

Abstract
During disease, infection, or trauma, the cytokine tumor necrosis factorα (TNFα) causes fever, fatigue, malaise, allodynia, anorexia, gastric stasis associated with nausea, and emesis via interactions with the central nervous system. Our studies have focused on how TNFα produces a profound gastric stasis by acting on vago-vagal reflex circuits in the brainstem. Sensory elements of this circuit (i.e., nucleus of the solitary tract [NST] and area postrema) are activated by TNFα. In response, the efferent elements (i.e., dorsal motor neurons of the vagus) cause gastroinhibition via their action on the gastric enteric plexus. We find that TNFα presynaptically modulates the release of glutamate from primary vagal afferents to the NST and can amplify vagal afferent responsiveness by sensitizing presynaptic intracellular calcium-release mechanisms. The constitutive presence of TNFα receptors on these afferents and their ability to amplify afferent signals may explain how TNFα can completely disrupt autonomic control of the gut.
Keywords: Immune-neural interactions, Gastric stasis, Visceral malaise, Hypersensitivity, Potentiation, Allodynia, Illness behavior
Immune system activation in response to infection or injury initiates a constellation of physiological and behavioral changes that include fever, fatigue, listlessness, loss of appetite, malaise, nausea, emesis, hypersensitivity to touch or pain, and significant changes in sleep patterns (Hermann, Holmes, and Rogers 2005). This Ȝsickness behaviorȝ represents an attempt by the host to reorganize its physiological priorities following injury or infection. For example with infection, an elevation in body temperature may help destroy the microorganisms, and the loss of appetite and emesis will minimize ingestion and cause elimination of pathogens and toxins. The reduction in activity caused by allodynia and increased sleep will act to immobilize the host, thus conserving available energy resources (Turrin and Plata-Salaman 2000).
These physiological changes are caused by the endocrine-like and paracrine-like actions of signal molecules called cytokines. Cytokines are released by immune effector cells as well as a variety of nonimmune cell types (e.g., Guzik and others 2006; Muller and Meineke 2007). The production of cytokines by the immune system in response to infection or injury plays a critical role in coordinating the host’s defense (e.g., Correa and others 2007; Kaisho and Akira 2006). The process is triggered by interactions with transmembrane proteins, Toll-like receptors that are mainly expressed on antigen-presenting cells such as macrophages or dendritic cells and provoke the release of proinflammatory cytokines (e.g., tumor necrosis factor–α, interleukin–1, interleukiu–6; Kaisho and Akira 2006). In addition to coordinating the immune attack, cytokines also cause the acute physiological reactions in the host, such as those considered part of “sickness behavior, ” to eliminate the “challenge” and restore homeostasis.
A number of noninfectious pathophysiological processes including autoimmune disorders (Chatzantoni and Mouzaki 2006), metastatic carcinoma (Kurzrock 2001), trauma (Beattie and others 2002), ischemia (Stoll, Jander, and Schroeter 2002), and radiation exposure (Muller and Meineke 2007) are also associated with elevated levels of TNFα. These disease processes are strongly associated with delayed digestive processes, visceral malaise, nausea, and emesis (e.g., Hermann, Holmes, and Rogers 2005; Kasner and others 2001; Turrin and Plata-Salaman 2000). In clinical trials in which TNFα levels have been elevated pharmacologically, profound nausea, cachexia, and emesis followed (e.g., Esper and Harb 2005; Moritz and others 1989). Recent work on the mechanism of action of TNFα within the CNS suggests that the severe disruption in autonomic control caused by this cytokine occurs as a result of direct effects on gastric regulatory circuitry in the caudal brainstem (Hermann, Holmes, and Rogers 2005; Rogers and others 2005; Rogers, Van Meter, and Hermann 2006).
Brainstem Visceral Autonomic Control
The dorsal vagal complex (DVC) constitutes the basic neural circuitry of the vago-vagal reflex control of gastrointestinal function (e.g., gastric motility, tone, and acid secretion). This dorsal hindbrain region is composed of the visceral sensory nuclei of the solitary tract (NST) and the area postrema (AP) and their interconnections with the dorsal motor nucleus of the vagus (DMV; Rogers and others 1995; Fig. 1). This region possesses vascular characteristics of a circumventricular organ with fenestrated capillaries and enlarged perivascular spaces (Gross and others 1990). Additionally, dendrites from neurons of the DVC have been seen to penetrate the ependymal layer and enter the floor of the fourth ventricle (Rogers and McCann 1993; Shapiro and Miselis 1985). Thus, this complex is in position to monitor blood-borne and cerebrospinal fluid–borne chemicals, hormones, peptides, cytokines, and toxins and evoke appropriate physiological responses (Rogers and others 1995).
Fig. 1.

Components of gastric vago-vagal reflexes. Top: Coronal histological section through caudal hindbrain. Components of the dorsal vagal complex have been high-lighted by the uptake of horseradish peroxidase via the vagus nerve. Bottom: Schematic describing essential details of vago-vagal gastric motility control reflexes. 1) Vagal mechanosensors from the esophagus, antrum, and duodenum are activated by contact with food. These afferents enter the brainstem and collect as the solitary tract. Vagal afferents terminate on neurons in the nucleus of the solitary tract (NST). 2) Neurons in the NST are activated by vagal afferents that release glutamate onto postsynaptic NST neurons. Different phenotypes of NST neurons direct different aspects of the efferent vagal response. 3) For example, inhibitory NST neurons (RED) use GABA or NE to inhibit tonically active and excitatory DMN neurons that normally act to increase gastric tone and motility. 4) Other excitatory NST neurons (GREEN: probably glutamatergic) activate an inhibitory (NANC) vagal efferent pathway to the stomach. 5) The net result is a vagally mediated inhibition of gastric motility. The esophageal-gastric relaxation reflex (Rogers and others 2005), for example, uses this essential circuitry. 6) This reflex circuitry is contained in a circumventricular region of the brainstem out-side the blood-brain barrier. Considerable evidence suggests that neural elements in this vago-vagal reflex network can be dramatically modulated by hormones, cytokines, and other chemical agents in the circulation that arrives via fenestrated capillaries (BLUE ARROWS). Not shown are the multitude of neural inputs from the brainstem and hypothalamus that adjust vagal reflex functions appropriate to changes in behavior and internal state, as well as parallel ascending projections from the NST associated with the control of feeding behavior (Berthoud 2002). Also not shown is the parallel reflex path connecting gastric distension with a vagally mediated increase in gastric acid output (Rogers and others 2005). AP = area postrema; DMN or DMV = dorsal motor nucleus of the vagus; NA = nucleus ambiguus; NST = nucleus of the solitary tract; ST = solitary tract of vagal fibers.
Nausea and vomiting are adaptive responses that have evolved to defend the body against toxins and pathogens incidentally ingested with food. However, both of these responses are inappropriate in clinical settings (such as during chemotherapy or radiation therapy) or noninfection pathologies such as metastasis or trauma, in that vomiting, under these circumstances, does not remove a “toxin.” Indeed, emesis and nausea significantly compromise clinical management of the primary disease process and often lead to strong aversions to further treatment (Andrews and Horn 2006).
Gastric hypomotility or stasis is commonly perceived as nausea and is a requisite for vomiting (Andrews and Horn 2006). It should not be surprising that the basic neural circuitry that regulates normal gastrointestinal function is also involved in generating emesis (Andrews and others 1990). Current theories of the mechanisms of nausea and emesis suggest that vagal afferents from the gut are hypersensitized by agonist substances such as serotonin and substance P (Andrews and Horn 2006).
Most vago-vagal control of gastric motility and tone is inhibitory. That is, activation of vagal afferents by distension of the stomach, intestine, or esophagus results in a marked reduction in gastric motility and tone (McCann and Rogers 1992; Zhang and others 1992); refer to the schematic in Figure 1. The predominant relationship is that activated vagal afferents excite NST neurons, which, in turn, inhibit spontaneously active neurons in the dorsal motor nucleus of the vagus (DMN). These spontaneously active DMN neurons, acting through the enteric plexus, normally provide the source of tonic cholinergic activation of the stomach. A second type of nonspontaneously active DMN neuron is activated by NST activation. This subgroup (referred to as the nonadrenergic, noncholinergic efferent pathway, or NANC) of DMN neurons normally provides gastric inhibition by also working through the enteric nervous system of the gut. The net result is that activation of NST neurons leads to potent inhibition of gastric function by coordinating the function of two parallel and antagonistic vagal pathways to the stomach (Hermann, Nasse, and Rogers 2005; Rogers and others 1999; Rogers and others 2003). An example of the normal function of this circuit is seen when a bolus of food is swallowed (i.e., causing esophageal distension). The fundus of the stomach transiently relaxes (i.e., drop in tone and/or motility) to allow the increase in gastric volume without an accompanying increase in pressure.
TNFα and Brainstem Control of Gastrointestinal Function
There is a strong correlation between pathological states that demonstrate gastric stasis, nausea, and emesis that also are associated with elevated, circulating TNFα levels (e.g., Beattie and others 2002; Chatzantoni and Mouzaki 2006; Hermann, Holmes, and Rogers 2005; Kasner and others 2001; Kurzrock 2001; Muller and Meineke 2007; Stoll, Jander, and Schroeter 2002; Turrin and Plata-Salaman 2000). These observations lead to the hypothesis that TNFα might act on the neural circuits that control gastrointestinal function.
Kinouchi and others (1991) published studies demonstrating that the CNS had binding sites for TNFα and that the brainstem had a particularly high density of such binding sites. Around this time period, it was demonstrated that TNFα could cross the blood-brain barrier via a specific and saturable transport system (Gutierrez and others 1993). Subsequently, Nadeau and Rivest (1999) reported the constitutive expression of messenger RNA for the type 1-TNF receptor (TNFR1) on circumventricular brain regions, although the residence of TNF-receptors on elements of the DVC had not been demonstrated.
Our initial studies demonstrated that microinjections of TNFα directly into the DVC produce a profound dose-related suppression of gastric motility. In these studies, strain gauges were secured to the antral portion of the stomach of anesthetized rats. Basal gastric motility and tone is minimal under anesthesia. Therefore, maximal gastric motility was evoked via vagal, cholinergic stimulation (i.e., application of 0.2 nanomoles of thyrotropin-releasing hormone [TRH] onto the floor of the fourth ventricle. TRH is normally released as a neurotransmitter within the DVC by afferent projections from the raphe nuclei. TRH directly activates DMN neurons that, in turn, excite the stomach, making this peptide a convenient tool for specifically activating vagal efferent pathways to the stomach [Rogers and others 2005]). This stimulated gastric motility could be rapidly suppressed by microinjections of TNFα directly into the DVC. This induction of gastric stasis had a threshold in the subfemtomolar range (0.02 femtomoles) and was dependent on intact vagal pathways (Hermann and Rogers 1995; Fig. 2).
Fig. 2.

Polygraph records of strain gauge activity monitoring gastric motility: Effects of TNFα. Basal gastric motility and tone of a food-deprived and anesthetized animal are minimal. Therefore, gastric motility was maximally stimulated by application of the peptide thyrotropin-releasing hormone (TRH; i.e., vagal, cholinergic stimulation) directly onto the exposed brainstem (TRH-icv). (A) Top trace: When stimulated gastric motility has plateaued (~ 5 to 10 minutes after TRH application), unilateral microinjection of 20nl of phosphate buffered saline (PBS; vehicle) was made into the left dorsal vagal complex (DVC). No change in motility was observed. Bottom trace: When the same volume of TNFα (0.02 femtomoles) was microinjected into the DVC, gastric motility was rapidly suppressed for prolonged periods of time (in excess of two hours in this example). (B) Central TNFα effects on gastric motility depend on intact vagal pathways. Polygraph tracing of gastric motility in a unilaterally vagotomized animal: Minimal basal gastric motility is maximally stimulated by application of TRH onto the brainstem surface (TRH-icv). Unilateral microinjection of the highest dose of TNFα (20 femtomoles) into the DVC of the vagotomized side is no longer effective in suppressing gastric motility. Adapted with permission from Hermann and Rogers (1995).
Mechanism of TNFα Action in the Brainstem to Cause Autonomic Dysfunction
Although the previously described studies were instructive about a possible site of action and dose range of TNFα to produce gastric stasis, it was necessary to demonstrate that endogenously and systemically produced TNFα could also evoke such effects. In this set of studies (Hermann and others 1999), endogenous production of TNFα (and other cytokines) was provoked by the systemic administration of lipopolysaccharide (LPS). Without the availability of a specific antagonist for TNFα, we relied on a TNFα-specific adsorbing construct, etanercept (Enbrel, Amgen), to suppress the effects of TNFα. hi this case, application of the TNF-adsorbant to the fourth ventricular surface (i.e., directly above the DVC) blocked both the TNF-induced activation of NST neurons (as demonstrated by the activation of cFOS; compare left and right panels of Fig. 3) and the induction of gastric stasis (Fig. 4) that was normally elicited by systemic LPS challenge (Hermann and others 2002; Hermann and others 2003). These data verified that peripherally generated, endogenous TNFα can gain access to neurons of the DVC and act there to inhibit gastric motility.
Fig. 3.

Endotoxin challenge provokes production and release of proinflammatory cytokines including TNFα into the systemic circulation. Pretreatment with the TNF-adsorbant construct (TNFR:Fc) directly within the dorsal vagal complex blocked cFOS-activation (right panel) of NST cells in response to systemic lipopolysaccharide (LPS) exposure. In contrast, pretreatment with only the Fc fragment did not prevent cFOS-induction (left panel). The middle panel outlines structures of interest in adjacent panels. AP = area postrema; DMN = dorsal motor nucleus of the vagus; NST = nucleus of the solitary tract; ST = solitary tract. Scale bar = 400 micron. Adapted with permission from Hermann and others (2003).
Fig. 4.

Raw polygraph records of strain gauge activity monitoring gastric motility. Both records were obtained at ~90 minutes after systemic (IV) administration of lipopolysaccharide (LPS) i.e., circulating proinflammatory cytokine levels are elevated. (A) This animal had received LPS (IV) and continuous ventricular perfusion (intracerebroventricular [ICV]) of the Fc fragment vehicle control. ICV application of thyrotropin-releasing hormone (TRH) is unable to override the suppressive effects of the endogenously produced TNFα. (B) This animal received LPS (IV) and continuous ventricular perfusion of the TNFR:Fc adsorbant construct (ICV). Presumably, the endogenously produced TNFα is adsorbed and effectively neutralized as it passes through the ventricular flow. In this case, application of TRH to the floor of the fourth ventricle is able to maximally stimulate gastric motility. Adapted with permission from Hermann and others (2002).
Up-regulation of the immediate early gene product, cFOS, serves as a marker for neuronal activation (Rinaman and others 1993). Activation of NST neurons by TNFα was demonstrable using immunohistological techniques to stain for cFOS generation (Fig. 3 and Fig. 5; Emch and others 2001). Given that the majority of afferents to the NST are glutamatergic (Torrealba and Muller 1999), we hypothe-sized that one means of activation of NST neurons by TNFα may be via its interaction with glutamatergic vagal afferents. To test this concept, we co-injected TNFα along with antagonists of glutamate (e.g., NBQX or MK-801) directly into the DVC and monitored cFOS activation. These glutamatergic antagonists suppressed TNFα-induced cFOS generation in NST neurons, suggesting that TNFα may act to alter vago-vagal reflex functions through presynaptic modulation of glutamate release from afferents (compare Fig. 5B with C or D; Emch and others 2001) or amplify the influence of glutamate through rapid trafficking of AMPA glutamate receptors on postsynaptic neurons (Pickering and others 2005).
Fig. 5.

Photomicrographs of coronal histological sections through the NST immunohistochemically processed for cFOS-activation. Immunoreactivity for cFOS-activation is characterized by darkly stained nuclei. (A) control injection of PBS (20nL) directly into the DVC results in few cFOS-(+) cells in the NST and DMN; (B) injection of TNFα (0.2 femtomoles) induces a significant increase in cFOS expression; (C) co-injection of the same amount of TNFα with the AMPA glutamate receptor antagonist, NBQX, suppresses cFOS production to control levels; (D) co-injection of TNFα with the NMDA antagonist, MK-801, also reduces cFOS activation. These data support the hypothesis that TNFα may excite NST neurons by increasing presynaptic (i.e., vagal afferents) glutamate neurotransmission. AP = area postrema; DMN = dorsal motor nucleus of the vagus; NST = nucleus of the solitary tract; ST = solitary tract. Adapted with permission from Emch and others (2001).
Recent studies of the mechanics of reflex control of the stomach have revealed that noradrenergic neurons in the NST and their connections with gastric vagal efferents are especially important to inhibitory vago-vagal control of the stomach (Rogers and others 1999; Rogers and others 2003). These same neurons may also be important to the development of toxin-induced or CCK-induced anorexia (Rinaman 2003). This confluence of observations showing that TNFα may regulate gastric function by acting within the NST and that noradrenergic neurons may be particularly important to the coordination of inhibitory reflex control lead us to consider that TNFα action within the NST may preferentially affect putative noradrenergic neurons.
Our preliminary double immunohistochemical studies (manuscript in preparation) looking at the phenotype of NST neurons that are activated (i.e., cFOS-positive) by exposure to TNFα reveal a significant and dose-dependent elevation in cFOS expression in NST neurons that also contain the enzyme tyrosine hydroxylase (i.e., indicative of adrenergic neurons; Fig. 6). In addition to involvement in inhibitory gastric reflex control (Rogers and others 1999; Rogers and others 2003), dorsal medullary noradrenergic neurons may be generally involved in processing anorexigenic stimuli that not only can affect gastric function but can support aversive conditioning and otherwise severely depress feeding behavior (Rinaman 2003). Together, these results suggest that TNFα action at vagal afferents may selectively activate catecholamine neurons in the NST that are involved in gastric reflex control as well as in the generation of anorexia and aversion conditioning.
Fig. 6.

TNFα activation of adrenergic cells in the nucleus of the solitary tract (NST). Preliminary data suggest that TNFα may specifically cause the activation of adrenergic neurons in the NST. This figure shows that microinjection of TNFα into the medial NST provokes cFOS activation (seen as brown nuclear staining) of virtually every adrenergic neuron (tyrosine hydroxylase [Th]–positive immunore-activity is seen as black cytoplasmic staining) in the area. These neurons do not express cFOS at rest nor fallowing vehicle (PBS) microinjection (not shown here, but see Fig. 5).
TNFα Potentiation of Vago-Vagal Reflex Responsiveness
Electrophysiological studies (Emch and others 2000; Emch and others 2002) have shown that nano-injection of TNFα into the DVC produces an activation of identified NST (sensory) neurons (Fig. 7) and an inhibition of identified DMN (motor) neurons (Fig. 8). According to the basic vago-vagal reflex circuit outlined in Figure 1, these neurophysiological observations are consistent with the physiological observations of gastroinhibition. That is, activation of NST neurons that form the afferent limb of vago-vagal gastric control circuitry causes a reflex gastroinhibition mediated by vagal efferents (Rogers and others 2005). Of particular interest was the observation that sensory neurons exposed to TNFα displayed a hypersensitivity to natural stimulation that out-lasted the exposure to the TNFα. That is, identified NST neurons (i.e., those neurons that were responsive to gastric distension) that were briefly exposed to small amounts of TNFα (0.03 femtomoles; 5-second exposure) were hyperresponsive to subsequent exposures to identical magnitudes of gastric distension (Emch and others 2000); compare Figure 7A and 7C. This prolonged potentiation of the NST response to stimulation after exposure to TNFα may provide a basis by which elevated circulating levels of TNFα (e.g., during illness) may amplify visceral sensory input to the brain. In the case of vago-vagal reflex circuits, this enhanced vagal afferent input to the NST would, ultimately, evoke gastroinhibition (refer to Fig. 1 and 2).
Fig. 7.

Effects of gastric distension, PBS, or TNFα microinjection on a single neuron of the nucleus of the solitary tract (NST). A–C are records from the same neuron. (A) Upper trace on left: Raw oscillograph record of the identification of a gastric distension related neuron in the NST. Distension of the antrum (lower trace) produces a brisk increase in the firing rate of the NST neuron; phase-locked to the stimulus. Upper trace on right: Integrated rate-meter record of the same event. Inset: 20 superimposed NST spikes; profile of identified NST neuron responding to gastric distension alone. (B) integrated record of neuronal activity. (Upper) Control injection of PBS (3nL delivered at arrow) has no effect on the neuron’s firing rate; same neuron as recorded in A. Inset shows the neuron’s signature spike profile. (Lower) TNFα (0.03 femtomoles) microinjected onto the same NST neuron causes activation of identified neuron. Scale bar = 1 minute. (C) Response to gastric distension of the same NST neuron depicted in A, now 30 minutes after exposure to and recovery from the TNFα injection seen in B. Note the greatly potentiated response to gastric distension that significantly outlasts the stimulus. Inset shows superimposed, individual NST spikes identifying this neuron as the same as that seen in A. Scale bars for insets: 200uV/2.5msec. Adapted with permission from Emch and others (2000).
Fig. 8.
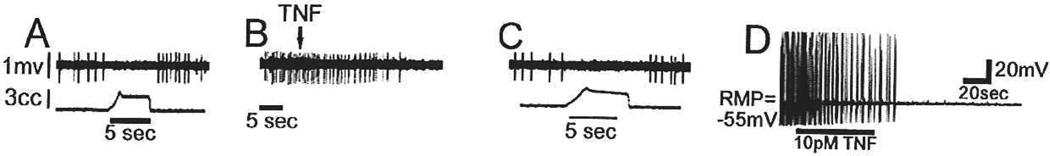
TNFα inhibits tonically active gastric dorsal vagal motor nucleus (DMN) neurons. (A) A raw oscilloscope spike record shows a reduction in neuronal activity (upper trace) during gastric balloon distension (lower trace), identifying the cell as a DMN neuron. (B) Response of the same DMN neuron to application of TNFα (0.03 femtomoles) at the arrow (note the difference in lime scales); spontaneous activity is inhibited. (C) One hour after TNFα application, DMN firing rate of the same neuron in response to gastric distension returns to normal. (D) Intracellular recordings of in vitro brain slices also demonstrate that spontaneous activity of DMN neurons is inhibited by TNFα (10pM). Adapted with permission from Emch and others (2001).
Constitutive Expression of TNF-Receptors on Vagal Afferents
There are two cell surface receptor types for TNFα: TNF receptor type-1 (TNFR1; also referred to as p55, p60, or CD 120a) and TNF receptor type-2 (TNFR2; also referred to as p75, p80, or CD 120b). The TNFR1 receptor possesses the domain responsible for the cascade of cellular signaling molecules that can affect a myriad of cellular functions through stress-activated kinase pathways. Under some circumstances, TNFR1 receptor activation also initiates apoptosis through caspase pathways (e.g., Shamash and others 2003). However, the TNFR2 receptor may contribute to the initiation of this cascade via a couple of mechanisms (Chan and Lenardo 2000). These type 2 receptors also exist in a soluble form (e.g., sTNFR2). As such, sTNFR2 is thought to function as a ligand-passing molecule (Tartaglia and others 1993), thus acting as a TNFα-transport molecule subserving TNFR1 activation. In the capacity of a TNFα-transport molecule, one can hypothesize a role for the sTNFR2 receptor to prolong the inflammatory response by the intermittent delivery and release of TNFα to TNFR1. There have also been reports that the TNFR2 receptor may play an important signaling role in chronic inflammatory conditions such as Crohn’s disease (Holtmann and others 2002).
Although there have been reports of high density of binding sites for TNFα within the brainstem (Kinouchi and others 1991) and constitutive expression of messenger RNA for the TNFR1 on circumventricular organs (Nadeau and Rivest 1999), specific details of the residence of TNF-receptors on elements of the DVC was only demonstrated recently.
The immunohistochemical demonstration of TNF receptors on vagal afferents (Hermann and others 2004) required the use of a heat-induced antigen retrieval technique. (Details about this immunohistochemical technique can be found in Hermann and others [2004] and its references.) This protocol allowed us to demonstrate that dense TNFR1-immunoreactivity (TNFR1-ir) is constitutively present on central (but not peripheral) vagal afferents in the solitary tract and nucleus (Fig. 9A–9C as well as on afferents entering the spinal trigeminal nucleus (Fig. 9F–9H). Fibers in cross-section present as fine filamentous strands of dark reaction product as they diverge to enter the medial solitary nucleus located dorsal to the DMN at the level of the AP. Adjacent histological sections from the same animal indicate that only TNFR1, and not TNFR2, is constitutively expressed in the fibers of the solitary tract as well as spinal trigeminal and AP afferents (refer to original manuscript—Hermann and others [2004]).
Fig. 9.

Constitutive expression of TNF receptor type 1 immunoreactivity (TNFR1-ir) on afferents in the medullary brainstem of the rat. (A) Ponto-medullary junction area illustrating the entry of vagal sensory nerve fibers staining positively for TNFR1-ir. Trigeminal afferents in the spinal nucleus of the trigeminal nerve are also TNFR1-ir positive. (B) Vagal afferent fibers in the solitary tract (ST) staining for TNFR1-ir. (C) Vagal afferent fibers diverging from the ST and entering the medial solitary nucleus (mNST). (D) TNFR1-ir positive vagal afferent fibers continue through the mNST and ramify widely through the area postrema (higher magnification in inset on left). (E) TNFR1-ir on afferents in the dorsal root entry zone (the section shown is the cervical spinal cord). (F) TNFR1-ir labeled trigeminal afferent fibers penetrate the surface of the inferior cerebellar peduncle (ICP) and diverge within the spinal trigeminal nucleus (SN V). (G) Higher power detail of TNFR1-ir–positive fibers on the brainstem surface penetrating the ICR (H) Higher power detail of TNFR1-ir afferent fibers in the SN V. Abbreviations: AP = area postrema; ICP = inferior cerebellar peduncle; mNST = medial solitary nucleus; SN V = spinal nucleus of trigeminal nerve; ST = solitary tract; X aff = vagal sensory nerve fibers. Scale bars: F = 1000 micron; A = 500 micron; B, C, G, H = 100 micron; D = 200 micron; E = 20 microns. Adapted with permission from Hermann and others (2004).
Curiously, TNFR1-immunoreactivity was not observed in the peripheral vagal trunks, even though it was observed in the nodose ganglion and the centripetal vagal fibers that leave the ganglion and proceed to the brainstem (see Hermann and others 2004). This observation suggests that peripherally generated TNFα does not require the peripheral vagus to activate neurons in the NST (this result coincides well with our previous physiological work: Hermann, Emch, and others 2001; Hermann and others 2002; Hermann and others 2003) but can readily access this part of the hindbrain.
Note that TNFR1 is also present in dorsal root afferents entering the dorsal horn of the spinal cord (Fig. 9E). Its presence there has been linked to the effect of TNFα to increase sensitivity to pain, touch, and temperature. Spinal afferents from the viscera also enter the dorsal horn, and these afferents relay data about overdistension, irritation, ischemia, and chemical irritation—that is, visceral pain (Beyak and others 2006). In view of the previous discussion, one would expect that TNFα might also sensitize visceral pain afferents and the spinal autonomic processes that regulate intestinal secretomotor function. Indeed, a couple of studies describe a co-incidence between elevated levels of TNFα and either a reduced threshold of distension-pain–induced contractions of the large intestine (Coelho and others 2000) or visceral-rectal hypersensitivity (Liebregts and others 2007).
The presence of receptors such as TNFR1 on afferent fibers and presumptive terminal endings could play a significant role in determining the “gain” or “volume” of normal incoming sensory information. A consequence of this enhanced dynamic range of sensory processing could explain hypersensitivity or elicit different-motor responses to otherwise similar stimuli.
Mechanism of Action of TNFα at Vagal Afferent Terminals in the NST
Agonist effects on central vagal afferents have been inferred by a combination of methods. lmmunohistochemical demonstration of agonist receptors on vagal afferents supports the hypothesis that agonists directly modulate vagal afferent neurotransmission. Electrical recordings and calcium imaging as well as histochemical studies of explant cultures of nodose ganglion cells (i.e., cell bodies of origin for central vagal afferents) reveal that these cells possess functional receptors for a number of potential signal molecules (e.g., Corp and others 1993; Doyle and others 2002; Ellacott and others 2002; Glass and Pickel 2002; Hermann and others 2004). However, results obtained from these studies could be subject to interpretive difficulties. For example, the receptor population in the nodose cell body may not accurately reflect the distribution or functionality elsewhere in the afferent fiber or terminal; the nodose receptor population may not be exposed to the agonist under physiological conditions; the transduction machinery in the terminal may not be the same as in the cell body; or the extraction and culture methods used to prepare nodose cells may significantly alter receptor population and functionality (e.g., Hermann and others 2004; Lancaster and others 2001).
Results from in vitro whole cell voltage and current clamp studies of NST neurons strongly imply that some agonists act directly on vagal afferents (Jin and others 2004). The essential effects of vagal input to NST neurons studied using these methods (i.e., the generation of glutamatergic postsynaptic currents) are not controversial. However, observations of the modulation of that process can be open to interpretation given that agonists and drugs acting on receptors and transduction processes in the presynaptic terminal may also act on the recorded postsynaptic cell.
An additional anatomical feature complicates the physiological analysis of vagal afferent inputs to NST neurons. Whereas other in vitro model systems such as the hippocampus, cerebellum, tectum, and cortex possess highly structured and laminar relationships between afferent inputs and neurons receiving those inputs, the vagal-NST relationship is diffuse (Doyle and others 2004). As a consequence, imaging and neurophysiological methods appropriate for study of en mass discharge properties of a whole field of well-structured presynaptic afferents must be replaced by methods capable of imaging, at a minimum, small clusters of varicosities. This may be accomplished by taking advantage of the ability to track the flux of calcium reporter dyes as an index of cellular activity.
The dominant factor controlling transmitter release from presynaptic terminals is cytoplasmic calcium (Fill and Copello 2002). The presynaptic calcium pool linked to vesicle exocytosis is under the control of several factors such as voltage-gated and ligand-gated calcium channels on the presynaptic membrane as well as intracellular calcium storage pools in the endoplasmic reticulum. The regulation of these sources of cytoplasmic calcium is responsible for short-term modulation of synaptic transmission (Fill and Copello 2002; Lelli and others 2003). Therefore, visualization of terminal and preterminal cytoplasmic calcium levels in vagal afferents in the NST, in situ, could provide a powerful tool to investigate the transduction mechanisms by which agonists affect change in vagal afferent synaptic transmission. Additionally, these calcium-imaging methods may be used in adult rodents, whereas in vitro whole-cell patch or intracellular recording methods are usually restricted to neonates and juveniles because of the overgrowth of the neuropil and glia. The development of methods for direct analysis of vagal afferent terminal calcium responses yielded a powerful tool for examining presynaptic regulatory mechanisms (Rogers, Nasse, and Hermann 2006).
To perform these studies, vagal afferent cell bodies in the nodose ganglion were prefilled with the calcium reporter dye calcium green 1-dextran. This dye is transported to the terminals in the NST within two days, at which time, medullary slices are prepared for in vitro live cell imaging (Rogers, Nasse, and Hermann 2006; Rogers, Van Meter, and Hermann 2006).
By applying calcium-imaging methods to vagal afferent terminal varicosities in vitro, we find that TNFα has a weak effect, at best, to directly increase the presynaptic calcium signal, hence, transmitter release. However, TNFα strongly amplifies terminal calcium signals produced by other agonists (such as ATP) that also have their site of action at the terminal (Fig. 10). ATP activates a transmembrane calcium flux by causing P2X3 ligand-gated cation channels to open. As a consequence, ATP causes glutamate release from vagal afferents onto second-order NST neurons (Jin and others 2004). Although the direct entry of calcium into the terminal region certainly activates the transmitter release mechanism, entering calcium can also serve as a trigger for the release of endoplasmic reticulum (ER)–stored calcium through the calcium-induced calcium release (CICR) mechanism (Bardo and others 2006); see Figure 11. Two parallel processes regulate this release of stored calcium: ryanodine channels and the lP3-calmodulin release mechanism (Hoesch and others 2002). Calcium entering the terminal interacts with binding sites on the ryanodine channel to provoke release of ER calcium stores. (Note the unusual naming of the ryanodine channel based on the name of the channel antagonist, ryanodine; this could be the basis for unnecessary confusion.) Our results suggest that TNFα operates predominantly via the ryanodine channel stores release mechanism (i.e., ryanodine blocks the TNFα effect on ATP-evoked calcium release). In contrast, U73122, an lP3 synthesis inhibitor, does not interfere with the TNFα effect. Whereas our immunohistochemical studies demonstrated that both receptor types are expressed on central vagal afferent fibers, our pharmacological studies indicated that the lP3 receptor did not play a role in TNFα modulation of the ATP-evoked calcium mobilization.
Fig. 10.

Modulation of vagal afferent terminals responses to ATP by exposure to TNFα. A low-power field brainstem slice contains a section of the nucleus of the solitary tract (NST); numerous calcium green-labeled central vagal afferent terminals are visible. A–D depict the exact same field of this brainstem slice under different perfusion conditions. A and B were taken before exposure of the slice to 1nM TNFα; C and D were taken just after 10-minute exposure to TNFα. (A) Varicosities at rest just before perfusion of the slice with ATP (100uM). (B) Varicosities in the same field as A showing the peak effect of ATP. (C) Varicosities at rest. (D) Varicosities in the same field as C showing the peak effect of ATP after the slice was exposed to TNFα. (E) Relative fluorescence plot of the initial response to ATP to elevate the calcium signal in the varicosity that is indicated by the arrow (see B) before TNFα exposure. (F) Relative fluorescence plot of the response to ATP to elevate the calcium signal in the same varicosity indicated by the arrow (see D) after TNFα exposure. This TNF amplification of the calcium signal is sensitive to ryanodine (see Fig. 11). Adapted with permission from Rogers, Van Meter, and Hermann (2006).
Fig. 11.

TNFα can amplify terminal calcium signaling by activating ryanodine channels. ATP acts at terminal ligand-gated (P2X3 receptor) cation channels to produce an increase in intraterminal calcium. This increase, in turn, activates calcium-induced calcium release (CICR) mechanisms. TNFα apparently amplifies this effect via the generation of cAOP ribose (cADPR), a known ryanodine channel agonist molecule. CD38 generates cADPR; TNFα drives CD38 transcription (Iqbal and others 2006) and may enhance CD38 trafficking. Adapted with permission from Rogers, Van Meter, and Hermann (2006).
The mechanisms by which agonist molecules affect the function of ryanodine receptors, hence, presynaptic potentiation and depression, are incompletely understood. However, studies of the relationship between TNFα and ryanodine receptor function in nonneural tissues strongly suggest that TNFα can increase the expression of the transmembrane ectoenzyme ADP-ribosyl-cyclase (also called CD38; Deshpande and others 2003). CD38 generates cADP ribose (cADPR). This product is a potent activator of the ryanodine channel. cADPR, acting on presynaptic ryanodine channels, may serve as a potent positive modulator of CNS neurotransmission (Barata and others 2004; Collin and others 2005). Our studies show that 8-Br-cADPR, a selective antagonist of cADPR, completely blocks the action of TNFα to augment calcium signaling in vagal afferents. Although this result suggests a connection between TNFα receptor activation and up-regulation of CD38-mediated cADPR production, the time course of the effect is problematic. Whereas CD38 transcription can be up-regulated by exposure to TNFα in as little as three hours (Iqbal and others 2006), the effects we observe here develop within minutes.
It has been suggested that the effectiveness of CD38 in regulating cytoplasmic calcium is a function of the delivery of substrate (NAD+) and the destination of the cADPR product. Connexin hemichannels or nucleoside transporters (De Flora and others 2004) may perform these functions. Trafficking of the CD38 molecule itself has also been suggested as a mechanism for controlling the production of cADPR, hence, the activity of the ryanodine channel (De Flora and others 2004). Although the connection between TNF receptor activation and the rapid regulation of CD38 is not presently clear, convincing evidence suggests that TNFα can potently affect neurotransmission through rapid control of receptor trafficking (Pickering and others 2005).
TNFα and Neurodegeneration
Although the principal thrust of this review has been the role of the TNF receptor to augment signaling in the dorsal vagal complex, a substantial literature suggests that under some circumstances, activation of the TNFR1 receptor is a key step in the initiation of neuronal apoptosis after injury (e.g., Beattie and others 2002; Hermann, Rogers, and others 2001; Shamash and others 2003; Stoll, Jander, and Myers 2002). Indeed, vagal efferent neurons are among the few that completely degenerate after peripheral axotomy (Laiwand and others 1987), and it is very likely that the TNPR1 receptor is involved in the process that eliminates these visceral motorneurons after damage.
Recall from our earlier discussion that constitutive TNFR1 immunostaining was predominantly on vagal afferent fibers and varicosities in the DVC. Under normal circumstances, immunostaining was not apparent above background levels in the neurons of the NST or DMN. However, damage to the DMN (via vagotomy) caused a substantial expression of the receptor that is correlated with the disappearance of DMN neurons over time.
The nodose ganglion (i.e., the source of the cell bodies of origin for vagal afferents) surrounds fibers of the efferent vagus. At the nodose ganglion, the vagus is already a mixed sensory-motor nerve. Cutting the vagus distal to the nodose ganglion leaves primary afferent vagal fibers entering the brainstem intact and connected with their respective cell bodies; cutting the vagus proximal to the nodose disconnects central vagal afferents from their cell bodies and causes them to degenerate. However, in either case (proximal or distal section), the efferent vagal fibers are cut. Supra-nodose vagotomy effectively disconnects the primary afferent cell bodies from central afferent fibers. Following such a vagotomy, there was a complete absence of TNFR1-ir in the afferent fibers of the solitary tract and in the solitary nucleus (Fig. 12).
Fig. 12.

Neurodegeneration after vagotomy. (A) Montage of micrographs illustrating the overall effects on TNF receptor type 1 immunoreactivity (TNFRI-ir) staining within the medulla two weeks after unilateral vagotomy (left side) at the supra-nodose level. Supra-nodose vagotomy effectively disconnects the primary afferent cell bodies from central afferent fibers. These unilateral vagotomies allowed each animal to serve as its own control; comparisons between left (vagotomized) and right (control) sides could be made directly. (Right) Control side: brainstem side contralateral to the vagus nerve section. Note the constitutive presence of TNFR1-ir vagal afferents in the solitary tract (ST) and tenth nerve (X) afferent pathway. Also note the lack of TNFR1-ir in the dorsal motor nucleus of the vagus (DMN) on this side of the brainstem. (Left) Side ipsilateral to the supra-nodose vagotomy. Note the complete absence of TNFR1-ir in the afferent fibers of the ST on this side. In contrast, the neurons in the DMN on this side show enhanced TNFR1-ir. Following vagotomy there is also an increase in staining of the efferent fibers leaving the DMN neurons (at *) relative to the control side. Scale bar = 0.5 mm. (B) Photomicrographs of medullary brainstem sections demonstrating the time course of the up regulation of TNFR1-ir in DMN neurons on the side ipsilateral to the vagotomy (left side) relative to the control (right) side. Within three days following unilateral vagotomy, neurons of the DMN (and nucleus ambiguus [NA], data not shown), ipsilateral to vagal section, demonstrated a dramatic increase in TNFR1-ir throughout the entire rostro-caudal extent of these nuclei. This increase in the number of neurons expressing TNFR1-ir was quantified across time postvagotomy; the number of neurons expressing TNFR1-ir in each nucleus peaked at one week and began to decline over time. The absolute number of DMN neurons on the vagotomized side also declined over time. Panels i–v illustrate changes in the appearance of TNFR1-ir in DMN neurons on the side of the vagotomy (left) versus control (right) over the course of time. Scale bar = 500 microns. (C) High-power views of the DMN overtime. Note Wallerian degeneration of the DMN as characterized by the large, eccentric nuclei. Scale = 100 micron. Adapted with permission from Hermann and others (2004).
Within three days after unilateral vagotomy, vagal efferent neurons ipsilateral to the vagotomy demonstrated a dramatic increase in TNFR1-ir throughout the entire rostro-caudal extent of these nuclei (i.e., in both the dorsal vagal motor nucleus [DMN; Fig. 12] and the nucleus ambiguus [NA]; refer to Hermann and others 2004). This increase in the number of neurons expressing TNFR1-ir was quantified across time postvagotomy. The number of neurons expressing TNFR1-ir peaked at one week and began to decline over time. This decline in the number of TNFR1-ir neurons corresponded to a drop in the total number of vagal motorneurons on the vagotomized side. At higher magnification, these neurons show signs of Wallerian degeneration (Shamash and others 2003) as characterized by the large, eccentric nuclei (Fig. 12: lower panel). This increase in TNFR1-ir in axotomized neurons may play a pivotal role in the connection between the occurrence of the injury and the initiation of processes (apoptotic or necrotic) resulting in elimination of damaged neurons (Hermann and others 2004).
The role of TNFα as a potential mediator of posttrauma neurodegeneration is now well established (e.g., Hermann and others 2004; Schafers and others 2002; Shubayev and Myers 2002; Stoll, Jander, and Myers 2002). TNFα levels (as well as receptor expression) rise in the vicinity of nerve fibers and their cell bodies following axon injury, probably as a consequence of the recruitment of cytokine-secreting macrophages into the immediate surround of the damaged cell (Griffin and others 1993). TNFα action through the TNFR1 receptor initiates a cascade of events resulting in the degradation of the axonal cytoskeleton and neuronal apoptosis (Shamash and others 2003). TNFα can accelerate macrophage-induced damage by up-regulating the expression of intercellular adhesion molecules, making macrophage-neuron interactions more efficient. This effect is mediated by the TNFR1 receptor (Schafers and others 2002). Our results suggest that degenerative changes in vagal motor neurons could be further potentiated after injury by a significant up-regulation of the TNFR1 receptor itself.
The details regarding the connection between cell injury and the up-regulation of the TNFR1 receptor is not clear, but a possible pathway may be inferred from recent investigations. It has been shown (Ji and others 2002) that vagotomy causes the rapid up-regulation of the NMDA—calcium-nNOS signal pathway in DMN neurons ipsilateral to vagus nerve section. This same signal pathway has, in turn, been implicated in the up-regulation of several apoptotic gene products including the TNFR1 receptor (Laabich and others 2001). It is now well known that TNFα binding with p55 (i.e., TNFR1) receptors can initiate apoptosis through the caspase family (Haviv and Stein 1998). Caspase-3 is also up-regulated in DMN neurons ipsilateral to the vagal trunk section (Ji and others 2002).
About the time that we began formulating the hypothesis that TNFα caused a sensitization of vagal afferent terminal glutamate release (Emch and others 2000; Emch and others 2001), we also observed the connection between damage to the peripheral vagus, expression of the TNFR1 receptor in the DMN, and the correlated elimination of vagal motor neurons. Given that TNFα levels are elevated after CNS injury (e.g., Beattie and others 2002; Griffin and others 1993), perhaps neurodegeneration is exacerbated by TNF-induced release of glutamate and damage-induced increased expression of TNFR1; in essence, another example of amplification. To test this hypothesis, we microinjected small amounts (3.5 femtomoles) of TNFα into the spinal cord and observed dramatic cFOS activation of neurons, astrocytes, and microglia in the grey matter surrounding the injection site (Hermann, Rogers, and others 2001). This same dose of TNFα provoked neurodegeneration within 90 minutes when paired with a benign dose (150 pmoles) of the glutamate agonist kainate. The neurodegeneration was blocked by AMPA-glutamate antagonists. These results supported the hypothesis that TNFα may act within the CNS to provoke presynaptic glutamate release, and perhaps, to increase the strength of postsynaptic glutamate signaling in normal and pathophysiological conditions (Pickering and others 2005). This effect would, of course, be accelerated by the overexpression of TNFR1 receptors that also occurs after neuronal trauma (Hermann and others 2004).
Overview of TNFα Effects on Visceral Sensory Processing
Factors that elicit TNFα release also cause a profound autonomically mediated hypersensitization of gastrointestinal control reflexes. At the level of the intact animal, the result is a dramatic suppression of gastric motility, tone, and transit that is accompanied by the perception of nausea, and, frequently, emesis. The actions of TNFα are directed principally at the central afferent projections of the vagus nerve, which terminate in the NST. TNFα acts at these afferent terminals to increase cytosolic calcium concentrations through action on ryanodine channels. The net effect of this change in calcium flux is the amplification of glutamate release from these terminals through sensitization of terminal calcium-induced calcium release (CICR) mechanisms. The propagation of this amplification of NST neurons’ responsiveness to other incoming information is reflected in the DVC reflex network to ultimately produce the observed effects on the stomach (e.g., gastric stasis, perception of malaise). TNFα can also sensitize (trigeminal and spinal somatosensory and visceral afferents, presumably through a similar mechanism. The end result for somatosensory sensation is allodynia. The effect on visceral sensation mediated by spinal-visceral afferents is probably a lowered threshold for visceral pain and for distension-induced contractions of the intestine. Although TNFα adsorbent compounds (e.g., Enbrel) show promise in relieving many of the CNS reflex-mediated ill effects of TNFα production, there are very few reports in the literature available on which an evaluation can be made.
Acknowledgments
The authors thank John and Erna Hermann, Richard and Lois Rogers, Karen Rogers Wadkins, and Kathleen Rogers for their encouragement and inspiration. Grant support: National Institutes of Health Grants DK52142, DK56373, and HD47643.
References
- Andrews PL, Davis CJ, Bingham S, Davidson HI, Hawthorn J, Maskell L. The abdominal visceral innervation and the emetic reflex: pathways, pharmacology, and plasticity. Can J Physiol Pharmacol. 1990;68:325–345. doi: 10.1139/y90-047. [DOI] [PubMed] [Google Scholar]
- Andrews PL, Horn CC. Signals for nausea and emesis: implications for models of upper gastrointestinal diseases. Auton Neurosci. 2006;125:100–115. doi: 10.1016/j.autneu.2006.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barata H, Thompson M, Zielinska W, Han YS, Mantilla CB, Prakash YS, et al. The role of cyclic-ADP-ribose-signaling pathway in oxytocin-induced Ca2+ transients in human myometrium cells. Endocrinology. 2004;145:881–889. doi: 10.1210/en.2003-0774. [DOI] [PubMed] [Google Scholar]
- Bardo S, Cavazzini MG, Emptage N. The role of the endoplasmic reticulum Ca2+ store in the plasticity of central neurons. Trends Pharmacol Sci. 2006;27:78–84. doi: 10.1016/j.tips.2005.12.008. [DOI] [PubMed] [Google Scholar]
- Beattie MS, Hermann GE, Rogers RC, Bresnahan JC. Cell death in models of spinal cord injury. Prog Brain Res. 2002;137:37–47. doi: 10.1016/s0079-6123(02)37006-7. [DOI] [PubMed] [Google Scholar]
- Berthoud HR. Multiple neural systems controlling food intake and body weight. Neurosci Biobehav Rev. 2002;26:393–428. doi: 10.1016/s0149-7634(02)00014-3. [DOI] [PubMed] [Google Scholar]
- Beyak MJ, Bulmer DCE, Jiang W, Keating C, Rong W, Grundy D. Extrinsic sensory afferent nerves innervating the gastrointestinal tract. In: Johnson LR, editor. Physiology of the gastrointestinal tract. 4th ed. San Diego (CA): Academic Press; 2006. pp. 685–726. [Google Scholar]
- Chan FK, Lenardo MJ. A crucial role for p80 TNF-R2 in amplifying p60 TNF-R1 apoptosis signals in T lymphocytes. Eur J Immunol. 2000;30:652–660. doi: 10.1002/1521-4141(200002)30:2<652::AID-IMMU652>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- Chatzantoni K, Mouzaki A. Anti-TNF-alpha antibody therapies in autoimmune diseases. Curr Top Med Chem. 2006;6:1707–1714. doi: 10.2174/156802606778194217. [DOI] [PubMed] [Google Scholar]
- Coelho A-M, Fioramonti J, Buéno L. Systemic lipopolysaccharide influences rectal sensitivity in rats: role of mast cells, cytokines, and vagus nerve. Am J Physiol Gastrointest Liver Physiol. 2000;279:G7819–G7890. doi: 10.1152/ajpgi.2000.279.4.G781. [DOI] [PubMed] [Google Scholar]
- Collin T, Marty A, Llano I. Presynaptic calcium stores and synaptic transmission. Curr Opin Neurobiol. 2005;15:275–281. doi: 10.1016/j.conb.2005.05.003. [DOI] [PubMed] [Google Scholar]
- Corp ES, McQuade J, Moran TH, Smith GP. Characterization of type A and type B CCK receptor binding sites in rat vagus nerve. Brain Res. 1993;623:161–166. doi: 10.1016/0006-8993(93)90024-h. [DOI] [PubMed] [Google Scholar]
- Correa SG, Maccioni M, Rivero VE, Iribarren P, Sotomayor CE, Riera CM. Cytokines and the immune-neuroendocrine network: what did we learn from infection and autoimmunity? Cytokine Growth Factor Rev. 2007;18:125–134. doi: 10.1016/j.cytogfr.2007.01.011. [DOI] [PubMed] [Google Scholar]
- De Flora A, Zocchi E, Guida L, Franco L, Bruzzone S. Autocrine and paracrine calcium signaling by the CD38/NAD+/cyclic ADP-ribose system. Ann N Y Acad Sci. 2004;1028:176–191. doi: 10.1196/annals.1322.021. [DOI] [PubMed] [Google Scholar]
- Deshpande DA, Walseth TF, Panettieri RA, Kannan MS. CD38/cyclic ADP-ribose-mediated Ca2+ signaling contributes to airway smooth muscle hyper-responsiveness. Faseb J. 2003;17:452–454. doi: 10.1096/fj.02-0450fje. [DOI] [PubMed] [Google Scholar]
- Doyle MW, Bailey TW, Jin YH, Andresen MC. Vanilloid receptors presynaptically modulate cranial visceral afferent synaptic transmission in nucleus tractus solitarius. J Neurosci. 2002;22:8222–8229. doi: 10.1523/JNEUROSCI.22-18-08222.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyle MW, Bailey TW, Jin YH, Appleyard SM, Low MJ, Andresen MC. Strategies for cellular identification in nucleus tractus solitarius slices. J Neurosci Methods. 2004;137:37–48. doi: 10.1016/j.jneumeth.2004.02.007. [DOI] [PubMed] [Google Scholar]
- Ellacott KL, Lawrence CB, Rothwell NJ, Luckman SM. PRL-releasing peptide interacts with leptin to reduce food intake and body weight. Endocrinology. 2002;143:368–374. doi: 10.1210/endo.143.2.8608. [DOI] [PubMed] [Google Scholar]
- Emch GS, Hermann GE, Rogers RC. TNF-alpha activates solitary nucleus neurons responsive to gastric distension. Am J Physiol Gastrointest Liver Physiol. 2000;279:G582–G586. doi: 10.1152/ajpgi.2000.279.3.G582. [DOI] [PubMed] [Google Scholar]
- Emch GS, Hermann GE, Rogers RC. TNF-alpha-induced c-Fos generation in the nucleus of the solitary tract is blocked by NBQX and MK-801. Am J Physiol Regul Integr Comp Physiol. 2001;281:R1394–R1400. doi: 10.1152/ajpregu.2001.281.5.R1394. [DOI] [PubMed] [Google Scholar]
- Emch GS, Hermann GE, Rogers RC. Tumor necrosis factor-alpha inhibits physiologically identified dorsal motor nucleus neurons in vivo. Brain Res. 2002;951:311–315. doi: 10.1016/s0006-8993(02)03178-5. [DOI] [PubMed] [Google Scholar]
- Esper DH, Harb WA. The cancer cachexia syndrome: a review of metabolic and clinical manifestations. Nutr Clin Pract. 2005;20:369–376. doi: 10.1177/0115426505020004369. [DOI] [PubMed] [Google Scholar]
- Fill M, Copello JA. Ryanodine receptor calcium release channels. Physiol Rev. 2002;82:893–922. doi: 10.1152/physrev.00013.2002. [DOI] [PubMed] [Google Scholar]
- Glass MJ, Pickel VM. Alpha(2A)-adrenergic receptors are present in mu-opioid receptor containing neurons in rat medial nucleus tractus solitarius. Synapse. 2002;43:208–218. doi: 10.1002/syn.10036. [DOI] [PubMed] [Google Scholar]
- Griffin JW, George R, Ho T. Macrophage systems in peripheral nerves. A review. J Neuropathol Exp Neurol. 1993;52:553–560. doi: 10.1097/00005072-199311000-00001. [DOI] [PubMed] [Google Scholar]
- Gross PM, Wall KM, Pang JJ, Shaver SW, Wainman DS. Microvascular specializations promoting rapid interstitial solute dispersion in nucleus tractus solitarius. Am J Physiol Regul Integr Comp Physiol. 1990;259(6 Pt 2):R1131–R1138. doi: 10.1152/ajpregu.1990.259.6.R1131. [DOI] [PubMed] [Google Scholar]
- Gutierrez EG, Banks WA, Kastin AJ. Murine tumor necrosis factor alpha is transported from blood to brain in the mouse. J Neuroimmunol. 1993;47:169–176. doi: 10.1016/0165-5728(93)90027-v. [DOI] [PubMed] [Google Scholar]
- Guzik TJ, Mangalat D, Korbut R. Adipocytokines—novel link between inflammation and vascular function? J Physiol Pharmacol. 2006;57:505–528. [PubMed] [Google Scholar]
- Haviv R, Stein R. The intracellular domain of p55 tumor necrosis factor receptor induces apoptosis which requires different caspases in naive and neuronal PC12 cells. J Neurosci Res. 1998;52:380–389. doi: 10.1002/(SICI)1097-4547(19980515)52:4<380::AID-JNR2>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- Hermann G, Rogers RC. Tumor necrosis factor-alpha in the dorsal vagal complex suppresses gastric motility. Neuroimmunomodulation. 1995;2:74–81. doi: 10.1159/000096874. [DOI] [PubMed] [Google Scholar]
- Hermann GE, Emch GS, Tovar CA, Rogers RC. c-Fos generation in the dorsal vagal complex after systemic endotoxin is not dependent on the vagus nerve. Am J Physiol Regul Integr Comp Physiol. 2001;280:R289–R299. doi: 10.1152/ajpregu.2001.280.1.R289. [DOI] [PubMed] [Google Scholar]
- Hermann GE, Hebert SL, Van Meter MJ, Holmes GM, Rogers RC. TNF(alpha)-p55 receptors: medullary brainstem immunocytochemical localization in normal and vagus nerve-transected rats. Brain Res. 2004;1004:156–166. doi: 10.1016/j.brainres.2003.11.078. [DOI] [PubMed] [Google Scholar]
- Hermann GE, Holmes GM, Rogers RC. TNF(alpha) modulation of visceral and spinal sensory processing. Curr Pharm Des. 2005;11:1391–1409. doi: 10.2174/1381612053507828. [DOI] [PubMed] [Google Scholar]
- Hermann GE, Nasse JS, Rogers RC. Alpha-1 adrenergic input to solitary nucleus neurones: calcium oscillations, excitation and gastric reflex control. J Physiol. 2005;562(Pt 2):553–568. doi: 10.1113/jphysiol.2004.076919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermann GE, Rogers RC, Bresnahan JC, Beattie MS. Tumor necrosis factor-alpha induces cFOS and strongly potentiates glutamate-mediated cell death in the rat spinal cord. Neurobiol Dis. 2001;8:590–599. doi: 10.1006/nbdi.2001.0414. [DOI] [PubMed] [Google Scholar]
- Hermann GE, Tovar CA, Rogers RC. Induction of endogenous tumor necrosis factor-alpha: suppression of centrally stimulated gastric mobility. Am J Physiol Regul Integr Comp Physiol. 1999;276(1 Pt 2):R59–R68. doi: 10.1152/ajpregu.1999.276.1.R59. [DOI] [PubMed] [Google Scholar]
- Hermann GE, Tovar CA, Rogers RC. LPS-induced suppression of gastric motility relieved by TNFR:Fc construct in dorsal vagal complex. Am J Physiol Gastrointest Liver Physiol. 2002;283:G634–G639. doi: 10.1152/ajpgi.00412.2001. [DOI] [PubMed] [Google Scholar]
- Hermann GE, Tovar CA, Rogers RC. TNFalpha-stimulation of cFos-activation of neurons in the solitary nucleus is suppressed by TNFR:Fc adsorbant construct in the dorsal vagal complex. Brain Res. 2003;976:69–74. doi: 10.1016/s0006-8993(03)02687-8. [DOI] [PubMed] [Google Scholar]
- Hoesch RB, Yienger K, Weinreich D, Kao JP. Coexistence of functional 1P(3) and ryanodine receptors in vagal sensory neurons and their activation by ATP. J Neurophysiol. 2002;88:1212–1219. doi: 10.1152/jn.2002.88.3.1212. [DOI] [PubMed] [Google Scholar]
- Holtmann MH, Schuchmann M, Zeller G, Galle PR, Neurath MF. The emerging distinct role of TNF-receptor 2 (p80) signaling in chronic inflammatory disorders. Arch Immunol Ther Exp (Warsz) 2002;50:279–288. [PubMed] [Google Scholar]
- Iqbal J, Kumar K, Sun L, Zaidi M. Selective upregulation of the ADPribosyl-cyclases CD38 and CD157 by TNF but not by RANK-L reveals differences in downstream signaling. Am J Physiol Renal Physiol. 2006;291:F557–F566. doi: 10.1152/ajprenal.00066.2006. [DOI] [PubMed] [Google Scholar]
- Ji J, Dheen ST, Tay SS. Molecular analysis of the vagal motoneuronal degeneration after right vagotomy. J Neurosci Res. 2002;69:406–417. doi: 10.1002/jnr.10300. [DOI] [PubMed] [Google Scholar]
- Jin YH, Bailey TW, Li BY, Schild JH, Andresen MC. Purinergic and vanilloid receptor activation releases glutamate from separate cranial afferent terminals in nucleus tractus solitarius. J Neurosci. 2004;24:4709–4717. doi: 10.1523/JNEUROSCI.0753-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaisho T, Akira S. Toll-like receptor function and signaling. J Allergy Clin Immunol. 2006;117:979–987. doi: 10.1016/j.jaci.2006.02.023. quiz 988. [DOI] [PubMed] [Google Scholar]
- Kasner SE, Demchuk AM, Berrouschot J, Schmutzhard H, Harms L, Verro P, et al. Predictors of fatal brain edema in massive hemispheric ischemic stroke. Stroke. 2001;32:2117–2123. doi: 10.1161/hs0901.095719. [DOI] [PubMed] [Google Scholar]
- Kinouchi K, Brown G, Pasternak G, Donner DB. Identification and characterization of receptors for tumor necrosis factor-alpha in the brain. Biochem Biophys Res Commun. 1991;181:1532–1538. doi: 10.1016/0006-291x(91)92113-x. [DOI] [PubMed] [Google Scholar]
- Kurzrock R. The role of cytokines in cancer-related fatigue. Cancer. 2001;92(6 Suppl):1684–1688. doi: 10.1002/1097-0142(20010915)92:6+<1684::aid-cncr1497>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- Laabich A, Li G, Cooper NG. Characterization of apoptosis-genes associated with NMDA mediated cell death in the adult rat retina. Brain Res Mol Brain Res. 2001;91:34–42. doi: 10.1016/s0169-328x(01)00116-4. [DOI] [PubMed] [Google Scholar]
- Laiwand R, Werman R, Yarom Y. Time course and distribution of motoneuronal loss in the dorsal motor vagal nucleus of guinea pig after cervical vagotomy. J Comp Neurol. 1987;256:527–537. doi: 10.1002/cne.902560405. [DOI] [PubMed] [Google Scholar]
- Lancaster E, Oh EJ, Weinreich D. Vagotomy decreases excitability in primary vagal afferent somata. J Neurophysiol. 2001;85:247–253. doi: 10.1152/jn.2001.85.1.247. [DOI] [PubMed] [Google Scholar]
- Lelli A, Perin P, Martini M, Ciubotaru CD, Prigioni I, Valli P, et al. Presynaptic calcium stores modulate afferent release in vestibular hair cells. J Neurosci. 2003;23:6894–6903. doi: 10.1523/JNEUROSCI.23-17-06894.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liebregts T, Adam B, Bredack C, Roth A, Heinzel S, Lester S, et al. Immune activation in patients with irritable bowel syndrome. Gastroenterology. 2007;132:913–920. doi: 10.1053/j.gastro.2007.01.046. [DOI] [PubMed] [Google Scholar]
- McCann MJ, Rogers RC. Impact of antral mechanoreceptor activation on the vago-vagal reflex in the rat: functional zonation of responses. J Physiol. 1992;453:401–411. doi: 10.1113/jphysiol.1992.sp019235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moritz T, Niederle N, Baumann J, May D, Kurschel E, Osieka R, et al. Phase I study of recombinant human tumor necrosis factor alpha in advanced malignant disease. Cancer Immunol Immunother. 1989;29:144–150. doi: 10.1007/BF00199290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller K, Meineke V. Radiation-induced alterations in cytokine production by skin cells. Exp Hematol. 2007;35(4) Suppl 1:96–104. doi: 10.1016/j.exphem.2007.01.017. [DOI] [PubMed] [Google Scholar]
- Nadeau S, Rivest S. Effects of circulating tumor necrosis factor on the neuronal activity and expression of the genes encoding the tumor necrosis factor receptors (p55 and p75) in the rat brain: a view from the blood-brain barrier. Neuroscience. 1999;93:1449–1464. doi: 10.1016/s0306-4522(99)00225-0. [DOI] [PubMed] [Google Scholar]
- Pickering M, Cumiskey D, O’Connor JJ. Actions of TNF-alpha on glutamatergic synaptic transmission in the central nervous system. Exp Physiol. 2005;90:663–670. doi: 10.1113/expphysiol.2005.030734. [DOI] [PubMed] [Google Scholar]
- Rinaman L. Hindbrain noradrenergic lesions attenuate anorexia and alter central cFos expression in rats after gastric viscerosensory stimulation. J Neurosci. 2003;23:10084–10092. doi: 10.1523/JNEUROSCI.23-31-10084.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinaman L, Verbalis JG, Stricker EM, Hoffman GE. Distribution and neurochemical phenotypes of caudal medullary neurons activated to express cFos following peripheral administration of cholecystokinin. J Comp Neurol. 1993;338:475–490. doi: 10.1002/cne.903380402. [DOI] [PubMed] [Google Scholar]
- Rogers RC, Hermann GE, Travagli RA. Brainstem pathways responsible for oesophageal control of gastric motility and tone in the rat. J Physiol. 1999;514(Pt 2):369–383. doi: 10.1111/j.1469-7793.1999.369ae.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers RC, Hermann GE, Travagli RA. Brainstem control of gastric function. In: Johnson LR, editor. Physiology of the gastrointestinal tract. 4th ed. San Diego (CA): Elsevier; 2005. pp. 851–875. [Google Scholar]
- Rogers RC, McCann MJ. Intramedullary connections of the gastric region in the solitary nucleus: a biocytin histochemical tracing study in the rat. J Auton Nerv Syst. 1993;42:119–130. doi: 10.1016/0165-1838(93)90043-t. [DOI] [PubMed] [Google Scholar]
- Rogers RC, McTigue DM, Hermann GE. Vagovagal reflex control of digestion: afferent modulation by neural and “endoneurocrine” factors. Am J Physiol Gastrointest Liver Physiol. 1995;268(1 Pt 1):G1–G10. doi: 10.1152/ajpgi.1995.268.1.G1. [DOI] [PubMed] [Google Scholar]
- Rogers RC, Nasse JS, Hermann GE. Live-cell imaging methods for the study of vagal afferents within the nucleus of the solitary tract. J Neurosci Methods. 2006;150:47–58. doi: 10.1016/j.jneumeth.2005.05.020. [DOI] [PubMed] [Google Scholar]
- Rogers RC, Travagli RA, Hermann GE. Noradrenergic neurons in the rat solitary nucleus participate in the esophageal-gastric relaxation reflex. Am J Physiol Regul Integr Comp Physiol. 2003;285:R479–R489. doi: 10.1152/ajpregu.00155.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers RC, Van Meter MJ, Hermann GE. Tumor necrosis factor potentiates central vagal afferent signaling by modulating ryanodine channels. J Neurosci. 2006;26:12642–12646. doi: 10.1523/JNEUROSCI.3530-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafers M, Schmidt C, Vogel C, Toyka KV, Sommer C. Tumor necrosis factor-alpha (TNF) regulates the expression of ICAM-1 predominantly through TNF receptor 1 after chronic constriction injury of mouse sciatic nerve. Acta Neuropathol (Berl) 2002;104:197–205. doi: 10.1007/s00401-002-0541-9. [DOI] [PubMed] [Google Scholar]
- Shamash S, Reichert F, Rotshenker S. The cytokine network of Wallerian degeneration: TNF-alpha, IL1-alpha and IL1-beta. J Neuroscience. 2003;22:3052–3060. doi: 10.1523/JNEUROSCI.22-08-03052.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro RE, Miselis RR. The central Organization of the vagus nerve innervating the stomach of the rat. J Comp Neurol. 1985;238:473–488. doi: 10.1002/cne.902380411. [DOI] [PubMed] [Google Scholar]
- Shubayev VI, Myers RR. Anterograde TNF alpha transport from rat dorsal root ganglion to spinal cord and injured sciatic nerve. Neurosci Lett. 2002;320:99–101. doi: 10.1016/s0304-3940(02)00010-1. [DOI] [PubMed] [Google Scholar]
- Stoll G, Jander S, Myers RR. Degeneration and regeneration of the peripheral nervous system: From Augustus Waller’s observations to neuroimflammation. J Peripheral Nerv Sys. 2002;7:13–27. doi: 10.1046/j.1529-8027.2002.02002.x. [DOI] [PubMed] [Google Scholar]
- Stoll G, Jander S, Schroeter M. Detrimental and beneficial effects of injury-induced inflammation and cytokine expression in the nervous system. Adv Exp Med Biol. 2002;513:87–113. doi: 10.1007/978-1-4615-0123-7_3. [DOI] [PubMed] [Google Scholar]
- Tartaglia LA, Pennica D, Goeddel DV. Ligand passing: the 75-kDa tumor necrosis factor (TNF) receptor recruits TNF for signaling by the 55-kDa TNF receptor. J Biol Chem. 1993;268:18542–18548. [PubMed] [Google Scholar]
- Torrealba F, Muller C. Ultrastructure of glutamate and GABA immunoreactive axon terminals of the rat nucleus tractus solitarius, with a note on infralimbic cortex afferents. Brain Res. 1999;820:20–30. doi: 10.1016/s0006-8993(98)01326-2. [DOI] [PubMed] [Google Scholar]
- Turrin NP, Plata-Salaman CR. Cytokine-cytokine interactions and the brain. Brain Res Bull. 2000;51:3–9. doi: 10.1016/s0361-9230(99)00203-8. [DOI] [PubMed] [Google Scholar]
- Zhang X, Fogel R, Renehan WE. Physiology and morphology of neurons in the dorsal motor nucleus of the vagus and the nucleus of the solitary tract that are sensitive to distension of the small intestine. J Comp Neurol. 1992;323:432–448. doi: 10.1002/cne.903230310. [DOI] [PubMed] [Google Scholar]