

Abstract
Bone remodeling is a very complex process. Homocysteine (Hcy) is known to modulate this process via several known mechanisms such as increase in osteoclast activity, decrease in osteoblast activity and direct action of Hcy on bone matrix. Evidence from previous studies further support a detrimental effect on bone via decrease in bone blood flow and an increase in matrix metalloproteinases (MMPs) that degrade extracellular bone matrix. Hcy binds directly to extracellular matrix and reduces bone strength. There are several bone markers that can be used as parameters to determine how high levels of plasma Hcy (hyperhomocysteinemia, HHcy) affect bone such as: hydroxyproline, N-terminal collagen 1 telopeptides. Mitochondrion serves an important role in generating reactive oxygen species (ROS). Mitochondrial abnormalities have been identified during HHcy. The mechanism of Hcy-induced bone remodeling via the mitochondrial pathway is largely unknown. Therefore, we propose a mitochondrial mechanism by which Hcy can contribute to alter bone properties. This may occur both through generations of ROS that activate MMPs and could be extruded into matrix to degrade bone matrix. However, there are contrasting reports on whether Hcy affects bone density, with some reports in favour and others not. Earlier studies also found an alteration in bone biomechanical properties with deficiencies of vitamin B12, folate and HHcy conditions. Moreover, existing data opens speculation that folate and vitamin therapy act not only via Hcy-dependent pathways but also via Hcy-independent pathways. However, more studies are needed to clarify he mechanistic role of Hcy during bone diseases.
Keywords: bone remodeling, collagen crosslinking, matrix metalloproteinases, oxidative stress
Introduction
Homocysteine (Hcy) is a molecule in the blood that has been shown to be an independent risk factor for heart disease as well as associated with kidney and brain disease [1–3]. Moreover, there are mixed results whether Hcy is an innocent bystander used to predict a state of organ damage or has a direct role in incurring this damage. Because we understand the detrimental effects that Hcy incurs on these systems, we sought to discover what effects Hcy may have on bone. Osteoporosis and injury from fracture has a vast social and economic impact worldwide. Out of 100 women, 40 are likely to have a fracture after the age of 50 years with 17.5% having a hip fracture as estimated by Lips [4]. The annual cost of osteoporosis in the USA is estimated to be 5–10 billion USD [5]. Hcy is proposed to cause a decrease in bone quality via an increase in reactive oxygen species (ROS) and activation of matrix metalloproteinases (MMPs) that can then degrade extracellular bone matrix.
Total Hcy (tHcy) has also been shown to alter blood flow by our laboratory, thereby depriving the bone of vital nutrients needed for bone maintenance and repair, and this probably occurs via the nitric oxide (NO) mechanism [6]. Herrmann et al. [7] have also discovered a direct role of the Hcy molecule on extracellular bone matrix, thereby reducing bone quality. Many markers are used as parameters to detect change in bone turnover density, quality, etc. This includes osteocalcin, osteoprotegerin, N-terminal telopeptides and hydroxyproline. These markers have been shown to be modulated with tHcy treatment as well as with vitamins such as folic acid and vitamin B12 that perform an antioxidant role.
In other diseases, it is known that hyperhomocysteinemia (HHcy) disturbs the mitochondrial membrane permeability transition and leads to activation of MMPs within mitochondria. MMPs then extrude from mitochondria and disturb the pathophysiological state. We have proposed similar mechanisms in the case of bone. These mechanisms involve intracellular and intramitochondrial ROS generation as well as translocation of MMPs. Some interesting results remain to be discovered in such upcoming trials as the B-PROOF (B-Vitamins for the Prevention of Osteoporotic Fractures) study that examines how administering vitamin B12 and folate will influence fracture risk [8].
Homocysteine and bone turnover, bone quality, collagen crosslinks
The process of bone remodeling occurs throughout life via cells such as osteoblasts (OBs) and osteoclasts (OCs), possibly modulated by the influence from OCs distributed throughout the bone matrix. OBs help to build bone by depositing collagen matrix, whereas OCs serve to degrade bone. OCs move to a surface of bone matrix and resorb the bone. Part of the reason why the body does this, is to make available a source of calcium needed for various life processes. Herrmann et al. [7] proposed four mechanisms by which tHcy is thought to alter bone remodeling: (i) increase in OC activity, (ii) decrease in OB activity, (iii) decrease in bone blood flow, and (iv) direct action of tHcy molecule on bone matrix.
In determining the effects of high levels of plasma tHcy on bone, there must also be measureable parameters by which tHcy acts to effect detrimental changes. Hence, several markers for biochemical bone turnover are used across a wide array of studies. The following markers for bone resorption were increased in HHcy rats: hydroxyproline and N-terminal collagen 1 telopeptides [9]. Osteocalcin is also used as a measure of bone formation and was found to decrease in one experiment of HHcy conditions by Ozdem et al. [9]. Osteocalcin is specific for OBs and osteocytes and it is the most bone specific non-collagenous matrix protein. It is localized next to OBs, osteoid and mineralized bone; moreover, it is also present in osteocyte lacunae in cement lines [10].
Herrmann et al. have observed the relationship between tHcy and markers of organic and inorganic bone resorption. The following markers were used: (i) bone mineral density at lumbar spine (BMD-LS), (ii) bone mineral density at total hip (BMD-HIP), (iii) fasting venous blood and urine sampling, (iv) osteocalcin measurement, (v) serum calcium (Ca), (vi) urinary deoxypyridinoline (DPD) crosslinks, (vii) osteoprotegerin (OPG), and (viii) soluble receptor activator of NF-κB ligand (sRANKL) [11]. There was a significant relationship found between Hcy and calcium measurements (r=0.170, p=0.045) as well as Hcy and DPD measurements (r=0.193, p=0.022); however, the other parameters mentioned did not have a significant relationship with Hcy.
Some further parameters measured in patients with femoral neck fractures such as: (i) collagen crosslink ratios, bone mineralization density distribution (BMD), (ii) Fourier transform infrared imaging (FTIRI), and (iii) quantitative backscattered electron imaging (qBEI). The collagen crosslink ratio was earlier used to measure pyridinoline/divalent crosslinks (pyr/divalent). Moreover, other parameters included: calcium (Ca) mean (weighted mean calcium concentration), Ca peak (the most frequent calcium concentration), Ca width (the width of the distribution – a measure of the mineralization homogeneity), Ca low (the percentage of bone area that is mineralized below the 5th percentile in the reference range), and Ca high (the percentage of bone that is mineralized above the 95th percentile reference range). This study showed a significant correlation between HHcy and collagen crosslink ratio (p<0.0001) in primary mineralized bone area, but not in trabecular bone surfaces undergoing resorption (p<0.05). Because there was no correlation obtained between HHcy and BMD (p<0.05) [12] and, hence, it could be possible that HHcy alters bone quality by interfering with collagen crosslinking along with no alteration in BMD. Further studies are required to prove a significant effect of HHcy on BMD because there are very few studies available in this area.
The importance of immunostaining for bone makes sense, especially when measuring the bimolecular markers of bone, and bone is such a hard material that is difficult to slice and manipulate. This is equally problematic for any scientist who has tried to purify bone samples for procedures such as zymography where quite a bit of grinding could destroy the matrix with release of matrix proteins [13]. An example of a protein that is stained is (i) osteonectin, which is produced by OBs along the basal membranes of blood vessels [10], (ii) osteopontin, which is found in osteocyte lacunae, osteoid, cement lines and in the mineralized bone [10], and (iii) collagen type 1, which is found around OBs, cement lines, osteoid, in mineralized matrix and resorption areas [14]. Staining for these proteins has been done previously and can continue to be used in future bone studies to determine modulation of bone properties by such cases as HHcy.
Another key protein marker that could be taken into consideration, for tHcy bone studies, is β-catenin. This protein marker has been observed to regulate bone homeostasis. This protein signals Wnt protein and plays an important role in bone metabolism by controlling the differentiation of OB and OC cells. For instance, a study by Kramer et al. found that a low-bone mass phenotype was found to have an increased OC number and activity, whereas OB cells remained the same. This was shown in osteocyte-specific β-catenin-deficient knockout mice. Hence, these proteins can also serve as markers in HHcy mice for future experiments [15].
Direct and indirect role of Hcy in bone
Hcy was discovered to have a direct effect on bone biomechanical properties as physical accumulation of tHcy in bone is associated with a reduction of cancellous bone (trabecular bone or spongy bone). Herrmann et al. [16] suggested that the elevated tHcy concentrations are mainly caused by folate, vitamin B12 and B6 deficiencies. More-over, the tHcy molecule binds to the proteins within the matrix via thiol group and an amino group. Because bone matrix is mainly collagen type 1; hence, tHcy is thought to bind to collagen. The study also investigated that 65% of tHcy binds to collagen in the extracellular matrix (ECM) and thus reduces bone strength [17].
An article by Liu et al. showed that tHcy inhibits lysyl oxidase (Lox gene), thereby interfering with post-translational modifications of collagen [18] and this contributes to decreased bone quality. It was shown that tHcy stimulated interleukin-6 (IL-6) synthesis in OBs, known to affect bone metabolism via OCs. IL-6 stimulation results from JAK2, FLI1 DNMT1 by methylation and this revealed a new mechanism affecting bone matrix formation [19].
Hcy and mitochondria
Mitochondria are involved in several processes in addition to producing ATP through oxidative phosphorylation. They have an established role in cellular death that includes release of such apoptotic molecules as cytochrome c [20–27]. In fact, mitochondria are also involved in the translocation and activation of several enzymes that occur through the outer mitochondrial membrane (OMM) and inner mitochondrial membrane (IMM). Mitochondria also serve an important role in both generating and detoxifying cellular reactive oxygen species (ROS) by utilizing the electron transport chain. One of the mechanisms by which MMPs are activated in mitochondria is by generation of hypochlorous acid (HOCl) from H2O2 by enzyme, myeloperoxidase [28]. The study showed that HOCl can regulate activity of MMP-7 in vitro as well as converting pro-MMP-7 to MMP-7 in vivo by converting thiol residue of cysteine switch to sulfinic acid [28]. An important mechanism for MMP-9 activation that involves mitochondria is via calpain-1 (Figure 1A). tHcy ligand has been shown to activate and translocate calpain-1 from cytosol to mitochondria, thereby increasing intramitochondrial oxidative stress resulting in MMP-9 activation within mitochondria. Moreover, proximal to this downstream process is increase of calcium and activation of MAPK [29]. The mechanism by which MMPs are extruded out of mitochondria occurs via the transitioning pore within mitochondria known as the mitochondrial pore transition [30]. This occurs in cardiomyocytes and microvascular endothelial cells (MVECs) via ligand receptors such as anti-N-methyl-D-aspartate receptor (NMDA-R) that increases calcium.
Figure 1.
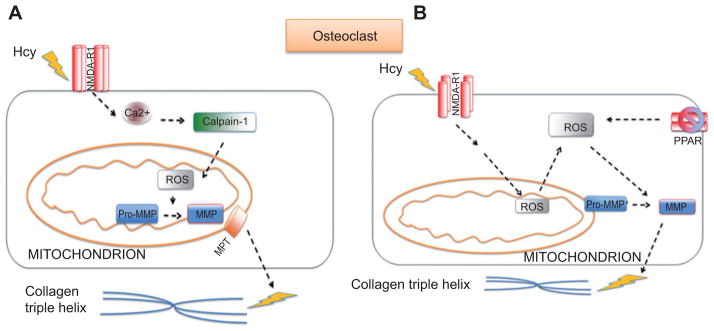
(A) The proposed mechanism of bone remodeling induced with HHcy. tHcy increases intracellular calcium by agonizing NMDA-R1. This increases expression of calcium-dependent calpain-1, thereby disrupting the mitochondrial membrane potential and electron transport chain. This increases the presence of ROS. An increase in ROS will activate MMPs; calpain will activate mitochondrial pore transition, resulting in MMPs exiting mitochondria and causing matrix degradation. (B) The suggested mechanism in bone remodeling engaged with elevated tHcy. tHcy increases intracellular calcium by agonizing NMDA-R1. This disrupts the electron transport chain and increases the presence of ROS. An increase in ROS will activate MMPs. ROS is also generated via a decreased expression of the PPAR receptor, allowing a greater presence of ROS that will activate MMPs within the cell and result in disruption of the extracellular matrix of bone.
The NMDA-R is also found on osteocytes [31]. tHcy binds to NMDA-R, thereby increasing intracellular calcium and increasing the expression of calpain-1; the mechanism has been reported in MVECs and we propose a similar mechanism in bone as illustrated in Figure 1 [29]. Calpain-1 is involved in directing mitochondrial membrane permeability transition (MPT), thereby allowing the extrusion of proteins through mitochondria [29]. The ROS from the calpain protein disrupt the mitochondrial MPT, which also activates MMPs within mitochondria. The MMPs can then be extruded from mitochondria, as similar in the case of MVECs [29].
Normally when OCs resorb bone, calcium is transported in the OC vesicles to be released into the blood stream and made available for various life processes [32]. In OCs, the cells accumulated calcium granules in the mitochondria to prevent the release of the granules in the cytoplasm during bone resorption. This perhaps occurs to prevent the signal for cell death [32]. It is unclear how it plays a role in further MMP activation because calcium is already known to play a role in MMP activation in other cells that are not osteocytes. Further studies are required by those who work with vesicular transport.
We propose that MMPs can enter the ECM via this mechanism that includes mitochondrial activation of MMPs. MMPs play a very important role in bone turnover and repair in addition to their well-known role in bone remodeling. If these enzymes are not working properly, bone also does not heal properly [33]. The presence of MMP-7 and MMP-12 in nonunion tissue (bone that did not heal properly after fracture) also confirmed this finding [33]. MMPs are thought to act as a good source of biomarkers because they are involved in pathological processes as well as regular maintenance processes of ECM [34]. One study has shown that MMP inhibitors (otherwise known as gelatinase inhibitors) gelatinase A (MMP-2) and gelatinase B (MMP-9) participate in bone resorption [35]. It is already known that cleavage of triple helices of collagen is the rate limiting step in type I collagen degradation [36].
There have been several recent experiments to discern under what conditions certain MMPs are expressed. For example, MMP-9 and MMP-13 have been shown to be expressed in cases of endochondral ossification by the following cells: OCs, endothelial cells and bone marrow stromal cells [37]. Moreover, OCs can also express the following MMPs: MMP-1, -2, -3, -10, -12, -13, -14. [37]. Specific MMPs have even been known to perform specific functions. MMP-13, for example, is known to be involved in cell migration through type I collagen that has not yet entirely mineralized. After this role, MMP-13 can continue with metabolizing this unmineralized tissue [38]. MMP-9 is also known to be required for OC invasion into cartilage. Excluding these functions, MMPs have also been ascertained to play regulatory roles, for example, MMP-14 regulates other MMPs such as MMP-2 and can promote osteogenesis from OBs [39].
Investigators have even gone so far as to profile which particular MMPs are elevated in diseased conditions. The following are some profiles: (i) rheumatoid arthritis – increase in MMP-1, -2, -3, -8, -9; (ii) inherited osteolysis – decrease in MMP-2; (iii) osteoporosis – increase in MMP-1, -2, -3; (iv) osteoarthritis – increase in MMP-1, -2, -9, -13; (v) ankylosing spondylitis – increase in MMP-3; and (vi) bone fracture healing effects – increase in MMP-1, -8, decrease in TIMP-1. In fact, MMPs are used in the clinical context to monitor these bone disorders over time, for example, MMP-9 as a marker of disease progression in multiple myeloma [40].
Hcy and oxidative stress
The cascade by which tHcy is known to increase ROS in cells such as MVECs [41] is also found to be present in OC cells that were differentiated from mouse bone marrow cells. These cascades include: cascade of p38 mitogen-activated protein kinase (MAPK), extracellular signal-regulated kinase (ERK), c-Jun N-terminal kinase (JNK), and NF-κB [42]. A study by Koh et al. found that tHcy can increase generation of intracellular ROS in OCs. The OC activity was measured by tartrate-resistant acid phosphatase (TRAP) activity. Moreover, the antioxidant, N-acetyl cysteine, blocked the negative changes in bone that tHcy incurred [42]. As tHcy is known to activate OC activity, it was examined whether tHcy also decreases OB activity [16]. The results were surprising because the authors found that tHcy also increases OB activity. However, the magnitude of activation was thought to be less than that of OCs, which suggested a disbalance between OB and OC activity with greater OC activity [16].
It is known that tHcy acts as a ligand on NMDA-R1, which is found on bone to regulate calcium intake [31]. Moreover, tHcy acts as a ligand on bone cells with PPAR-γ receptor [43]. There is some controversy on how activation of PPAR-γ might affect bone resorption. It was previously thought that tHcy binds and downregulates PPAR-γ and this may provide an oxidizing environment [43]. However, it is well known that mere activation of PPAR-γ results in resorption [43]. Perhaps not the activation, but the repeated activation and resultant downregulation of the receptor are the mechanism by which PPAR-γ acts to provide a detrimental oxidizing environment to the bone.
Intracellular calcium from NMDA-R1 can increase ROS. ROS can be extruded from the cell and MMPs are activated from pro-MMP to MMP. Activation of PPAR-γ may also play a part and this proposed mechanism in bone is illustrated in Figure 1B.
Nuclear transcription factors (NFs) are another source of ROS that are involved in the oxidative/reductive environment of the cell. For instance, tHcy is known to increase NF-κB, which creates an oxidized environment [44]. tHcy has been shown to act as a ligand on PPAR-γ to promote a reducing environment [45]. However, with increasing concentrations of tHcy, there is a reduction in the available PPAR-γ receptor for binding. Hence, its antioxidant effects are diminished under tHcy exposure [46]. Normally, the activation of the PPAR receptor increases superoxide dismutase (SOD) and catalase while decreasing NAD/NADPH oxidase; this reduces radicals and acts as a beneficial process [47]. This beneficial process is reduced by tHcy, thereby creating a detrimental scenario for the oxidation/ reduction balance.
A study by Yilmaz and Eren [48] analyzed BMD in relation to total antioxidant status, or balance of oxidation and reduction in patients. It was found that plasma tHcy, urine deoxypyridinoline and plasma total peroxide were all found to be higher in low BMD patients as compared to normal BMD individuals. This study concluded that slightly elevated tHcy may be a contributing factor for increasing total peroxide and reducing total antioxidant status in low BMD patients. Moreover, there was a weak and negative correlation found between total antioxidant status and BMD (p<0.035) [48].
In an earlier study, Herrmann et al. showed that increased tHcy concentration stimulated OC activity in vitro, suggesting a direct role of the tHcy molecule on the cells. OC activity was measured via detection of TRAP where concentrations as little as 10 or 20 μ/L stimulated TRAP activity. Cathepsin K activity was also measured, showing bone resorption at concentrations of 50 and 100 μ/L. Moreover, cysteine and glutathione significantly decreased TRAP and cathepsin-K activity. It is logical to consider how glutathione can decrease the presence of free radicals that are known to be produced by tHcy [49].
It was suggested that as tHcy is known to cause apoptosis via generation of ROS via the mitochondrial mechanism in cells (MVECs), this same process occurs in OB cells leading to osteoporosis. Human bone marrow stromal cells (hbMSCs) and the HS-5 cell line (human bone marrow stromal cell line) were used to examine the above mechanisms. It was found that tHcy induced apoptosis in these cell lines via the caspase mechanism and mitochondrial mechanism involving the release of cytochrome c. This provides evidence that tHcy may contribute to the development of osteoporosis by reducing bone formation [50].
Hcy and osteoporosis
Physical activity is one factor that is very important in the maintenance of the skeleton because mechanotransduction plays as important role in this process. In fact, stress imposed on the skeleton during life processes become signals to retain skeletal mass and even to build and deposit further matrix. This is a dynamic process that occurs throughout life [51]. The process described is in accordance with Wolff’s law that occurs in bone similar to other parts of the body and results in adaptation so that it can better accommodate the stress placed on it [51]. Of course, the risk of overtraining or imposing too much stress on the skeleton may frequently lead to maladaptive consequences such as osteoarthritis or stress fractures. Hence, it has been suggested that cyclical or intermittent stress provides the skeleton with adequate recovery time compared to continuous daily stress [51]. Extrapolated into the practical context, lifting significantly heavy weights every day or engaging in a long duration of exercise without adequate recovery time can be detrimental to the skeleton. Osteocytes can act as sensory cells, whereas OBs and OCs perform metabolic activities of remodeling the ECM [52].
An interesting article by McLean et al. found that physical activity or stimulation of the skeleton and muscles actually served to decrease levels of tHcy and this relationship was independent of intake of vitamin supplements, fruits and vegetables. It was concluded that although nutritional status is important, tHcy levels are also largely modulated by physical activity [53].
In a study of women aged between 70 and 85 years, it was found that the highest tertile of tHcy was associated with a greater hip BMD loss over 4 years (–2.8%) compared to middle (– 1.6%) and lowest tertiles (– 1.2%). However, higher levels of tHcy did not have an effect on prevalence or incidence of fracture. Hence, it was concluded that increased tHcy in elderly patients is associated with significant hip bone loss [54].
In another study, it was shown that fracture risk was higher in women with tHcy in the highest quartile, but not when adjusted for age [55]. The authors concluded that tHcy may be a marker of frailty that can be modulated by factors such as nutritional state, physical activity and renal impairment. They also argued that age is the main determinant of plasma tHcy, and that when fracture risk was adjusted for age tHcy was not found to be an independent risk factor [55]. However, a study by Claes et al. showed that HHcy leads to impaired fracture repair. Mice were fed on Hcy for 3 weeks, a closed femoral fracture was induced, and biomechanical parameters were observed after 4 weeks of healing. The following parameters were weakened under HHcy rats with fracture: (i) decreased bending stiffness of femora and (ii) smaller callus diameter with no difference in tissue composition. This leads to decreased bone quality and impaired fracture repair [56].
It is known that estrogen receptors are important for regulation of OB function. This is especially pertinent to postmenopausal women who have decreased levels of estrogen [57, 58], because at menopause there are hormonal changes that lead to a decrease in estrogen levels.
A study by Aaron et al. [58] explored methylation of estrogen receptor α and levels of tHcy. The in vivo and in vitro data indicated that tHcy can promote hypermethylation of promoter A region, thereby reducing estrogen receptor α mRNA transcription. This was proposed to be a possible mechanism for the pathogenesis of postmenopausal osteoporosis. Hence, tHcy may play an estrogen receptor-mediated role in deteriorating biomechanical properties of bone. Estrogen receptor transcription is downregulated in bone and all the beneficial osteogenic effects also become downregulated with high levels of tHcy.
In trying to develop some possible therapeutic options to high plasma tHcy levels that may cause detrimental effects of bone, strontium ranelate 2 g/day was used. This is a substance found to significantly decrease plasma tHcy levels in osteoporotic women and was proposed to be used as a means to decrease fracture risk by lowering tHcy levels [59]. Strontium ranelate was chosen from its known effect of decreasing bone resorption in addition to encouraging bone production [60, 61]. It is possible that reduction in tHcy levels is responsible for allowing the known effects of strontium ranelate to occur.
If Hcy is an important mechanism whereby altered bone biomechanical properties are thought to occur and hormone replacement therapy is known to improve bone quality, it was hypothesized that hormone replacement therapy (HRT) would improve bone quality via lowering of tHcy levels. In fact, the authors did find evidence that HRT lowered Hcy levels with the most pronounced effect occurring in those who had the highest levels of tHcy [62].
In a smaller study on 94 males and females who underwent hip arthroplasty for osteoarthritis, serum tHcy levels were measured along with vitamin levels (folate, B6 and B12) in order to find the relationship among these factors and osteoarthritis. However, this study could not find a significant relationship between circulating tHcy and morphological or biomechanical properties of bone as assessed by DEXA and histomorphometry [63].
High levels of tHcy seem to affect fracture risk and bone quality [59], but there have been no distinctive changes in bone density as observed in most studies. However, some studies show a trend towards decreased density without being clinically significant. In a very small case control study of 30 patients, there was no demonstration of a correlation between plasma tHcy and increased levels of osteoporosis. The bone of interest here was the lumbar spine as well as femoral neck [64]. In this study, the patients were examined based on densitometry criteria for osteoporosis in femoral neck or lumbar spine along with blood tests to check for serum tHcy [64]. However, this study is not conclusive, owing to the limited number of subjects and considering multiple factors for osteoporosis risk.
Herrmann et al. found that 3 months of HHcy condition caused increased bone fragility of the femoral neck by 18% in methionine-fed rats, and twice in Hcy-fed rats. This two-fold relationship was also seen for Bar/Tar at distal femur in methionine-fed rats versus Hcy-fed rats. Femoral neck and lumbar spine also showed decrease of biomechanical property but by even >2-fold in methionine- versus Hcy-fed rats [65].
Hcy and vasculature
One of the proposed mechanisms by which tHcy affects bone biomechanics is via a decrease in bone blood flow. Blood flow in bones is affected by several mechanisms such as sympathetic activity and vasoconstriction/relaxation, local effects of metabolites such as NO, as well as circulating hormones. Bone blood flow is important as bones are living tissues composed of cells that require nutrients for maintenance and support of life processes [66]. Fleming et al. found that bone arterioles are sensitive to circulating hormones and less so to endothelium-mediated cellular processes [66]. There is an association between atherosclerosis and arterioles whereby vessels are partially occluded by the intrusion of the inner layer of vessel into the main circuit of flow. This limits the ability to replenish the bone by decreasing bone blood flow and the stream of nutrients that go along with that [67]. One study demonstrated that when there is a different distribution of cardiovascular pressure along with flow changes, a difference in bone density was observed [68].
HRT has been shown to enhance NO production in postmenopausal women [69]. Estrogen is known to exert its beneficial effects due to the NO mechanism [69]. NO is known to modulate the activity of OBs and OCs, and an increase in NO provides a conducive environment for bone growth [70]. NO inhibitors are known to inhibit bone growth and are pro-resorptive [71]. For example, amino-guanidine (also known as L-NAME) can lead to a process of osteopenia and mice deficient in endothelial nitric oxide synthase (eNOS) do not develop bone properly. Several studies have even shown that the process of angiogenesis begins before osteogenesis in situations that involve bone repair. This emphasizes the crucial role that vasculature plays for the maintenance and support of bone.
Our laboratory is very interested in how bone flow would be affected by HHcy. In our experimentation with bone, we found that bone blood flow is decreased in HHcy conditions. Mice were fed Hcy (0.67 g/L) in drinking water for 8 weeks. The tibia blood flow index of the control group was significantly higher compared with the Hcy-treated group. Moreover, tibia bone density was unchanged in Hcy-treated groups [72]. Hence, we propose a mechanism by which superoxide anion created by HHcy may react with NO and reduce NO bioavailability [73]. An increase in oxidative stress that reduces NO bioavailability could also decrease bone blood flow and lead to osteoporosis [74]. The proposed mechanism is illustrated in Figure 2.
Figure 2. The decreased bone blood flow in CBS +/− mice with HHcy.

An increased expression of NOX-4, iNOS and MMP-9, as well as a decreased expression of Trx-1 and eNOS, may lead to decreased NO bioavailability and concomitant bone blood flow [6].
Bone flow is so important and it adjusts to meet the metabolic needs of the healing tissue [75]. We did not note a change in bone density in Hcy-treated groups of rats. In a follow-up experiment with CBS mice, mice lacking the cystathionine β-synthase (CBS) gene involved in tHcy clearance, we further confirmed a decrease in tibia blood flow. Moreover, there was an increase in NADPH oxidase-4 (NOX-4), inducible nitric oxide synthase (iNOS), MMP-9 protein, as well as MMP-9 activity in CBS mice with a decrease in thioredxion (Trx-1), eNOS protein levels, thereby decreasing NO bioavailability [6]. The detrimental protein expression profile and flow effects were improved with folic acid, which suggested that folic acid may play a role in therapy. Moreover, this experiment demonstrated a decrease in bone density.
Effects of vitamin B12 and folate on biomechanical properties
Some individuals have tried to discern whether the increase in bone biomechanical properties is due to the deficiencies in the vitamins themselves as they alter bone metabolism or whether tHcy is simply a bystander in this process. No significant change was noted in BMD of postmenopausal women deficient in vitamin B12 or with elevated tHcy levels. However, there was a decrease in BMD with folate indicating its important role in preserving BMD. Folate is thought to play a direct role in bone metabolism via the following mechanisms: protein and nucleic acid methylation and nitric oxide synthesis [76, 77].
Risk of vitamin deficiencies is more possible in the elderly population and developing countries whereby vitamin B12 deficiency can lead to increase tHcy and methylmalonic acid (MMA). This also stresses the importance of an adequate diet to prevent chronic problems such as osteoporosis and its complications. It was shown that decreased vitamin B12 did not have an effect on osteoclastogenesis in vitro using human mesenchymal stem cells; however, tHcy and MMA resulted in dose-dependent osteoclastogenesis. One study looked at whether vitamin B12 deficiency itself incurs biomechanical problems with bone or if subsequent increases in tHcy and MMA were responsible [78].
Another study found that tHcy is a predictor of BMD and HHcy is associated with lower BMD in Slovak women on a vegetarian diet, owing to low levels of vitamin B12 and folate which lead to higher tHcy levels [79]. The study used controls for variables such as calcium, age, weight, etc. This study was especially pertinent considering that these people had been vegetarians for quite a long while, and it demonstrated a prolonged effect of diet on bone biomechanics.
Herrmann et al. analyzed the effects of a vitamin B12 and folate-deficient diet on bone biomechanical properties as well as markers of bone turnover in rats. The study found that rats with a vitamin B12 and folate-deficient diet developed HHcy. The biomechanical properties of bone did not change which included: (i) maximal force of axial compression (Fmax), (ii) bone area/total area, (iii) osteocalcin, and (iv) collagen I C-terminal cross-laps. Although the serum levels of tHcy had changed, the bone concentrations of tHcy, S-adenosylhomocysteine (SAH) and S-adeno-sylmethionine (SAM) remained the same. Unchanged tHcy metabolism within bone tissue was concluded to be the reason for the unchanged effects of bone quality in this experiment, although over a 12-week period [80].
Interestingly, vitamin D metabolism, which is critical to bone metabolism and calcium absorption, was also found to be modulated by tHcy. A study by Kriebitzsch et al. showed the tHcy metabolism was affected by the presence of vitamin D. It was shown that vitamin D upregulates the CBS enzyme, allowing clearance of tHcy. Hence, vitamin D deficiency may act via the tHcy mechanism in addition to other roles it plays with OBs and OCs [81].
To determine some therapeutic approaches for the negative effect caused by higher tHcy levels on bone biomechanics, current clinical trials are being performed in patients aged 65 years or older with elevated tHcy levels (>12 μM/L). The study is known with the acronym B-PROOF. Patients are given 500 μg of B12, 400μg of folate, and 15 μg (600 IU) of vitamin D. It will be examined how vitamin supplementation in humans will influence osteoporotic fractures and the results are awaited in 2013 [8].
A study by Dhonukshe-Rutten et al. has linked higher levels of tHcy >15 and vitamin B12 levels to bone turn-over, broadband ultrasound attenuation, and fractures in healthy elderly people. It was found that women with vitamin B12 levels <200 pM and tHcy >15 μM had lower broadband ultrasound attenuation, higher serum osteocalcin concentrations than those with normal levels of tHcy and vitamin B12. There was a 2.8-fold increased relative risk of fracture for women having increased tHcy levels and decreased vitamin B12. In men, however, there was no correlation between different tHcy levels and vitamin B12 categories in the means of broadband ultrasound attenuation and osteocalcin [82].
Morris et al. [83] discovered that BMD decreased and osteoporosis increased significantly with increasing serum MMA (p<0.01). Moreover, serum vitamin B12 was also related to BMD in dose-dependent manner up to 200 pmol/L and subjects with tHcy ≥20 μmol/L had significantly lower BMD than those subjects with tHcy <20 μM/L [83]. Another study found that HHcy that resulted from folate deficiency was independently associated with low BMD, thereby contributing to the pathogenicity of osteoporosis in postmenopausal Iranian women [84]. Interestingly, this association did not result from methylenetetrahydrofolate reductase polymorphism. This kind of result was also found for pyridoxine deficiency, whereby there was a significant increase in osteoporotic fractures with pyridoxine deficiency [85].
It was found that B-vitamin supplementation in osteoporotic subjects increased BMD at lumbar spine (t-score, −2.7 to −1.7) and decreased around 50% osteoclast activity [86]. This was not true for patients that were not HHcy [86]. There are some studies that looked at whether a particular vitamin actually altered bone mechanics specifically. For instance, cell culture experiments confirmed a vitamin B12 dependency of human OB progenitor cells [87]. The data showed an increased incidence of hip fracture in smokers who had vitamin B6 deficiencies, and another study showed patients with hip fractures with reduced intake of vitamin B6 and folate [88, 89].
In an older study concerning the mechanistic roles of vitamin cofactors, it was discovered that there was a reduced alkaline phosphatase activity and osteocalcin level in vitamin B12-deficient patients, but that this was reversible after supplementing with vitamin B12 [90].
In Crohn’s disease, patients who have a decreased ability to absorb vitamin B12 have higher tHcy levels and a higher risk for fracture [91]. McLean et al. have found an increased risk of hip fracture in men (4-fold) and also in women, but with a more moderate (1.9×) risk [92].
A very large study was conducted on 93,676 patients from whom 400 incident cases of hip fracture were selected along with 400 controls (matched with age, ethnicity and blood draw date). The incident of hip fractures was measured among these individuals and it was found that the risk of hip fracture increased 1.38-fold [95% confidence interval (CI): 1.14, 1.66] for each SD increase in serum Hcy level. However, the association between tHcy and hip fracture was not affected by adjustment for dietary folate, B6 or B12 intake [93].
Conclusion
There is conclusive evidence that tHcy does affect biomechanical properties of bone. The results of studies are mixed on HHcy and its effect on bone density. Moreover, vitamin supplementation that ameliorates HHcy conditions has been shown to be helpful in several studies. Further studies are required on specific mechanisms by which tHcy acts to affect bone although it appears to be multifactorial. We have investigated how alteration in bone blood flow, as a consequence of NO bioavailability, alters bone. Moreover, we are interested in a mitochondrial mechanism of MMP activation and alteration of bone matrix. We also welcome further studies in the proposed area.
Acknowledgments
This work was supported by National Institutes of Health grants: HL-107640 to N.T. and HL-71010 to S.C.T.
Biographies

Thomas P. Vacek received his MD degree from the University of Louisville following his MS in Physiology and Biophysics at he same university and a BS in General Biology from Cornell University, New York. Presently he is a PGY1 Resident in Internal Medicine at the University of Toledo Medical Center. His research area of interest is genetic, molecular, cellular and physiological mechanisms that contribute to hypertension, diabetes, heart failure and osteoporosis.

Anuradha Kalani received her PhD in Biochemistry from the Central Drug Research Institute, Lucknow, India in 2011. She worked as a Research Associate at the Sanjay Gandhi Post Graduate Institute of Medical Sciences, Lucknow, India on the early biomarker search for diabetic nephropathy using exosomes. Currently, she is working as a Research Associate at the Physiology and Biophysics Department, University of Louisville. She is working on stroke pathology, epigenetic mechanisms, cerebrovascular and bone remodeling. Her research interests include the molecular, and proteomic characterization of biomolecules, cerebrovascular and bone remodeling.

Michael J. Voor received his PhD in Biomedical Engineering in 1992 following his BS in Mechanical Engineering in 1988 and his Master’s Degree in Biomedical Engineering in 1990 all from Tulane University. He was appointed Assistant Professor of Orthopaedic Surgery at the University of Louisville in 1992 when he founded the Orthopaedic Bioengineering Laboratory on the Health Sciences Campus. He is now Associate Professor and Director of Research for the Department. He is also on the associate faculty of Mechanical Engineering, Anatomical Sciences and Neurobiology, Neurological Surgery (Kentucky Spinal Cord Injury Research Center), and Bioengineering. His research interests include the design, testing, and biomechanical behavior of orthopedic implants for fracture fixation, total joint arthroplasty, and spine fusion; micro-computed tomography; bone grafting and bone graft substitute materials; as well as basic bone mechanics studies centered on cancellous bone remodeling response to disease and treatment for osteoporosis.

Suresh C.Tyagi, M.Phil., PhD, FAHA, FAPS, earned his PhD in Biophysics at the University of Aligarh, India. He completed postgraduate work in Bioinorganic Chemistry at University College, Cork, Ireland and the State University of New York at Stony Brook, in Pharmacology and Pathology. He was an Associate Professor at the University of Mississippi Medical Center before joining the University of Louisville in 2003 as a Professor. His research is supported by the National Institutes of Health. He is the recipient of numerous awards and honors including a medal of merit from the International Society for Heart Research. His research area of interest is genetic, molecular and cellular mechanisms that contribute to hypertension diabetes and heart failure. In particular, he studies homocysteine, and amino acid by-product that plays a critical role in artery damage and may be responsible for many heart attacks in individuals who do not display traditional cardiac risk factors. His work may lead to new treatments for these conditions, which afflict millions.
Tyagi has published more than 156 peer-reviewed articles, invited papers, and reviews. He has served on editorial boards and committees for the American Journal of Physiology, Clinical & Experimental Hypertension, Molecular Cellular Biochemistry and the Journal of Molecular and Cellular Physiology. He has also been a member of multiple study sections with the American Heart Association and the National Institutes of Health. Presently he is the Stodghill Endowed Professorship in Biomedical Research.

Neetu Tyagi received her PhD in Zoology from the Central Drug Research Institute, Lucknow, India in 2002 following her MS in Biotechnology. She was appointed as Senior Research Associate at the Physiology and Biophysics Department, University of Louisville, in 2004. She is now Assistant Professor. Her research focuses on vascular biology related to disease complications associated with hyperhomocysteinemia. Her research interests include the molecular biology; physiology; mitochondrial pathology; bone remodeling response to disease and treatment for osteoporosis.
Footnotes
Conflict of interest statement
Authors’ conflict of interest disclosure: The authors stated that there are no conflicts of interest regarding the publication of this article.
Research funding: None declared.
Employment or leadership: None declared.
Honorarium: None declared.
References
- 1.Homocysteine Studies Collaboration. Homocysteine and risk of ischemic heart disease and stroke–a meta-analysis. J Am Med Assoc. 2002;288:2015–22. doi: 10.1001/jama.288.16.2015. [DOI] [PubMed] [Google Scholar]
- 2.Sen U, Basu P, Abe OA, Givvimani S, Tyagi N, Metreveli N, et al. Hydrogen sulfide ameliorates hyperhomocysteinemia-associated chronic renal failure. Am J Physiol Renal Physiol. 2009;297:410–9. doi: 10.1152/ajprenal.00145.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lominadze D, Roberts AM, Tyagi N, Moshal KS, Tyagi SC. Homocysteine causes cerebrovascular leakage in mice. Am J Physiol Heart Cir Physiol. 2006;290:1206–13. doi: 10.1152/ajpheart.00376.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lips P. Epidemiology and predictors of fractures associated with osteoporosis. Am J Med. 1997;103:3S–8S. doi: 10.1016/s0002-9343(97)90021-8. [DOI] [PubMed] [Google Scholar]
- 5.Riggs BL, Melton LJ., 3rd The worldwide problem of osteoporosis: insights afforded by epidemiology. Bone. 1995;17:505–11. doi: 10.1016/8756-3282(95)00258-4. [DOI] [PubMed] [Google Scholar]
- 6.Tyagi N, Kandel M, Munjal C, Qipshidze N, Vacek JC, Pushpakumar SB, et al. Homocysteine mediated decrease in bone blood flow and remodeling: role of folic acid. J Orthop Res. 2011;29:1511–6. doi: 10.1002/jor.21415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Herrmann M, Widmann T, Herrmann W. Homocysteine–a newly recognised risk factor for osteoporosis. Clin Chem Lab Med. 2005;43:1111–7. doi: 10.1515/CCLM.2005.194. [DOI] [PubMed] [Google Scholar]
- 8.van Wijngaarden JP, Dhonukshe-Rutten RA, van Schoor NM, van der Velde N, Swart KM, Enneman AW, et al. Rationale and design of the B-PROOF study, a randomized controlled trial on the effect of supplemental intake of vitamin B12 and folic acid on fracture incidence. BMC Geriatr. 2011;11:80. doi: 10.1186/1471-2318-11-80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ozdem S, Samanci S, Tasatargil A, Yildiz A, Sadan G, Donmez L, et al. Experimental hyperhomocysteinemia disturbs bone metabolism in rats. Scand J Clin Lab Invest. 2007;67:748–56. doi: 10.1080/00365510701342088. [DOI] [PubMed] [Google Scholar]
- 10.Derkx P, Nigg AL, Bosman FT, Birkenhäger-Frenkel DH, Houtsmuller AB, Pols HA. Immunolocalization and quantification of noncollagenous bone matrix proteins in methylmethacrylate-embedded adult human bone in combination with histomorphometry. Bone. 1998;22:367–73. doi: 10.1016/s8756-3282(97)00299-8. [DOI] [PubMed] [Google Scholar]
- 11.Herrmann M, Kraenzlin M, Pape G, Sand-Hill M, Herrmann W. Relation between homocysteine and biochemical bone turnover markers and bone mineral density in peri- and postmenopausal women. Clin Chem Lab Med. 2005;43:1118–23. doi: 10.1515/CCLM.2005.195. [DOI] [PubMed] [Google Scholar]
- 12.Blouin S, Thaler HW, Korninger C, Schmid R, Hofstaetter JG, Zoehrer R, et al. Bone matrix quality and plasma homocysteine levels. Bone. 2009;44:959–64. doi: 10.1016/j.bone.2008.12.023. [DOI] [PubMed] [Google Scholar]
- 13.Wittenburg G, Volkel C, Mai R, Lauer G. Immunohistochemical comparison of differentiation markers on paraffin and plastic embedded human bone samples. J Physiol Pharmacol. 2009;60:43–9. [PubMed] [Google Scholar]
- 14.Yang R, Davies CM, Archer CW, Richards RG. Immunohistochemistry of matrix markers in Technovit 9100 new-embedded undecalcified bone sections. Eur Cell Mater. 2003;6:57–61. doi: 10.22203/ecm.v006a06. [DOI] [PubMed] [Google Scholar]
- 15.Kramer I, Halleux C, Keller H, Pegurri M, Gooi JH, Weber PB, et al. Osteocyte Wnt/β-catenin signaling is required for normal bone homeostasis. Mol Cell Biol. 2010;30:3071–5. doi: 10.1128/MCB.01428-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Herrmann M, Umanskaya N, Wildemann B, Colaianni G, Widmann T, Zallone A, et al. Stimulation of osteoblast activity by homocysteine. J Cell Mol Med. 2008;12:1205–10. doi: 10.1111/j.1582-4934.2008.00104.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Herrmann M, Tami A, Wildemann B, Wolny M, Wagner A, Schorr H, et al. Hyperhomocysteinemia induces a tissue specific accumulation of homocysteine in bone by collagen binding and adversely affects bone. Bone. 2009;44:467–75. doi: 10.1016/j.bone.2008.10.051. [DOI] [PubMed] [Google Scholar]
- 18.Liu G, Nellaiappan K, Kagan HM. Irreversible inhibition of lysyl oxidase by homocysteine thiolactone and its selenium and oxygen analogues. Implications for homocystinuria. J Biol Chem. 1997;272:32370–7. doi: 10.1074/jbc.272.51.32370. [DOI] [PubMed] [Google Scholar]
- 19.Thaler R, Agsten M, Spitzer S, Paschalis EP, Karlic H, Klaushofer K, et al. Homocysteine suppresses the expression of the collagen cross-linker lysyl oxidase involving IL-6, Fli1, and epigenetic DNA methylation. J Biol Chem. 2011;286:5578–88. doi: 10.1074/jbc.M110.166181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Du C, Fang M, Li Y, Li L, Wang X. Smac, a mitochondrial protein that promotes cytochrome c-dependent caspase activation by eliminating IAP inhibition. Cell. 2000;102:33–42. doi: 10.1016/s0092-8674(00)00008-8. [DOI] [PubMed] [Google Scholar]
- 21.Kroemer G, Reed JC. Mitochondrial control of cell death. Nat Med. 2000;6:513–9. doi: 10.1038/74994. [DOI] [PubMed] [Google Scholar]
- 22.Li LY, Luo X, Wang X. Endonuclease G is an apoptotic DNase when released from mitochondria. Nature. 2001;412:95–9. doi: 10.1038/35083620. [DOI] [PubMed] [Google Scholar]
- 23.Liu X, Kim CN, Yang J, Jemmerson R, Wang X. Induction of apoptotic program in cell-free extracts: requirement for dATP and cytochrome c. Cell. 1996;86:147–57. doi: 10.1016/s0092-8674(00)80085-9. [DOI] [PubMed] [Google Scholar]
- 24.Spiess C, Beil A, Ehrmann M. A temperature-dependent switch from chaperone to protease in a widely conserved heat shock protein. Cell. 1999;97:339–47. doi: 10.1016/s0092-8674(00)80743-6. [DOI] [PubMed] [Google Scholar]
- 25.Susin SA, Lorenzo HK, Zamzami N, Marzo I, Snow BE, Brothers GM, et al. Molecular characterization of mitochondrial apoptosis-inducing factor. Nature. 1999;397:441–6. doi: 10.1038/17135. [DOI] [PubMed] [Google Scholar]
- 26.van Loo G, Schotte P, van Gurp M, Demol H, Hoorelbeke B, Gevaert K, et al. Endonuclease G: a mitochondrial protein released in apoptosis and involved in caspase-independent DNA degradation. Cell Death Differ. 2001;8:1136–42. doi: 10.1038/sj.cdd.4400944. [DOI] [PubMed] [Google Scholar]
- 27.Verhagen AM, Ekert PG, Pakusch M, Silke J, Connolly LM, Reid GE, et al. Identification of DIABLO, a mammalian protein that promotes apoptosis by binding to and antagonizing IAP proteins. Cell. 2000;102:43–53. doi: 10.1016/s0092-8674(00)00009-x. [DOI] [PubMed] [Google Scholar]
- 28.Fu X, Kassim SY, Parks WC, Heinecke JW. Hypochlorous acid oxygenates the cysteine switch domain of pro-matrilysin (MMP-7). A mechanism for matrix metalloproteinase activation and atherosclerotic plaque rupture by myeloperoxidase. J Biol Chem. 2001;276:41279–87. doi: 10.1074/jbc.M106958200. [DOI] [PubMed] [Google Scholar]
- 29.Moshal KS, Singh M, Sen U, Rosenberger DS, Henderson B, Tyagi N, et al. Homocysteine-mediated activation and mitochondrial translocation of calpain regulates MMP-9 in MVEC. Am J Physiol Heart Circ Physiol. 2006;291:2825–35. doi: 10.1152/ajpheart.00377.2006. [DOI] [PubMed] [Google Scholar]
- 30.Moshal KS, Metreveli N, Frank I, Tyagi SC. Mitochondrial MMP activation, dysfunction and arrhythmogenesis in hyperhomocysteinemia. Curr Vasc Pharmacol. 2008;6:84–92. doi: 10.2174/157016108783955301. [DOI] [PubMed] [Google Scholar]
- 31.Itzstein C, Cheynel H, Burt-Pichat B, Merle B, Espinosa L, Delmas PD, et al. Molecular identification of NMDA glutamate receptors expressed in bone cells. J Cell Biochem. 2001;82:134–44. doi: 10.1002/jcb.1114. [DOI] [PubMed] [Google Scholar]
- 32.Kawahara I, Koide M, Tadokoro O, Udagawa N, Nakamura H, Takahashi N, et al. The relationship between calcium accumulation in osteoclast mitochondrial granules and bone resorption. Bone. 2009;45:980–6. doi: 10.1016/j.bone.2009.07.010. [DOI] [PubMed] [Google Scholar]
- 33.Fajardo M, Liu CJ, Ilalov K, Egol KA. Matrix metalloproteinases that associate with and cleave bone morphogenetic protein-2 in vitro are elevated in hypertrophic fracture nonunion tissue. J Orthop Trauma. 2010;24:557–63. doi: 10.1097/BOT.0b013e3181ed294c. [DOI] [PubMed] [Google Scholar]
- 34.Galliera E, Banfi G, Corsi MM. Human bone disorders: pathological role and diagnostic potential of matrix metalloproteinases. Int J Biochem Cell Biol. 2010;42:1590–3. doi: 10.1016/j.biocel.2010.06.015. [DOI] [PubMed] [Google Scholar]
- 35.Hill PA, Murphy G, Docherty AJ, Hembry RM, Millican TA, Reynolds JJ, et al. The effects of selective inhibitors of matrix metalloproteinases (MMPs) on bone resorption and the identification of MMPs and TIMP-1 in isolated osteoclasts. J Cell Sci. 1994;107:3055–64. doi: 10.1242/jcs.107.11.3055. [DOI] [PubMed] [Google Scholar]
- 36.Murphy G, Reynolds JJ. Progress towards understanding the resorption of connective tissues. Bioessays. 1985;2:55–60. [Google Scholar]
- 37.Pasternak B, Aspenberg P. Metalloproteinases and their inhibitors–diagnostic and therapeutic opportunities in orthopedics. Acta Orthop. 2009;80:693–703. doi: 10.3109/17453670903448257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nathalie O, Danielle B, Dominique S, Zena W. How proteases regulate bone morphogenesis. An N Y Acad Sci. 2003;995:109–16. doi: 10.1111/j.1749-6632.2003.tb03214.x. [DOI] [PubMed] [Google Scholar]
- 39.Sato H, Takino T, Okada Y, Cao J, Shinagawa A, Yamamoto E, et al. A matrix metalloproteinase expressed on the surface of invasive tumour cells. Nature. 1994;370:61–5. doi: 10.1038/370061a0. [DOI] [PubMed] [Google Scholar]
- 40.Alexandrakis MG, Sfiridaki A, Miyakis S, Pappa C, Kandidaki E, Alegakis A, et al. Relationship between serum levels of vascular endothelial growth factor, hepatocyte growth factor and matrix metalloproteinase-9 with biochemical markers of bone disease in multiple myeloma. Clin Chim Acta. 2007;379:31–5. doi: 10.1016/j.cca.2006.11.024. [DOI] [PubMed] [Google Scholar]
- 41.Tyagi N, Sedoris KC, Steed M, Ovechkin AV, Moshal KS, Tyagi SC. Mechanisms of homocysteine-induced oxidative stress. Am J Physiol Heart Circ Physiol. 2005;289:2649–56. doi: 10.1152/ajpheart.00548.2005. [DOI] [PubMed] [Google Scholar]
- 42.Koh JM, Lee YS, Kim YS, Kim DJ, Kim HH, Park JY, et al. Homocysteine enhances bone resorption by stimulation of osteoclast formation and activity through increased intracellular ROS generation. J Bone Miner Res. 2006;21:1003–11. doi: 10.1359/jbmr.060406. [DOI] [PubMed] [Google Scholar]
- 43.Stunes AK, Westbroek I, Gustafsson BI, Fossmark R, Waarsing JH, Eriksen EF, et al. The peroxisome proliferator-activated receptor (PPAR) α agonist fenofibrate maintains bone mass, while the PPARγ agonist pioglitazone exaggerates bone loss, in ovariectomized rats. BMC Endocr Disord. 2011;11:11. doi: 10.1186/1472-6823-11-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang L, Jin M, Hu XS, Zhu JH. Homocysteine stimulates nuclear factor κB activity and interleukin-6 expression in rat vascular smooth muscle cells. Cell Biol Int. 2006;30:592–7. doi: 10.1016/j.cellbi.2006.03.007. [DOI] [PubMed] [Google Scholar]
- 45.Brude IR, Finstad HS, Seljeflot I, Drevon CA, Solvoll K, Sandstad B, et al. Plasma homocysteine concentration related to diet, endothelial function and mononuclear cell gene expression among male hyperlipidaemic smokers. Eur J Clin Invest. 1999;29:100–8. doi: 10.1046/j.1365-2362.1999.00419.x. [DOI] [PubMed] [Google Scholar]
- 46.Inoue I, Noji S, Awata T, Takahashi K, Nakajima T, Sonoda M, et al. Bezafibrate has an antioxidant effect: peroxisome proliferator-activated receptor α is associated with Cu 2+, Zn2+ superoxide dismutase in the liver. Life Sci. 1998;63:135–144. doi: 10.1016/s0024-3205(98)00249-5. [DOI] [PubMed] [Google Scholar]
- 47.Poynter ME, Daynes RA. Peroxisome proliferator-activated receptor α activation modulates cellular redox status, represses nuclear factor κB signaling, and reduces inflammatory cytokine production in aging. J Biol Chem. 1998;273:32833–41. doi: 10.1074/jbc.273.49.32833. [DOI] [PubMed] [Google Scholar]
- 48.Yilmaz N, Eren E. Homocysteine oxidative stress and relation to bone mineral density in post-menopausal osteoporosis. Aging Clin Exp Res. 2009;21:353–7. doi: 10.1007/BF03324927. [DOI] [PubMed] [Google Scholar]
- 49.Herrmann M, Widmann T, Colaianni G, Colucci S, Zallone A, Herrmann W. Increased osteoclast activity in the presence of increased homocysteine concentrations. Clin Chem. 2005;51:2348–53. doi: 10.1373/clinchem.2005.053363. [DOI] [PubMed] [Google Scholar]
- 50.Kim DJ, Koh JM, Lee O, Kim NJ, Lee YS, Kim YS, et al. Homocysteine enhances apoptosis in human bone marrow stromal cells. Bone. 2006;39:582–90. doi: 10.1016/j.bone.2006.03.004. [DOI] [PubMed] [Google Scholar]
- 51.Huang C, Ogawa R. Mechanotransduction in bone repair and regeneration. FASEB J. 2010;24:3625–32. doi: 10.1096/fj.10-157370. [DOI] [PubMed] [Google Scholar]
- 52.Mi LY, Basu M, Fritton SP, Cowin SC. Analysis of avian bone response to mechanical loading. Part two: development of a computational connected cellular network to study bone intercellular communication. Biomech Model Mechanobiol. 2005;4:132–6. doi: 10.1007/s10237-004-0066-3. [DOI] [PubMed] [Google Scholar]
- 53.McLean RR, Jacques PF, Selhub J, Tucker KL, Samelson EJ, Broe KE, et al. Homocysteine as a predictive factor for hip fracture in older persons. N Engl J Med. 2004;351:2042–9. doi: 10.1056/NEJMoa032739. [DOI] [PubMed] [Google Scholar]
- 54.Zhu K, Beilby J, Dick IM, Devine A, Soós M, Prince RL. The effects of homocysteine and MTHFR genotype on hip bone loss and fracture risk in elderly women. Osteoporos Int. 2009;20:1183–91. doi: 10.1007/s00198-008-0804-y. [DOI] [PubMed] [Google Scholar]
- 55.Périer MA, Gineyts E, Munoz F, Sornay-Rendu E, Delmas PD. Homocysteine and fracture risk in postmenopausal women: the OFELY study. Osteoporos Int. 2007;18:1329–36. doi: 10.1007/s00198-007-0393-1. [DOI] [PubMed] [Google Scholar]
- 56.Claes L, Schmalenbach J, Herrmann M, Olkü I, Garcia P, Histing T, et al. Hyperhomocysteinemia is associated with impaired fracture healing in mice. Calcif Tissue Int. 2009;85:17–21. doi: 10.1007/s00223-009-9262-6. [DOI] [PubMed] [Google Scholar]
- 57.Levin ER. Plasma membrane estrogen receptors. Trends Endocrinol Metab. 2009;20:477–82. doi: 10.1016/j.tem.2009.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Aaron W, Alexander A, Brugmann SA, Xu Y, Carre AL, Leucht P, et al. Estrogen/estrogen receptor α signaling in mouse postero-frontal cranial suture fusion. PLoS One. 2009;4:e7120. doi: 10.1371/journal.pone.0007120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bayhan I, Uygur D, Ugurlu N, Ozaksit G. Strontium ranelate decreases plasma homocysteine levels in postmenopausal osteoporotic women. Rheumatol Int. 2009;29:263–6. doi: 10.1007/s00296-008-0693-6. [DOI] [PubMed] [Google Scholar]
- 60.Marie PJ, Ammann P, Boivin G, Rey C. Mechanisms of action and therapeutic potential of strontium in bone. Calcif Tissue Int. 2001;69:121–9. doi: 10.1007/s002230010055. [DOI] [PubMed] [Google Scholar]
- 61.Buehler J, Chappuis P, Saffar JL, Tsouderos Y, Vignery A. Strontium ranelate inhibits bone resorption while maintaining bone formation in alveolar bone in monkeys (Macaca fascicularis) Bone. 2001;29:176–9. doi: 10.1016/s8756-3282(01)00484-7. [DOI] [PubMed] [Google Scholar]
- 62.Mijatovic V, van der Mooren MJ. Homocysteine in postmen-opausal women and the importance of hormone replacement therapy. Clin Chem Lab Med. 2001;39:764–7. doi: 10.1515/CCLM.2001.127. [DOI] [PubMed] [Google Scholar]
- 63.Holstein JH, Herrmann M, Splett C, Herrmann W, Garcia P, Histing T, et al. Hyperhomocysteinemia is not associated with reduced bone quality in humans with hip osteoarthritis. Clin Chem Lab Med. 2010;48:821–7. doi: 10.1515/CCLM.2010.155. [DOI] [PubMed] [Google Scholar]
- 64.Mobasseri M, Aliasgharzadeh A, Ebrahimzadeh Z, Najafipoor F, Niafar M, Aghamohammadzadeh N, et al. Hyperhomocysteinemia and bone mineral density: a case-control study. Pak J Med Sci. 2009;25:91–6. [Google Scholar]
- 65.Herrmann M, Wildemann B, Claes L, Klohs S, Ohnmacht M, Taban-Shomal O, et al. Experimental hyperhomocysteinemia reduces bone quality in rats. Clin Chem. 2007;53:1455–61. doi: 10.1373/clinchem.2007.086272. [DOI] [PubMed] [Google Scholar]
- 66.Fleming JT, Barati MT, Beck DJ, Dodds JC, Malkani AL, Parameswaran D, et al. Bone blood flow and vascular reactivity. Cells Tissues Organs. 2001;169:279–84. doi: 10.1159/000047892. [DOI] [PubMed] [Google Scholar]
- 67.Ness J, Aronow WS. Comparison of prevalence of atherosclerotic vascular disease in postmenopausal women with osteoporosis or osteopenia versus without osteoporosis or osteopenia. Am J Cardiol. 2006;97:1427–8. doi: 10.1016/j.amjcard.2005.12.033. [DOI] [PubMed] [Google Scholar]
- 68.McCarthy ID. Fluid shifts due to microgravity and their effects on bone: a review of current knowledge. Ann Biomed Eng. 2005;33:95–103. doi: 10.1007/s10439-005-8967-6. [DOI] [PubMed] [Google Scholar]
- 69.Wimalawansa SJ. Nitric oxide and bone. Ann NY Acad Sci. 2010;1192:391–403. doi: 10.1111/j.1749-6632.2009.05230.x. [DOI] [PubMed] [Google Scholar]
- 70.Studer RK, Decker K, Melhem S, Georgescu H. Nitric oxide inhibition of IGF-1 stimulated proteoglycan synthesis: role of cGMP. J Orthop Res. 2003;21:914–21. doi: 10.1016/S0736-0266(03)00029-9. [DOI] [PubMed] [Google Scholar]
- 71.Sunyer T, Rothe L, Kirsch D, Jiang X, Anderson F, Osdoby P, et al. Ca 2+ or phorbol ester but not inflammatory stimuli elevate inducible nitric oxide synthase messenger ribonucleic acid and nitric oxide (NO) release in avian osteoclasts: autocrine NO mediates Ca 2+-inhibited bone resorption. Endocrinology. 1997;138:2148–62. doi: 10.1210/endo.138.5.5144. [DOI] [PubMed] [Google Scholar]
- 72.Tyagi N, Vacek TP, Fleming JT, Vacek JC, Tyagi SC. Hyperhomocysteinemia decreases bone blood flow. Vasc Health Risk Manag. 2011;7:31–5. doi: 10.2147/VHRM.S15844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Banfi G, Iorio EL, Corsi MM. Oxidative stress, free radicals and bone remodeling. Clin Chem Lab Med. 2008;46:1550–5. doi: 10.1515/CCLM.2008.302. [DOI] [PubMed] [Google Scholar]
- 74.Sánchez-Rodríguez MA, Ruiz-Ramos M, Correa-Muñoz E, Mendoza-Núñez VM. Oxidative stress as a risk factor for osteoporosis in elderly Mexicans as characterized by antioxidant enzymes. BMC Musculoskel Disord. 2007;19:124. doi: 10.1186/1471-2474-8-124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Fleming JT, Barati MT, Beck DJ, Dodds JC, Malkani AL, Parameswaran D, et al. Bone blood flow and vascular reactivity. Cells Tissues Organs. 2001;169:279–84. doi: 10.1159/000047892. [DOI] [PubMed] [Google Scholar]
- 76.Lucock M. Folic acid: nutritional biochemistry, molecular biology, and role in disease processes. Mol Genet Metab. 2000;71:121–8. doi: 10.1006/mgme.2000.3027. [DOI] [PubMed] [Google Scholar]
- 77.Das UN. Folic acid says NO to vascular diseases. Nutrition. 2003;19:686–92. doi: 10.1016/s0899-9007(02)01044-4. [DOI] [PubMed] [Google Scholar]
- 78.Vaes BL, Lute C, Blom HJ, Bravenboer N, de Vries TJ, Everts V, et al. Vitamin B(12) deficiency stimulates osteoclastogenesis via increased homocysteine and methylmalonic acid. Calc Tissue Int. 2009;84:413–22. doi: 10.1007/s00223-009-9244-8. [DOI] [PubMed] [Google Scholar]
- 79.Krivosíková Z, Krajcovicová-Kudlácková M, Spustová V, Stefíková K, Valachovicová M, Blazícek P, et al. The association between high plasma homocysteine levels and lower bone mineral density in Slovak women: the impact of vegetarian diet. Eur J Nutr. 2010;49:147–53. doi: 10.1007/s00394-009-0059-1. [DOI] [PubMed] [Google Scholar]
- 80.Herrmann M, Wildemann B, Wagner A, Wolny M, Schorr H, Taban-Shomal O, et al. Experimental folate and vitamin B12 deficiency does not alter bone quality in rats. J Bone Miner Res. 2009;24:589–96. doi: 10.1359/jbmr.081211. [DOI] [PubMed] [Google Scholar]
- 81.Kriebitzsch C, Verlinden L, Eelen G, van Schoor NM, Swart K, Lips P, et al. 1,25-Dihydroxyvitamin D3 influences cellular homocysteine levels in murine preosteoblastic MC3T3-E1 cells by direct regulation of cystathionine β-synthase. J Bone Miner Res. 2011;26:2991–3000. doi: 10.1002/jbmr.493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Dhonukshe-Rutten RA, Pluijm SM, de Groot LC, Lips P, Smit JH, van Staveren WA, et al. Homocysteine and vitamin B12 status relate to bone turnover markers, broadband ultrasound attenuation, and fractures in healthy elderly people. J Bone Miner Res. 2005;20:921–9. doi: 10.1359/JBMR.050202. [DOI] [PubMed] [Google Scholar]
- 83.Morris MS, Jacques PF, Selhub J. Relation between homocysteine and B-vitamin status indicators and bone mineral density in older Americans. Bone. 2005;37:234–42. doi: 10.1016/j.bone.2005.04.017. [DOI] [PubMed] [Google Scholar]
- 84.Golbahar J, Hamidi A, Aminzadeh MA, Omrani GR. Association of plasma folate, plasma total homocysteine, but not methyl-enetetrahydrofolate reductase C667T polymorphism, with bone mineral density in postmenopausal Iranian women: a cross-sectional study. Bone. 2004;35:760–5. doi: 10.1016/j.bone.2004.04.018. [DOI] [PubMed] [Google Scholar]
- 85.Yazdanpanah N, Zillikens MC, Rivadeneira F, de Jong R, Lindemans J, Uitterlinden AG, et al. Effect of dietary B vitamins on BMD and risk of fracture in elderly men and women: the Rotterdam study. Bone. 2007;41:987–94. doi: 10.1016/j.bone.2007.08.021. [DOI] [PubMed] [Google Scholar]
- 86.Herrmann M, Umanskaya N, Traber L, Schmidt-Gayk H, Menke W, Lanzer G, et al. The effect of B-vitamins on biochemical bone turnover markers and bone mineral density in osteoporotic patients: a 1-year double blind placebo controlled trial. Clin Chem Lab Med. 2007;45:1785–92. doi: 10.1515/CCLM.2007.352. [DOI] [PubMed] [Google Scholar]
- 87.Kim GS, Kim CH, Park JY, Lee KU, Park CS. Effects of vitamin B12 on cell proliferation and cellular alkaline phosphatase activity in human bone marrow stromal osteoprogenitor cells and UMR106 osteoblastic cells. Metabolism. 1996;45:1443–6. doi: 10.1016/s0026-0495(96)90171-7. [DOI] [PubMed] [Google Scholar]
- 88.Melhus H, Michaëlsson K, Holmberg L, Wolk A, Ljunghall S. Smoking, antioxidant vitamins, and the risk of hip fracture. J Bone Miner Res. 1999;14:129–35. doi: 10.1359/jbmr.1999.14.1.129. [DOI] [PubMed] [Google Scholar]
- 89.Lumbers M, New SA, Gibson S, Murphy MC. Nutritional status in elderly female hip fracture patients: comparison with an age-matched home living group attending day centres. Br J Nutr. 2001;85:733–40. doi: 10.1079/bjn2001350. [DOI] [PubMed] [Google Scholar]
- 90.Carmel R, Lau KH, Baylink DJ, Saxena S, Singer FR. Cobalamin and osteoblast-specific proteins. N Engl J Med. 1988;319:70–5. doi: 10.1056/NEJM198807143190202. [DOI] [PubMed] [Google Scholar]
- 91.Roblin X, Phelip JM, Genevois M, Ducros V, Bonaz B. Hyperhomo-cysteinaemia is associated with osteoporosis in patients with Crohn’s disease. Aliment Pharmacol Ther. 2007;25:797–804. doi: 10.1111/j.1365-2036.2007.03260.x. [DOI] [PubMed] [Google Scholar]
- 92.McLean RR, Jacques PF, Selhub J, Tucker KL, Samelson EJ, Broe KE, et al. Homocysteine as a predictive factor for hip fracture in older persons. N Eng J Med. 2004;350:2042–9. doi: 10.1056/NEJMoa032739. [DOI] [PubMed] [Google Scholar]
- 93.Leboff MS, Narweker R, LaCroix A, Wu L, Jackson R, Lee J, et al. Homocysteine levels and risk of hip fracture in postmenopausal women. J Clin Endocrinol Metab. 2009;94:1207–13. doi: 10.1210/jc.2008-1777. [DOI] [PMC free article] [PubMed] [Google Scholar]