

Abstract
Current treatments for allergies include epinephrine and anti-histamines which treat the symptoms after an allergic response has taken place, steroids that result in local and systemic immune suppression and IgE-depleting therapies which can only be used for a narrow range of clinical IgE titers. The limitations of current treatments motivated the design of a heterobivalent inhibitor (HBI) of IgE-mediated allergic responses that selectively inhibits allergen-IgE interactions, thereby preventing IgE clustering and mast cell degranulation. The HBI was designed to simultaneously target the allergen binding site and the adjacent conserved nucleotide binding site (NBS) found on the Fab of IgE antibodies. The bivalent targeting was accomplished by linking a hapten to an NBS ligand with an ethylene glycol linker. The hapten moiety of HBI enables selective targeting of a specific IgE while the NBS ligand enhances the avidity for the IgE. Simultaneous bivalent binding to both sites provided HBI with 120 fold enhancement in avidity for the target IgE compared to the monovalent hapten. The increased avidity for IgE made HBI a potent inhibitor of mast cell degranulation in the rat basophilic leukemia (RBL) mast cell model, in the passive cutaneous anaphylaxis (PCA) mouse model of allergy, and in mice sensitized to the model allergen. Additionally, HBI did not have any observable systemic toxic effects even at elevated doses. Taken together, these results establish the HBI design as a broadly applicable platform with therapeutic potential for the targeted and selective inhibition of IgE-mediated allergic responses including food, environmental, and drug allergies.
Introduction
Type-1 hypersensitivity results from allergen induced cross-linking of IgE antibodies, bound to the high affinity receptor FcεRI, on the surface of mast cells (1–3). Cross-linking of the IgE antibodies initiates an intracellular signaling cascade resulting in degranulation, which causes the release of preformed mediators stored in cytoplasmic granules including vasoactive amines, neutral proteases, proteoglycans, cytokines, and chemokines (4). Current therapies for allergic responses include epinephrine, anti-histamines, steroids, and IgE-depleting therapies such as omalizumab. Epinephrine and anti-histamines only treat the symptoms of IgE-mediated hypersensitivity after allergen exposure and do not prevent an allergic response. Steroids and IgE-depleting therapies can reduce the severity of an allergic response but have limitations. The use of steroids results in local and systemic immune suppression with numerous side effects while IgE-depleting therapies (i.e. omalizumab) can only be used for a narrow range of clinical IgE titers (5–7). The limitations of current therapies require the design of more specific treatments of IgE-mediated allergic reactions that would not result in local or systemic suppression of the immune system.
In this study, we engineered heterobivalent inhibitors (HBI) by utilizing the conserved nucleotide binding site (NBS) found on the Fab domain of IgE antibodies to competitively inhibit allergen binding to the IgE antibodies, thereby inhibiting mast cell degranulation. The HBI was designed to simultaneously bind to the NBS as well as the antigen binding site that are found in proximity in the Fab domain of antibodies (8–12). This was specifically accomplished by conjugating a hapten, to model an IgE epitope, to an NBS ligand that targets the NBS. In our bivalent design, the hapten enabled selective targeting of an IgE, while the NBS ligand increased the avidity of HBI for the target IgE allowing for the competitive inhibition of allergen-IgE binding interactions. The bivalent targeting provided HBI with the enhanced avidity for IgE required to competitively inhibit allergen-IgE interactions thereby preventing IgE clustering and mast cell degranulation. This study establishes the HBI design as a novel approach for the selective targeting of IgE antibodies in an allergen specific manner with the therapeutic potential to selectively inhibit allergic responses.
Materials and Methods
Synthesis of Ligands
All ligands were synthesized as previously described in detail (11). Briefly, all ligands were synthesized using fluorenylmethyloxycarbonyl (Fmoc) chemistry on a solid support. Residues were activated with O-Benzotriazole-N,N,N′,N′-tetramethyl-uronium-hexafluoro-phosphate (HBTU) and N,N-Diisopropylethylamine (DIEA) in DMF for 3 min and coupling completion was monitored with Kaiser tests. The Fmoc protected residues were deprotected by 3 exposures to 20% piperidine in DMF for 3 min. The ligands were cleaved from the solid support by 2 exposures to 92/4/4: trifluoroacetic acid/H2O/triisopropylsilane for 30 min and were purified using RP-HPLC on an Agilent 1200 series system with a semi-preparative Zorbax C18 column (9.4 mm x 250 mm), using linear solvent gradients of 2.5% min-1 increments in acetonitrile concentration at 4.0 ml/min flow rate. We monitored the column eluent with a diode array detector allowing a spectrum from 200 to 400 nm to be analyzed. The purified product was characterized using a Bruker micrOTOF II mass spectrometer. The purity of all synthesized ligands was estimated to be >97% by an analytical injection using the above described HPLC with a Zorbax C18 analytical column (4.6mm x 150 mm). The calculated exact mass of monovalent NF (C24H41N5O11) was 575.2803 Da; found 576.2884 Da. The calculated exact mass of monovalent IBA (C37H50N6O7S) was 722.3462 Da; found 723.3416 Da. The calculated exact mass of HBI (C38H56N6O13) was 804.3905 Da; found 805.4017 Da.
Synthesis of BSA Conjugates
BSA conjugates were synthesized and purified as previously described in detail (11). The average number of additions of each hapten to BSA was determined spectroscopically by analyzing the ratios of absorbance at 280 nm (tryptophan residues in BSA and OVA) to 315 nm for NF and 335 nm for dansyl. NF-BSA had an average of 12 haptens, NF-OVA had an average of 6 haptens and dansyl-BSA had an average of 14 haptens per BSA.
Fluorescence Quenching Assay
The binding constants of monovalent IBA, monovalent NF, and HBI to IgEDNP were determined as previously described in detail (11). Briefly, NF and dansyl quench the fluorescence from the IgEDNP tryptophan residues, occurring at 335 nm, only when the two molecules are in proximity to each other (<10 nm). The ligands were titrated into a 96 well plate containing a 200 μL solution of 10 nM IgEDNP in PBS. All experiments were repeated in at least triplicates.
RBL Degranulation Assay
RBL cells and IgEDNP were kindly provided by Dr. Wilson (University of New Mexico) and IgEdansyl (clone 27–74) was purchased from BD Biosciences. RBL cells were maintained as described previously (11). For the degranulation assays, 100 μL of cells were plated at 0.5 x 106 cells/mL in a 96 well plate, and were incubated for 24 hours followed by a two hour incubation with the indicated IgE antibody. Cells were washed immediately before experiments and were stimulated with the indicated concentrations of allergen for 60 minutes. When testing the allergy inhibitors, the inhibitor was added to cells 30 minutes prior to the allergen. Degranulation was detected spectroscopically by measuring the activity of the granule stored enzyme β-hexosaminidase secreted into the supernatant on the substrate p-nitrophenyl-N-actyl-β-O-glucosamine. All degranulation assays were repeated in at least triplicates. In all experiments the IgE concentration was 1 μg/mL.
Animals
C57BL/6 female mice (7–8 weeks) were obtained from Harlan Biosciences (Indianapolis, IN). Mice were maintained in pathogen-free conditions, and studies were approved by the Indiana University Institutional Animal Care and Use Committee.
Passive Cutaneous Anaphylaxis (PCA)
PCA was performed as previously described (13). Briefly, mice were injected via intradermal with PBS (left ear) and 100 ng of IgEDNP (right ear). After 24 hours, mice were injected intravenously with NF-BSA (100 μg) and the indicated amount of HBI. The thickness of ears was measured before NF-BSA (0 hour), and at 2 and 4 hour after challenge. The data was expressed as change in tissue thickness compared with values before NF-BSA administration. Ear tissue was collected 4 hours after challenge for histological examination.
Sensitization with NF-OVA
C57BL/6 mice were sensitized with two intraperitoneal injections (0.5 ml) of NF-OVA/alum (20 μg/2 mg) at one-week interval. Seven days after the last sensitization, mice were challenged via intradermal injection with 20 μg NF-OVA, 20 μg NF-OVA with 10 nmols HBI, 20 μg NF-BSA, or 20 μg NF-BSA with 10 nmols HBI. The thickness of ears were measured before and at 2 hours after challenge. The data was expressed as change in thickness compared with values before NF-OVA or NF-BSA inoculation. Ear tissue was collected 2 hours after challenge for histological examination.
Ear histological analysis
Ear biopsies from PCA mice were placed in 10% formalin for 24 hours. Routine histological techniques were used to paraffin embed ears, and 5-μm sections of whole ears were stained with H&E. Quantitative digital morphometric analysis of ear thickness was performed using the application program ImageJ. A minimum of 10 measurements was analyzed. The same area for each ear was captured and dermal thickness was calculated in inches per pixel.
Statistics
Statistical significance was determined using the ANOVA followed by Tukey’s test for multiple comparisons. P ≤ 0.05 was considered statistically significant. Calculations were performed using the Prism 6.0 software program (GraphPad Software).
Results
Design and Characterization of the HBI
We engineered the HBI to simultaneously bind to the antigen binding site and the adjacent NBS on an IgE Fab (Figure 1). This was specifically accomplished by conjugating a hapten, to model an IgE epitope, to an NBS ligand. The hapten portion of HBI enabled targeting of a specific IgE while the NBS ligand provided increased avidity of HBI for the target antibody, thereby allowing for the competitive inhibition of allergen-IgE binding interactions. Additionally, the HBI design targets two nearby sites on the same Fab arm of a single IgE ensuring that the HBI would not cross-link multiple IgEs and stimulate a degranulation response. In our previous work we extensively characterized the NBS using a combination of molecular modeling and experimental approaches and demonstrated that this under-utilized site is conserved across all immunoglobulins despite being proximal to the hypervariable region (Figure 1) (9–12). Additionally, our analysis of the NBS revealed that the conformation of the four residues that make up this site, two tyrosine residues on the light chain and one tyrosine and one tryptophan on the heavy chain, are also highly conserved (9, 11). The conservation of the NBS site in immunoglobulins provides a universal site that can be used to increase the avidity of an appropriately designed HBI to IgE for competitive inhibition of allergen-IgE interactions.
FIGURE 1.

Heterobivalent targeting strategy. Representative crystal structure of an IgG antibody (to model IgE) and an enlargement of the Fab region with the antigen binding site and the nucleotide binding site (NBS) identified. The heterobivalent inhibitor (HBI) was designed to simultaneously bind to the antigen binding site and the NBS on an IgE.
For an effective HBI design, the affinity of the NBS ligand is a critical factor. If the NBS ligand has too weak an affinity for the NBS, it will not provide the required enhancement in avidity for HBI to IgE. Conversely, if the affinity is too high, HBI will bind non-specifically to all immunoglobulins since the NBS is present on all antibodies. Therefore we predicted that a moderate affinity of ~1 μM, which is common for interactions between immune components, would be best suited for this application. In a previous study, we identified indole-3-butyric acid (IBA) as an NBS ligand by screening a library of ligands and determined that IBA has an affinity of 1–8 μM for the NBS site depending on the antibody (11, 12). Therefore, we selected IBA as the NBS ligand in our approach.
The most commonly used allergen-IgE experimental model is the dinitrophenyl/anti-dinitrophenyl IgE (DNP/IgEDNP) pair (14). In this system mast cells are primed with monoclonal IgEDNP and stimulated with a multivalent synthetic allergen typically synthesized by conjugating DNP to BSA (DNP-BSA). The drawback of this system is that DNP binds to IgEDNP with an atypically high monovalent affinity. Therefore to identify an alternate hapten to be used as the synthetic allergen as well as the hapten moiety in our HBI design, we screened a large number of molecules that bind to IgEDNP with a range of affinities and selected NF as the hapten to develop the HBI and test our hypothesis. NF has an affinity of 45 μM to IgEDNP providing a hapten with similar affinity as IBA to IgE.
To synthesize the HBI we conjugated NF to IBA with an ethylene glycol linker (Figure 2A). In our previous studies we have used ethylene glycol extensively as a linker in multivalent targeting due to its numerous favorable properties including hydrophilicity to improve solubility, and flexibility to allow multivalent binding without steric constraints (15–20). Moreover, ethylene glycol does not form non-specific interactions with proteins. The binding affinities of monovalent IBA, monovalent NF, and HBI for IgEDNP were determined using a fluorescence quenching technique as described in the Materials and Methods. NF quenches the fluorescence of tryptophan residues in the IgE antibody when in close proximity (<10 nm) allowing the affinity of NF and HBI to be determined directly. IBA does not quench tryptophan fluorescence so the monovalent IBA ligand was synthesized incorporating the dansyl moiety which quenches tryptophan fluorescence (Figure 2A). In a control experiment we demonstrated that dansyl does not bind to IgEDNP (data not shown). The binding affinities of monovalent IBA, monovalent NF, and HBI for IgEDNP were found to be 4.3 ± 0.6 μM, 45 ± 5 μM, and 0.375 ± 0.04 μM respectively (Figure 2B). The bivalent targeting strategy provided HBI with a 120 fold increase in avidity for IgEDNP compared to monovalent NF and greater than 11 fold increase in avidity for IgEDNP compared to monovalent IBA (Figure 2B). Combined these results indicate the potential of HBI to inhibit allergen binding and prevent mast cell degranulation.
FIGURE 2.

Chemical structures and affinities for IgEDNP. (A) Structures of monovalent nitro furan (NF), monovalent indole butyric acid (IBA), and heterobivalent inhibitor (HBI). (B) Binding curves for HBI, monovalent IBA, and monovalent NF to IgEDNP. Binding was observed by monitoring the fluorescence quenching of the tryptophan residues in the IgE antibodies caused by NF or dansyl. Values for the binding constants are Kd,HBI = 0.375 ± 0.04 μM, Kd,IBA = 4.3 ± 0.6 μM, and Kd,NF = 45 ± 5 μM. Data represents the mean ± standard deviation of triplicate experiments.
Evaluation of HBI using the in vitro RBL Mast Cell Model
Multivalent allergens mediate clustering of FcεR1-IgE protein complexes on the surface of mast cells stimulating an allergic response. A multivalent allergen was modeled by conjugating multiple copies of NF to BSA resulting in the synthetic allergen NF-BSA with an average of 14 NF moieties per BSA. The RBL-2H3 cell line is the most commonly used cell line in inflammation, allergy, and immunological research and has been shown to be a suitable model system for mast cell degranulation (14, 21–23). Thus, to assess the potential of HBI to inhibit mast cell degranulation we first utilized the well-established, histamine releasing, RBL-2H3 cell line as our mast cell model. First the cells were incubated with IgEDNP to allow the antibody to bind to the FcεR1 receptor on the surface of the cells. Next the cells were exposed to increasing concentrations of NF-BSA to crosslink the surface bound IgEDNP and stimulate degranulation. NF-BSA stimulated a dose dependent response from 1 μg/mL to 100 μg/mL with near maximal response occurring at 10 μg/mL (Figure 3A). The ability of NF-BSA to stimulate degranulation was significant. The most commonly used synthetic allergen is DNP conjugated to BSA (DNP-BSA). The hapten DNP has a high affinity for IgEDNP while NF has a relatively weak affinity for the same antibody. Consequently, degranulation stimulated by NF-BSA demonstrated the significance of low affinity epitopes in allergy while at the same time providing a suitable experimental model to evaluate the HBI design.
FIGURE 3.
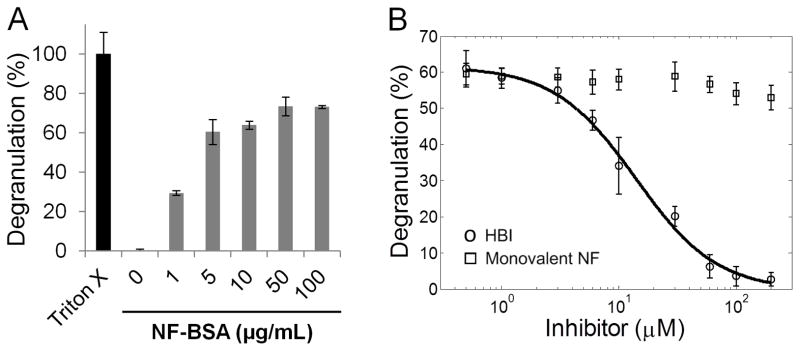
Effect of HBI on RBL mast cell degranulation. (A) Dose response of RBL mast cell degranulation in response to increasing concentrations of NF-BSA. (B) Inhibition of RBL mast cell degranulation by HBI (○) and monovalent NF (□). The indicated concentration of inhibitor was added to the cells 30 minutes prior to the addition of 10 μg/mL NF-BSA. Triton X-100 (1%) was used to lyse cells and determine maximum response. Data represents the mean ± standard deviation of triplicate experiments.
Next, HBI was evaluated by exposing RBL cells primed with IgEDNP to 10 μg/mL NF-BSA with increasing concentrations of HBI. Based on these results, HBI completely inhibited the degranulation response with an IC50 of 13.6 μM (Figure 3B). To demonstrate the significance and necessity of the heterobivalent targeting strategy to inhibit allergen binding, we repeated the same assay using monovalent NF or monovalent IBA in place of HBI. Neither monovalent NF nor monovalent IBA were able to inhibit degranulation at concentrations up to 200 μM (Figure 3B, Figure S1). These results highlight the importance of the enhancement in avidity for IgE obtained with the heterobivalent design. It is important to note that maximum degranulation can occur with as little as 1%–10% of the surface bound IgE cross-linked (24). This means that greater than 90% of the allergen IgE interactions must be inhibited before there is a decrease in the degranulation response thus explaining an IC50 greater than the Kd. In a separate control experiment we synthesized and tested a molecule identical to HBI except the IBA moiety was replaced with dansyl, a structurally similar molecule to IBA, but has no detectable affinity for the NBS. As expected, this molecule did not inhibit mast cell degranulation (Figure S1). Combined these results demonstrated that both the NF and IBA moieties were necessary for efficient inhibition of mast cell degranulation.
Finally to demonstrate the specificity of HBI for IgEDNP, the RBL cells were primed with IgEdansyl then stimulated with the allergen dansyl-BSA. Dansyl-BSA, similar to NF-BSA, stimulated a dose dependent degranulation response (Figure S2A). RBL cells, primed with IgEdansyl were incubated with dansyl-BSA and increasing concentrations of HBI. As expected HBI did not inhibit degranulation stimulated by dansyl-BSA (Figure S2B) highlighting the specificity of HBI for IgEDNP.
Assessment of HBI Toxicity in vivo
As the next step in evaluating the therapeutic potential of the HBI design, we evaluated the toxic effects of the HBI on C57BL/6 mice. The acute and chronic systemic toxicity of HBI was assessed by a single intravenous injection of HBI at 1, 10, and 30 nmol per mouse at 3 days and 14 days after administration, respectively. The dose of HBI, 1, 10, and 30 nmol per mouse were selected based on the in vitro inhibition data and correspond to initial blood concentrations of 1 – 30 μM (1 mL per mouse) approximating the concentration required to inhibit mast cell degranulation in vitro. The body weights of mice receiving the HBI injection were monitored every 2 or 3 days for 14 days and did not vary significantly from the control group (Figure 4A). Additionally, mice were sacrificed at 3 and 14 days after HBI injection and organ weight and blood counts were analyzed to determine any specific toxicity. As shown in Figure 4, even the highest dose of HBI did not cause any change in liver, kidney, spleen, heart, liver, lung, or brain organ weights at 3 or 14 days post injection. Additionally, the white blood cell count, red blood cell counts, and platelets remained unchanged from the control group both 3 and 14 days post HBI injection. Combined these results demonstrate the HBI to be well tolerated in vivo thus demonstrating the potential of the HBI design for the specific inhibition of IgE-mediated allergic responses.
FIGURE 4.

Assessment of HBI toxicity in vivo. (A) Percentage of body weight of C57BL/6 mice intravenously injected with HBI as a measure of systemic toxicity. (B) Organ weight of mice sacrificed 3 or 14 days after receiving a single intravenous injection of HBI as a measure of specific toxicity. (C) Blood counts from mice receiving a single intravenous injection of HBI. Data represents the mean ± SEM of 6 mice per group.
Evaluation of HBI using in vivo PCA Mouse Model of Allergy
To determine whether HBI has an in vivo inhibitory effect on mast cell degranulation, we selected the well-established passive cutaneous anaphylaxis (PCA) mouse model of allergy (13). Before evaluating the HBI, it was first necessary to determine if the weak affinity allergen, NF-BSA, was capable of stimulating a degranulation response in vivo. To assess NF-BSA, C57BL/6 mice were injected intradermally with PBS (left ear) and 100 ng of IgEDNP (right ear), and after 24 hours, mice were challenged intravenously with 100 μg of NF-BSA. The severity of the allergic reaction was quantified by comparing the tissue thickness of the ear that received the IgE injection to the ear that received the PBS injection using both micrometer measurements and histology. As observed in Figure 5A, NF-BSA significantly increased tissue swelling at 2 and 4 hours after challenge, when compared with the control group. Mice treated with DNP-BSA were used as a control and showed a similar increase in ear thickness (data not shown). Next we investigated the inflammatory response induced by NF-BSA. Histopathological analysis of ear sections did not show an increase in inflammatory infiltrate after NF-BSA injection indicating that the observed edema is caused by the release of inflammatory mediators from mast cells in the tissue. Morphometric analysis of ear sections confirmed the effects of NF-BSA on ear swelling as indicated in Figure 5B and C. The in vivo mast cell degranulation response elicited by NF-BSA mirrored the in vitro degranulation results and demonstrated the suitability of the PCA model for the evaluation of HBI.
FIGURE 5.

Effects of HBI on passive cutaneous anaphylaxis in C57BL/6 mice. (A) Ear swelling during PCA was determined after 2 and 4 hours of systemic NF-BSA challenge in mice receiving no treatment or 0.5, 5, and 10 nmol of HBI. (B) Ear tissue was stained with H&E at 4 hours after systemic challenge. Representation of dermal thickness in control and NF-BSA challenged mice is shown at original magnification, x100. (C) Morphometric analysis of changes in dermal thickness was performed. Data represents the mean ± SEM of 5 mice from two independent experiments. *p<0.05.
Next, HBI was evaluated using the described PCA mouse model by challenging C57BL/6 mice intravenously with 100 μg of NF-BSA with the indicated amount of HBI. The amounts of HBI, 0.5, 5, and 10 nmols per mouse were selected based on the in vitro inhibition data and correspond to initial blood concentrations of 0.5 – 10 μM (1 mL per mouse) approximating the range where inhibition was observed in vitro. At 2 hours after challenge, 5 and 10 nmol of HBI significantly inhibited ear swelling when compared to non-treated mice. Four hours after challenge, the highest concentration of HBI significantly inhibited ear edema, while a partial decrease in ear thickness was observed with 0.5 and 5 nmol of HBI treatment (Figure 5A). The decreased edema observed with HBI treatment was confirmed by analyzing the inflammatory response by histopathological and morphometric analysis of ear sections. Ear sections from mice treated with 10 nmol of HBI showed significantly decreased ear thickness while mice treated with 0.5 and 5 nmol showed partially decreased ear thickness (Figure 5B and C). Thus, these results indicate that HBI blocks the effects of NF-BSA on tissue inflammation and edema induced by mast cell degranulation and demonstrates the therapeutic potential of the HBI design.
HBI Decreases Immediate Hypersensitivity Following Allergen Sensitization
In an active allergic response polyclonal IgE antibodies bind to multiple epitopes on the allergen with a wide range of affinities. In addition, there may be allergen specific IgG and IgA antibodies that could bind and sequester the HBI, prevent it from reaching its target IgE antibody and inhibiting the degranulation response. Therefore, to demonstrate the clinical relevance of HBI design, mice were sensitized with NF conjugated OVA (NF-OVA) adsorbed to alum (Figure 6A). NF-OVA sensitized mice were challenged intradermally with NF-OVA, NF-OVA + HBI, NF-BSA, or NF-BSA + HBI and ear swelling was determined 2 hours after challenge. Mice challenged with NF-OVA experienced a significant increase in ear swelling which was significantly reduced when NF-OVA was co-administered with HBI (Figure 6B). The reduction in ear swelling, as a result of inhibition by HBI, was not as dramatic as in the PCA allergy model where complete inhibition was observed. In the PCA allergy model, mice were sensitized via intradermal injection with IgEDNP ensuring high specificity to the NF hapten whereas in the active allergic response it is likely that there were IgE antibodies specific for epitopes on the surface of OVA that did not include the NF hapten. HBI would not inhibit IgE antibodies from binding to these epitopes as it was specifically designed to inhibit the NF-IgE interactions.
FIGURE 6.

Effects of HBI in an active allergic response in C57BL/6 mice. (A) C57BL/6 mice were sensitized twice with two intraperitoneal injections of NF-OVA absorbed to alum at one-week intervals. Seven days after the last sensitization, mice were challenged via intradermal injections. (B) Ear swelling was measured two hours after challenge with 20 μg NF-OVA, 20 μg NF-OVA + 10 nmol HBI, 20 μg NF-BSA, or 20 μg NF-BSA + 10 nmol HBI and compared to values prior to the administration of the synthetic allergens. (C) Ear tissue was stained with H&E at 2 hours after challenge and the scale bar represents 200 μm top row and 50 μm bottom row. Arrows indicate cells with morphological characteristics of eosinophils. Data represents the mean ± SEM of 5 mice from 3 independent experiments. *p<0.05.
Interestingly, NF-BSA caused only a minimal increase in ear swelling (Figure 6B). This result indicates that the IgE epitopes likely include residues of OVA adjacent to the conjugation sites of NF in addition to the NF hapten such that the IgE antibodies had a very low affinity for the NF hapten alone and due to this low affinity no significant response was observed. As expected, mice treated with NF-BSA + HBI did not display any significant allergic response. Since mice treated with NF-BSA alone or in combination with HBI did not experience increased edema this demonstrated that HBI alone did not cause any damage to the ear tissue. Histopathological analysis of the ear tissue revealed an inflammatory infiltrate composed of cells with the morphological characteristics of eosinophils in mice challenged with NF-OVA (Figure 6C). NF-OVA also resulted in increased dermal and epidermal thickening when compared to mice challenged with NF-BSA (Figure 6C). Co-injection of NF-OVA with the HBI limited the inflammatory cell infiltrate and the dermal and epidermal thickening (Figure 6C). Combined, these results demonstrate that an injection of an HBI specific for an allergen epitope can block an active allergic response in vivo.
Discussion
In this study we designed HBIs to simultaneously target the antigen binding site and the NBS on an IgE Fab to competitively inhibit allergen-IgE interaction on the mast cell surface, thereby preventing mast cell degranulation. The bivalent targeting strategy provided HBI with 120 and 11 fold increased avidity for IgEDNP compared to monovalent NF and monovalent IBA, respectively. The increased avidity of HBI competitively inhibited allergen binding and prevented mast cell degranulation in vitro and in passive and active in vivo allergy models, establishing the HBI design as a potential therapeutic approach in selective inhibition of IgE-mediated allergic responses.
There are a number of important considerations in moving the HBI design forward as a potential therapeutic strategy. We observed no indication of systemic toxicity over the concentration range required for effective inhibition of the degranulation response, though further studies would be required to eliminate more subtle effects on specific organ systems. Although the short time frame for these studies precludes the generation of an immune response to the HBI, a therapeutic application may require repeated administration and would therefore have the potential for an anti-drug response by the recipient. However, the main component of the HBI is an ethylene glycol linker and ethylene glycol is commonly used in liposomal drug delivery to provide “stealth” from the recipient immune system. Thus, we do not expect a significant response to develop, even after repeated exposure. The half-life of the HBI will also be important to consider. The HBI design enables bivalent targeting for allergen-specific IgE antibodies with high affinity and specificity. However, the NBS is present on all immunoglobulins causing the HBI to interact weakly with all antibodies and this weak association may act to increase the circulation half-life of the HBI. Further studies are required to determine what effect this will have on the circulation half-life of the HBI.
The non-specific interaction of the HBI to all immunoglobulins through the NBS could prevent the HBI from binding to the target IgE. However our design circumvented this problem by using a NBS ligand with moderate affinity for the NBS (~1 μM). If the NBS ligand bound too tightly to this site, the majority of the HBI would be sequestered by other immunoglobulins present in the physiological system and would not reach the target IgE. Conversely, if the NBS ligand bound too weakly, it would not bring the significant enhancement in avidity required for the competitive inhibition of allergen-IgE interactions. We attribute the enhanced avidity and selectivity of HBI to result from the increased effective concentration of the binding moieties (11, 25, 26). When the first component of HBI binds to IgE, either the hapten or the NBS ligand, the second component is held in close proximity to the IgE raising the effective ligand concentration resulting in increased avidity. This principle ensures that the HBI will selectively and exclusively bind to the target IgE with a high affinity, in an allergen specific manner, as the target IgE is the only macromolecule present in biological systems with both the NBS and the specific antigen binding site. Our results confirmed our prediction that an NBS ligand with a moderate affinity would be best suited for the HBI design as was demonstrated by the efficient inhibition of mast cell degranulation in vivo. Additionally, the HBI was well tolerated and no systemic toxicity was observed in vivo over the concentration range required for effective inhibition of a degranulation response.
This study demonstrates the potential of the HBI design to selectively inhibit allergen-IgE interactions, which is the key event in initiating an allergic response. Current treatments for allergies either only treat the symptoms following allergen exposure, cause local or systemic suppression of the immune system to decrease the severity of an allergic response, or are only available to patients with a narrow range of IgE titers and are very expensive. The results presented in this study demonstrate that the HBI design has the potential to address the limitations of current therapies by selectively inhibiting mast cell degranulation in an allergen specific manner. In addition, the concepts presented in this study establish a utility for the conserved NBS found on immunoglobulins in selective inhibition of IgE-mediated allergic responses. Importantly, this approach has broad implications in inhibiting drug allergies, food allergies, environmental allergies, and asthmas where the allergy epitopes are known. As an example β-lactam antibiotics and the class of sulfonamide drugs are well-defined small molecules, which frequently cause allergic reactions. These small molecules can be readily incorporated in the HBI design for use as the hapten portion of HBI thus allowing the specific inhibition of drug allergies. Food and environmental allergens are structurally more complex, however, increasing amounts of knowledge on these allergens are becoming available due to technological advances in epitope mapping techniques and the establishment of centralized databases such as the Immune Epitope Database (27). For these types of allergens, short peptide sequences that mimic the IgE epitope can be used as the hapten portion of the HBI design allowing for the specific and widespread use of the HBI design to prevent IgE-mediated allergy.
Supplementary Material
Acknowledgments
This work was supported by the NIH-NIAID (National Institutes of Health National Institute of Allergy and Infectious Diseases) [grant number R03 AI085485 (to B.B.) and R01 AI095282 (to M.H.K.)].
We thank Dr. Bridget Wilson (University of New Mexico, Albuquerque, NM, U.S.A) for generously providing us with IgEDNP and the RBL-2H3 cell line, Dr. Paul Bryce (Northwestern University) for providing protocols and advice on the PCA model and Dr. Bill Boggess at the Mass Spectrometry and Proteomics Facility in the University of Notre Dame for the use of MS instrumentation. We also wish to thank the expert technical assistance of Jayne Silver and Kacie Peterman in the In Vivo Therapeutics Core at the Indiana University Simon Cancer Center.
Abbreviations used in this article
- RBL
rat basophilic leukemia cells
- PCA
passive cutaneous anaphylaxis
- HBI
heterobivalent inhibitor
- NBS
nucleotide binding site
- NF
nitrofuran
- IBA
indole-3-butyric acid
- DNP
dinitrophenyl
Footnotes
Disclosures
The authors have no financial conflicts of interest.
References
- 1.Alber G, Kent UM, Metzger H. Functional comparison of Fc-Epsilon-RI, Fc-Gamma-RII, and Fc-Gamma-RIII in mast-cells. J Immunol. 1992;149:2428–2436. [PubMed] [Google Scholar]
- 2.Turner H, Kinet JP. Signalling through the high-affinity IgE receptor Fc epsilon RI. Nature. 1999;402:B24–B30. doi: 10.1038/35037021. [DOI] [PubMed] [Google Scholar]
- 3.Galli SJ, Tsai M, Piliponsky AM. The development of allergic inflammation. Nature. 2008;454:445–454. doi: 10.1038/nature07204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Blank U, Rivera J. Assays for regulated exocytosis of mast cell granules. Curr Protoc Cell Biol. 2006;Chapter 15(Unit 15.11) doi: 10.1002/0471143030.cb1511s32. [DOI] [PubMed] [Google Scholar]
- 5.Buchman A. Side effects of corticosteroid therapy. J Clin Gastroenterol. 2001;33:289–294. doi: 10.1097/00004836-200110000-00006. [DOI] [PubMed] [Google Scholar]
- 6.Corren J, Casale TB, Lanier B, Buhl R, Holgate S, Jimenez P. Safety and tolerability of omalizumab. Clinical and Experimental Allergy. 2009;39:788–797. doi: 10.1111/j.1365-2222.2009.03214.x. [DOI] [PubMed] [Google Scholar]
- 7.Deniz V, Gupta N. Safety and tolerability of omalizumab (Xolair (R)), a recombinant humanized monoclonal anti-IgE antibody. Clin Rev Allergy Immunol. 2005;29:31–48. doi: 10.1385/criai:29:1:031. [DOI] [PubMed] [Google Scholar]
- 8.Rajagopalan K, Pavlinkova G, Levy S, Pokkuluri PR, Schiffer M, Haley BE, Kohler H. Novel unconventional binding site in the variable region of immunoglobulins. Proc Natl Acad Sci U S A. 1996;93:6019–6024. doi: 10.1073/pnas.93.12.6019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Alves NJ, Champion MM, Stefanick JF, Handlogten MW, Moustakas DT, Shi Y, Shaw BF, Navari RM, Kiziltepe T, Bilgicer B. Selective photocrosslinking of functional ligands to antibodies via the conserved nucleotide binding site. Biomaterials. 2013;34:5700–5710. doi: 10.1016/j.biomaterials.2013.03.082. [DOI] [PubMed] [Google Scholar]
- 10.Alves NJ, Stimple SD, Handlogten MW, Ashley JD, Kiziltepe T, Bilgicer B. A Small Molecule Based Affinity Chromatography Method for Antibody Purification via Nucleotide Binding Site Targeting. Anal Chem. 2012;84:7721–7728. doi: 10.1021/ac300952r. [DOI] [PubMed] [Google Scholar]
- 11.Handlogten MW, Kiziltepe T, Moustakas DT, Bilgicer B. Design of a heterobivalent ligand to inhibit IgE clustering on mast cells. Chem Biol. 2011;18:1179–1188. doi: 10.1016/j.chembiol.2011.06.012. [DOI] [PubMed] [Google Scholar]
- 12.Handlogten MW, Kiziltepe T, Serezani AP, Kaplan MH, Bilgicer B. Inhibition of weak-affinity epitope-IgE interactions prevents mast cell degranulation. Nat Chem Biol. 2013;9:789–795. doi: 10.1038/nchembio.1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hsu C, Neilsen CV, Bryce PJ. IL-33 Is Produced by Mast Cells and Regulates IgE-Dependent Inflammation. Plos One. 2010;5:e11944. doi: 10.1371/journal.pone.0011944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Passante E, Frankish N. The RBL-2H3 cell line: its provenance and suitability as a model for the mast cell. Inflammation Res. 2009;58:737–745. doi: 10.1007/s00011-009-0074-y. [DOI] [PubMed] [Google Scholar]
- 15.Handlogten MW, Kiziltepe T, Bilgicer B. Design of a heterotetravalent synthetic allergen that reflects epitope heterogeneity and IgE antibody variability to study mast cell degranulation. Biochem J. 2013;449:91–99. doi: 10.1042/BJ20121088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Handlogten MW, Stefanick JF, Alves NJ, Bilgicer B. Nonchromatographic affinity precipitation method for the purification of bivalently active pharmaceutical antibodies from biological fluids. 2013 doi: 10.1021/ac4008286. [DOI] [PubMed] [Google Scholar]
- 17.Stefanick JF, Kiziltepe T, Handlogten MW, Alves NJ, Bilgicer B. Enhancement of Antibody Selectivity via Bicyclic Complex Formation. J Phys Chem Lett. 2012;3:598–602. [Google Scholar]
- 18.Handlogten MW, Kiziltepe T, Alves NJ, Bilgicer B. Synthetic allergen design reveals the significance of moderate affinity epitopes in mast cell degranulation. ACS Chem Biol. 2012;7:1796–1801. doi: 10.1021/cb300193f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bilgicer B, Thomas SW, Shaw BF, Kaufman GK, Krishnamurthy VM, Estroff LA, Yang J, Whitesides GM. A non-chromatographic method for the purification of a bivalently active monoclonal IgG antibody from biological fluids. J Am Chem Soc. 2009;131:9361–9367. doi: 10.1021/ja9023836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bilgicer B, Moustakas DT, Whitesides GM. A synthetic trivalent hapten that aggregates anti-2,4-DNP IgG into bicyclic trimers. J Am Chem Soc. 2007;129:3722–3728. doi: 10.1021/ja067159h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ludowyke RI, Elgundi Z, Kranenburg T, Stehn JR, Schmitz-Peiffer C, Hughes WE, Biden TJ. Phosphorylation of nonmuscle myosin heavy chain IIA on Ser(1917) is mediated by protein kinase C beta II and coincides with the onset of stimulated degranulation of RBL-2H3 mast cells. J Immunol. 2006;177:1492–1499. doi: 10.4049/jimmunol.177.3.1492. [DOI] [PubMed] [Google Scholar]
- 22.Choi O, Kim J, Kinet J. Calcium mobilization via sphingosine kinase in signalling by the Fc epsilon RI antigen receptor. Nature. 1996;380:634–636. doi: 10.1038/380634a0. [DOI] [PubMed] [Google Scholar]
- 23.Andrews NL, Pfeiffer JR, Martinez AM, Haaland DM, Davis RW, Kawakami T, Oliver JM, Wilson BS, Lidke DS. Small, mobile Fc epsilon R1 receptor aggregates are signaling competent. Immunity. 2009;31:469–479. doi: 10.1016/j.immuni.2009.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Holowka D, Baird B. Antigen-mediated IgE receptor aggregation and signaling: A window on cell surface structure and dynamics. Annu Rev Biophys Biomolec Struct. 1996;25:79–112. doi: 10.1146/annurev.bb.25.060196.000455. [DOI] [PubMed] [Google Scholar]
- 25.Kiessling L, Gestwicki J, Strong L. Synthetic multivalent ligands as probes of signal transduction. Angewandte Chemie-International Edition. 2006;45:2348–2368. doi: 10.1002/anie.200502794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mammen M, Choi SK, Whitesides GM. Polyvalent interactions in biological systems: Implications for design and use of multivalent ligands and inhibitors. Angew Chem -Int Edit. 1998;37:2755–2794. doi: 10.1002/(SICI)1521-3773(19981102)37:20<2754::AID-ANIE2754>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 27.Vita R, Zarebski L, Greenbaum JA, Emami H, Hoof I, Salimi N, Damle R, Sette A, Peters B. The Immune Epitope Database 2.0. Nucleic Acids Res. 2010;38:D854–D862. doi: 10.1093/nar/gkp1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.