

Abstract
This letter describes the development and SAR of a novel series of GlyT1 inhibitors derived from a scaffold hopping approach that provided a robust intellectual property position, in lieu of a traditional, expensive HTS campaign. Members within this new [3.1.0]-based series displayed excellent GlyT1 potency, selectivity, free fraction, CNS penetration and efficacy in a preclinical model of schizophrenia (prepulse inhibition).
Keywords: GlyT1, Scaffold hopping, transporter, schizophrenia
Significant efforts are currently focused on non-dopaminergic strategies to address the unmet medical needs in schizophrenia, and targeting N-methyl-D-aspartate (NMDA) receptor hypofunction has garnered a great deal of attention.1–3 Elevation of synaptic glycine levels near NMDA-containing synapses, by inhibition of the glycine transporter type 1 (GlyT1), has proven to be a viable mechanism for achieving efficacy in multiple preclinical models of schizophrenia with a diverse array of GlyT1 chemotypes.4–9 More recently, Roche reported Phase II clinical efficacy with their GlyT1 inhibitor (RG6178) in improving the negative symptoms in patients with schizophrenia,10 a symptom cluster largely unmet with available antipsychotic agents, that re-ignited the field. Furthermore, new data suggests roles for GlyT1 inhibition in alcohol dependence, addiction and pain.11
Based on these data, the late-stage clinical status of GlyT1, and the crowded intellectual property space,4–9 we elected to attempt to develop novel chemical space by scaffold hopping, a strategy we previously employed successfully for T-Type calcium channel inhibitors,12 to accelerate a fast follower GlyT1 inhibitor program. For this exercise, we were attracted to the GlyT1 inhibitors reported from both Merck, represented by 1 and 2,13–15 and Pfizer’s 316 (Fig. 1), as they possessed potent GlyT1 inhibition with good DMPK profiles and efficacy in preclinical models;13–16 moreover, homology and overlap was noted between these otherwise disparate chemotypes.
Figure 1.
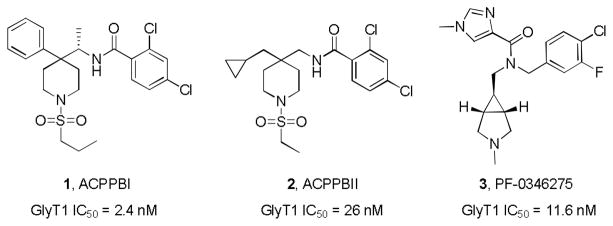
Structures and activities of GlyT1 inhibitors disclosed by Merck (1 and 2) and Pfizer (3) utilized in subsequent scaffold-hopping to access new chemical space.
Our initial approach was to replace the central piperidine core of 1 and 213–15 with the [3.1.0] bicyclic ring system found in 3,16 to arrive at analogs such as 4 (Fig. 2). Synthetically, analogs 4 were arrived at via a seven step route in low (~4%) overall yield. Starting from commercially available (1R,5S,6r)-3-tert-butyl 6-ethyl 3-azabicyclo[3.1.0]hexane-3,6-dicarboxylate 5, two step conversion to the primary carboxamide 6 proceeded smoothly, followed by treatment with cyanuric chloride to afford the nitrile 7 (Scheme 1). Deprotonation with KHMDS and alkylation with cyclopropyl methylbromide in toluene at 0 °C provided 8 in 20% yield, as a single diastereomer (steroechemical assignment based on literature precedent17 and nOe studies). TFA-mediated removal of the Boc group, followed by sulfonylation with various sulfonyl chlorides, generated congeners 9. Finally, ‘Raney’ Ni reduction of the nitrile and subsequent acylation with 2,4-dichlorobenzoyl chloride delivered analogs 4.
Figure 2.
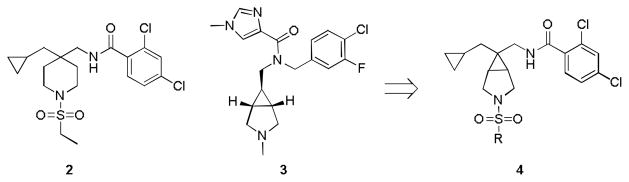
Initial approach to hybridize 2 and 3 to afford novel GlyT1 [3.1.0] scaffold 4.
Scheme 1.
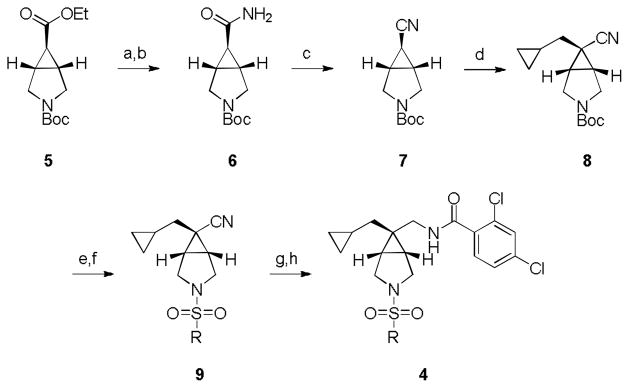
Reagents and conditions. (a) LiOH, THF, MeOH, rt; (b) NH4Cl, EDC, HOBt, DIEPA, DMF, 0 °C; (c) cyanuric chloride, DMF, 0 °C; (d) KHMDS, toluene, 0 °C, cyclopropyl methylbromide; (e) TFA, CH2Cl2, 0 °C; (f) RSO2Cl, CH2Cl2, DIEPA, 0 °C; (g) Raney Ni, H2 (45 psi), NH4OH, MeOH; (h) 2,4-dichlorobenzoyl chloride, CH2Cl2, DIEPA, 0 °C.
The initial 10-membered library of analogs 4 displayed somewhat unexpected SAR, with a significant diminution in potency relative to the piperdine-based GlyT1 inhibitors 1 and 2 (Table 1).13–15 For example, 4a (GlyT1 IC50 = 1.3 μM), the direct analog of 2 (GlyT1 IC50 = 26 nM), lost 50-fold in potency, and 4b (GlyT1 IC50 = 360 nM), the n-propyl congener of 1 (GlyT1 IC50 = 2.4 nM), lost ~150-fold. A trend towards increased activity resulted with a cyclopropyl methyl sulfonamide 4d (GlyT1 IC50 = 230 nM), but all other modifications, including aryl (4i and 4j) and heteroaryl sulfonamides (4g and 4h) led to significant loss in GlyT1 potency. However, all analogs 4 retained high selectivity versus GlyT2 (IC50 > 30 μM) and preliminary in vitro DMPK profiling indicated that this core retained the favorable disposition properties of 1–3 (fu 2–4%, IC50 >10 μM vs. CYP3A4, 2D6, 1A2 and 2C9). Substitution of the 6-cyclopropyl methyl group in 4 with either phenyl or 2-pyridyl moieties, also led to inactive analogs (GlyT1 IC50 > 30 μM) and represented a divergence from the SAR of the piperidine series 1 and 2.13–15 Analogs of 4 where the optimal sulfonamides were maintained (4b and 4d), but the substitution on the benzamide moiety was varied, once again led to a large diminution in GlyT1 potency (data not shown).
Table 1.
Structures and activities of [3.1.0] analogs 4.
| |||
|---|---|---|---|
| Compound | R | GlyT1 IC50 (μM)a | GlyT2 IC50 (μM)a |
| 4a |
|
1.3 | >30 |
| 4b |
|
0.36 | >30 |
| 4c |
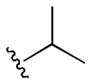
|
3.4 | >30 |
| 4d |
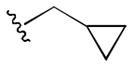
|
0.23 | >30 |
| 4e |
|
5.5 | >30 |
| 4f |
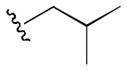
|
2.9 | >30 |
| 4g |
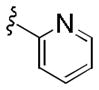
|
1.0 | >30 |
| 4h |
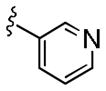
|
0.89 | >30 |
| 4i |
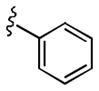
|
>10 | >30 |
| 4j |
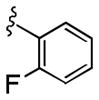
|
>10 | >30 |
IC50s represent single determinations performed in duplicate
As we began to assess and consider the data generated thus far, we were attracted to the N-methyl imidazole moiety of Pfizer’s 3,16 and with simple models could achieve an orientation in which the N-methyl imidazole moiety could fill the same space as the alkyl sulfonamides in 4b and 4d. Thus, we hypothesized that N-methylimidazole sulfonamide congeners might enhance GlyT1 potency in analogs 4.
To test this concept, we took advantage of our large supply of various 2-pyridyl containing [3.1.0] cores (inactive with alkyl or aryl sulfonamides) and prepared the N-methylimidazole sulfonamide analogs 10 and 11 (Fig. 3), as work form Merck demnstarted that the 2-pyridyl moiety was superior to the orignal 4-phenyl moiety in 1.14,15 These analogs all displayed sub-micromolar potency at GlyT1 (IC50s of 247 nM for 10 and 185 nM for 11), clogPs of 2.3, large fraction unbound in plasma (fu 7–14%), clean CYP profiles (IC50 >30 μM) and in oral brain tissue distribution studies, Kp ([brain]/[plasma]) ratios of 1.1. Interestingly, replacement of the N-methylimidazole with either imidazole or an N-methyl pyrazole led to a complete loss of GlyT1 activity. As incorporation of the N-methyl imidazole sulfonamide increased potency in the 2-pyridyl [3.1.0] core from IC50s >30 μM to IC50s < 250 nM, we were excited to see the impact of this modification to the already very potent cyclopropyl methyl [3.1.0] core of 4.
Figure 3.
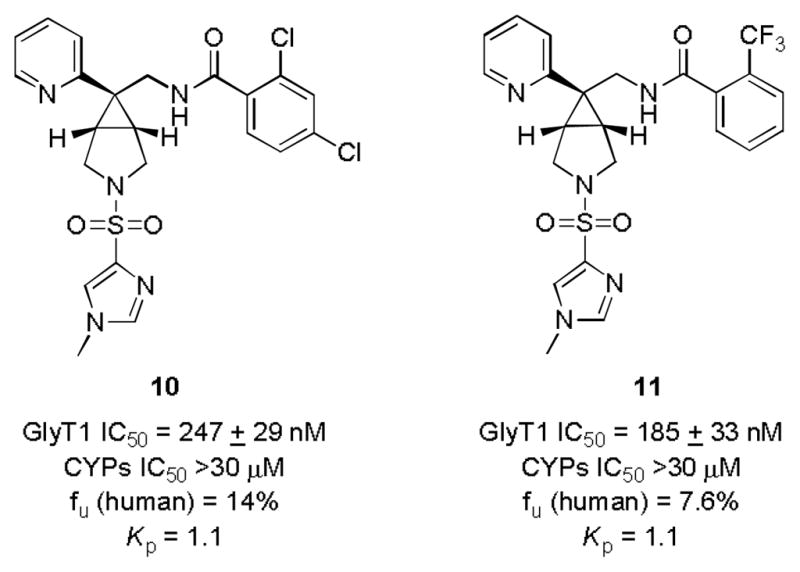
Impact of incorporation of an N-methyl imidazole sulfonamide into the [3.1.0] scaffold to afford 10 and 11.
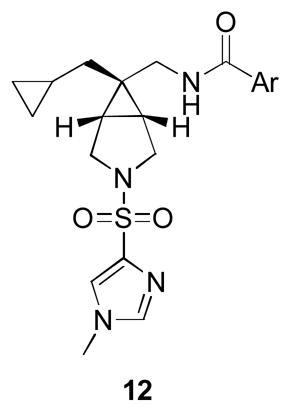
With a slight modification of the chemistry in Scheme 1, we were able to readily prepare a library of analogs 12, where the benzamide moiety was varied in the context of the cyclopropyl methyl [3.1.0] core containing an N-methyl imidazole sulfonamide. As shown in Table 2, this endeavor was very productive, affording potent (IC50s from 4 to 119 nM) and selective GlyT1 inhibitors with excellent DMPK profiles and CNS penetration in rat with oral dosing (Kp of 0.3 to 0.8). While many analogs displayed excellent potency at GlyT1, such as 12a, 12d and 12f, analog 12d stood out as having the best balance of potency (GlyT1 IC50 = 5 nM), fraction unbound in plasma (fu 7%), and in an oral brain tissue distribution study, a Kp ([brain]/[plasma]) ratio of 0.8.
Table 2.
Stuctures and activities of [3.1.0] analogs 12.
| |||||
|---|---|---|---|---|---|
| Cmpd | Ar | GlyT1 IC50 (nM)a | clogP | fu (hum) | Kpb (rat PO) |
| 12a | 2,4-diClPh | 4 | 2.3 | 5% | 0.3 |
| 12b | 2,4-diFPh | 44 | 1.9 | 8% | 0.5 |
| 12c | 2,6-dFPh | 28 | 1.9 | 8% | 0.5 |
| 12d | 2-CF3Ph | 5 | 2.5 | 7% | 0.8 |
| 12e | 3-CF3Ph | 64 | 2.5 | 4% | 0.7 |
| 12f | 2-ClPh | 19 | 2.6 | 4% | 0.4 |
| 12g | 3,4-diClPh | 82 | 3.2 | 2% | 0.3 |
| 12h | 2-ClPh | 119 | 2.7 | 3% | 0.4 |
| 12i | 3,5-diClPh | 100 | 3.3 | 2% | 0.3 |
| 12j | 4-ClPh | 32 | 2.7 | 3% | 0.3 |
IC50s represent single determinations performed in dupliate.
All analogs were inactive on GlyT2 (IC50 > 30 μM).
Kp = partitioning coefficient = [brain]/[plasma]
After assessing ancillary pharmacology in a Eurofin Lead Profiling panel of radioligand binding assays against 68 GPCRs, ion channels and transporters, our focus narrowed on 12d, as it displayed no inhibition >50%@10 μM against any target in the panel, yet was a potent and selective GlyT1 inhibitor (GlyT1 IC50 = 5±0.5 nM (N=3), GlyT2 IC50 >30 μM). Eadie-Hoffstee plots, where the reaction rate is plotted versus the ratio of the reaction rate and substrate concentration, provide useful insight into the mechanism of enzymatic inhibition, with competitive and noncompetitive enzymatic inhibition demonstrating distinct patterns. An Eadie-Hoffstee plot (Fig. 5) of the effect of this series, represented by 12c, on the enzyme kinetics of [14C]-glycine transport showed that this series is competitive with respect to glycine, in accordance with the known mechanism of action for 1 and 213–15, and distinct from the non-competitive mechanism of action of the sarcosine-derived GlyT1 inhibitors, such as NFPS.5–8 In addition, 12d possessed a clean CYP profile (IC50 >30 μM against 1A2, 2C9 and 2D6; 14.9 μM against 3A4) and good unbound fraction in both human (fu = 6.8%) and rat (fu = 44.8%). We assessed stability in rat plasma for 4 hours at 37 °C, and 12d was stable, indicating the free fraction in rat plasma is truly high. In vitro intrinsic clearance experiments suggest 12d posssesses moderate to high predicted clearance for both human (ClHEP = 17.7 mL/min/kg) and rat (ClHEP = 43.0 mL/min/kg). A rat IV PK study was conducted with 12d and it displayed moderate in vivo clearance (0.5 mg/kg) (Clp = 31 mL/min/kg) with a short half-life (t1/2 = 14.5 min). This is in-line with data reported for Merck’s 2, which was moderate to high clearance in rat, but low clearance in dog. We evaluated 12d in two separate rat brain tissue distribution studies: one with subcutaneous (10 mg/kg s.c. in 10% tween 80) dosing and one with oral (10 mg/kg p.o. in 0.5% methocellulose) dosing. Both dosing routes exhibited good exposure (s.c. plasma AUC0-6hr: 976 nM*h; s.c brain, AUC0-6hr: 431 nM*h (or Kp = 0.44); p.o. plasma AUC0-6hr: 1156 nM*hr, p.o. brain, AUC0-6hr: 956 nM*h (or Kp = 0.83)) with unbound concentrations above the GlyT1 IC50 at 6 hr. Due to the higher exposure, for an in vivo proof of concept study in a preclinical model of schizophrenia, we proceeded with oral dosing.
Figure 5.

(A) Saturation [14C]-glycine transport in the presence of vehicle (red squares) or 40 nM 12c (blue triangles). (B) An Eadie-Hoffstee diagram for 12c and [14C]-glycine.
Based on the precendent with other GlyT1 inhibitors such as 1,13 we evaluated both 215 and 12d for their ability to enhance prepulse inhibition (PPI) of the rodent acoustic startle response, a measure of sensorimotor gating known to be deficient in schizophrenic patients.18,19 In this study (Fig. 6), both 2 and 12d were dosed orally at 30 mg/kg (a dose known to engender >90% occupancy for 2),15,20,21 and evaluated against four increasing prepulse intensities (70–88 dB). Both 2 and 12d showed a statistically significant enhancement in prepulse inhibiton at the 82 and 88 dB prepulse intensities, with no effect on basal startle amplitude during no-stimulus trials. Thus, 12d (VU0240391), derived from a scaffold-hopping exercise employing 2 and 3, led to a novel [3.1.0]- based GlyT1 inhibitor with in vitro and in vivo properties comparable to other advanced GlyT1 inhbitors in short order, and for which a U.S. patent was issued.22
Figure 6.
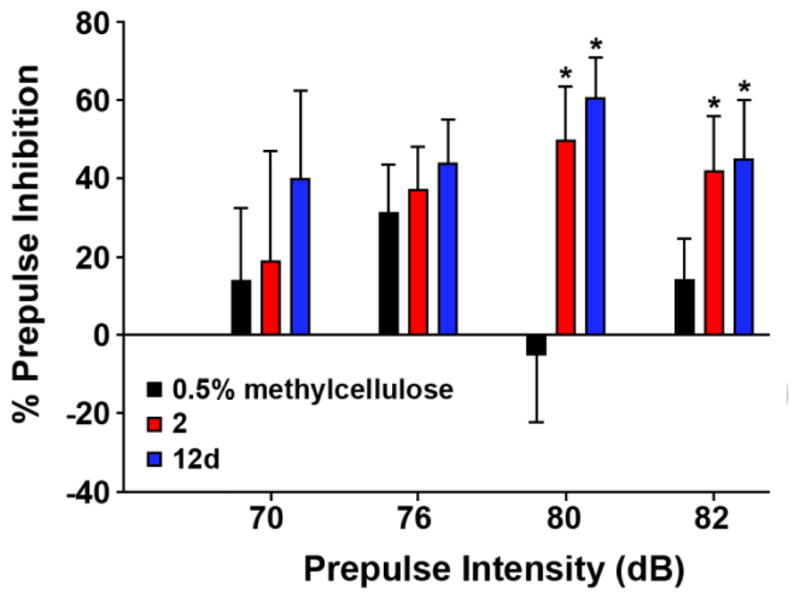
The effect of vehicle, 2 and 12d on PPI in rat at 30 mg/kg oral (p.o.) dosing in 0.5% methylcellulose. *p<0.05 in comparison to vehicle by Dunnett’s test (n=6–7).
In conclusion, we were able to scaffold hop and merge elements from both the Merck piperidine-based series of GlyT1 inhibitors, represented by 1 and 2,13–15 and Pfizer’s 3,16 into a novel, patented series of [3.1.0]-based N-methylimidazole sulfonamides 12. Members of this series displayed exceptional GlyT1 potency, DMPK profiles, CNS penetration and comparable in vivo efficacy to advanced GlyT1 inhibitors without the need for an HTS to enable a fast-follower program. Additional scaffolds developed during the course of this scaffold-hopping program will be reported in due course.
Figure 4.

Concentration response curves (N=3) for glycine and 12d in the [14C]-glycine uptake assay.
Acknowledgments
This work was supported by the NIH/NIMH under a National Cooperative Drug Discovery and Development grant U01 MH08795. DJS is a recipient of a National Alliance for Research on Schizophrenia and Depression (NARSAD)–Dylan Tauber Young Investigator Award. Vanderbilt is a member of the MLPCN and houses the Vanderbilt Specialized Chemistry Center for Accelerated Probe Development supported by U54 MH084659. The support of William K. Warren, Jr. who funded the William K. Warren, Jr. Chair in Medicine (to C.W.L.) is gratefully acknowledged.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Lindsley CW, Shipe WD, Wolkenberg SE, Theberge CR, Williams DL, Jr, Sur C, Kinney GG. Curr Topics in Med Chem. 2006;8:771. doi: 10.2174/156802606777057599. [DOI] [PubMed] [Google Scholar]
- 2.Menniti FS, Lindsley CW, Conn PJ, Pandit J, Zagouras P, Volkmann RA. Curr Topics in Med Chem. 2013;13:26. doi: 10.2174/1568026611313010005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Olney JW, Newcomer JW, Farber NB. J Psychiatry Res. 1999;33:523. doi: 10.1016/s0022-3956(99)00029-1. [DOI] [PubMed] [Google Scholar]
- 4.Coyle JT. Cell Mol Neurobiol. 2006;26:365. doi: 10.1007/s10571-006-9062-8. [DOI] [PubMed] [Google Scholar]
- 5.Kinney GG, Sur C. Curr Neuropharmacology. 2005;3:35. [Google Scholar]
- 6.Bridges TM, Williams R, Lindsley CW. Curr Opin Mol Ther. 2008;10:591. [PubMed] [Google Scholar]
- 7.Lindsley CW, Wolkenberg SE, Kinney GG. Curr Topics in Med Chem. 2006;6:1883. doi: 10.2174/156802606778249784. [DOI] [PubMed] [Google Scholar]
- 8.Wolkenberg SE, Sur C. Curr Topics in Med Chem. 2010;10:170. doi: 10.2174/156802610790410974. [DOI] [PubMed] [Google Scholar]
- 9.Hashimoto K. Curr Pharm Des. 2011;17:112. doi: 10.2174/138161211795049598. [DOI] [PubMed] [Google Scholar]
- 10.For information on RG1678, please see: www.roche.com
- 11.Harvey RJ, Yee BK. Nature Rev Drug Discov. 2013;12:866. doi: 10.1038/nrd3893. [DOI] [PubMed] [Google Scholar]
- 12.Xiang Z, Thompson AD, Brogan JT, Schulte ML, Mi D, Lewis LM, Yang L, Zhou B, Melancon BJ, Morrison R, Santomango T, Byers F, Brewer K, Aldrich JS, Yu H, Dawson ES, Li M, McManus O, Jones CK, Daniels JS, Conn PJ, Xie X, Weaver CD, Lindsley CW. ACS Chem Neurosci. 2011;2:730. doi: 10.1021/cn200090z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lindsley CW, Zhao Z, Leister WH, O’Brien JA, Lemiare W, Williams DL, Jr, Chen TB, Chang RSL, Burno M, Jacobson MA, Sur C, Kinney GG, Pettibone DJ, Tiller PR, Smith S, Tsou NN, Duggan ME, Conn PJ, Hartman GD. Chem Med Chem. 2006;1:807. doi: 10.1002/cmdc.200600097. [DOI] [PubMed] [Google Scholar]
- 14.Zhao Z, Leister WH, O’Brien JA, Lemiare W, Williams DL, Jr, Jacobson MA, Sur C, Kinney GG, Pettibone DJ, Tiller PR, Smith S, Hartman GD, Lindsley CW, Wolkenberg SE. Bioorg Med Chem Lett. 2009;19:1488. doi: 10.1016/j.bmcl.2008.12.115. [DOI] [PubMed] [Google Scholar]
- 15.Wolkenberg SE, Zhao Z, Wisnoski DD, Leister WH, O’Brien JA, Lemiare W, Williams DL, Jr, Jacobson MA, Sur C, Kinney GG, Pettibone DJ, Tiller PR, Smith S, Gibson C, Ma BK, Polsky-Fisher SL, Lindsley CW, Hartman GD. Bioorg Med Chem Lett. 2009;19:1492. doi: 10.1016/j.bmcl.2009.01.015. [DOI] [PubMed] [Google Scholar]
- 16.Lowe JA, III, Hou X, Schmidt C, Tingley FD, III, McHardy S, Kalman M, DeNinno S, Sanner M, Ward K, Lebel L, Tunucci D, Valnetine J. Bioorg Med Chem Lett. 2009;19:2974. doi: 10.1016/j.bmcl.2009.04.035. [DOI] [PubMed] [Google Scholar]
- 17.Klapars A, Waldman JH, Campos KR, Jensen MS, McLaughlin M, Chung JYL, Cvetovich RJ, Chen CY. J Org Chem. 2005;70:10186–1-189. doi: 10.1021/jo051737f. [DOI] [PubMed] [Google Scholar]
- 18.Geyer MA, Krebs-Thomson K, Braff DL, Swerdlow NR. Psychopharmacology. 2001;156:117. doi: 10.1007/s002130100811. [DOI] [PubMed] [Google Scholar]
- 19.Braff DL, Geyer MA, Swerdlow NR. Psychopharmacology. 2001;156:234. doi: 10.1007/s002130100810. [DOI] [PubMed] [Google Scholar]
- 20.Zeng Z, O’Brien JA, Leimare W, O’Malley SS, Miller PJ, Zhao Z, Wallace MA, Raab C, Lindsley CW, Sur C, Williams DL., Jr Nuc Med and Bio. 2008;35:315. doi: 10.1016/j.nucmedbio.2007.12.002. [DOI] [PubMed] [Google Scholar]
- 21.Hamill TG, Eng W, Jennings ASR, Lewis RT, Thomas S, Wood S, Street LJ, Wisnoski DD, Wolkenberg SW, Lindsley CW, Sanabria-Bohorquez SM, Patel S, Ryan C, Riffel K, Cook J, Sur C, Burns HD, Hargreaves R. Synapse. 2011;65:261. doi: 10.1002/syn.20842. [DOI] [PubMed] [Google Scholar]
- 22.Lindsley CW, Conn PJ, Williams R, Jones CK, Sheffler DJ. 8,497,289. US. 2013