

Abstract
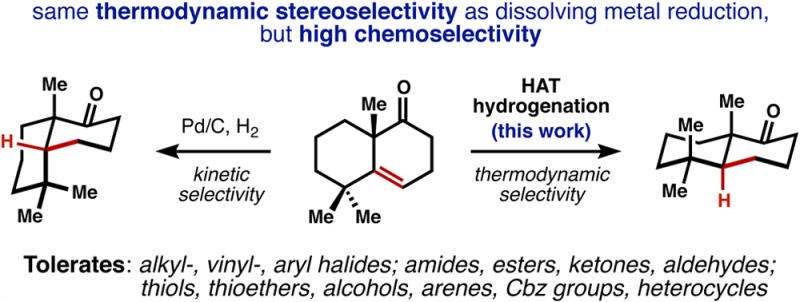
Few methods permit the hydrogenation of alkenes to a thermodynamically favored configuration when steric effects dictate the alternative trajectory of hydrogen delivery. Dissolving metal reduction achieves this control, but with extremely low functional group tolerance. Here we demonstrate a catalytic hydrogenation of alkenes that affords the thermodynamic alkane products with remarkably broad functional group compatibility and rapid reaction rates at standard temperature and pressure.
Hydrogenation of alkenes is among the cardinal reactions available to synthetic chemists. Applied retrosynthetically, this transform adds points of unsaturation to carbon skeletons, which can be used to dissect complex bond networks into simple fragments.1 However, a long-standing challenge in complex molecule synthesis is hydrogenation of alkenes to a thermodynamically favored configuration2 when steric constraints of the substrate favor hydrogenation to a nonthermodynamic (kinetic) alkane product (see 1→cis-2, Figure 1a). Dissolving metal reduction provides a means to achieve thermodynamic control and can reduce conjugated3 or electron-poor alkenes4 at low temperature (see 1→trans-2), but requires ambient or elevated temperature to reduce electron-neutral alkenes.5 These latter substrates are therefore seldom employed because chemoselectivity is unsatisfactory: most other functional groups are reduced preferentially to electron-neutral alkenes. This problem is especially evident in chemical syntheses of terpenoid secondary metabolites, which include the FDA approved steroid, taxoid, artemisinin, and ingenoid classes. Thus, many terpenes that contain equatorial methyl groups, for instance 3–5, have proven difficult to access, since hydrogenation of the corresponding exomethylene using standard methods yields either the wrong epimer or an equimolar mixture of two epimers (Figure 1b).6−8 The origin of the poor chemoselectivity associated with dissolving metal reduction is the low reduction potential of electron neutral alkenes (Ered < –3 V, Pb cathode),9 which form high-energy radical anions (6→7, Figure 1c) prior to protonation to lower energy tertiary radicals (8) and further reduction, protonation to alkanes (9). We thought that circumvention of radical anion 7 via direct hydrogen atom transfer (HAT)10 might increase chemoselectivity but lead to the same stereochemical preferences as dissolving metal reduction. Here we show that manganese and cobalt catalysts can effect this stepwise radical hydrogenation of electron-neutral alkenes and exhibit the same stereochemical preferences as dissolving metals but spare a variety of reactive functional groups that are normally reduced.
Figure 1.
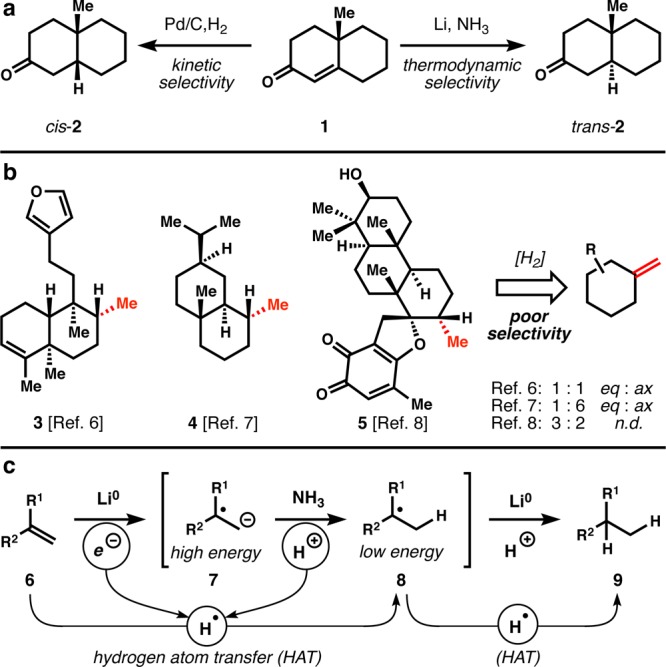
Kinetic versus thermodynamic hydrogenation. (a) Example of stereodivergent hydrogenation; (b) examples where kinetic hydrogenation yields the incorrect stereoisomer; (c) poor chemoselectivity of dissolving metal reduction might be circumvented by HAT hydrogenation.
As a representative model system for the terpenes shown in Figure 1b and as a starting point to probe the concept and consequences of HAT hydrogenation, we chose 4-tert-butylmethylenecyclohexane 10, since extensive data on its hydrogenation are available (Table 1).11,12 Whereas hydrogenation of 10 with Wilkinson’s12 or Adam’s catalyst13 delivers primarily 12 (cis), bearing an axial methyl, and diimide reduction gives a 1:1 mixture,14 dissolving metal reduction instead provides high selectivity for 11 (95:5).15 It seemed reasonable that hydrogenation via a tertiary carbon radical might lead to high selectivity for an equatorial methyl group, since methyl-substituted cyclohexyl radicals are known to trap tributyltin deuteride with high axial selectivity for C–D bond formation, owing to the nonplanar ground state16 and transition state pyramidalization of tertiary radicals.17 We considered the redox hydration conditions of Mukaiyama18 as a good starting point for an iterative HAT hydrogenation of alkenes since tertiary radicals are readily generated from electron-neutral alkenes, and the cobalt or manganese catalysts19 are inexpensive and air-stable. However, the oxidizing conditions required for this transformation appeared to be an ostensible barrier to implementation of a reductive reaction.20 Even so, Carreira demonstrated that tert-butyl hydroperoxide can serve as a replacement activator and/or reoxidant in the context of cobalt-catalyzed hydroazidation19 and hydrocyanation of alkenes.21 Similarly, we found that if TBHP is added in stoichiometric quantities to a mixture of alkene 10, phenylsilane, and a manganese catalyst in the absence of heteroatom radical traps, the reaction rapidly produces 12 (trans) with high selectivity, albeit in poor yield. Higher conversion to hydrogenated products is highly ligand and solvent dependent; the dipivaloyl-methane (dpm) ligand and isopropanol solvent were found to be optimal.22 Most appealingly, the experimental procedure is simple, conversion occurs usually within 1 h, and no hydrogen atmosphere is required. Nonanhydrous conditions are tolerated, however excess water does inhibit conversion (10 equiv = 5 M water in i-PrOH, 33% conversion at 1 h; see Supporting Information (SI) for an optimization table and corresponding observations). Co(dpm)2 induces equally high levels of stereoselectivity and also allows nonpolar solvents and hydrophobic substrates to be used (vide infra), however Mn(dpm)3-catalyzed reactions are generally higher yielding.
Table 1. Comparison to Existing Methods.
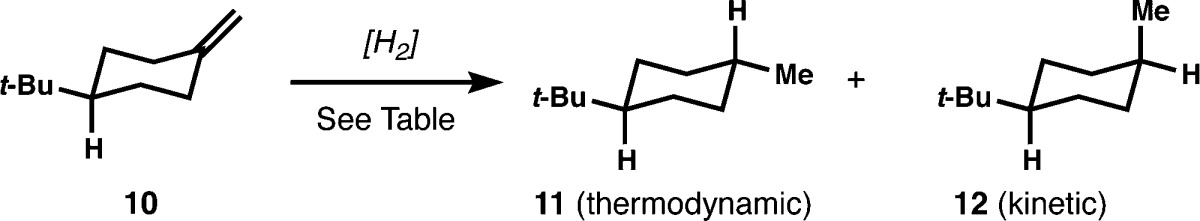
| conditions | yield | 11:12 |
|---|---|---|
| homogeneous: 5 mol % RhCl(PPh3)3, 1 atm H2, PhH, 18 °C | 100% | 32:68 |
| heterogeneous: 9 mol % PtO2, 2 atm H2, AcOH | 100% | 21:79 |
| pericyclic: N2H4, O2, EtOH, 55 °C | ND | 49:51 |
| dissolving metal: Li0, EDA, 35 °C | ND | 95:5 |
| radical: 10 mol % Mn(dpm)3, 1.0 equiv PhSiH3, 1.5 equiv TBHP, i-PrOH (0.5 M), 22 °C, 1 h | 86% | 84:16 |
| or: 10 mol % Co(dpm)2, 1.0 equiv PhSiH3, 1.5 equiv TBHP, i-PrOH (0.5 M), 22 °C, 1 h | 69% | 86:14 |
As illustrated by Table 2a, trans-selective hydrogenation of cyclohexenes is difficult to achieve by standard methods. For instance 13a and b afford poor stereoselectivity using Pd/C catalysis, and only marginally better ratios using Crabtree’s catalyst, due to the poor directivity afforded by sterically encumbered substituents.23 However, using HAT hydrogenation, where directivity is irrelevant, trans-substitution with high stereoselectivity is favored in both cases.
Table 2. Initial Survey of Method’s Utility.
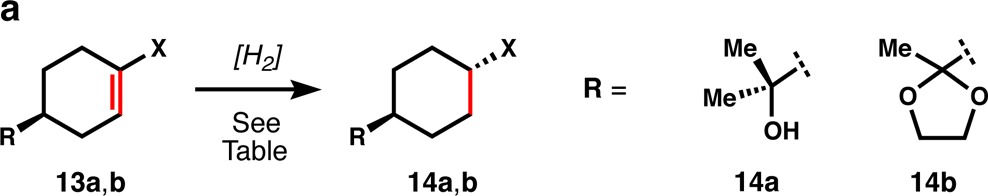
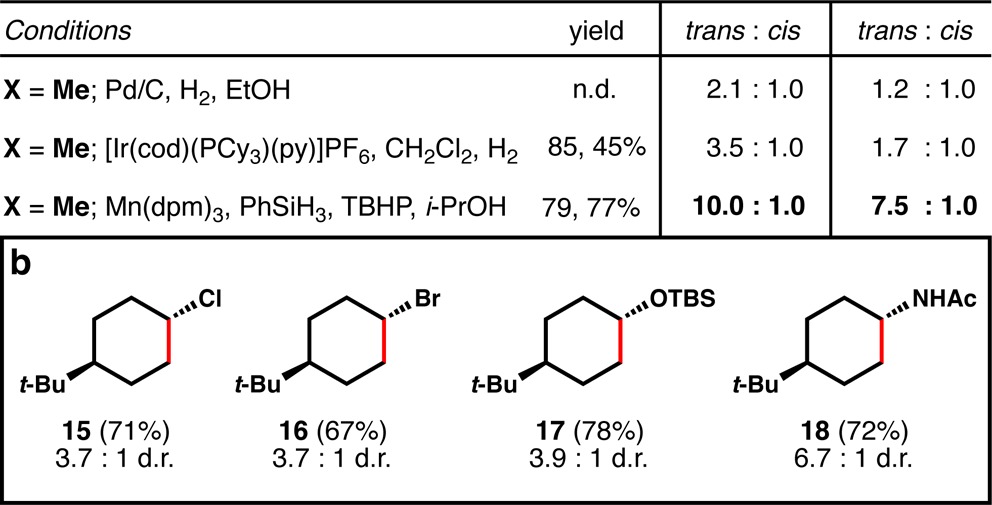
As a result, diversely substituted cyclohexenes now can be hydrogenated to the thermodynamically preferred trans-isomers (Table 2b) in the absence of any directing groups.23b Remarkably, chloro- and bromoalkenes, which generally do not tolerate dissolving metal conditions and are challenging substrates even with standard hydrogenation methods, are efficiently reduced to equatorial halo-cyclohexanes 15 and 16. Electron-releasing siloxy (enolsilane) and acetamide (enamide) groups are also efficiently reduced to the trans-isomers 17 and 18, illustrating the electronic flexibility of the method.
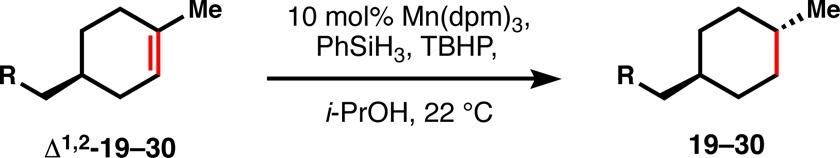
As suggested by substrates 15–18, the functional group compatibility of our method is significantly better than dissolving metal reduction and a fuller scope is illustrated by Table 3. For instance, not only alcohols (19) but also their corresponding alkyliodides (20) are viable substrates for this transformation. Selectivity against the reduction of aromatic rings is excellent, and therefore carboxybenzyl (Cbz) groups (21) are spared from reduction, as are phenyl ethers (22), aryl chlorides and fluorides (23), aryl bromides (24), and aryl iodides (25), although some deiodination was observed by GCMS (∼5%). Trifluoromethyl ethers (26), electron-rich arenes (27), phenyl thioethers (28), trifluoromethylarenes (29), and heterocycles like imidazole 30 are all well tolerated, although some rate decrease is observed with this last entry, likely due to equilibrium coordination/deactivation of the catalyst.
Table 3. Functional Group Tolerance.
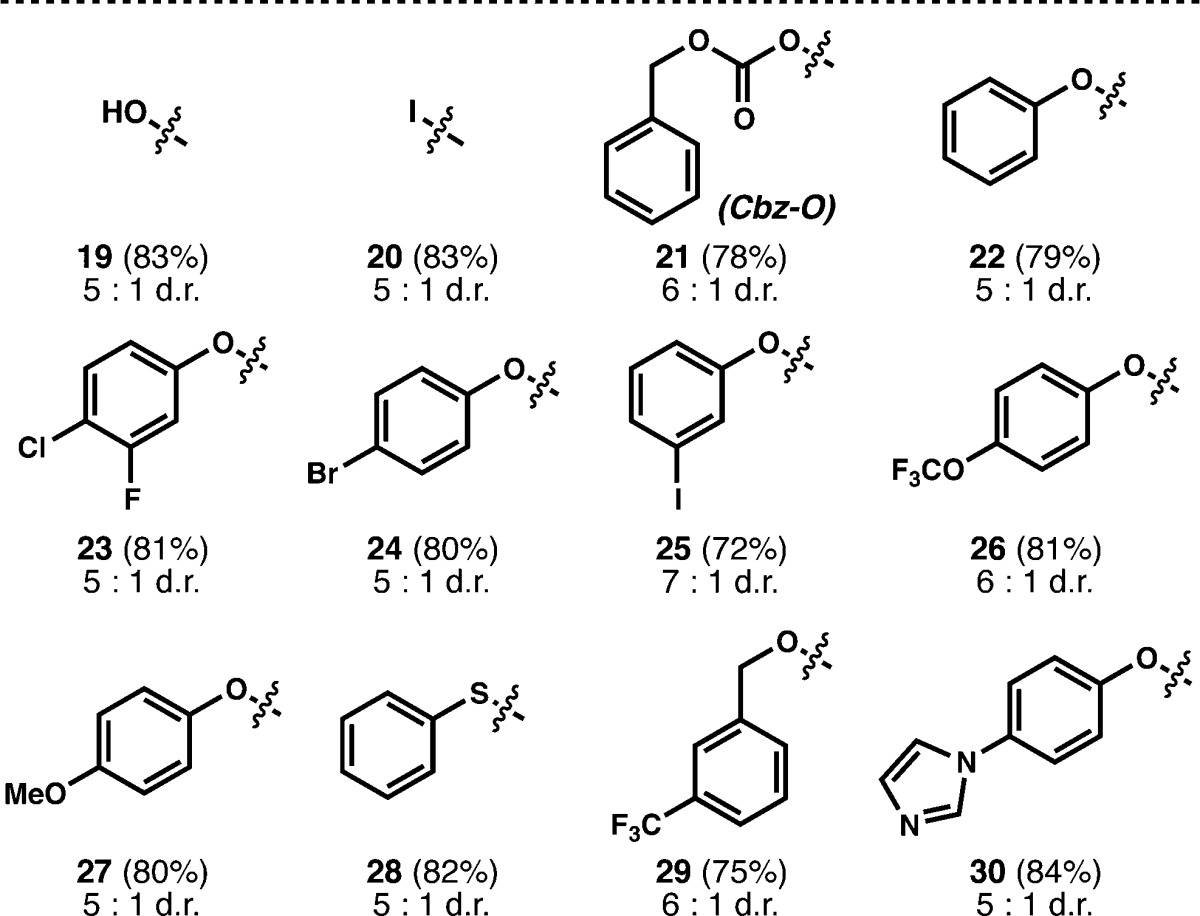
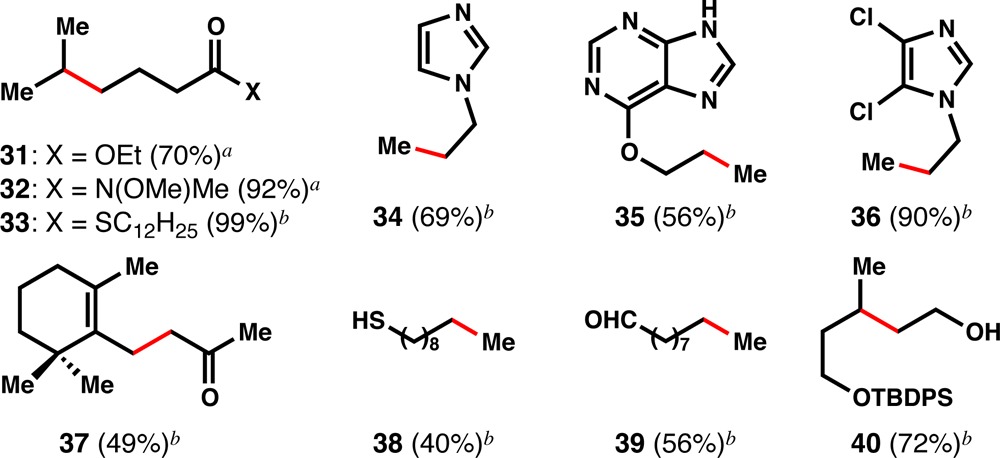
Weinreb amides, which suffer N–O bond cleavage under dissolving metal conditions,24 do not react competitively with the reduction of trisubstituted alkenes (32, Table 4). Thioesters undergo facile reduction to aldehydes using tributylstannyl radical reduction (Bu3SnH, AIBN) or palladium catalysis (Pd/C, Et3SiH),25 but using our method, saturated thioester 33 is produced and no aldehyde or alcohol is observed. When the alkene is allylic to a potentially labile C-heteroatom bond, simple to complex heterocycles (34–36) are tolerated and no scission of the allylic bond is observed, which might argue against the intermediacy of a carbon–metal bond.18,19 No Minisci addition products were observed using these substrates. Interestingly, β-ionone can be selectively reduced at the α,β-positions (37),20b in contrast to the selectivity observed using Mukaiyama’s hydration.20 Unsaturated thiols, aldehydes, and allylic alcohols (38–40) are also chemoselectively reduced; the thiol is oxidized in situ to the disulfide which can be cleaved on workup.
Table 4. Diverse Unsaturated Substrates are Reduced.
Using Co(dpm)2 conditions,
Using Mn(dpm)3 conditions.
Polycyclic systems can also be predictably hydrogenated to the thermodynamically stable diastereomer in preference to the normally observed kinetic stereochemistry (Scheme 1). For instance, Δ9,10-octalin (41) is preferentially hydrogenated with iridium catalysis to cis-decalin (42a) via syn-hydrogenation of the alkene.26 However, using HAT hydrogenation, a formal anti-addition of hydrogen is observed to produce the lower-energy trans-decalin (42b) with nearly the same selectivity as dissolving metal conditions.5 Similarly, 1,2-dimethyl cyclohexene (43) is hydrogenated to the cis-stereoisomer 44a using iridium catalysis,27 whereas our method produces trans-1,2-dimethylcyclohexane (44b), albeit with lower selectivity. Nevertheless, this stereochemical dichotomy between existing hydrogenation methods and the title reaction can be most vividly illustrated using problems already encountered in terpene syntheses. For example, en route to the putative structure of a sesquiterpene isolated from Cistus creticus, Katerinopoulos could not directly access a targeted trans-decalone framework since hydrogenation of their intermediate ketone 45 using heterogeneous catalysis produced cis-decalone 46a with high selectivity.28 In contrast, HAT hydrogenation can directly access this thermodynamically preferred but kinetically disfavored configuration (46b). Additionally, the terpene-derived petroleum biomarker drimane (48b) could not be directly accessed via hydrogenation of drimene 47, since use of Adam’s catalyst delivers the kinetically favored axial methyl substituent in 48a, and a four-step work-around was devised instead.29 In contrast, our method directly yields drimane (48b), which bears the thermodynamically favored equatorial methyl.
Scheme 1. Examples of Divergent Stereocontrol.
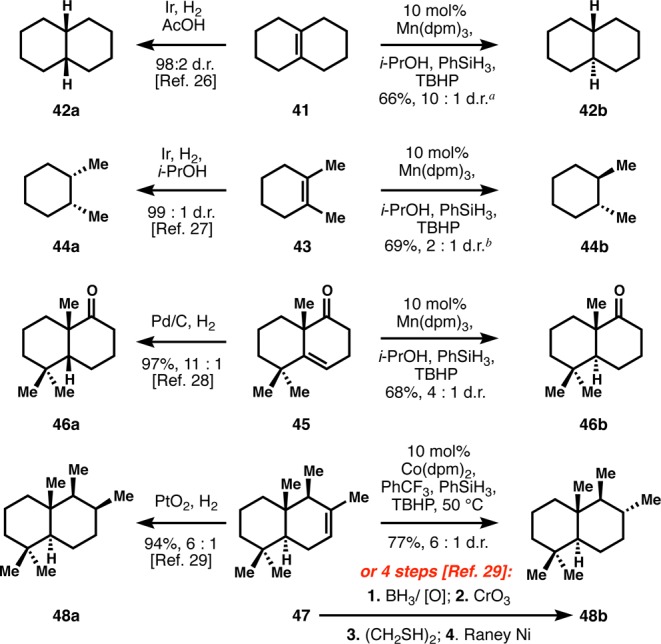
41 contained 16% of the 1,9-isomer.
43 contained 26% of the 1,6-isomer.
An unusual aspect of this hydrogenation is its general disregard for alkene substitution or electronic patterning (Scheme 2a). Unlike dissolving metal reduction, which exhibits profound rate acceleration when the alkene is conjugated to an electron acceptor,5 various substitution patterns are readily reduced by our HAT method with little influence by directly attached functionality (see SI for competition experiments). These minor effects of electron-modulating groups may indicate a direct hydrogen atom transfer to generate a carbon-centered radical,30b rather than the intermediacy of a carbon–metal bond en route to carbon–metal bond homolysis, as often proposed.18,19,22 Based on competition experiments, some trends are observed: increased substitution decreases the rate of consumption; electron-withdrawing groups have a minor accelerating effect on reaction rate; and electron-donating groups are weakly deactivating. Consequently, simple reductive cyclizations of polyenes are possible (Scheme 2b).30 For instance, diene 49 is readily cyclized to cyclopentane 50, which bears vicinal all-carbon quaternary centers, highlighting the ability of carbon-centered radicals to overcome severe steric clash via early transition states.31 This preliminary example paves the way for a general approach to reductive mono- and polycyclizations of unconjugated polyenes and a “traceless” approach to fully saturated molecules like terpenes.32
Scheme 2. Observed Reactivity Trends and Cyclizations.

The demonstrated utility of this new method for the thermodynamic hydrogenation of alkenes illustrates its potential to solve current and future problems in complex molecule synthesis, especially the reduction of halogenated alkenes (see Table 2b).33 Given the frequency with which hydrogenations are employed and their breadth of applications in chemistry, a new tool is advantageous. We expect that the experimental ease, broad scope, and orthogonal stereoselectivity of HAT hydrogenation will lead to its extensive use.
Acknowledgments
Financial support for this work was provided by the NIH (GM104180), the NSF GRFP (K.K.W.; DGE-1346837), and the Italian Ministry for Education and Research (fellowship to A.O.). We thank Professors Dale L. Boger, Phil S. Baran, and Donna G. Blackmond for helpful conversations. We are grateful to the Scripps Research Institute, Eli Lilly, Boehringer Ingelheim, Amgen, and the Baxter Foundation for additional financial support.
Supporting Information Available
Experimental procedures and spectroscopic data. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Present Address
† Department of Chemistry, Tohoku University, 6-3 Aoba-ku, Sendai, 980-8578, Japan.
Author Contributions
‡ These authors contributed equally.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- For example:Corey E. J.; Cheng X.-M.. The Logic of Chemical Synthesis; Wiley: New York, 1995; pp 47–57. [Google Scholar]
- Barton D. H. R.; Robinson C. H. J. Chem. Soc. 1954, 3045. [Google Scholar]
- Johnson W. S.; Bannister B.; Bloom B. M.; Kemp A. D.; Pappo R.; Rogier E. R.; Szmuszkovicz J. J. Am. Chem. Soc. 1953, 75, 2275. [Google Scholar]
- Stork G.; Darling S. D. J. Am. Chem. Soc. 1960, 82, 1512. [Google Scholar]
- Whitesides G. M.; Ehmann W. J. J. Org. Chem. 1970, 35, 3565. [Google Scholar]
- Takahashi S.; Kusumi T.; Kakisawa H. Chem. Lett. 1979, 515. [Google Scholar]
- Alexander R.; Kagi R.; Noble R. J. Chem. Soc., Chem. Comm. 1983, 226. [Google Scholar]
- Justicia J.; Rosales A.; Buñuel E.; Oller-López J. L.; Valdivia M.; Haïdour A.; Oltra J. E.; Barrero A. F.; Cárdenas D. J.; Cuerva J. M. Chem.—Eur. J. 2004, 10, 1778. [DOI] [PubMed] [Google Scholar]
- Budnikova Y. G.; Krasnov S. A. In Developments in Electrochemistry;Chun J. H., Ed.; InTech: Rijeka, 2012; Chapter 5, pp 101–124. [Google Scholar]
- Seminal study of a hydrogenation where HAT is implicated:Sweany R. L.; Halpern J. J. Am. Chem. Soc. 1977, 99, 8335. [Google Scholar]
- Imaizumi S.; Murayama H.; Ishiyama J.; Senda Y. Bull. Chem. Soc. Jpn. 1985, 58, 1071. [Google Scholar]
- Mitchell T. R. B. J. Chem. Soc. B 1970, 823. [Google Scholar]
- Siegel S.; Dmuchovsky B. J. Am. Chem. Soc. 1962, 84, 3132. [Google Scholar]
- van Tamelen E. E.; Timmons R. J. J. Am. Chem. Soc. 1962, 84, 1067. [Google Scholar]
- Ficini J.; Francillette J.; Touzin A. M. J. Chem. Research (S) 1979, 150. [Google Scholar]
- Lloyd R. V.; Williams R. V. J. Phys. Chem. 1985, 89, 5379. [Google Scholar]
- Damm W.; Giese B.; Hartung J.; Hasskerl T.; Houk K. N.; Hüter O.; Zipse H. J. Am. Chem. Soc. 1992, 114, 4067. [Google Scholar]
- Review and references to early work:Mukaiyama T.; Yamada T. Bull. Chem. Soc. Jpn. 1995, 68, 17. [Google Scholar]
- Waser J.; Gaspar B.; Nambu H.; Carreira E. M. J. Am. Chem. Soc. 2006, 128, 11693. [DOI] [PubMed] [Google Scholar]
- a Magnus P.; Payne A. H.; Waring M. J.; Scott D. A.; Lynch V. Tetrahedron Lett. 2000, 41, 9725. [Google Scholar]; b Magnus P.; Waring M. J.; Scott D. A. Tetrahedron Lett. 2000, 41, 9731.In Ref (20b), the catalyst turns over by protonation of a manganese enolate. [Google Scholar]
- Carreira’s pioneering work in this area:; a Gaspar B.; Waser J.; Carreira E. M. Org. Syn. 2010, 87, 88. [Google Scholar]; b Gaspar B.; Carreira E. M. J. Am. Chem. Soc. 2009, 131, 13214. [DOI] [PubMed] [Google Scholar]; c Gaspar B.; Carreira E. M. Angew. Chem., Int. Ed. 2008, 47, 5758. [DOI] [PubMed] [Google Scholar]; d Gaspar B.; Waser J.; Carreira E. M. Synthesis 2007, 3839. [Google Scholar]; e Gaspar B.; Carreira E. M. Angew. Chem., Int. Ed. 2007, 46, 4519. [DOI] [PubMed] [Google Scholar]; f Waser J.; González-Gómez J. C.; Nambu H.; Huber P.; Carreira E. M. Org. Lett. 2005, 7, 4249. [DOI] [PubMed] [Google Scholar]; g Waser J.; Nambu H.; Carreira E. M. J. Am. Chem. Soc. 2005, 127, 8294. [DOI] [PubMed] [Google Scholar]; h Waser J.; Carreira E. M. Angew. Chem., Int. Ed. 2004, 43, 4099. [DOI] [PubMed] [Google Scholar]; i Waser J.; Carreira E. M. J. Am. Chem. Soc. 2004, 126, 5676. [DOI] [PubMed] [Google Scholar]
- Inoki S.; Kato K.; Isayama S.; Mukaiyama T. Chem. Lett. 1990, 1869. [Google Scholar]
- a Crabtree R. H.; Davis M. W. J. Org. Chem. 1986, 51, 2655. [Google Scholar]; b Excellent review on directed homogenous hydrogenation:Brown J. M. Angew. Chem., Int. Ed. 1987, 26, 190. [Google Scholar]
- Taillier C.; Bellosta V.; Meyer C.; Cossy J. Org. Lett. 2004, 6, 2145. [DOI] [PubMed] [Google Scholar]
- Fukuyma T.; Tokuyama H. Ald. Acta 2004, 37, 87. [Google Scholar]
- Weitkamp A. W. J. Catal. 1966, 6, 431.Notably, many other metal catalysts competitively isomerize 41 to Δ1,9-octalin, which is hydrogenated to trans-decalin 42b. However, this thermodynamic control is not general. [Google Scholar]
- Nishimura S.; Mochizuki F.; Kobayakawa S. Bull. Chem. Soc. Jpn. 1970, 43, 1919. [Google Scholar]
- Hatzellis K.; Pagona G.; Spyros A.; Demetzos C.; Katerinopoulos H. E. J. Nat. Prod. 2004, 67, 1996. [DOI] [PubMed] [Google Scholar]
- González-Sierra M.; Laborde M. A.; Rúveda E. A. Synth. Commun. 1987, 17, 431. [Google Scholar]
- a Wang L.-C.; Jang H.-Y.; Roh Y.; Lynch V.; Schultz A. J.; Wang X.; Krische M. J. J. Am. Chem. Soc. 2002, 124, 9448. [DOI] [PubMed] [Google Scholar]; b Hartung J.; Pulling M. E.; Smith D. M.; Yang D. X.; Norton J. R. Tetrahedron 2008, 64, 11822. [Google Scholar]; c Leggans E. K.; Barker T. J.; Duncan K. K.; Boger D. L. Org. Lett. 2012, 14, 1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lackner G. L.; Quasdorf K. W.; Overman L. E. J. Am. Chem. Soc. 2013, 135, 15342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mundal D. A.; Avetta C. T. Jr.; Thomson R. J. Nature Chem 2010, 2, 294. [DOI] [PubMed] [Google Scholar]
- Syntheses of complex halogenated molecules:; a King S. M.; Calandra N. A.; Herzon S. B. Angew. Chem., Int. Ed. 2013, 52, 3642. [DOI] [PubMed] [Google Scholar]; b Nilewski C.; Carreira E. M. Eur. J. Org. Chem. 2012, 1685. [Google Scholar]; c Chung W.-J.; Vanderwal C. D.. Acc. Chem. Res.; in press. [DOI] [PMC free article] [PubMed]; d Snyder S. A.; Brucks A. P.; Treitler D. S.; Moga I. J. Am. Chem. Soc. 2012, 134, 17714. [DOI] [PubMed] [Google Scholar]; Recent methods to synthesize chiral nonracemic organohalides:; e Hu D. X.; Shibuya G. M.; Burns N. Z. J. Am. Chem. Soc. 2013, 135, 12960. [DOI] [PubMed] [Google Scholar]; f Nicolaou K. C.; Simmons N. L.; Ying Y.; Heretsch P. M.; Chen J. S. J. Am. Chem. Soc. 2011, 133, 8134. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.