

Abstract
This letter describes the development and SAR of a novel series of GlyT1 inhibitors derived from a scaffold hopping approach, in lieu of an HTS campaign, which provided intellectual property position. Members within this new [3.3.0]-based series displayed excellent GlyT1 potency, selectivity, free fraction, and modest CNS penetration. Moreover, enantioselective GlyT1 inhibition was observed, within this novel series and a number of other piperidine bioisosteric cores.
Keywords: GlyT1, Scaffold hopping, transporter, schizophrenia
Scaffold hopping has emerged as an attractive approach to rapidly access new chemical space and enable fast-follower programs without the need for expensive and time-consuming HTS campaigns.1–4 As the negative symptom cluster in schizophrenia remains a critical unmet medical need,5–7 and GlyT1 inhibition has been shown to be affective toward negative symptoms in Phase II clicnial trials,8–14 we initiated scaffold hopping efforts to expediently develop novel GlyT1 inhibitors within a crowded intellectual property (IP) space. In a recent Letter, we reported on our preliminary scaffold hopping exercise (Fig. 1) employing GlyT1 inhibitors from Merck and Pfizer, 1 and 2, respectivley, that generated a novel, patented series exemplified by 3.15 Notably, 3 was a potent GlyT1 inhibitor with an exceptional DMPK profile, high CNS penetration and robust efficacy in preclinical models of schizophrenia.15
Figure 1.
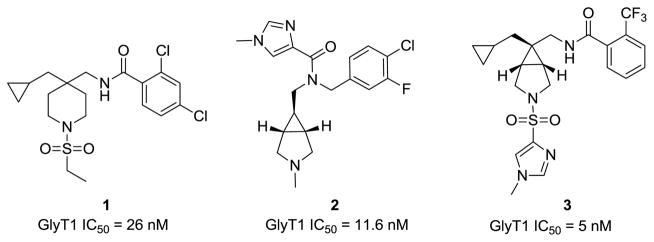
Reported GlyT1 inhibitors 1 (Merck) and 2 (Pfizer), and the novel series 3 (VU0240391), derived from scaffold hopping.
Based on work from our labs with mGlu1 NAMs, and the ability of [3.3.0] systems, such as the octahydropyrrolo[3,4-c]pyrrole, to effectively mimic piperazines,16 we focused our attention on the potential bioisoteric replacement of the [3.1.0] system of 2 and 3, as well as the piperidine of 1, with a [3.3.0] system, an octahydrocyclopenta[c]pyrrole, 5, and effectively scaffold hop from analogs 4 (Fig. 2). If successful at maintaining GlyT1 inhibitory activity, this would represent a major strucutral change, eliminating the pendant cyclopropylmethyl moeity while introducing an additional chiral center (providing an opportunity for enantioselective activity).
Figure 2.
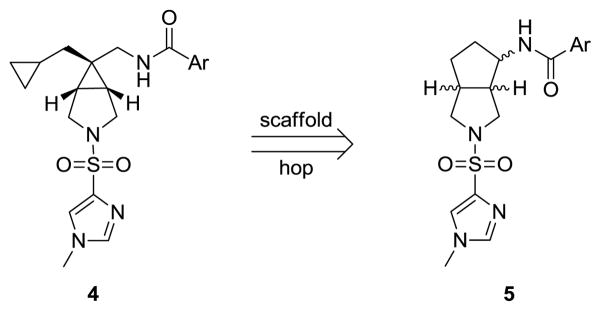
Envisioned scaffold hopping from the novel series 4 to a [3.3.0]-core, an octahydrocyclopenta[c]pyrrole, 5.
Synthetically, analogs 10 were initially prepared as racemates via a six step route that proceeded in ~22% overall yield (Scheme 1). Commercial racemic, 90% cis-benzylhexahydrocyclopenta[c]pyrrol-4(2H)-one 6 was subjected to hydrogenation conditions to deprotect the benyl moiety in the presence of Boc2O to provide 7. Conversion of the ketone to the oxime, followed by ‘Raney’ nickel reduction generated the racemic primary amine 8, which was subsequently acylated with a variety of benzoyl chlorides to deliver analogs 9. Finally, the Boc moiety was removed with HCl, and the secondary pyrrolidine nitrogen capped with various sulfonyl chlorides to afford analogs 10.
Scheme 1.

Reagents and conditions. (a) Boc2O, Pd(OH)2/C, H2 (50 psi), EtOH, rt; (b) NH2OH, MeOH, 100 °C; (c) ‘Raney’ Ni, H2 (50 psi), rt; (d) ArCOCl, DIEPA, CH2Cl2, 0 °C; (e) 4 N HCl/dioxane, rt; (f) RSO2Cl, DIEPA, CH2Cl2, rt. Overall yields range from 10–34%.
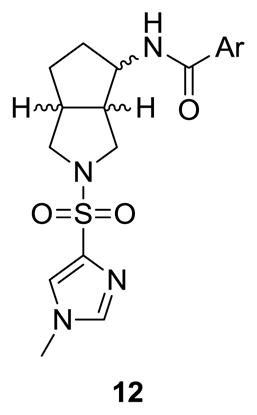
Initially, we held the 2,4-dichlorobenzamide constant and surveyed a wide-range of sulfonamides in analogs 11 (Table 1). Unlike the piperdine 1 and [3.1.0] series 3, few sulfonamide moieties were tolerated. Ethyl (11a) and propyl congeners (11b) that were very potent in the piperidine series 1, afforded inactive compounds (GlyT1 IC50 > 10 μM). Aryl and heteroaryl analogs, such as 11d-11f, were also devoid of GlyT1 activity. Only the N-methyl imidazole (11g) and the N-methyl triazole (11h) derivative were active, 15 both displayed low nanomolar potency (GlyT1 IC50s of 25 nM and 15 nM, respectively) and were selective versus GlyT2 (IC50 > 30 μM). Based on the disposition previously noted for the N-methyl imidazole sulfonamide in 3, we prepared a second library held the N-methyl imidazole sulfonamide moiety constant, and surveyed a broader range of amides in analogs 12 (Table 2). The SAR was far more shallow than in the case of 3,15 with the 2,4-dichlorobenzamide (11g/12a) possessing optimal potency. Other analogs such as the 2-triflouromethylbenzamide (12b) and the 2-chlorobenzamide (12c) were respectable, with GlyT1 IC50s of 112 ± 6 nM and 115 ± 18 nM, respectively. The vast majority of other substitution patterns afforded a considerable loss in potency (GlyT1 IC50s from 631 nM to 10 μM), as did a cyclohexyl amide congener 12l (GlyT1 IC50 = 617 nM). To ensure that the major structural change in scaffold hopping from 1 to 3 to 12 did not alter the competitive mechanism of action of GlyT1 inhibition, we evaluated the affect of 12b on enzyme kinetics of [14C]-glycine transport. As shown in an Eadie-Hoffstee plot (Fig. 3), this [3.3.0] series, represented by 12b, competitively inhibits the enzyme kinetics of [14C]-glycine transport. Thus, this series is competitive with respect to glycine, in accordance with the known mechanism of action for 1-3.15,17–20
Table 1.
Structures and activities of analogs 11.
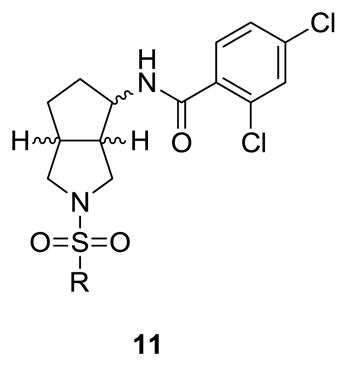
| |||
|---|---|---|---|
| Compound | R | GlyT1 IC50 (μM)a | GlyT2 IC50 (μM)a |
| 11a |
|
>10 | >30 |
| 11b |
|
>10 | >30 |
| 11c |
|
>10 | >30 |
| 11d |
|
>10 | >30 |
| 11e |
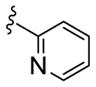
|
>10 | >30 |
| 11f |
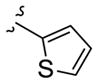
|
>10 | >30 |
| 11g |
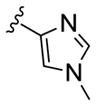
|
0.025 | >30 |
| 11h |
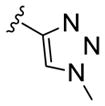
|
0.015 | >30 |
IC50s represent single determinations performed in duplicate
Table 2.
Structures and activities of analogs 12.
| |||
|---|---|---|---|
| Compound | Ar | GlyT1 IC50 (nM)a | GlyT2 IC50 (μM)a |
| 12a (11g) | 2,4-diCIPh | 25 | >30 |
| 12b | 2-CF3Ph | 112* | >30 |
| 12c | 2-CIPh | 115* | >30 |
| 12d | 2,4-diFPh | 926 | >30 |
| 12e | 2,6-diFPh | 631 | >30 |
| 12f | 2-FPh | 1,815 | >30 |
| 12g | 3-FPh | >10,000 | >30 |
| 12h | 4-FPh | 2,215 | >30 |
| 12i | 3,4-diFPh | 1569 | >30 |
| 12j | 4-CIPh | 1,029 | >30 |
| 12k | 3,4-diCIPh | 891 | >30 |
| 121 |
|
617 | >30 |
IC50s represent single determinations performed in duplicate or *the average of four determinations performed in duplicate
Figure 3.
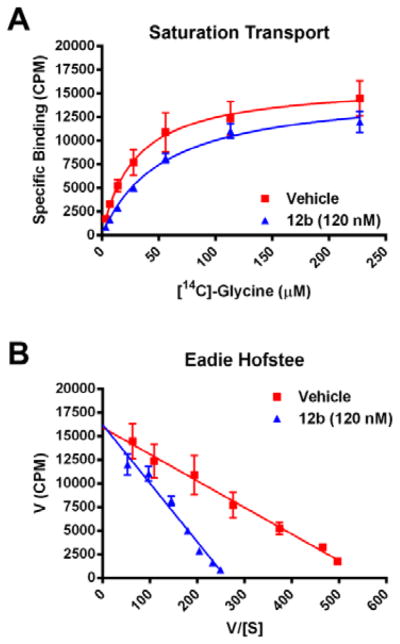
(A) Saturation [14C]-glycine transport in the presence of vehicle (red squares) or 120 nM 12b (blue triangles). (B) An Eadie-Hoffstee diagram for 12b and [14C]-glycine.
Racemic 12a, the most potent of the [3.3.0]-series, possessed a favorable DMPK profile, with a good unbound fraction in rat (fu = 8.1%), clean CYP profile (IC50s >10 μM), and reasonable microsomal stability (30% remaining at 90 minutes in fortified rat liver microsomes). An oral plasma:brain level (PBL) study with oral dosing (10 mg/kg p.o. in 0.5% methocellulose) of 12a afforded a low BrainAUC:PlasmaAUC of 0.15. This preliminary data was encouraging, and since 12a was a racemate (90% cis at the bridgehead), and thus a mixture of 8 compounds, we then attempted to separate the mixture by chiral SFC. We were able to separate three peaks off the SFC, two as single species (GlyT1 IC50s > 10 μM), and one as a mixture (GlyT1 IC50 = 34 ± 2 nM); however, we were unable to definitively assign the absolute stereochemistry. An enantioselective synthetic route (Scheme 2) was employed to quickly access the pure cis-(3a,6a)-enantiomers 20a and 20b (Fig. 4).21 Following the work of Beebe,21 azomethine ylid precursor 13 underwent a dipolar cycloaddition reaction with cyclopentenone to give the key racemic ketone 14, with cis-stereochemistry at the ring junction. Enantiomeric resolution via the (R)-tert-butyl sulfonamide provided (3aS,6aR)-15 and (3aR,6aS)-15, which were subsequently separated by silica gel chromatography, in accord with literature precedent.21 Scheme 2 shows the complete route to 20a, employing (3aR,6aS)-15. Here, reduction with NaBH4 delivered 16, followed by deprotection under acidic conditions to the primary amine 17. Acylation, removal of the benzyl protecting group and sulfonylation provided 20a, the (3aR,4R,6aS) isomer. By employing (S)-tert-butyl sulfinamide, the other cis-(3a,6a)-enantiomers, (3aR,4S,6aS) and (3aS,4R,6aR) could not be accessed.21 For the isomers that could be obtained, enantiospecific inhibition was noted with 20a, possessing a GlyT1 IC50 of 433 nM, while the other isomer 20b was inactive (GlyT1 IC50 >10 μM). Interestingly, these analogs were weak to inactive relative to racemic 12a, and suggests that the active isomer(s) are either the trans-(3a,6a)-isomers or the other cis congeners, and synthetic efforts to access both are underway. Thus, 12b, derived from a scaffold-hopping exercise employing 1-3, led to a novel [3.3.0]-based GlyT1 inhibitor with in vitro properties comparable to other advanced GlyT1 inhbitors in short order, and for which a U.S. patent was issued.22
Scheme 2.

Reagents and conditions. (a) cyclopentenone, TFA, CH2Cl2, 0 °C, 16h; (b) (R)-tert-butylsulfinamide, Ti(OEt)4, THF, 0 °C, 16 h, chromatographic separation of diastereomers; (c) NaBH4, MeOH, −78 °C to rt, 3 h; (d) 2 N HCl (aq), MeOH, rt, 16 h; (e) ArCOCl, CH2Cl2, rt, 16 h; (f) chloroethyl chloroformate, Et3N, ClCH2CH2Cl, MeOH, rt, 20 h; (g) N-methyl imidazole sulfonyl chloride, Et3N, CH2Cl2, rt, 12 h. Overall yields range 5–22%.
Figure 4.
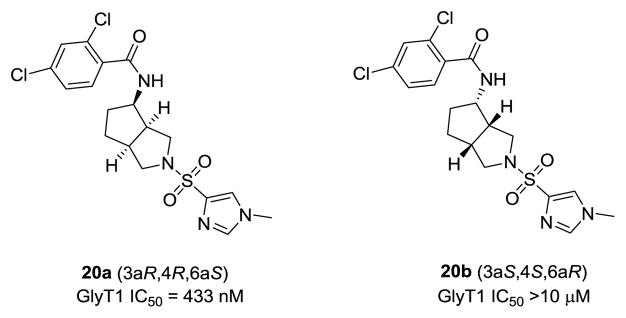
Structures and activities of cis-(3a,6a)-enantiomers 20a and 20b.
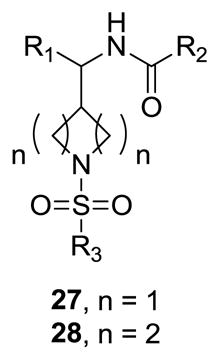
In parallel, we were also preparing and evaluating other piperidine bioisosteres and modifications to 1-3 to further access additional novel intellectual property (IP) space. Modeling work suggested that 4-position homologated piperidines, as well as 3-position homologated azetidines overlapped favorably with 1-3, 12 and 20. Thus, chemistry was quickly developed to access these cores (Scheme 3). Starting from commercially available N-Boc-azetidine-3-carboxylic acid 21 or N-Boc-piperidine-4-carboxylic acid 22, conversion to the Weinreb amide and treatment with an aryl, heteroaryl or aliphatic Gringard reagent provided 23 and 24, respectively. Condensation with hydroxylamine, reduction and acylation afforded amides 25 and 26. Finally, removal of the Boc moiety and sulfonylation of the secondary amine led to putative, racemic GlyT1 inhibitor series 27 and 28.
Scheme 3.

Reagents and conditions. (a) N,O-dimethylhydroxylamine, EDC, HOBt, DIPEA, DMF, rt; (b) Ar(Het)MgX or R1MgX, THF, −78 °C; (c) NH2OH, MeOH, 50 °C; (d) Raney Ni, H2, (45 psi), MeOH; (e) RCOCl, DIEPA, CH2Cl2, rt; (f) 4 N HCl, dioxane, rt; (g) RSO2Cl, DIEPA, CH2Cl2, rt.
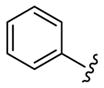
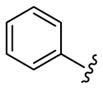
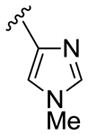
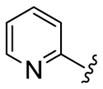
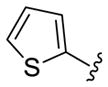
As shown in Table 3, the homologated azetidine-based analogs 27 were uniformly more potent than the corresponding homologated piperidine-based analogs 28, affording GlyT1 inhibitors with low nanomolar potency. While the 2,4-dichlorobenzamide was the most potent congener, other benzamides displayed a wide range of GlyT1 potency (GlyT1 IC50s from 80 nM to 7 μM). Moreover, in the azetidine series 27, the aryl/heteroaryl R1 moieties could be replaced with aliphatic groups and retain potency (R1 = iPr, GlyT1 IC50 = 394 nM; R1 = n-Pr, GlyT1 IC50 = 185 nM; R1 = cycPr, GlyT1 IC50 = 253 nM), whereas the corresponding analogs in the piperidine series 28 were inactive.
Table 3.
Structures and activities of analogs 27 and 28.
| ||||
|---|---|---|---|---|
| Cmpd | R1 | R2 | R3 | GlyT1 IC50 (nM)a |
|
27a 28a |
|
2,4-diCIPh |
|
627 1,500 |
|
27b 28b |
|
2,4-diCIPh |
|
39 201 |
|
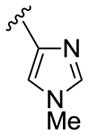
27c 28c |
|
2,4-diCIPh |
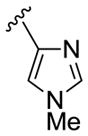
|
68 46 |
|
27d 28d |
|
2,4-diCIPh |
|
40 374 |
IC50s represent single determinations performed in triplicate.
Representative members from both 27 and 28 were evaluated for their effect on enzyme kinetics of [14C]-glycine transport, and both were shown to be competitive with glycine, as well as selective versus GlyT2 (IC50 >30 μM). Initial evaluation in our in vitro DMPK assays demonstrated that 27c was stable in fortified rat liver microsomes (75% parent remaining at 90 minutes), possessed a good unbound fraction in rat (fu = 14%) and clean CYP profile (IC50s >10 μM). An oral plasma:brain level (PBL) study with oral dosing (10 mg/kg p.o. in 0.5% methocellulose) of 27c afforded a low BrainAUC:PlasmaAUC of 0.11. SCF separation of the 27c enantiomers led to the isolation of the two pure enantiomers, and one was quite active (IC50 = 39 nM) while the other proved much weaker (IC50 = 900 nM). In consultation with the Johnston group, they developed an asymmetric synthesis of 27c, via chiral proton catalysis of a secondary nitroalkane addition to an azomethine, and we were able to elucidate that the potent enantiomer had the (S)-configuration.23 Overall, the low brain:plasma ratios of these series, 11, 12, 27 and 28 diminished enthusiasm; however, the scaffold hopping strategy again secured robust IP position for both the 27 and 28 series of GlyT1 inhibitors.24,25
In summary, we were able to successfully further scaffold hop from 3, originally derived at from a scaffold hopping exercise from 1 and 2, and develop three new series for which US patents were granted without the need for an HTS campaign. This was critical, as the time required to perform a SPA-based HTS campaign and identify/optimize the hits would have required far more time and uncertain IP position in a highly crowded and competitive space. These new series retained the potency and selectivity of the advanced compounds from which they were derived, but did suffer from only modest CNS exposure. Finally, all of these new series displayed enantioselective inhibition of the GlyT1 transporter. Further refinements are in progress and will be reported in due course.
Acknowledgments
This work was supported by the NIH/NIMH under a National Cooperative Drug Discovery and Development grant U01 MH08795. DJS is a recipient of a National Alliance for Research on Schizophrenia and Depression (NARSAD)–Dylan Tauber Young Investigator Award. Vanderbilt is a member of the MLPCN and houses the Vanderbilt Specialized Chemistry Center for Accelerated Probe Development supported by U54 MH084659. The support of William K. Warren, Jr. who funded the William K. Warren, Jr. Chair in Medicine (to C.W.L.) is gratefully acknowledged.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Langdon SR, Ertl P, Brown N. Mol Inf. 2010;29:366. doi: 10.1002/minf.201000019. [DOI] [PubMed] [Google Scholar]
- 2.Sun H, Tawa G, Wallqvist A. Drug Discov Today. 2012;17:310. doi: 10.1016/j.drudis.2011.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Martin YC, Muchmore S. QSAR Comb Sci. 2009;28:797. [Google Scholar]
- 4.Xiang Z, Thompson AD, Brogan JT, Schulte ML, Mi D, Lewis LM, Yang L, Zhou B, Melancon BJ, Morrison R, Santomango T, Byers F, Brewer K, Aldrich JS, Yu H, Dawson ES, Li M, McManus O, Jones CK, Daniels JS, Conn PJ, Xie X, Weaver CD, Lindsley CW. ACS Chem Neurosci. 2011;2:730. doi: 10.1021/cn200090z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lindsley CW, Shipe WD, Wolkenberg SE, Theberge CR, Williams DL, Jr, Sur C, Kinney GG. Curr Topics in Med Chem. 2006;8:771. doi: 10.2174/156802606777057599. [DOI] [PubMed] [Google Scholar]
- 6.Menniti FS, Lindsley CW, Conn PJ, Pandit J, Zagouras P, Volkmann RA. Curr Topics in Med Chem. 2013;13:26. doi: 10.2174/1568026611313010005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Olney JW, Newcomer JW, Farber NB. J Psychiatry Res. 1999;33:523. doi: 10.1016/s0022-3956(99)00029-1. [DOI] [PubMed] [Google Scholar]
- 8.Coyle JT. Cell Mol Neurobiol. 2006;26:365. doi: 10.1007/s10571-006-9062-8. [DOI] [PubMed] [Google Scholar]
- 9.Kinney GG, Sur C. Curr Neuropharmacology. 2005;3:35. [Google Scholar]
- 10.Bridges TM, Williams R, Lindsley CW. Curr Opin Mol Ther. 2008;10:591. [PubMed] [Google Scholar]
- 11.Lindsley CW, Wolkenberg SE, Kinney GG. Curr Topics in Med Chem. 2006;6:1883. doi: 10.2174/156802606778249784. [DOI] [PubMed] [Google Scholar]
- 12.Wolkenberg SE, Sur C. Curr Topics in Med Chem. 2010;10:170. doi: 10.2174/156802610790410974. [DOI] [PubMed] [Google Scholar]
- 13.Hashimoto K. Curr Pharm Des. 2011;17:112. doi: 10.2174/138161211795049598. [DOI] [PubMed] [Google Scholar]
- 14.For information on RG1678, please see: www.roche.com
- 15.Jones CK, Sheffler DJ, Williams R, Jadhav SB, Felts AS, Morrison RD, Niswender CM, Daniels JS, Conn PJ, Lindsley CW. Bioorg Med Chem Lett. doi: 10.1016/j.bmcl.2014.01.013. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Manka JT, Rodriguez AL, Venable DF, Morrison RD, Venable DF, Plumley HC, Blobaum AL, Daniels JS, Niswender CM, Conn PJ, Lindsley CW, Emmitte KA. Bioorg Med Chem Lett. 2013;23:5091. doi: 10.1016/j.bmcl.2013.07.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lindsley CW, Zhao Z, Leister WH, O’Brien JA, Lemiare W, Williams DL, Jr, Chen TB, Chang RSL, Burno M, Jacobson MA, Sur C, Kinney GG, Pettibone DJ, Tiller PR, Smith S, Tsou NN, Duggan ME, Conn PJ, Hartman GD. Chem Med Chem. 2006;1:807. doi: 10.1002/cmdc.200600097. [DOI] [PubMed] [Google Scholar]
- 18.Zhao Z, Leister WH, O’Brien JA, Lemiare W, Williams DL, Jr, Jacobson MA, Sur C, Kinney GG, Pettibone DJ, Tiller PR, Smith S, Hartman GD, Lindsley CW, Wolkenberg SE. Bioorg Med Chem Lett. 2009;19:1488. doi: 10.1016/j.bmcl.2008.12.115. [DOI] [PubMed] [Google Scholar]
- 19.Wolkenberg SE, Zhao Z, Wisnoski DD, Leister WH, O’Brien JA, Lemiare W, Williams DL, Jr, Jacobson MA, Sur C, Kinney GG, Pettibone DJ, Tiller PR, Smith S, Gibson C, Ma BK, Polsky-Fisher SL, Lindsley CW, Hartman GD. Bioorg Med Chem Lett. 2009;19:1492. doi: 10.1016/j.bmcl.2009.01.015. [DOI] [PubMed] [Google Scholar]
- 20.Lowe JA, III, Hou X, Schmidt C, Tingley FD, III, McHardy S, Kalman M, DeNinno S, Sanner M, Ward K, Lebel L, Tunucci D, Valnetine J. Bioorg Med Chem Lett. 2009;19:2974. doi: 10.1016/j.bmcl.2009.04.035. [DOI] [PubMed] [Google Scholar]
- 21.Beebe X, Darczak D, Henry RF, Vortherms T, Janis R, Namovic M, Donnelly-Roberts D, Kage KL, Surowy C, Milicic I, Niforatos W, Swensen A, Marsh KC, Wetter JM, Franklin P, Baker S, Zhong C, Simler G, Gomez E, Boyce-Rustay JM, Zhu CZ, Stewart AO, Jarvis MF, Scott VE. Bioorg Med Chem. 2012;20:4128. doi: 10.1016/j.bmc.2012.04.057. [DOI] [PubMed] [Google Scholar]
- 22.Lindsley CW, Conn PJ, Williams R, Sheffler DJ. 8,211,933. US. 2012
- 23.Davis TA, Danneman MW, Johnston JN. Chem Commun. 2012;48:5578. doi: 10.1039/c2cc32225k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lindsley CW, Conn PJ, Williams R, Jones CK, Sheffler DJ. 8,207,155. US. 2012
- 25.Lindsley CW, Conn PJ, Williams R, Jones CK, Sheffler DJ. 8,436,019. US. 2013