google Scholar]
- 62.Mossman KD, Campi G, Groves JT, Dustin ML. Altered TCR signaling from geometrically repatterned immunological synapses. Science. 2005;310(5751):1191–1193. doi: 10.1126/science.1119238. [DOI] [PubMed] [Google Scholar]
- 63.Pike LJ. Rafts defined: a report on the Keystone Symposium on Lipid Rafts and Cell Function. J Lipid Res. 2006;47(7):1597–1598. doi: 10.1194/jlr.E600002-JLR200. [DOI] [PubMed] [Google Scholar]
- 64.Lingwood D, Simons K. Lipid rafts as a membrane-organizing principle. Science. 2010;327(5961):46–50. doi: 10.1126/science.1174621. [DOI] [PubMed] [Google Scholar]
- 65.Hancock JF. Lipid rafts: contentious only from simplistic standpoints. Nat Rev Mol Cell Biol. 2006;7(6):456–462. doi: 10.1038/nrm1925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Nicolau DV, Burrage K, Parton RG, Hancock JF. Identifying optimal lipid raft characteristics required to promote nanoscale protein–protein interactions on the plasma membrane. Mol Cell Biol. 2006;26(1):313–323. doi: 10.1128/MCB.26.1.313-323.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Vogt AB, Spindeldreher S, Kropshofer H. Clustering of MHC–peptide complexes prior to their engagement in the immunological synapse: lipid raft and tetraspan microdomains. Immunol Rev. 2002;189:136–151. doi: 10.1034/j.1600-065x.2002.18912.x. [DOI] [PubMed] [Google Scholar]
- 68.Anderson HA, Hiltbold EM, Roche PA. Concentration of MHC class II molecules in lipid rafts facilitates antigen presentation. Nat Immunol. 2000;1(2):156–162. doi: 10.1038/77842. [DOI] [PubMed] [Google Scholar]
- 69.Zappasodi R, Di Nicola M, Carlo-Stella C, et al. The effect of artificial antigen-presenting cells with preclustered anti-CD28/-CD3/-LFA-1 monoclonal antibodies on the induction of ex vivo expansion of functional human antitumor T cells. Haematologica. 2008;93(10):1523–1534. doi: 10.3324/haematol.12521. [DOI] [PubMed] [Google Scholar]
- 70.Tan S, Li X, Guo Y, Zhang Z. Lipid-enveloped hybrid nanoparticles for drug delivery. Nanoscale. 2013;5(3):860–872. doi: 10.1039/c2nr32880a. [DOI] [PubMed] [Google Scholar]
- 71.Mossman KD, Campi G, Groves JT, Dustin ML. Altered TCR signaling from geometrically repatterned immunological synapses. Science. 2005;310(5751):1191–1193. doi: 10.1126/science.1119238. [DOI] [PubMed] [Google Scholar]
- 72.Jin T, Pennefather P, Lee PI. Lipobeads: a hydrogel anchored lipid vesicle system. FEBS Lett. 1996;397(1):70–74. doi: 10.1016/s0014-5793(96)01021-6. [DOI] [PubMed] [Google Scholar]
- 73.Liu J, Jiang X, Ashley C, Brinker CJ. Electrostatically mediated liposome fusion and lipid exchange with a nanoparticle-supported bilayer for control of surface charge, drug containment, and delivery. J Am Chem Soc. 2009;131(22):7567–7569. doi: 10.1021/ja902039y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Mornet S, Lambert O, Duguet E, Brisson A. The formation of supported lipid bilayers on silica nanoparticles revealed by cryoelectron microscopy. Nano Lett. 2005;5(2):281–285. doi: 10.1021/nl048153y. [DOI] [PubMed] [Google Scholar]
- 75.Hu CM, Zhang L, Aryal S, Cheung C, Fang RH. Erythrocyte membrane-camouflaged polymeric nanoparticles as a biomimetic delivery platform. Proc Natl Acad Sci USA. 2011;108(27):10980–10985. doi: 10.1073/pnas.1106634108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76▪▪.Ashley CE, Carnes EC, Phillips GK, et al. The targeted delivery of multicomponent cargos to cancer cells by nanoporous particle-supported lipid bilayers. Nat Mater. 2011;10(5):389–397. doi: 10.1038/nmat2992. Demonstrated supported lipid bilayers on nanoparticles with conjugation of targeting ligands. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Porotto M, Yi F, Moscona A, LaVan DA. Synthetic protocells interact with viral nanomachinery and inactivate pathogenic human virus. PLoS One. 2011;6(3):e16874. doi: 10.1371/journal.pone.0016874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Parodi A, Quattrocchi N, van de Ven AL, et al. Synthetic nanoparticles functionalized with biomimetic leukocyte membranes possess cell-like functions. Nat Nanotechnol. 2013;8(1):61–68. doi: 10.1038/nnano.2012.212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Fletcher DA, Mullins RD. Cell mechanics and the cytoskeleton. Nature. 2010;463(7280):485–492. doi: 10.1038/nature08908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Merkel TJ, Herlihy KP, Nunes J, Orgel RM, Rolland JP, DeSimone JM. Scalable, shape-specific, top-down fabrication methods for the synthesis of engineered colloidal particles. Langmuir. 2010;26(16):13086–13096. doi: 10.1021/la903890h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Glotzer SC, Solomon MJ. Anisotropy of building blocks and their assembly into complex structures. Nat Mater. 2007;6(8):557–562. doi: 10.1038/nmat1949. [DOI] [PubMed] [Google Scholar]
- 82.Merkel TJ, Jones SW, Herlihy KP, et al. Using mechanobiological mimicry of red blood cells to extend circulation times of hydrogel microparticles. Proc Natl Acad Sci USA. 2011;108(2):586–591. doi: 10.1073/pnas.1010013108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Champion JA, Katare YK, Mitragotri S. Particle shape: a new design parameter for micro- and nanoscale drug delivery carriers. J Control Release. 2007;121(1–2):3–9. doi: 10.1016/j.jconrel.2007.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84▪.Champion JA, Mitragotri S. Role of target geometry in phagocytosis. Proc Natl Acad Sci USA. 2006;103(13):4930–4934. doi: 10.1073/pnas.0600997103. Method to produce particles with diverse shape without extensive hardware requirements. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Champion JA, Mitragotri S. Shape induced inhibition of phagocytosis of polymer particles. Pharm Res. 2009;26(1):244–249. doi: 10.1007/s11095-008-9626-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Sharma G, Valenta DT, Altman Y, et al. Polymer particle shape independently influences binding and internalization by macrophages. J Control Release. 2010;147(3):408–412. doi: 10.1016/j.jconrel.2010.07.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Muro S, Garnacho C, Champion JA, et al. Control of endothelial targeting and intracellular delivery of therapeutic enzymes by modulating the size and shape of ICAM-1-targeted carriers. Mol Ther. 2008;16(8):1450–1458. doi: 10.1038/mt.2008.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Geng Y, Dalhaimer P, Cai S, et al. Shape effects of filaments versus spherical particles in flow and drug delivery. Nat Nanotechnol. 2007;2(4):249–255. doi: 10.1038/nnano.2007.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89▪.Barua S, Yoo JW, Kolhar P, Wakankar A, Gokarn YR, Mitragotri S. Particle shape enhances specificity of antibody-displaying nanoparticles. Proc Natl Acad Sci USA. 2013;110(9):3270–3275. doi: 10.1073/pnas.1216893110. Demonstrates the functional effect of the shape of nanoparticles on binding/unbinding events. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Fadel TR, Steenblock ER, Stern E, et al. Enhanced cellular activation with single walled carbon nanotube bundles presenting antibody stimuli. Nano Lett. 2008;8(7):2070–2076. doi: 10.1021/nl080332i. [DOI] [PubMed] [Google Scholar]
- 91.Fadel TR, Look M, Staffier PA, Haller GL, Pfefferle LD, Fahmy TM. Clustering of stimuli on single-walled carbon nanotube bundles enhances cellular activation. Langmuir. 2010;26(8):5645–5654. doi: 10.1021/la902068z. [DOI] [PubMed] [Google Scholar]
- 92.Ho CC, Keller A, Odell JA, Ottewill RH. Preparation of monodisperse ellipsoidal polystyrene particles. Colloid Polym Sci. 1993;271(5):469–479. [Google Scholar]
- 93.Champion JA, Katare YK, Mitragotri S. Making polymeric micro- and nanoparticles of complex shapes. Proc Natl Acad Sci USA. 2007;104(29):11901–11904. doi: 10.1073/pnas.0705326104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Yoo JW, Mitragotri S. Polymer particles that switch shape in response to a stimulus. Proc Natl Acad Sci USA. 2010;107(25):11205–11210. doi: 10.1073/pnas.1000346107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Sunshine JC, Perica K, Schneck JP, Green JJ. Non-spherical artificial antigen presenting cells for tumor immunotherapy. Presented at: Society For Biomaterials Annual Meeting 2013; Boston, MA, USA. 10–13 April 2013. [Google Scholar]
- 96.Rolland JP, Maynor BW, Euliss LE, Exner AE, Denison GM, DeSimone JM. Direct fabrication and harvesting of monodisperse, shape-specific nanobiomaterials. J Am Chem Soc. 2005;127(28):10096–10100. doi: 10.1021/ja051977c. [DOI] [PubMed] [Google Scholar]
- 97.Kelly JY, DeSimone JM. Shape-specific, monodisperse nano-molding of protein particles. J Am Chem Soc. 2008;130(16):5438–5439. doi: 10.1021/ja8014428. [DOI] [PubMed] [Google Scholar]
- 98.Wang Y, Merkel TJ, Chen K, Fromen CA, Betts DE, DeSimone JM. Generation of a library of particles having controlled sizes and shapes via the mechanical elongation of master templates. Langmuir. 2011;27(2):524–528. doi: 10.1021/la1045095. [DOI] [PubMed] [Google Scholar]
- 99.Dendukuri D, Pregibon DC, Collins J, Hatton TA, Doyle PS. Continuous-flow lithography for high-throughput microparticle synthesis. Nat Mater. 2006;5(5):365–369. doi: 10.1038/nmat1617. [DOI] [PubMed] [Google Scholar]
- 100.Yu Y, Ai B, Mohwald H, Zhou ZW, Zhang G, Yang B. Fabrication of binary and ternary hybrid particles based on colloidal lithography. Chem Mater. 2012;24(23):4549–4555. [Google Scholar]
- 101.Castellino F, Huang AY, Altan-Bonnet G, Stoll S, Scheinecker C, Germain RN. Chemokines enhance immunity by guiding naive CD8+ T cells to sites of CD4+ T cell-dendritic cell interaction. Nature. 2006;440(7086):890–895. doi: 10.1038/nature04651. [DOI] [PubMed] [Google Scholar]
- 102.Banchereau J, Steinman RM. Dendritic cells and the control of immunity. Nature. 1998;392(6673):245–252. doi: 10.1038/32588. [DOI] [PubMed] [Google Scholar]
- 103.Dustin ML, Groves JT. Receptor signaling clusters in the immune synapse. Annu Rev Biophys. 2012;41:543–556. doi: 10.1146/annurev-biophys-042910-155238. [DOI] [PMC free article] [PubMed] [Google Scholar]