

Abstract
Pilocytic astrocytoma, the most common childhood brain tumor1, is typically associated with mitogen-activated protein kinase (MAPK) pathway alterations2. Surgically inaccessible midline tumors are therapeutically challenging, showing sustained tendency for progression3 and often becoming a chronic disease with substantial morbidities4.
Here we describe whole-genome sequencing of 96 pilocytic astrocytomas, with matched RNA sequencing (n=73), conducted by the International Cancer Genome Consortium (ICGC) PedBrain Tumor Project. We identified recurrent activating mutations in FGFR1 and PTPN11 and novel NTRK2 fusion genes in non-cerebellar tumors. New BRAF activating changes were also observed. MAPK pathway alterations affected 100% of tumors analyzed, with no other significant mutations, indicating pilocytic astrocytoma as predominantly a single-pathway disease.
Notably, we identified the same FGFR1 mutations in a subset of H3F3A-mutated pediatric glioblastoma with additional alterations in NF15. Our findings thus identify new potential therapeutic targets in distinct subsets of pilocytic astrocytoma and childhood glioblastoma.
Pilocytic astrocytoma is the most common central nervous system (CNS) neoplasm in childhood, accounting for ~20% of all pediatric brain tumors. Tumor locations in our cohort reflect the fact that pilocytic astrocytomas occur throughout the CNS, with about half arising outside the cerebellum (Supplementary Figure 1). Extra-cerebellar tumors are often surgically inaccessible, leading to a chronic disease with multiple recurrences, visual and neurological impairment, and/or side-effects of therapy1,4. Genetic alterations within the MAPK signaling pathway are a hallmark of this tumor, with KIAA1549:BRAF fusion being the most frequent event6–8. A smaller number harbor BRAF or KRAS point mutations, alternative BRAF/RAF1 fusions, or germline NF1 mutations2. Pilocytic astrocytoma has therefore been hypothesized to represent a single-pathway disease2. Previously, however, no MAPK pathway changes were identifiable in 15–20% of tumors (mostly non-cerebellar).
To investigate the full range of genetic alterations in pilocytic astrocytoma, we performed whole-genome sequencing of tumor and blood DNA from 96 patients (Supplementary Table 1). Corresponding RNA sequencing data and larger insert mate-pair sequencing (for enhanced structural rearrangement detection) were generated for 73 and 68 samples, respectively. The average somatic mutation rate was extremely low (<0.1/Mb), with a mean of 1.6 non-synonymous single nucleotide variants (SNVs) per tumor (range 0–9; Supplementary Table 1) – similar to the rate described in NF1-associated pilocytic astrocytomas9. This is markedly lower than recently reported for the malignant pediatric brain tumor medulloblastoma10–12, and several other pediatric solid tumors13. The average number of small insertions/deletions (InDels) affecting coding sequences was <1 per case. All coding somatic SNVs/InDels are listed in Supplementary Table 2.
In line with other tumor types10,14,15, genome-wide mutation rates positively correlated with patient age (r = 0.42, P = 2.3 × 10−5, Pearson’s product-moment correlation; Supplementary Figure 2a). The observed mutations were predominantly C-to-T transitions at CpG sites (likely deamination of methylated cytosines), suggesting that the age-dependent increase may largely be due to background processes occurring in progenitor cells prior to tumorigenesis, as recently reported in leukemia15 (Supplementary Figure 2b).
Most of the known MAPK pathway activating events were also found in this series, including KIAA1549:BRAF fusion variants (70 cases), a FAM131B:BRAF fusion16, four BRAFV600E mutations and one BRAFins599T (Supplementary Table 1). Three tumors were associated with Neurofibromatosis Type 1. This is fewer than would be expected for prospective cohorts (5–10%), since material for biological studies from these typically optic pathway-associated tumors is limited. NF1 has been reported to follow a classical tumor suppressor model in pilocytic astrocytoma, with a somatic second hit in addition to the germline alteration9. This also held true in our series (Supplementary Table 1).
Analysis of copy number and structural alterations using DNA and RNA sequencing revealed four novel BRAF fusions (Figure 1, Supplementary Figure 3). As expected, all variants resulted in loss of the amino-terminal regulatory region of BRAF. An RNF130:BRAF fusion derived from a reciprocal t(5;7)(q35;q34) translocation was seen in two cases (Figure 1a), with single examples of CLCN6:BRAF, MKRN1:BRAF and GNAI1:BRAF fusions (Supplementary Figure 3a–c). Thus, non-KIAA1549:BRAF fusions comprise a significant minority of activating events, with BRAF apparently being a promiscuous fusion partner.
Figure 1. Novel BRAF alterations in pilocytic astrocytoma.

a, Schematic representation of the RNF130:BRAF fusion gene in ICGC_PA112 resulting from a translocation between chromosomes 5 and 7. A similar fusion was observed in ICGC_PA96. The cDNA sequence at the fusion breakpoint and resulting exon and protein structures are indicated. A reciprocal fusion between RUN and FYVE domain containing 1 (RUFY1) and transmembrane protein 178B (TMEM178B) on the derivative chromosome 5 in ICGC_PA112 was also found to be expressed by RNA-seq analysis (not shown). RPM, reads per million; KD, kinase domain.
b, Computational modeling of two BRAF monomers (light/dark grey) with a VLR insertion (blue/magenta) between Arg506 and Lys507 (green), as identified in ICGC_PA65. The PDB structure 4E26 was used as a template. Val600, a mutational hotspot, is shown in yellow. A novel dimer interface is formed between the protomers, with hydrogen bonds from the new arginine side chains (dashed lines) and a hydrophobic interaction between the leucine side chains (magenta).
c, Western blot analysis of NIH3T3 cells transfected with empty vector (EV), BRAFWT, BRAFV600E and BRAFinsVLR. The newly identified insVLR mutant results in elevation of ERK1/2 phosphorylation (pERK1/2) at a similar level to that seen with the known oncogenic V600E form.
d, Co-immunoprecipation assay with pull-down of HA-tagged BRAFinsVLR, demonstrating that this novel mutant forms homodimers with co-transfected AU1-tagged insVLR, but does not appear to form a strong heterodimer with wild-type BRAF.
Another new BRAF alteration was identified in ICGC_PA65, resulting in a three amino acid insertion (p.R506_insVLR) in the interdomain cleft of BRAF - a structural region linked to its activity17 and homodimerization18. Protein modeling predicted that these residues stabilize a dimeric form of BRAF (known to be active independent of RAS stimulation19) (Figure 1b). Homodimerization was confirmed by immunoprecipitation, and the BRAFinsVLR mutant increased ERK phosphorylation as effectively as BRAFV600E (Figure 1c,d).
Novel alterations in KRAS were also observed. ICGC_PA117 and ICGC_PA142 both showed two distinct mutations (E63K/R73M and L19F/Q22K, respectively). DNA and RNA-seq data confirmed that both alterations affected the same allele (Supplementary Figure 4). Whilst there are reports of double KRAS mutations in entities such as colon cancer20, these typically involve at least one hotspot residue (codons 12, 13 or 61), and often represent heterogeneous tumor subclones rather than two hits in one allele (although this has also been described, e.g. 21). The alterations seen in our tumors do not encompass classical hotspots, suggesting that further characterization of downstream effects is warranted.
All but one of the cerebellar tumors in our series harbored a BRAF fusion, with the other displaying a KRAS alteration. 9/48 (19%) of the non-cerebellar tumors, however, lacked any of the above alterations. Further assessment of structural rearrangements revealed two novel gene fusions in a total of 3 samples, involving the kinase domain of NTRK2 (also known as TrkB) - an oncogene implicated in the tumorigenesis of neuroblastoma, amongst others22,23. The related NTRK1 and NTRK3 genes have previously been shown to be activated by fusion events (e.g. TPM3:NTRK1 in papillary thyroid cancer24 and ETV6:NTRK3 in multiple tumors25). The QKI:NTRK2 and NACC2:NTRK2 fusions identified here were verified by PCR (Figure 2, Supplementary Figure 3d). Both 5′ partners contain dimerization domains, and are therefore predicted to induce ligand-independent dimerization. Interestingly, N-terminal TrkB truncation has recently been shown to induce transformation of neural crest cells26. The downstream effects of TrkB activation are mediated at least in part via MAPK pathway activation27.
Figure 2. NTRK2 is a new gene fusion target in pilocytic astrocytoma.

Schematic representation of the QKI:NTRK2 fusion gene in ICGC_PA159 resulting from a translocation between chromosomes 6 and 9. A similar fusion was observed in ICGC_PA82. The cDNA sequence at the fusion breakpoint and resulting exon and protein structures are indicated. RPM, reads per million; KD, kinase domain; QUA1, Qua1 dimerization domain.
A second new recurrent alteration, namely mutation of two hotspot residues (N546 & K656) in the kinase domain of FGFR1, was seen in five tumors (Supplementary Table 3 and Figure 3a). FGFR1 is more commonly activated through amplification, in tumors such as breast28 and lung29,30 cancer. Occasional FGFR1 mutations have been observed in adult glioblastoma (GBM)31,32, a highly malignant astrocytoma, as have FGFR1:TACC1 or FGFR3:TACC3 fusion genes33. Mutations in homologous codons in FGFR2 and FGFR3 are commonly found in other tumor types, particularly bladder, skin and endometrial cancers (see the COSMIC database34). Both mutations result in midbrain hyperproliferation in developing chick embryos35. The N546K mutation alters FGFR1 auto-phosphorylation, resulting in increased kinase activity and transforming potential36, while the K656E variant is also transforming in vitro37. Interestingly, the latter study suggested that FGF2 (bFGF) ligand was necessary in addition to FGFR1 mutation to maintain neurosphere formation in vitro. Gene expression array data of 118 pilocytic astrocytomas, including 66 from the present series, revealed significantly increased FGF2 expression in pilocytic astrocytoma compared with 158 other astrocytic tumors38,39, or normal tissues40. This increase was not restricted to only FGFR1-mutant or wild-type tumors, suggesting that ligand-mediated pathway activation may play a general role in tumorigenesis (Figure 3b). Immunohistochemical detection of phospho-FGFR1 revealed strong, diffuse positivity in all seven pilocytic astrocytomas harboring an FGFR1 mutation for which material was available. No positivity was observed in eleven FGFR1WT tumors. All samples displayed strong phospho-ERK staining (Supplementary Figure 5). Interestingly, ICGC_PA89 harbored an alternative alteration in FGFR1, consisting of a ~4.5kb internal tandem duplication (ITD) of the portion of the gene encoding the kinase domain, reminiscent of the activating internal tandem duplications of the FLT3 kinase observed in acute myeloid leukemia41 (Figure 3c).
Figure 3. FGF pathway signaling molecules are recurrently altered in pilocytic astrocytoma.

a, Schematic of the domain structure of FGFR1, indicating the position and frequency of the hotspot mutations in pilocytic astrocytomas sequenced in the present study (including replication cases).
b, Gene expression data for FGF2 indicating significantly elevated expression in pilocytic astrocytomas (red) compared with other astrocytic tumors (blue), normal cerebellum (black) or other normal tissues (green); P <0.001, two-sided t-test. The pilocytic astrocytomas with expression data which harbor FGFR1 alterations (four mutants plus FGFR1-ITD) are highlighted.
c, Schematic of an additional alteration in FGFR1 identified in ICGC_PA89, comprising an internal tandem duplication (ITD) of part of intron 10, exons 11–17 and part of exon 18. The duplicated amino acids are aa478–820 (numbered according to the alpha A1 isoform), with an additional 40 amino acid linker sequence encoded by part of intron 10. The whole kinase domain is therefore duplicated in the resulting predicted protein (TK1′ and TK2′).
d, Schematic of the structure of PTPN11 (SHP-2), indicating the position and frequency of mutations in pilocytic astrocytomas sequenced in the present study.
e, Gene expression data for PTPN11 indicating significantly elevated expression in pilocytic astrocytomas compared with other groups, as in (b); P <0.001, two-sided t-test. The pilocytic astrocytomas with expression data which harbor FGFR1 alterations (four mutants plus FGFR1-ITD) are highlighted.
Ig, immunoglobulin-like domain; TM, transmembrane domain; TK, tyrosine kinase domain; SH2, src homology 2 domain; PTP, protein tyrosine phosphatase domain; DA, WHO Grade II diffuse astrocytoma; AA, anaplastic astrocytoma; K27/G34/IDH1 GBM, glioblastoma carrying a mutation at K27 or G34 in H3F3A or in IDH1; ped., pediatric; CBM, cerebellum, CNS, central nervous system.
Further recurrent alterations were found in a RAS/MAPK-related adaptor protein, the phosphatase PTPN11 (also called SHP-2, Figure 3c). Both alterations (E69K and E76A) were previously reported in juvenile monomyelocytic leukemia, which is frequently associated with SHP-2 activation42,43. Interestingly, both were found in FGFR1-mutant tumors (ICGC_PA84 & ICGC_PA166), suggesting a cooperative role in tumorigenesis (Supplementary Table 3). Overexpression of mutant SHP-2 alone did not elevate pERK levels in vitro, while the two FGFR1 mutants, either alone or in combination with mutant SHP-2, upregulated pERK (Supplementary Figure 6). This would support the hypothesis that PTPN11 mutation alone is insufficient for pilocytic astrocytoma development, but that it may play a modifying role in FGFR1-mutant tumors. Of note, PTPN11/SHP-2 expression was increased in pilocytic astrocytomas when compared with other astrocytomas or normal tissues (Figure 3d), suggesting a broader role in the biology of this entity. An additional cohort of 45 non-cerebellar pilocytic astrocytomas, negative for KIAA1549:BRAF, was screened for FGFR1 (exons 12 & 14) and PTPN11 (exon 3) mutations. Nine cases harbored FGFR1 mutations at N546 or K656, and one additionally carried a PTPN11/SHP-2 E69K change (Supplementary Table 3), confirming our whole-genome sequencing findings. Germline PTPN11 mutations are one of the causes of the hereditary developmental disorders Noonan syndrome (NS)44 and LEOPARD syndrome (LS)45. A few case reports have described pilocytic astrocytomas occurring in NS and LS patients46–49. Together with NF1, this makes three known ‘RASopathies’, characterized by germline MAPK pathway mutations50, linked with pilocytic astrocytoma tumorigenesis. In our germline sequencing data, however, NF1 was the only RASopathy gene disrupted at a higher frequency than in the 1000 Genomes Project (URL below).
Strikingly, all of the pilocytic astrocytomas in our cohort harbored a MAPK pathway alteration. BRAF, FGFR1, KRAS and NF1 were the only genes found to be significantly mutated using the Genome MuSiC algorithm (URL below, Supplementary Table 4). With the exception of FGFR1 and PTPN11, each case typically harbored only one pathway alteration (P < 0.0001, permutation test, Figure 4). Together with the finding that BRAF kinase activation alone is sufficient to induce pilocytic astrocytomas in mice51,52, these data strongly support the concept of pilocytic astrocytoma as a prototypic single-pathway disease driven by a limited number of oncogenic hits (possibly only one in some cases; Supplementary Figure 7).
Figure 4. Summary of MAPK pathway alterations in pilocytic astrocytoma.
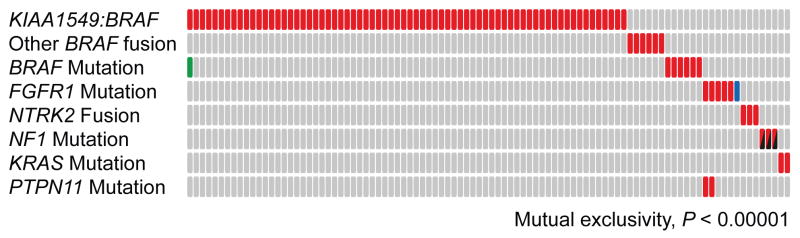
An overview of MAPK pathway alterations identified in the 96 whole-genome sequencing cases included in the present study, indicating the mutual exclusivity of the majority of these hits (with the exception of FGFR1 and PTPN11); P <0.0001, permutation test on 10,000 iterations. Each column represents one tumor sample. Red filled boxes indicate that a given alteration is present in this sample. The blue filled box represents FGFR1-ITD rather than a point mutation; the green box indicates a BRAFE451D mutation in a case with a KIAA1549:BRAF fusion (unknown functional significance, but included in the exclusivity testing); the black/red boxes indicate that the NF1 alterations comprise one germline and one somatic hit per case.
Interestingly, one of the FGFR1-mutant tumors (ICGC_PA69) also displayed an H3F3A K27M mutation and somatic alterations of NF1 – both of which are more commonly encountered in pediatric glioblastoma (GBM)5. Three experienced neuropathologists (AK, GR and JF) reported pilocytic astrocytoma histology for this case, although a diagnosis of GBM cannot be conclusively excluded due to limited material. By examining previous exome sequencing data for pediatric GBM5, we identified 3/48 samples (6%) with an FGFR1 mutation. Strikingly, all three harbored the same constellation of H3F3A K27M, somatic NF1 alteration, and FGFR1 activation (Supplementary Table 3). They were also wild-type for TP53, which is mutated in most K27M-mutant GBM or diffuse intrinsic pontine gliomas5,53. One tumor reported in a targeted sequencing cohort of medulloblastoma10 had a similar triplet, with H3F3A K27M, NF1 mutation and an FGFR2 K659E mutation (homologous to FGFR1 K656E), making a total of 5 cases with this triple-mutation combination. Gene expression analysis indicated that this is likely a GBM previously mis-classified as medulloblastoma. It is not currently clear why these mutations occur in concert, and additional work will be required to assess their roles. One possibility is that NF1 mutation may mimic elevated PTPN11 expression, since activation of PTPN11/SHP-2 inhibits the recruitment of Ras GTPase-activating proteins (RasGAPs, including NF1) to the plasma membrane54.
All FGFR1-mutant tumors were extra-cerebellar, mostly in midline locations (Supplementary Table 3), suggesting a link between cell of origin and/or microenvironment with FGFR1-driven tumorigenesis. The H3F3A K27M mutation is also associated with midline GBM39. Notably, FGFR1 plays a role in neural stem cell self-renewal55, and is essential for midline glial cell development56. This spatial clustering may also reflect differential sensitivity of distinct neural precursors to activating stimuli, particularly NF1 loss57,58. The type and timing of second hits (H3F3A/NF1 mutation) and/or the differentiation stage of the cell of origin may contribute to determining a fate of oncogene-induced senescence and slow growth (pilocytic astrocytoma)59,60 versus aggressive malignancy with poor outcome (GBM).
In summary, this study has identified new insights into the tumorigenesis of pilocytic astrocytoma. Each tumor harbored very few mutations, in keeping with a generally benign behavior. Importantly, our findings confirm the concept that pilocytic astrocytomas are predominantly driven by aberrant activation of the MAPK pathway. Most strikingly, however, we report novel recurrent alterations in NTRK2, FGFR1 and PTPN11, which were mutually exclusive with other RAF/RAS alterations. Combined with the observation of FGF2 and PTPN11 overexpression, these results indicate upstream contributors to MAPK pathway activation in this entity. Emerging preclinical data suggest that BRAF inhibitors may trigger paradoxical activation in tumors harboring KIAA1549:BRAF, i.e. the majority of pilocytic astrocytomas61. Single-drug or combination therapy with FGFR, NTRK2 and/or MEK inhibitors, several of which are currently in preclinical and clinical trials62–64, may therefore represent rational treatment options. BRAFV600E-specific agents may also be a logical choice for ~5% of patients. Finally, the identification of recurrent FGFR1 mutations in a subset of pediatric GBM provide an opportunity for therapeutic targeting of FGFR signaling in these clinically challenging brain tumors.
Methods
Sample collection
Informed consent and an ethical vote (Ethics Committee of the Medical Faculty of Heidelberg) were obtained according to ICGC guidelines (http://www.icgc.org). Tumor tissues were subjected to neuropathological review to confirm histology and tumor cell content.
DNA Sequencing
Paired-end library preparation was conducted using Illumina, Inc. v2 protocols. 1–5μg of genomic DNA were fragmented to ~300 bp insert-size with a Covaris device, followed by size selection through agarose gel excision. Mate-pair (long-range paired-end mapping) DNA library preparation was carried out using the v2 protocol from Illumina. 10ug of genomic DNA were fragmented to 4.5kb with a Hydroshear device, followed by size selection. Deep sequencing was carried out with Illumina HiSeq2000 instruments.
RNA Sequencing
Twenty-three RNAseq libraries were prepared with purified polyA+ RNA fractions using strand-specific methods, following dUTP-based protocols as described in65, featuring cDNA fragmentation after mRNA priming with random hexamers (dN)6 and oligo(dT) primers. Six libraries were constructed with a modified protocol whereby the polyA+ fraction was fragmented using RNA fragmentation reagents (Ambion, Cat. #AM8740); first strand synthesis was then performed with random hexamers only (cDNA fragmentation was omitted). Fifty RNAseq libraries were prepared with a ribosomal RNA-depleted fraction. In brief, 0.2 μg of total RNA was prepared using the RiboZero™ Gold kit (Epicentre). The resulting RNA was further processed following the library preparation protocol described in 66, starting at the fragmentation step (2nd step). Sequencing (2×51bp) was carried out on the HiSeq2000.
Mapping and analysis
Sequencing reads were mapped and aligned to the hg19 reference assembly as previously described10, using BWA67 (version 0.5), and processed with samtools68 (version 0.1.17) and Picard tools (version 1.61).
An in-house analysis pipeline based on samtools mpileup and bcftools68 was used to detect single nucleotide variants (SNV) and small insertions or deletions (InDels). In addition to previously described filters to remove artefacts10, we excluded variants located in regions of low mappability or overlapping with the hiSeqDepthTop10Pct, Encode DAC Blacklisted Regions, or Duke Excluded Regions tracks from UCSC genome browser. High confidence somatic SNVs were not allowed to overlap with any two of the following features: tandem repeats, simple repeats, low complexity, satellite repeats, or segmental duplications. In addition, following heuristic criteria were applied: (1) at least 5 tumor reads at the position; (2) more than one variant read per strand, or at least 5 variant reads in total and variant allele fraction > 0.1; (3) at least 12 reads at the position in the matching control; (4) less than 1/30 of the control reads supporting the variant; (5) less than 300 reads at the corresponding position in the control; (6) no non-reference, non-variant bases at the corresponding position in the control.
InDels were called with samtools mpileup and bcftools on reads with mapping quality >20 and scored in a similar way as SNVs. Overlap with tandem or simple repeats, however, was not penalized, since these elements are prone to InDels due to polymerase slippage. Since InDel alignments in the matched control can be slightly shifted in comparison to the tumor, or mismatches preferred over gaps, germline events can be falsely called as somatic. We therefore required not more than one mismatch or InDel in the control within 20bp of the tumor InDel.
Tumor and matched control samples were also analyzed with Pindel (version 0.2.4h)69. Events in the tumor were only considered when supported by at least five reads and if the number of supporting reads divided by the maximum of the read depth at the left and right breakpoints was >0.05. The matched control sample was also analyzed by samtools mpileup at tumor InDel positions and 10bp up- and downstream. Variants were classified as somatic if both Pindel and samtools mpileup did not call a multibase variant in the control sample. Only additional InDels in “RASopathy” genes were reported from the Pindel analysis (due to a high false positive rate), all other InDels were called with samtools as described above.
SNVs and InDels were functionally annotated with RefSeq gene annotations using Annovar70 and Oncotator (http://www.broadinstitute.org/oncotator/). For the identification of significantly mutated genes we used the high confidence somatic SNVs and InDels as input for Genome MuSiC71 (version 0.3), setting max-fdr to 0.05 in the genome music smg module. Substitution patterns of SNVs were evaluated in a sequence context of all 96 possible trinucleotides to assess mutational signatures72.
Integration of SNVs and InDels with RNAseq data
Gene expression levels were calculated per-exon according to reads per kilobase of exon model per million mapped reads (RPKM) using BedTools73 and custom Perl scripts. Where available, candidate DNA variant positions were annotated with RNA information by generating a pileup of the DNA variant position in the RNA BAM file (Supplementary Table 2).
Structural rearrangement detection & verification
Rearrangements based on paired-end data were identified using read-depth analysis, CREST74, DELLY75, and manual inspection of sequencing reads. Rearrangements based on mate-pair data were identified using DELLY and manual inspection of sequencing reads, as previously described10.
Structural variants were verified by PCR (conditions and primer sequences available upon request). PCR products were sent to GATC Biotech (Germany) for Sanger sequencing to confirm breakpoints.
Fusion transcript detection & verification
Fastq files from transcriptome sequencing were used for de novo annotation of fusion transcripts using the TopHat-Fusion76 and deFuse77 algorithms with standard parameters. Where neither algorithm detected fusions, but whole-genome sequencing supported a fusion, we extracted corresponding transcriptome reads matching the theoretical sequence surrounding the predicted fusion border and then counted as fusion reads those where the entire 51bp sequences were derived from the predicted fused exons.
Primers for amplification of neighboring exons in normal (unfused) transcripts were tested in RT-PCR using total RNA from normal cerebellum (BioChain, lot number B307003). Validated primers were used to amplify the normal transcripts (control) and fusion transcripts from tumor RNA using the SuperScript III One-Step RT-PCR System with Platinum Taq DNA Polymerase (Invitrogen). PCR conditions and primer sequences are available upon request.
Computational protein modeling
A dimeric model of mutant BRAFinsVLR was produced with Modeller78 using the PDB structure 4E26 as a template. Ten models were produced, with the one having the lowest DOPE score shown in main Figure 1b.
Expression Array Analysis
Affymetrix U133 Plus2.0 expression array data for genes of interest were extracted from publicly-available datasets via the R2 software tool (http://r2.amc.nl), and for additional cases on an early-access basis through collaboration with the Microarray Department of the University of Amsterdam, the Netherlands. The MAS5.0 algorithm of the GCOS program (Affymetrix Inc) was used for normalization and assignment of detection p-values. Array quality was ensured by inspection of beta-actin and GAPDH 5′-3′ ratios as well as the percentage of present calls.
Verification of SNVs/InDels
All SNVs/InDels in FGFR1, PTPN11, BRAF, KRAS, NF1 and H3F3A were verified by PCR followed by capillary (Sanger) sequencing. Additional variants were also verified in this way, as listed in Supplementary Table 2. Verification rates for SNVs was >98%, and for InDels was >70%. Alterations determined to be false are not included in Supplementary Table 2.
In vitro and protein assays
Coding sequences of BRAF, PTPN11 and FGFR1 were cloned from normal brain cDNA (Stratagene) into a pcDNA3.1 vector with HA, FLAG or AU1 epitope tags. Site-directed mutagenesis (QuikChangeII XL, Agilent Technologies) was used to generate BRAFV600E, BRAFinsVLR, PTPN11E69K, PTPN11E76A, FGFR1N546K and FGFR1K656E.
NIH3T3 murine fibroblasts (Leibniz Institute DSMZ, Germany; mycoplasma tested) were cultured in DMEM (Life Technologies), 10% FBS (Life Technologies) and penicillin/streptomycin at 37°C in 5% CO2. Cells were transfected using Lipofectamine® 2000 diluted in Opti-MEM® (Invitrogen), and switched to serum-free DMEM 24h after transfection. After a further 24h, cells were lysed in either RIPA buffer or RLT buffer (Qiagen). Protein electrophoresis was performed using 4–12% gradient NuPAGE Bis-Tris Precast Gels (Life Technologies) with transfer to a PVDF membrane. Antibodies for Western blotting, with detection using ECL (GE Healthcare Life Sciences), were as follows: anti-ERK1/2 (rabbit polyclonal, Cell Signaling Technology #9102), anti-phospho-ERK1/2 (rabbit monoclonal 20G11, Cell Signaling Technology #4376), anti-HA (rabbit monoclonal C29F4, Cell Signaling Technology #3724), anti-AU1 (rabbit polyclonal, Abcam ab3401), anti-DYKDDDDK (FLAG®) (rabbit polyclonal, Cell Signaling Technology #2368), HRP-conjugated goat anti-Rabbit IgG (Santa Cruz Biotechnology, sc-2004), HRP-conjugated goat anti-mouse Ig (Santa Cruz Biotechnology, sc-2005).
For co-immunoprecipitation (Co-IP) to assess BRAF dimerization, cells were washed twice in ice-cold PBS, scraped, pelleted, and then lysed on ice in 5 pellet-volumes of lysis buffer (50mM HEPES, pH7.5; 250mM NaCl; 1mM EDTA; 2,5 mM EGTA; 10% Glycerol; 0.1% Triton X-100; protease inhibitors) with regular vortexing. Lysates were run through a QiaShredder column (Qiagen), centrifuged, and transferred to a new tube. 8μl of anti-HA agarose slurry (Thermo Scientific) were washed 3 times then resuspended in 20μl lysis buffer. 50μg protein extract was added to the anti-HA slurry and rotated overnight at 4°C. Finally, beads were pelleted, washed seven times in lysis buffer, and resuspended in SDS sample buffer for Western blotting.
Immunohistochemistry (IHC)
IHC was performed with an automated stainer (Benchmark XT; Ventana). Phospho-FGFR (Y653/Y654) was assessed using antibody PA5-12594 (Thermo Scientific), diluted 1:50. Phospho-ERK1/2 (T202/Y204) was assessed using antibody #9101 (Cell Signaling Technology), diluted 1:100.
Supplementary Material
Acknowledgments
For technical support and expertise we thank: Bettina Haase, Dinko Pavlinic, and Bianka Baying (EMBL genomics core facility); Michael Wahlers and Rupert Lück (EMBL high-performance computing facility); the DKFZ Genomics and Proteomics Core Facility; Malaika Knopf (NCT Heidelberg); Karin Schlangen, Macha Metsger, Kerstin Schulz, Asja Nürnberger, Alexander Kovacsovics, and Matthias Linser (Max Planck Institute for Molecular Genetics); Susanne Peetz-Dienhart and Yvonne Floer (University Hospital Münster); Danita M. Pearson (University of Cambridge) and Bingding Huang, Gideon Zipprich, Michael Heinold, Rolf Kabbe, Andrea Wittmann, Laura Sieber and Linda Linke (DKFZ). Walter Stummer (Münster), Bernd Hoffmann (Münster), Burkhard Rama, (Osnabrück), Heinrich Ebel (Hamm), Hans Axel Trost (Bayreuth) and Uwe Wildförster (Gelsenkirchen) provided detailed clinical information. We also thank GATC Biotech AG for sequencing services.
This work was principally supported by the PedBrain Tumor Project contributing to the International Cancer Genome Consortium, funded by German Cancer Aid (109252) and by the German Federal Ministry of Education and Research (BMBF, grants #01KU1201A, MedSys #0315416C and NGFNplus #01GS0883). Additional support came from the German Cancer Research Center – Heidelberg Center for Personalized Oncology (DKFZ-HIPO), the Max Planck Society, Genome Canada and the Canadian Institute for Health Research (CIHR) with co-funding from Genome BC, Genome Quebec, CIHR-ICR (Institute for Cancer Research) and C17 (N. Jabado), Ian’s Friend Foundation (M.A.K.), NIH grant numbers RO1CA105607 and P30HD018655 (S.L.P.), the Dutch Cancer Foundations KWF (2010-4713) and KIKA (M.Ko.), the Brain Tumour Charity (S.R.L. and V.P.C.) and the Pediatric Low Grade Astrocytoma Foundation (M.W.K. and K.L.L.).
Footnotes
URLs
1000 Genomes Project: http://www.1000genomes.org/
GenomeMuSiC: http://gmt.genome.wustl.edu/genome-music/0.2/index.html
Accession codes
Sequencing data have been deposited at the European Genome-phenome Archive, which is hosted by the European Bioinformatics Institute (EBI), under accession EGAS00001000381.
Supplementary Information is available in the online version of the paper.
Author Contributions
D.T.W.J., S.R.L., D.A.K.Q., A.M.F., H-J.W., A.M.S., S.H., M.Zu., J.G., S.Sch., H.Ş -C., H.W., S.B., E.P., S.St., B.R., D.F., C.C.B., C.v.K., P.v.S, R.Ve., M.Su., S.W., M.H., and J.F. performed and/or coordinated experimental work.
B.H., N. Jäger, D.T.W.J., M.Ko., H-J.W., T.Z., B.L., P.A.N., V.H., J.S., J.M., M.Za., M.Sch., C.L.W., C.D.V., S.R., C.L., P.v.S., J.K., R.Vo, and M.Ra. performed data analysis.
A.K., M.Ry., C.M., B.W., A.U., C.H-M., T.M., A.E.K., M.E., M.U.S., Y-J.C., S.L.P., A.v.D., O.W., M.H., M.A.K., C.G.E., W.S., K.L.L., M.W.K., V.P.C., and N.Jabado collected data and provided patient materials.
D.T.W.J., B.H., N. Jäger, D.S., N. Jabado, R.E., P.L. and S.M.P. prepared the initial manuscript and figures.
A.K., U.D.W., M.D.T., J.O.K., H.L., M-L.Y., B.B., G.R., V.P.C., N. Jabado, R.E., P.L. and S.M.P. provided project leadership.
All authors contributed to the final manuscript
The authors declare no competing financial interests.
Reprints and permissions information is available at www.nature.com/reprints/index.html.
References
- 1.Central Brain Tumor Registry of the United States. Statistical Report: Primary Brain and Central Nervous System Tumors Diagnosed in the United States, 2004–2008. CBTRUS; Hinsdale, IL: 2012. [Google Scholar]
- 2.Jones DTW, Gronych J, Lichter P, Witt O, Pfister SM. MAPK pathway activation in pilocytic astrocytoma. Cell Mol Life Sci. 2012;69:1799–811. doi: 10.1007/s00018-011-0898-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gnekow AK, et al. Long-term follow-up of the multicenter, multidisciplinary treatment study HIT-LGG-1996 for low-grade glioma in children and adolescents of the German Speaking Society of Pediatric Oncology and Hematology. Neuro Oncol. 2012 doi: 10.1093/neuonc/nos202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Armstrong GT, et al. Survival and long-term health and cognitive outcomes after low-grade glioma. Neuro Oncol. 2011;13:223–34. doi: 10.1093/neuonc/noq178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schwartzentruber J, et al. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature. 2012;482:226–31. doi: 10.1038/nature10833. [DOI] [PubMed] [Google Scholar]
- 6.Jones DTW, et al. Tandem duplication producing a novel oncogenic BRAF fusion gene defines the majority of pilocytic astrocytomas. Cancer Res. 2008;68:8673–7. doi: 10.1158/0008-5472.CAN-08-2097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jones DTW, et al. Oncogenic RAF1 rearrangement and a novel BRAF mutation as alternatives to KIAA1549:BRAF fusion in activating the MAPK pathway in pilocytic astrocytoma. Oncogene. 2009;28:2119–23. doi: 10.1038/onc.2009.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pfister S, et al. BRAF gene duplication constitutes a mechanism of MAPK pathway activation in low-grade astrocytomas. J Clin Invest. 2008 doi: 10.1172/JCI33656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gutmann DH, et al. Somatic neurofibromatosis type 1 (NF1) inactivation characterizes NF1-associated pilocytic astrocytoma. Genome Res. 2013;23:431–9. doi: 10.1101/gr.142604.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jones DTW, et al. Dissecting the genomic complexity underlying medulloblastoma. Nature. 2012;488:100–5. doi: 10.1038/nature11284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pugh TJ, et al. Medulloblastoma exome sequencing uncovers subtype-specific somatic mutations. Nature. 2012;488:106–10. doi: 10.1038/nature11329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Robinson G, et al. Novel mutations target distinct subgroups of medulloblastoma. Nature. 2012;488:43–8. doi: 10.1038/nature11213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Downing JR, et al. The Pediatric Cancer Genome Project. Nat Genet. 2012;44:619–22. doi: 10.1038/ng.2287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stephens PJ, et al. The landscape of cancer genes and mutational processes in breast cancer. Nature. 2012;486:400–4. doi: 10.1038/nature11017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Welch JS, et al. The origin and evolution of mutations in acute myeloid leukemia. Cell. 2012;150:264–78. doi: 10.1016/j.cell.2012.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cin H, et al. Oncogenic FAM131B-BRAF fusion resulting from 7q34 deletion comprises an alternative mechanism of MAPK pathway activation in pilocytic astrocytoma. Acta Neuropathol. 2011;121:763–74. doi: 10.1007/s00401-011-0817-z. [DOI] [PubMed] [Google Scholar]
- 17.Wan PT, et al. Mechanism of activation of the RAF-ERK signaling pathway by oncogenic mutations of B-RAF. Cell. 2004;116:855–67. doi: 10.1016/s0092-8674(04)00215-6. [DOI] [PubMed] [Google Scholar]
- 18.Rushworth LK, Hindley AD, O’Neill E, Kolch W. Regulation and role of Raf-1/B-Raf heterodimerization. Mol Cell Biol. 2006;26:2262–72. doi: 10.1128/MCB.26.6.2262-2272.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Terai K, Matsuda M. The amino-terminal B-Raf-specific region mediates calcium-dependent homo- and hetero-dimerization of Raf. EMBO J. 2006;25:3556–64. doi: 10.1038/sj.emboj.7601241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Macedo MP, et al. Multiple mutations in the Kras gene in colorectal cancer: review of the literature with two case reports. Int J Colorectal Dis. 2011;26:1241–8. doi: 10.1007/s00384-011-1238-0. [DOI] [PubMed] [Google Scholar]
- 21.Naguib A, Wilson CH, Adams DJ, Arends MJ. Activation of K-RAS by co-mutation of codons 19 and 20 is transforming. J Mol Signal. 2011;6:2. doi: 10.1186/1750-2187-6-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schramm A, et al. Biological effects of TrkA and TrkB receptor signaling in neuroblastoma. Cancer Lett. 2005;228:143–53. doi: 10.1016/j.canlet.2005.02.051. [DOI] [PubMed] [Google Scholar]
- 23.Thiele CJ, Li Z, McKee AE. On Trk--the TrkB signal transduction pathway is an increasingly important target in cancer biology. Clin Cancer Res. 2009;15:5962–7. doi: 10.1158/1078-0432.CCR-08-0651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Greco A, Miranda C, Pierotti MA. Rearrangements of NTRK1 gene in papillary thyroid carcinoma. Mol Cell Endocrinol. 2010;321:44–9. doi: 10.1016/j.mce.2009.10.009. [DOI] [PubMed] [Google Scholar]
- 25.Lannon CL, Sorensen PH. ETV6-NTRK3: a chimeric protein tyrosine kinase with transformation activity in multiple cell lineages. Semin Cancer Biol. 2005;15:215–23. doi: 10.1016/j.semcancer.2005.01.003. [DOI] [PubMed] [Google Scholar]
- 26.Dewitt J, et al. Constitutively active TrkB confers an aggressive transformed phenotype to a neural crest-derived cell line. Oncogene. 2013 doi: 10.1038/onc.2013.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kaplan DR, Miller FD. Neurotrophin signal transduction in the nervous system. Curr Opin Neurobiol. 2000;10:381–91. doi: 10.1016/s0959-4388(00)00092-1. [DOI] [PubMed] [Google Scholar]
- 28.Theillet C, et al. FGFRI and PLAT genes and DNA amplification at 8p12 in breast and ovarian cancers. Genes Chromosomes Cancer. 1993;7:219–26. doi: 10.1002/gcc.2870070407. [DOI] [PubMed] [Google Scholar]
- 29.Dutt A, et al. Inhibitor-sensitive FGFR1 amplification in human non-small cell lung cancer. PLoS One. 2011;6:e20351. doi: 10.1371/journal.pone.0020351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Weiss J, et al. Frequent and focal FGFR1 amplification associates with therapeutically tractable FGFR1 dependency in squamous cell lung cancer. Sci Transl Med. 2010;2:62ra93. doi: 10.1126/scitranslmed.3001451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rand V, et al. Sequence survey of receptor tyrosine kinases reveals mutations in glioblastomas. Proc Natl Acad Sci U S A. 2005;102:14344–9. doi: 10.1073/pnas.0507200102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.TCGA. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455:1061–8. doi: 10.1038/nature07385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Singh D, et al. Transforming fusions of FGFR and TACC genes in human glioblastoma. Science. 2012;337:1231–5. doi: 10.1126/science.1220834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Forbes SA, et al. COSMIC: mining complete cancer genomes in the Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res. 2011;39:D945–50. doi: 10.1093/nar/gkq929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu A, et al. FGF17b and FGF18 have different midbrain regulatory properties from FGF8b or activated FGF receptors. Development. 2003;130:6175–85. doi: 10.1242/dev.00845. [DOI] [PubMed] [Google Scholar]
- 36.Lew ED, Furdui CM, Anderson KS, Schlessinger J. The precise sequence of FGF receptor autophosphorylation is kinetically driven and is disrupted by oncogenic mutations. Sci Signal. 2009;2:ra6. doi: 10.1126/scisignal.2000021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yoon K, et al. Fibroblast growth factor receptor signaling promotes radial glial identity and interacts with Notch1 signaling in telencephalic progenitors. J Neurosci. 2004;24:9497–506. doi: 10.1523/JNEUROSCI.0993-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gravendeel LA, et al. Intrinsic gene expression profiles of gliomas are a better predictor of survival than histology. Cancer Res. 2009;69:9065–72. doi: 10.1158/0008-5472.CAN-09-2307. [DOI] [PubMed] [Google Scholar]
- 39.Sturm D, et al. Hotspot Mutations in H3F3A and IDH1 Define Distinct Epigenetic and Biological Subgroups of Glioblastoma. Cancer Cell. 2012;22:425–37. doi: 10.1016/j.ccr.2012.08.024. [DOI] [PubMed] [Google Scholar]
- 40.Roth RB, et al. Gene expression analyses reveal molecular relationships among 20 regions of the human CNS. Neurogenetics. 2006;7:67–80. doi: 10.1007/s10048-006-0032-6. [DOI] [PubMed] [Google Scholar]
- 41.Breitenbuecher F, et al. Identification of a novel type of ITD mutations located in nonjuxtamembrane domains of the FLT3 tyrosine kinase receptor. Blood. 2009;113:4074–7. doi: 10.1182/blood-2007-11-125476. [DOI] [PubMed] [Google Scholar]
- 42.Tartaglia M, et al. Somatic mutations in PTPN11 in juvenile myelomonocytic leukemia, myelodysplastic syndromes and acute myeloid leukemia. Nat Genet. 2003;34:148–50. doi: 10.1038/ng1156. [DOI] [PubMed] [Google Scholar]
- 43.Chan G, Kalaitzidis D, Neel B. The tyrosine phosphatase Shp2 (PTPN11) in cancer. Cancer and Metastasis Reviews. 2008;27:179–192. doi: 10.1007/s10555-008-9126-y. [DOI] [PubMed] [Google Scholar]
- 44.Romano AA, et al. Noonan syndrome: clinical features, diagnosis, and management guidelines. Pediatrics. 2010;126:746–59. doi: 10.1542/peds.2009-3207. [DOI] [PubMed] [Google Scholar]
- 45.Tartaglia M, Gelb BD. Noonan syndrome and related disorders: genetics and pathogenesis. Annu Rev Genomics Hum Genet. 2005;6:45–68. doi: 10.1146/annurev.genom.6.080604.162305. [DOI] [PubMed] [Google Scholar]
- 46.Fryssira H, et al. Tumor development in three patients with Noonan syndrome. Eur J Pediatr. 2008;167:1025–31. doi: 10.1007/s00431-007-0636-3. [DOI] [PubMed] [Google Scholar]
- 47.Sanford RA, Bowman R, Tomita T, De Leon G, Palka P. A 16-year-old male with Noonan’s syndrome develops progressive scoliosis and deteriorating gait. Pediatr Neurosurg. 1999;30:47–52. doi: 10.1159/000028761. [DOI] [PubMed] [Google Scholar]
- 48.Schuettpelz LG, et al. Pilocytic astrocytoma in a child with Noonan syndrome. Pediatr Blood Cancer. 2009;53:1147–9. doi: 10.1002/pbc.22193. [DOI] [PubMed] [Google Scholar]
- 49.Vulpoi C, et al. LEOPARD syndrome and pilocytic astrocytome: a random association? Endocrine Abstracts. 2009;20 [Google Scholar]
- 50.Zenker M. Clinical manifestations of mutations in RAS and related intracellular signal transduction factors. Curr Opin Pediatr. 2011;23:443–51. doi: 10.1097/MOP.0b013e32834881dd. [DOI] [PubMed] [Google Scholar]
- 51.Gronych J, et al. An activated mutant BRAF kinase domain is sufficient to induce pilocytic astrocytoma in mice. J Clin Invest. 2011;121:1344–8. doi: 10.1172/JCI44656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kaul A, Chen YH, Emnett RJ, Dahiya S, Gutmann DH. Pediatric glioma-associated KIAA1549:BRAF expression regulates neuroglial cell growth in a cell type-specific and mTOR-dependent manner. Genes Dev. 2012;26:2561–6. doi: 10.1101/gad.200907.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Khuong-Quang DA, et al. K27M mutation in histone H3.3 defines clinically and biologically distinct subgroups of pediatric diffuse intrinsic pontine gliomas. Acta Neuropathol. 2012;124:439–47. doi: 10.1007/s00401-012-0998-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Agazie YM, Hayman MJ. Molecular mechanism for a role of SHP2 in epidermal growth factor receptor signaling. Mol Cell Biol. 2003;23:7875–86. doi: 10.1128/MCB.23.21.7875-7886.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ma DK, Ponnusamy K, Song MR, Ming GL, Song H. Molecular genetic analysis of FGFR1 signalling reveals distinct roles of MAPK and PLCgamma1 activation for self-renewal of adult neural stem cells. Mol Brain. 2009;2:16. doi: 10.1186/1756-6606-2-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tole S, Gutin G, Bhatnagar L, Remedios R, Hebert JM. Development of midline cell types and commissural axon tracts requires Fgfr1 in the cerebrum. Dev Biol. 2006;289:141–51. doi: 10.1016/j.ydbio.2005.10.020. [DOI] [PubMed] [Google Scholar]
- 57.Lee DY, Gianino SM, Gutmann DH. Innate neural stem cell heterogeneity determines the patterning of glioma formation in children. Cancer Cell. 2012;22:131–8. doi: 10.1016/j.ccr.2012.05.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lee DY, Yeh TH, Emnett RJ, White CR, Gutmann DH. Neurofibromatosis-1 regulates neuroglial progenitor proliferation and glial differentiation in a brain region-specific manner. Genes Dev. 2010;24:2317–29. doi: 10.1101/gad.1957110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jacob K, et al. Genetic aberrations leading to MAPK pathway activation mediate oncogene-induced senescence in sporadic pilocytic astrocytomas. Clin Cancer Res. 2011 doi: 10.1158/1078-0432.CCR-11-0127. [DOI] [PubMed] [Google Scholar]
- 60.Raabe EH, et al. BRAF Activation Induces Transformation and Then Senescence in Human Neural Stem Cells: A Pilocytic Astrocytoma Model. Clin Cancer Res. 2011;17:3590–9. doi: 10.1158/1078-0432.CCR-10-3349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sievert AJ, et al. Paradoxical activation and RAF inhibitor resistance of BRAF protein kinase fusions characterizing pediatric astrocytomas. Proc Natl Acad Sci U S A. 2013;110:5957–62. doi: 10.1073/pnas.1219232110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Dieci MV, Arnedos M, Andre F, Soria JC. Fibroblast growth factor receptor inhibitors as a cancer treatment: from a biologic rationale to medical perspectives. Cancer Discov. 2013;3:264–79. doi: 10.1158/2159-8290.CD-12-0362. [DOI] [PubMed] [Google Scholar]
- 63.Iyer R, et al. AZ64 inhibits TrkB and enhances the efficacy of chemotherapy and local radiation in neuroblastoma xenografts. Cancer Chemother Pharmacol. 2012;70:477–86. doi: 10.1007/s00280-012-1879-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Rusconi P, Caiola E, Broggini M. RAS/RAF/MEK inhibitors in oncology. Curr Med Chem. 2012;19:1164–76. doi: 10.2174/092986712799320510. [DOI] [PubMed] [Google Scholar]
- 65.Parkhomchuk D, et al. Transcriptome analysis by strand-specific sequencing of complementary DNA. Nucleic Acids Res. 2009;37(18):e123. doi: 10.1093/nar/gkp596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sultan M, et al. A simple strand-specific RNA-Seq library preparation protocol combining the Illumina TruSeq RNA and the dUTP methods. Biochem Biophys Res Commun. 2012;422(4):643–6. doi: 10.1016/j.bbrc.2012.05.043. [DOI] [PubMed] [Google Scholar]
- 67.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25 (14):1754–60. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Li H, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25 (16):2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ye K, Schulz MH, Long Q, Apweiler R, Ning Z. Pindel: a pattern growth approach to detect break points of large deletions and medium sized insertions from paired-end short reads. Bioinformatics. 2009;25:2865–2871. doi: 10.1093/bioinformatics/btp394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wang K, et al. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38 (16):e164. doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Dees ND, et al. MuSiC: identifying mutational significance in cancer genomes. Genome Res. 2012;22:1589–98. doi: 10.1101/gr.134635.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Nik-Zainal S, et al. Mutational Processes Molding the Genomes of 21 Breast Cancers. Cell. 2012;149:979–993. doi: 10.1016/j.cell.2012.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–842. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wang J, et al. CREST maps somatic structural variation in cancer genomes with base-pair resolution. Nat Methods. 2011;8(8):652–4. doi: 10.1038/nmeth.1628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Rausch T, et al. DELLY: structural variant discovery by integrated paired-end and split-read analysis. Bioinformatics. 2012;28 (18):i333–i339. doi: 10.1093/bioinformatics/bts378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kim D, Salzberg SL. TopHat-Fusion: an algorithm for discovery of novel fusion transcripts. Genome Biol. 2011;12(8):R72. doi: 10.1186/gb-2011-12-8-r72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.McPherson A, et al. deFuse: an algorithm for gene fusion discovery in tumor RNA-Seq data. PLoS Comput Biol. 2011;7(5):e1001138. doi: 10.1371/journal.pcbi.1001138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Sali A, Blundell TL. Comparative protein modelling by satisfaction of spatial restraints. J Mol Biol. 1993;234:779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.