

Abstract
The present microreview summarizes our progress over the last few years in defining regioselective reductive cross-coupling reactions of unsymmetrical alkynes with terminal- and internal alkynes, aldehydes, and imines. We begin with a brief historical perspective of metal-mediated reductive dimerization reactions of aromatic alkynes and discuss the challenges associated with “crossed” versions of this mode of reactivity. Next, a collection of available methods that allow for regioselective reductive cross-coupling of internal alkynes with terminal and internal alkynes, aldehydes, and imines is summarized. After an examination of the requirements for regioselectivity in these cases, the logic behind our design of alkoxide-directed titanium-mediated reductive cross-coupling reactions is presented. A nomenclature is introduced to delineate the presumed mechanistic origin of regioselection associated with each reaction design, and a presentation of alkoxide-directed regioselective reductive cross-coupling reactions of alkynes follows. Throughout, principal issues related to reactivity and selectivity are discussed to assess scope and limitations of available methods and to describe the broad challenges that exist for defining complex fragment union reactions based on reductive cross-coupling chemistry.
Keywords: reductive coupling, carbometalation, alkyne, titanium, alkene synthesis
1. Introduction
Bimolecular C–C bond formation is central to the field of organic synthesis. In addition to their evident necessity in molecular assembly, convergent coupling reactions that proceed by the formation of one, or multiple, C–C bonds often define processes that greatly impact the efficiency with which complex molecules can be prepared.[1] Given the principal role of convergent coupling reactions in chemical synthesis, it is striking how few modes of chemical reactivity are exploited in the highly selective bimolecular C–C bond forming reactions typically employed in complex molecule synthesis. While “classic” bond constructions include those afforded by nucleophilic substitution, nucleophilic addition to polarized π-bonds, or cycloaddition, “modern” strategies include metal-catalyzed cross-coupling and crossed-metathesis. Within these various classes of broad chemical reactivity reside numerous reactions of great utility in stereoselective organic synthesis. That said, the narrow range of chemical reactivity associated with these reaction classes is particularly surprising given the so-called “mature” nature of organic chemistry as a scientific discipline.[2]
In the search to define a mechanistically unique and broadly useful class of reactions to complement well-established methods for bimolecular C–C bond formation, we speculated that reductive cross-coupling chemistry could be particularly powerful. As illustrated in Figure 1, a range of functionalized products could derive from regioselective reductive cross-coupling of substrates bearing simple π-unsaturation (alkenes, alkynes, allenes, and carbonyl functionality; eqs 1-7). Due to the vast abundance of such functionality in commercial starting materials and complex synthetic intermediates alike, development of this broad area of chemical reactivity has the potential to define new paradigms for molecular construction.
Figure 1.

A small subset of possible bimolecular C–C bond forming processes based on reductive cross-coupling.
While the general reactivity pattern common to reductive coupling processes encompasses a rather broad array of potential fragment union processes (i.e. Figure 1), this Microreview will only highlight a subset of these. Specifically, we discuss advances made that have culminated in the discovery of a variety of highly selective reductive cross-coupling reactions of unsymmetrical alkynes with: 1) terminal alkynes, 2) aldehydes, 3) internal alkynes, and 4) imines.
2. Background and Challenges
A suitable starting point for the discussion of reductive cross-coupling chemistry is the Reppe reaction, where the metal catalyzed trimerization of alkynes delivers substituted benzenes (Figure 2).[3] Fifteen years following this report, Vol’pin and Kursanov described the titanium-mediated dimerization of diphenylacetylene via the formation of a titanacyclopentadiene – a process that, after protonation, delivers the product of reductive homodimerization of diphenylacetylene.[4] Since these early discoveries, a large variety of chemical methods have emerged from the basic reactivity pattern central to these seminal observations: formal [2+2+1] between two reactive π-systems and a metal center.[5]
Figure 2.
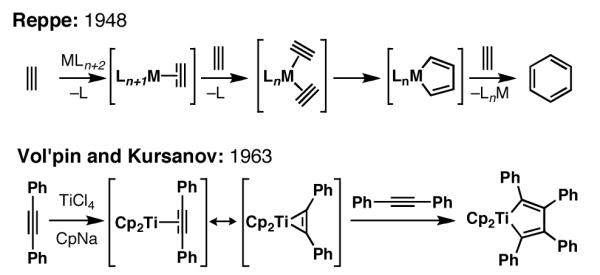
Early examples of metallacycle-mediated coupling reactions of alkynes.
Consistent with the focused nature of this Microreview, we consider only bimolecular C–C bond formation by the regioselective reductive cross-coupling of alkynes – the most thoroughly investigated coupling partner studied in this broad area of reaction methodology. With a focus on regio- and stereoselective bimolecular bond construction, the general discussion that follows is presented from a perspective that is independent of the nature and stoichiometries of the metal employed.
Despite the fact that metallacycle-mediated processes have been a topic of considerable interest in organic and organometallic chemistry, there exist relatively few highly site-selective bimolecular C–C bond forming processes within this class that functionalize unsymmetrical alkynes. In fact, most highly regioselective reductive coupling processes of unsymmetrical alkynes employ either conjugated- or TMS-substituted alkynes (vide infra). From a perspective of complex molecule synthesis, this restriction in alkyne-substitution greatly limits the potential impact of this class of reductive cross-coupling. While representing only a small subset of the possible reductive cross-coupling reactions depicted in Figure 1, reductive cross-coupling reactions of alkynes are the most well-developed. While related chemistry of alkenes and allenes lags far behind, the challenges associated with the control of reactivity and regiochemistry in alkyne-based reductive cross-coupling reactions will serve as a foundation to discuss: 1) the state-of-the-art in reductive cross-coupling technology, and 2) the eventual elucidation of a collection of complex alkoxide-directed regioselective reductive cross-coupling reactions of unsymmetrical alkynes.
2.1. The Basic Challenges
In bimolecular settings, challenges associated with the control of reactivity and selectivity emerge as central concerns that complicate the use of metallacyle-mediated reductive coupling processes in complex bond construction. A large variety of such reactions, and mechanistically related C–C bond forming processes, have been successfully demonstrated in intramolecular settings.[5] Here, the many challenges associated with control of reactivity and selectivity are addressed by the forced proximity of the reacting π-systems.
To achieve selective cross-coupling based on reductive coupling chemistry, one must overcome the typically poor reactivity of substituted and electronically unactivated π-systems in reactions with metal–π complexes.[5] Additional challenges reside in the control of: 1) cross-coupling vs. homo-dimerization, 2) regioselectivity (site selectivity), and 3) stereoselectivity (Figure 3). A brief summary of the advances made with respect to each of these issues follows.
Figure 3.

Challenges in the control of metallacycle-mediated reductive cross-coupling reactions of alkynes.
2.1.1 Cross-Coupling vs. Homo-Dimerization
The challenge of controlling the partition between cross-coupling and homo-dimerization is a complex issue in metallacycle-mediated C–C bond formation. The most general procedure to favor the formation of “crossed” products has been based on stoichiometric metal-mediated coupling reactions, where preformation of an activated metallacyclopropene is followed by exposure to the second coupling partner. Examples of this type of control can be seen in early demonstrations of Pauson–Khand chemistry, Bercaw’s study of the reactivity of (Cp*)2Ti–C2H4, and Buchwald’s Zr-mediated alkyne–alkyne coupling process (Figure 4).[6]
Figure 4.

Examples of metallacycle-mediated cross-coupling reactions.
Thus, preformation of a metal–π complex provides one opportunity to enforce cross-coupling over homo-dimerization in a subset of coupling processes. For this operationally simple solution to be broadly effective in diverse reductive cross-coupling chemistry, thermodynamic equilibration of the intermediate metallacyclopentene must not lead to scrambling of metallacycle composition.[7]
In catalytic methods that proceed by the coupling of two unsaturated reaction partners via the intermediacy of a metallacyclopentene, the competition between homo- and hetero-coupling is ever present, as the reactive metal center is continually exposed to both reactive π-systems. As such, it is not surprising that a general catalytic method has not emerged for the cross-coupling of a diverse array of unsaturated coupling partners. Nevertheless, unique solutions in this area have appeared for: 1) coupling of polarized π-systems (i.e. carbonyl-based systems) to alkynes and terminal alkenes (vide infra), and 2) for the coupling of terminal- or 1,1-disubstituted alkenes with alkynes (via Alder-Ene chemistry).[8]
2.1.2. Regioselectivity
The control of regioselectivity is a major problem in bimolecular metallacycle-mediated C–C bond forming chemistry. While significant advances have been made in a subset of coupling processes, the dominant means for control in the cross-coupling of disubstituted alkynes with carbonyl-based π-systems or terminal alkynes is based on the use of trialkylsilyl-substituted, or conjugated alkynes (Figure 5).
Figure 5.
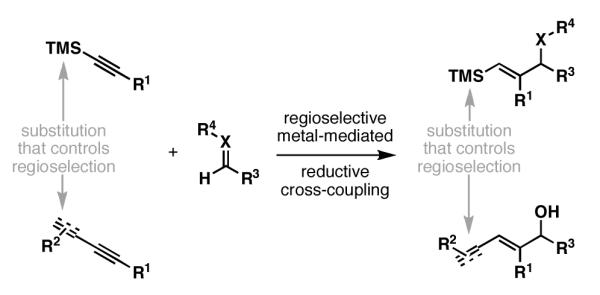
Substitution typically required for the control of site-selective C–C bond formation in alkyne–carbonyl-based reductive cross-coupling.
Coupling of Internal Alkynes with Carbonyl-Based π-Systems
Among the first substrate classes to be identified for the regioselective reductive cross-coupling of internal alkynes with carbonyl-based systems was TMS-substitututed alkynes. As illustrated in Figure 6, Zr- and Ti-mediated reductive cross-coupling reactions of TMS alkynes favor formation of products where C–C bond formation occurs distal to the TMS substituent (eqs 8 and 9).[9, 10]
Figure 6.

Methods for the control of regioselection in metallacycle-mediated coupling of disubstituted alkynes with carbonyl electrophiles.
With the advent of catalytic methods to address this class of reductive cross-coupling came interesting observations. For example, in recently reported Ni-catalyzed reductive coupling reactions of TMS-alkynes with aldehydes (eq 10), the regiochemical course of the reaction is opposite to that previously observed in the related coupling reactions promoted by group IV metals.[11] Here, the major product derives from C–C bond formation proximal to the TMS substituent.
Most recently, π-conjugation has emerged as a powerful structural feature that controls regioselection in a variety of metal-catalyzed reductive cross-coupling reactions.[12, 13] As depicted in eqs 11-15, high levels of regioselection are typically observed where C–C bond formation predominates at the site distal to the conjugated system. Examples include Ni, Rh, Ir and Ru-catalyzed reactions of alkynes conjugated to aromatics, heteroaromatics, alkenes and alkynes, with aldehydes and activated imines.[14-16]
Only recently have promising results been observed in the regioselective cross-coupling of internal alkynes that lack TMS-substitution or π-conjugation (Figure 7). As depicted in eq 16, Montgomery has reported a regioselective alkyne–aldehyde coupling with a simple internal alkyne. Here, selectivities were observed up to 6:1, favoring the formation of a product where C–C bond formation occurs at the least hindered position of the alkyne (Me vs. substituted Et).[17] Similarly, in Ir-catalyzed coupling of related alkynes with electron deficient sulfonyl imines, Krische has reported selectivities of approximately 10:1 (eq 17).[18] Notably, in this Ir-catalyzed process, exquisite levels of regioselectivity were observed in the functionalization of an alkyne bearing Me- vs. branched alkyl substitution. Finally, in parallel with our studies in alkoxide-directed reductive cross-coupling chemistry, Jamison recently described a Ni-catalyzed, alkene-directed coupling of non-conjugated enynes with aldehydes (eq 18).[19]
Figure 7.
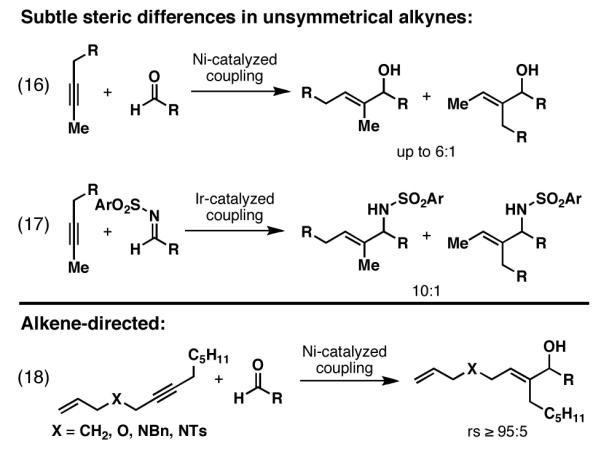
Examples of regioselective reductive cross-coupling reactions of alkynes with carbonyl electrophiles where TMS-substitution or π-conjugation do not play a role in selectivity.
Aside from these interesting cases, the most general means of attaining site-selectivity in the reductive cross-coupling of disubstituted alkynes with carbonyl-based π-systems requires alkynes that possess π-conjugation or TMS-substitution (Figure 7).[20] Therefore, despite the significant progress made in rendering this subset of reductive cross-coupling chemistry catalytic in the active metal component, substantial limitations in regiochemical control still exist that broadly restrain the impact that such bond constructions play in organic synthesis.
Coupling of Internal Alkynes with Terminal Alkynes
For the reductive cross-coupling reaction of internal alkynes with relatively non-polarized “all carbon”-based π-systems, the challenge associated with regioselection is more complex than related cross-coupling reactions with carbonyl-based systems. The additional complexity arises from the need to control regioselection with respect to both coupling partners. As illustrated in Figure 8, the reductive cross-coupling of an internal alkyne with a disubstituted alkene or alkyne has the potential to deliver four distinct regioisomeric products.
Figure 8.
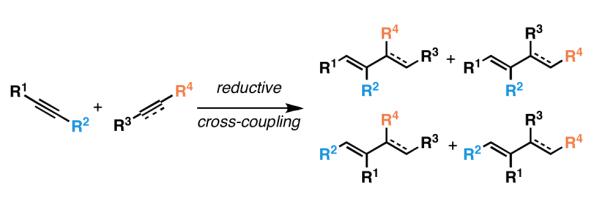
Regiochemical possibilities in the reductive cross-coupling reaction of internal alkynes with other all carbon-based π-systems.
While few reductive cross-coupling reactions of this complexity have been described, due to the often low reactivity profile of disubstituted alkene or alkyne coupling partners, the reductive union of internal alkynes with terminal alkynes has been reported (Figure 9). In 1989, Buchwald reported a Zr-mediated coupling reaction that, after equilibration of a mixture of isomeric intermediate metallacyclopentadienes, provides 1,3-dienes 22 with very high levels of regioselection (rs ≥ 98:2).[7d] Here, site-selective functionalization of the terminal alkyne, at the least substituted carbon, was coupled to regioselective functionalization of a TMS-substituted alkyne. As discussed previously in the related coupling reaction of TMS-alkynes with carbonyl systems, the site of C–C bond formation in the present case is also distal to the TMS-substituent.
Figure 9.

Methods for the control of regioselection in the metallacycle-mediated coupling of disubstituted alkynes with terminal alkynes.
Subsequent to these studies, in 1999, Sato described a Ti-mediated reductive cross-coupling reaction of internal alkynes with terminal alkynes, where high levels of regioselectivity were attained without the need for an equilibration step.[21] Again, π-conjugation or TMS-substitution was essential for obtaining good levels of regioselection in this reductive cross-coupling reaction (Figure 9; 23 and 24).
In conclusion, regioselection in these diene-forming processes have been accomplished in a similar fashion to the reductive cross-coupling of alkynes with carbonyl electrophiles. Again, TMS-substitution or π-conjugation has been employed to control site-selective functionalization of the internal alkyne.
2.1.3. Stereoselection
In addition to the aforementioned issues related to homo-coupling, regioselection, and reactivity, the control of stereoselection arises as a fourth significant issue in a subset of reductive cross-coupling reactions of alkynes. To date, the most progress in this area has been made in coupling of alkynes with carbonyl electrophiles. Success has been documented for reductive cross-coupling reactions of alkynes with α-alkoxy and α-silyloxy aldehydes (Figure 10). Initial studies reported that the Ti-mediated reductive coupling of glyceraldehyde acetonide with a range of internal alkynes proceeds in a diastereoselective manner.[22] In general, good selectivities are observed for formation of the anti-products (eq 19). In stereochemically related coupling processes, highly diastereoselective Ni-catalyzed reductive cross-coupling reactions of alkynes with α-alkoxy and α-silyloxy-substituted aldehydes have also appeared (eqs 20 and 21).[23]
Figure 10.

Diastereoselective reductive cross-coupling reactions of alkynes with chiral aldehydes.
Over the last few years there has been an explosion of reports that describe enantioselective versions of reductive cross-coupling reactions between alkynes and carbonyl-based systems (aldehydes, glyoxalates, electronically activated ketones and imines). This aspect of reductive cross-coupling chemistry is quite interesting and has been reviewed elsewhere.[24] That said, even with the significant accomplishments made in rendering this class of reductive cross-coupling chemistry enantioselective, the central bond constructions possible with this mode of reactivity are all subject to the limitations in regioselection previously described.
2.1.4. Conclusion regarding the background and associated general challenges
While significant advances have been made in metallacycle-mediated cross-coupling of disubstituted alkynes, it is worth reflecting on the current state of the field. Independent of the nature of the metal employed, or whether that metal is used in a catalytic or stoichiometric fashion, significant barriers related to general reactivity and regioselectivity have greatly impeded the development of diverse cross-coupling methods within this area. While the general mode of reactivity seems superficially suitable for the cross-coupling of a variety of common π-systems encountered in chemical synthesis (substituted alkenes, alkynes, allenes, and carbonyl-based systems), state-of-the-art methods allow for the selective cross-coupling of only a handful of these systems. Finally, even within known classes of reductive cross-coupling, regioselective processes proceed for only a small substrate scope (typically with carbonyl-based systems).
Overall, the lingering challenges that have prevented broad development of reductive cross-coupling for the types of bond constructions depicted in Figure 1 include: 1) reaction course – cross-coupling vs. homo-dimerization, 2) regiochemical control, 3) low reactivity of a variety of substituted π-systems (i.e. disubstituted alkenes, hindered alkynes), and 4) stereoselection. It is the merging of these challenges that complicate the design, discovery and development of all methods for complex fragment union by metallacycle-mediated reductive cross-coupling.
3. Design of Alkoxide-directed Reductive Cross-Coupling Reactions of Internal Alkynes
3.1. Our Goal
In the design of a general process to overcome the limitations of pre-existing methods in reductive cross-coupling technology, we targeted a metal-based system that could be used for the stoichiometric pre-activation of one of the coupling partners – a strategy that would provide a broad solution to the control of cross-coupling over homo-dimerization. Given this initial design criteria, the metal-based system needed to be: 1) inexpensive, 2) non-toxic, and 3) result in byproducts that are both non-toxic and easy to remove from the products of interest. In addition, we favored the selection of a metallic system that had the potential of being compatible with a range of Lewis-basic functionality, as we aimed to develop coupling reactions of utility in complex molecule synthesis (i.e. natural product synthesis). Furthermore, we desired a system that would be capable of forging C-C bonds in the presence of unprotected heteroatom-based functionality. Finally, we set out to devise a suite of heteroatom-directed reductive cross-coupling reactions where the control of reactivity and selectivity would be possible based on the strategic placement of a suitable directing group.[25]
3.2. The selection of a titanium alkoxide-based system
Given our goal of providing chemical methods useful for the convergent synthesis of complex natural products, we desired a system capable of being directed by functionality commonly found embedded in the backbone of such systems. Based on the prevalence of oxygen and nitrogen functionality in natural products of biomedical relevance, we focused our efforts on defining a suite of directed reductive cross-coupling methods where such functionality could serve to direct C-C bond formation - in preference to functionality based on phosphorous, sulfur, aromatic heterocycles or simple π-unsaturation (i.e. a remote alkene).[26-28]
To identify a suitable system based on these requirements, one needs to consider only a handful of relevant precedent. First, the ability of Ti alkoxides to undergo rapid and reversible ligand exchange has been well-showcased in the Sharpless epoxidation.[29] Second, Cp2Ti–π complexes are known to participate in reductive coupling chemistry.[6c-d, 30] In fact, this type of system was first demonstrated in the reductive dimerization of diphenylacetylene,[4] later studied by Bercaw,[6c-d] and subsequently employed in a variety of intramolecular reductive coupling reactions. Finally, the pioneering studies of Kulinkovich and Rothwell demonstrated that titanium aryloxides and titanium alkoxides could be employed to access similar reactivity seen with Cp2Ti–π complexes.[31] Subsequent investigation of the chemistry of Ti-alkoxides in metallacycle-mediated C–C bond forming processes, primarily in the laboratories of Professors Sato and Cha, has led to the discovery and development of a wide range of novel reactions.[32,33] While the contributions to date have established a solid foundation of Ti-mediated metallacycle-based bond constructions in organic chemistry, the previously described barriers related to the control of reactivity and selectivity have remained firmly in place.
As we further considered the use of Ti(OiPr)4 as a stoichiometric component of our reaction design, issues of cost and toxicity were at the forefront of our thoughts. First, the byproducts from aqueous work-up of stoichiometric Ti(OiPr)4-mediated coupling reactions (i.e. the Kulinkovich reaction) are iPrOH and TiO2. While iPrOH is a relatively benign solvent, TiO2 is a species encountered daily in most of our lives, as it is a component of products like toothpaste, sunscreen and paint. The accompanying reducing metal typically employed alongside the titanium(IV) alkoxide is a Grignard reagent, the byproducts of which are simple magnesium salts and hydrocarbons. Although we will describe Ti(OiPr)4-based reductive cross-coupling methods that provide a means to couple π-systems not currently possible with catalytic methods based on Ni, Rh, Ir or Ru, a cost analysis between projected use of stoichiometric Ti(OiPr)4 vs known catalysts for reductive cross-coupling of alkynes with carbonyl systems is informative (Figure 11). Based on a hypothetical reductive coupling reaction run on one mole scale, the cost of stoichiometric Ti(OiPr)4 is significantly less than known catalytic systems based on Ni, Rh, Ir or Ru.[34, 35] Future process optimization may lead to decreases in required catalyst loadings for such processes, however, even a potential 100-fold lower catalyst loadings (eg. 0.05 mol% for Rh-, Ir- and Ru- catalysts depicted) still render the stoichiometric Ti(OiPr)4-mediated method highly competitive on a cost bases for all but the Ni(COD)2-based reactions.
Figure 11.

Analysis of cost for a generic reductive cross-coupling process as a function of the metal employed.
The challenges associated with the development of metal-catalyzed C–C bond formation is a significant and popular concern in current organic chemistry. While many intellectual advances need to be made to enable a wealth of more complex metal-catalyzed reductive cross-coupling chemistry, and much scientific inquiry has embraced this pursuit, our interests were focused on defining a broad class of reductive cross-coupling reactions that extend beyond the basic bond-constructions afforded by current methods. Given the considerations described above that are based on reaction design, cost, toxicity, and the ease with which Ti(OiPr)4 can be handled,[36] we embraced a program aimed at defining novel alkoxide-directed reductive cross-coupling reactions mediated by stoichiometric Ti(OiPr)4.
3.3. The design of three “classes” of alkoxide-directed reductive cross-coupling
At the outset of our investigations, we aimed to formalize distinct reaction designs to accomplish alkoxide-directed metallacycle-mediated reductive cross-coupling. As depicted in Figure 12, three modes of alkoxide directed processes were conceived, each of which defines a unique means of overcoming the predefined barriers associated with the control of reactivity and selectivity associated with metallacycle-mediated cross-coupling. Our definition of each “class” of directed coupling process follows, along with a brief discussion to highlight the primary mechanistic distinctions between them.
Figure 12.

Design of alkoxide-directed reductive cross-coupling reactions.
Class I
Initial formation of a bicyclic metallacyclopropane is followed by bimolecular carbometalation.[37] Here, regioselective functionalization of component 25 is anticipated by selective reaction of a presumed bicyclic intermediate 26, generated by Ti–alkyne complex formation and association of a tethered alkoxide to the titanium center. Site selectivity in the bimolecular C–C bond forming event is then anticipated by carbometalation through a pathway that delivers a fused bicyclic metallacyclopentene 28 in preference to a bridged bicyclic isomer (not shown). While potentially defining a regioselective functionalization of unsaturated alkoxide 25, we anticipated that this reaction design would be limited to reductive coupling reactions with substrates (27) that are sufficiently reactive to participate in the bimolecular carbometalation event (26 + 27 → 28).
Class II
Initial formation of a monocyclic metallacyclopropane 30 is followed by introduction of an unsaturated alkoxide 25. Rapid and reversible ligand exchange (30 + 25 → 31) sets up an intramolecular carbometalation event to deliver the fused bicyclic metallacyclopentane 32.[38] Here, high regioselectivity in functionalization of 25 is anticipated based on: 1) a presumed faster rate of carbometalation by way of the mixed titanate ester 31 in comparison to bimolecular carbometalation via reaction of 30 with the alkyne of 25, and 2) the preference for generation of a fused bicyclic metallacyclopentane, rather than a bridged bicyclic isomer, in the intramolecular carbometalation event (31 →32).
Overall, regioselective functionalization of substrate 29 will result on simple steric grounds whereas regioselective functionalization of 25 should be based on the position of the tethered alkoxide. While defining a potentially powerful means to control regioselection in the functionalization of unsaturated alkoxides, a predicted virtue of this reaction design is the ability to render the carbometalation event intramolecular. This facet of the reaction design is anticipated to enable the participation of reaction partners that are known to exhibit poor levels of reactivity in bimolecular [2+2+1] chemistry by rendering the C–C bond forming event intramolecular.[39]
Class III
Merging the central features of the reaction designs designated as “Class I” and “II” directed carbometalation, a third unique process was envisioned as a pathway for the site-selective coupling of two unsaturated alkoxides. Here, the features that control the selectivity of “Class I” processes will be used to affect site selectivity in the functionalization of substrate 25, while the features that influence selectivity in “Class II” processes will lead to simultaneous selective carbometalation of substrate 33. As such, formation of a fused bicyclic metallacyclopropene 26, followed by rapid and reversible ligand exchange with 33 will provide access to the transient intermediate 34. Intramolecular carbometalation by way of 34 is then predicted to deliver the fused tricyclic metallacyclopentadiene 35. In this process, the steric differentiation of 26 and the intramolecularity of the carbometalation are anticipated to provide a means for selective functionalization of each unsaturated alkoxide (25 and 33) through a process that establishes one central C–C bond, two Ti–C bonds and two Ti–O bonds, while completely encapsulating the metal center in 35.
4. Results for New Alkoxide-Directed Regioselective Reductive Cross-Coupling of Disubstituted Alkynes
4.1. Synthesis of trisubstituted 1,3-dienes via Class I alkoxide-directed coupling of internal alkynes with terminal alkynes
The presence of trisubstituted (E,E)-1,3-dienes embedded in the backbone of complex natural products of biological significance prompted our study of a suitable alkoxide-directed reductive cross-coupling to access this stereodefined structural motif (Figure 13). While stepwise carbonyl olefination or Pd-catalyzed cross-coupling represent state-of-the-art methods to access 1,3-dienes,[40, 41] our familiarity with the multi-step nature of sequential carbonyl olefination, and the numerous stoichiometric functionalization reactions typically required to prepare the stereodefined substrates for Pd-mediated cross-coupling, led us to target a reductive coupling reaction between alkynes to access this class of 1,3-dienes. Here, we expected to develop a fragment coupling process of great value that proceeds by coupling two non-stereogenic π-systems (alkynes) and simultaneously establishes the stereochemistry of each alkene of the 1,3-diene. Overall, such a convergent assembly process would represent an important methodological advance for application to natural product synthesis.[42]
Figure 13.
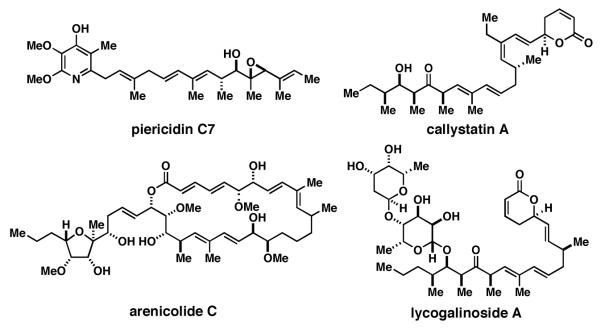
Complex natural products containing stereodefined (E,E)-trisubstituted 1,3-dienes.
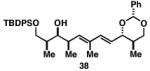
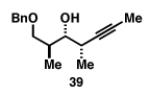
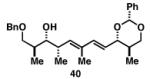
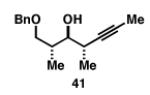
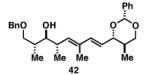
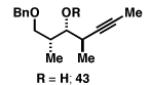
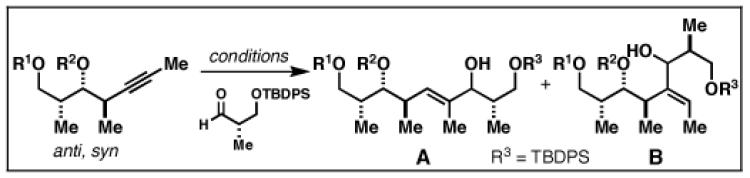
Our initial investigation of the Class I reductive cross-coupling between internal alkynes and terminal alkynes led to the identification of complex structure–selectivity relationships (Table 1).[43] As depicted in entries 1-4, the reductive cross-coupling of stereochemically isomeric homopropargylic alcohols with the chiral alkyne 36 led to a range of results. Overall, the product ratio (A:B:C) changed as a function of the homopropargylic alcohol employed. While high levels of selectivity were observed in some cases, entry 4 highlights the uniquely poor behavior of the anti-syn isomer 43. Interestingly, when the hydroxy substituent was protected in the anti,syn-isomer, selectivity for the formation of isomer A was enhanced (3:1; entry 5). This simple modification that consisted of removing the proximal alkoxide, similarly affected the regiochemical course of related coupling processes (entries 6 and 7).
Table 1.

| entry | internal alkyne |
yield | rr (A:B:C) |
major regioisomer |
|---|---|---|---|---|
| 1 |
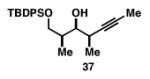
|
70 | 28:1:1 |
|
| 2 |
|
50 | 15:2:1 |
|
| 3 |
|
52 | 4:0:1 |
|
| 4 |
|
63 | 1.0:1.5:1.0 |
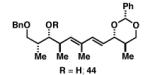
|
| 5 | R=PMB;45 | 82 | 3:1:0 | R=PMB;46 |
| 6 |
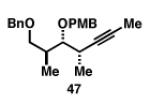
|
87 | 5:1:0 |
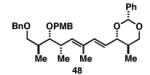
|
| 7 |

|
70 | 6:1:0 |
|
Note: For entries 5-7, no deprotonation step was necessary.
The variation of regioselection as a function of stereochemistry of the internal alkyne component was quite interesting, but ultimately disappointing as the proximal alkoxide was not the sole factor in the control of regioselection in these reductive cross-coupling reactions. In contemplating the potential complexity associated with the attempted alkoxide-directed reductive cross-coupling reactions depicted in entries 1-4 of Table 1, a simple hypothesis emerged. As depicted in Figure 14, alkoxide-directed reductive cross-coupling has the potential to proceed by reaction of a variety of titanium–alkyne complexes (A-C). Although it is not yet possible to understand the precise nature of the transition states operative in these processes, simple considerations of strain and the well-known behavior of Ti–alkoxides guided our mechanistic thoughts.
Figure 14.

Empirical model for regioselectivity.
First, the proposed bicyclic metallacycle A would likely be destabilized by significant ring strain, leading to the speculation that simple monocyclic Ti–alkyne complexes B may play a role in the transition state for reductive coupling. That said, the known ability of Ti–alkoxides to form oligomers leads to a further complexity due to the potential participation of oligomeric Ti–alkyne complexes C in the transition state for C–C bond formation. Making the analysis more complex, the likelihood of a reaction proceeding by way of any of these type of species in the transition state will also be a function of the relative stereochemistry of the starting material that houses the internal alkyne.
With the goal of forcing the reaction down a path that was “directed” by a proximal alkoxide, we reasoned that increasing the distance between the internal alkyne and the tethered alkoxide may result in a greater preference for bicyclic metallacyclopropene D. If such a species were possible, and it did play a role in the transition state for reductive cross-coupling, we would then anticipate high levels of regioselection whereby carbometalation would deliver a fused bicyclic metallacyclopentadiene E.
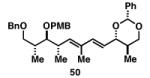
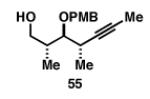
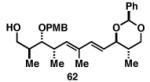
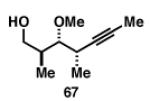
Gratifyingly, the reductive cross-coupling of internal alkynes bearing more remote alkoxide substitution led to uniform and highly regioselective coupling processes independent of stereochemistry (Table 2).[44] As depicted in entries 1-8, reductive cross-coupling of each diastereomer of the internal alkyne component with each enantiomer of the terminal alkyne was highly selective (rs ≥ 17:1). Interestingly, the nature of the protecting group at the homopropargylic position also plays a role in regioselection here. As depicted in entry 9, a drop to 10:1 regioselection was observed with methyl ether 67.
Table 2.

| entry | internal alkyne |
terminal alkyne |
yield a | rrb | major regioisomer |
|---|---|---|---|---|---|
| 1 |
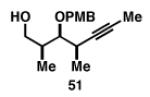
|
36 | 60 | 17:1 |
|
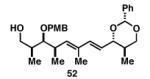
| 2 |
|
36 | 57 | ≥ 20:1 |
|
| 3 |
|
36 | 47 | ≥ 20:1 |

|
| 4 |
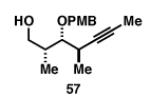
|
36 | 53 | ≥ 20:1 |

|
| 5 |
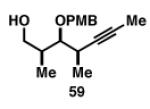
|
ent-36 | 64 | ≥ 20:1 |
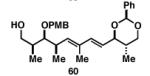
|
| 6 |

|
ent-36 | 63 | ≥ 20:1 |
|
| 7 |
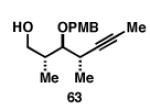
|
ent-36 | 64 | ≥ 20:1 |

|
| 8 |
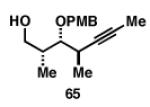
|
ent-36 | 68 | ≥ 20:1 |
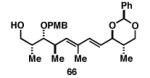
|
| 9 |
|
36 | 78 | 10:1 |
|
Yield based on terminal alkyne
Regioisomeric ratio with respect to functionalization of the internal alkyne (A/B – defined in Table 1). In a few cases, observable quantities of the minor regioisomer “C” were observed (entry 4 = 12:1, entry 6 = 14:1, entry 7 = 17:1).
While were delighted to achieve high levels of regioselection in a coupling reaction of potential utility in natural product synthesis, discovering that subtle structural features significantly influence regioselection was somewhat disappointing. Additional studies further indicated that branching at the propargylic position of the internal alkyne was an essential structural feature for attaining high levels of regioselection. Although these clear limitations dampened our hopes of realizing a suite of highly selective Class I alkoxide-directed reductive cross-coupling reactions, a potentially useful alkoxide-directed reductive cross-coupling reaction for the synthesis of stereodefined 1,3-dienes had emerged.
Subsequent studies have investigated the utility of this C–C bond forming process for the coupling of terminal alkynes bearing a variety of functionality that extends beyond that typically seen in natural products of polyketide biosynthetic origin. As depicted in Figure 15, aromatic and aliphatic heterocycles, fluorous tags and alkynylsilanes are all tolerated in this highly regioselective alkoxide-directed reductive cross-coupling reaction.[44]
Figure 15.

Some functional group compatibility in the Class I alkoxide-directed reductive cross-coupling reaction of internal alkynes with terminal alkynes.
4.2. Synthesis of ene-1,5-diols via Class I alkoxide-directed coupling of internal alkynes with aldehydes
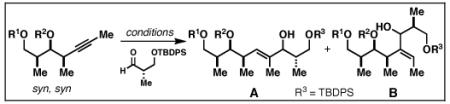
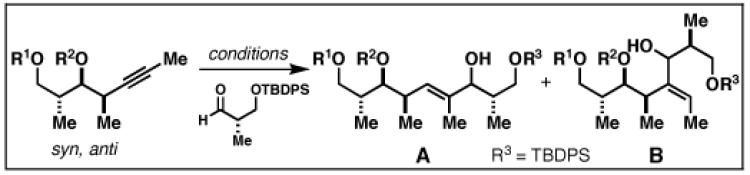
The basic control element for regioselection in the coupling of internal alkynes with terminal alkynes is also effective in the reductive cross-coupling of internal alkynes with aldehydes. Here, regioselection is an issue only with respect to the functionalization of the internal alkyne, but challenges with respect to stereoselection complicate this subset of Class I alkoxide-directed cross-coupling.
As illustrated in Table 3, regioselection was found to be a function of the relative stereochemistry of the alkyne and the presence and position of a tethered alkoxide.[45] Within each stereoisomeric series (syn-syn, syn-anti, anti-anti and anti-syn), highest levels of regioselection were observed with the substrates that contained a free hydroxy substituent δ-to the alkyne (77, 80, 83 and 86).
Table 3.
| entry | internal alkyne |
conditionsa | yield | regioselection A:B |
|---|---|---|---|---|
| 1 | 77; R1 = H | A | 65 b | 8:1 |
| R2 = PMB | ||||
| 2 | 78; R1 = TBDPS | A | 68 b | 4:1 |
| R2 = H | ||||
| 3 | 79; R1 = TBDPS | B | 42 | 1.3:1 |
| R2 = PMB | ||||
| ||||
| 4 | 80; R1 = H | A | 78 b | 18:1 |
| R2 = Bn | ||||
| 5 | 81; R1 = Bn | A | 58 b | 10:1 |
| R2 = H | ||||
| 6 | 82; R1 = Bn | B | 59b | 4:1 |
| R2 = Me | ||||

| ||||
| 7 | 83; R1 = H | A | 68 | 16:1 |
| R2 = Bn | ||||
| 8 | 84; R1 = Bn | A | 68 | 10:1 |
| R2 = H | ||||
| 9 | 85; R1 = Bn | B | 64 | 4:1 |
| R2 = Me | ||||
| ||||
| 10 | 86; R1 = H | A | 69 | 20:1 |
| 11 | 87; R1 = TBS | A | 61 | 4:1 |
| R2 = H | ||||
| 12 | 88; R1 = TBS | B | 61 | 2:1 |
| R2 = PMB | ||||
Reaction Conditions: A) nBuLi, PhMe, −78 °C; then ClTi(OiPr)3, cC5H9MgCl, −78 to −40 °C; then BF3•OEt2, −78 °C and aldehyde; B) ClTi(Oi-Pr)3, cC5H9MgCl, −78 to −40 °C; then BF3•OEt2, −78 °C and aldehyde.
Yield reported is for isomer A.
While we were quite pleased to observe very high levels of regioselection in these reductive cross-coupling reactions, diastereoselectivity remains a challenging hurdle. Here, products were generally produced as mixtures of isomers slightly favoring the product expected from net “Felkin” addition to the carbonyl (dr typically 2-3:1). As depicted in Figure 16, diastereoselection is marginally enhanced with the α-silyloxy propionaldehyde 89 (dr = 5:1; rs ≥ 20:1).
Figure 16.

Diastereoselective Class I alkoxide-directed Ti-mediated reductive alkyne–aldehyde cross-coupling.
Concerning the potential utility of this process in organic synthesis, it is important to consider its standing with respect to well established stereoselective convergent C–C bond forming reactions. While convergent aldol-based bond constructions typically proceed with higher levels of diastereoselection, the type of bond construction provided here defines a complimentary convergent coupling process.[46] Instead of delivering a β-hydroxy ketone, this process delivers a complex allylic alcohol, the functionalization of which by hydrogenation, dihydroxylation, or epoxidation delivers structural motifs not readily accessible with aldol technology. Finally, due to the wealth of highly stereoselective carbonyl reduction methods available, simple redox chemistry may define a temporary solution to the modest levels of stereoselection observed in this regioselective coupling process.[47] It is important to point out that no such solutions are available for reactions that do not proceed in a regioselective fashion.
4.3. Class II alkoxide-directed coupling reactions of internal alkynes
Unlike the alkoxide-directed reactions described in sections 4.1 and 4.2, that are limited in scope with respect to the reactivity profile of Ti–alkyne complexes in bimolecular C–C bond forming processes, Class II alkoxide-directed coupling reactions provide a unique means to control selectivity and overcome barriers associated with low reactivity in bimolecular metallacycle-mediated bond construction.
In Class II alkoxide-directed reductive coupling reactions, regioselective C–C bond formation is based on a sequence of events that sets up an intramolecular carbometalation via a fleeting intermediate (31→32), Figure 17. As a result, the wealth of transformations available in intramolecular reductive coupling processes have the potential of becoming available in these convergent coupling reactions. While we have employed this unique design for the regio- and stereoselective union of a variety of coupling partners, we will focus our attention here only on processes that have enabled the highly regioselective coupling of internal alkynes.
Figure 17.

A highly regioselective reductive cross-coupling reaction between internal alkynes.
In an attempt to validate the “Class II” alkoxide-directed reaction design, we focused our attention on an unprecedented reductive cross-coupling reaction for the union of unsymmetrically substituted internal alkynes with other internal alkynes. While seemingly representing an extension of our Class I alkoxide-directed alkyne coupling chemistry, the present reaction is significantly more challenging. In addition to the difficulties associated with control of regioselection in the functionalization of both internal alkynes, typically low levels of reactivity have stood as a significant barrier to the proposed convergent coupling process.
As depicted in Figure 17, initial studies with simple alkynes validated the basic merits of the “Class II” reaction design. Here, preformation of a Ti–alkyne complex with a symmetrically substituted alkyne was followed by exposure to an appropriately functionalized unsaturated alkoxide. After protonation of the presumed intermediate titanacyclopentadiene, stereodefined 1,3-dienes were produced. Initial studies demonstrated that the tethered hydroxy group indeed had an impressive effect on both reactivity and selectivity.[48] As depicted, the steric environment proximal to the site of C–C bond formation played little role in regioselection: Products 89-92 were all formed as single regioisomers, where C–C bond formation had occurred distal to the tethered hydroxy group, independent of the steric environment. Moving on from diphyenylacetylene, symmetrical alkynes that are not conjugated are also suitable substrates (93). Finally, extending the position of the tethered hydroxy group with respect to the internal alkyne was possible. In this case, coupling of a bishomopropargylic alcohol led to highly regioselective coupling (i.e. 94). Interestingly, if the hydroxy substituent is protected as the corresponding TBS-ether, or if it is not located in a suitable position for Class II directed carbometalation (i.e. propargylic and tris-homopropargylic alcohols), a complex product mixture results.
Finally, in the most complex examples investigated, reductive cross-coupling of internal alkynes 95 and 98 with 96 and 99 proceed with exquisite regioselection (Figure 18). Here, Ti–alkyne complexes of 96 and 99 were exposed to the preformed lithium alkoxides of 95 and 98, respectively. As anticipated, very high regioselection was observed in the functionalization of 95 and 98, likely as a result of the proposed sequence of ligand exchange followed by intramolecular carbometalation. What was most surprising in these coupling reactions was the very high levels of regioselection observed in the functionalization of alkynes 96 and 99. Here, the only criteria for selectivity should be based on the unique steric environment about each carbon of these alkynes. Unlike other reports that document the poor ability of simple alkyl branching to affect regioselection in related reductive cross-coupling reactions of internal alkynes with carbonyl electrophiles, here selectivity appears complete.[48]
Figure 18.

“Class II” alkoxide-directed regioselective reductive cross-coupling reactions of two unsymmetrically substituted disubstituted alkynes.
It is conceivable that the transition state for these reductive cross-coupling reactions are more product-like than that for related alkyne–aldehyde coupling reactions due to the anticipated difference in exothermicity of these two general processes.[49] In the transition state operative for the Class II alkoxide-directed reductive coupling processes depicted in Figure 18, it is possible that the metal–alkyne complex appears more olefin-like than in related alkyne–aldehyde coupling reactions. Based on expectations associated with minimization of A-1,3 strain,[50] and the subsequent result that this conformational consideration has on the steric environment about the metallacyclopropene, it is conceivable that significant preference results for the formation of the observed isomer, where C–C bond formation occurs at the site distal to the α-branch (Figure 19).
Figure 19.
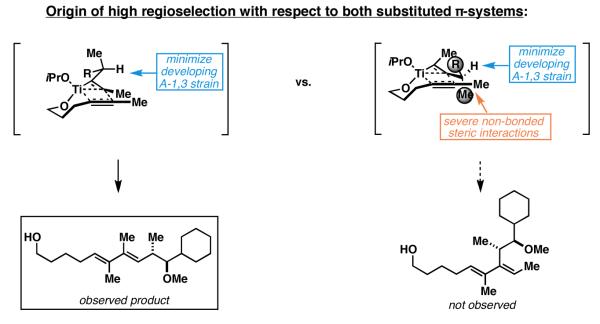
High regioselectivity in the functionalization of the unsymmetrically substituted Ti–alkyne complex in Class II alkoxide-directed reductive cross-coupling reactions.
4.4. Class II alkoxide-directed reductive cross-coupling reactions of internal alkynes with imines
Application of Class II alkoxide-directed coupling processes for the union of internal alkynes with imines requires the preformation of an azatitanacyclopropane, followed by introduction of an appropriately functionalized internal alkyne. Using the established procedure for generating such complexes from aromatic imines,[51] highly selective reductive cross-coupling reactions have been accomplished (Table 4).[52] As observed in the related reductive cross-coupling reaction of internal alkynes discussed in section 4.3, the high levels of regioselection observed here were independent of the steric environment at the distal position of the alkyne (entries 1-4, rs ≥ 20:1; Table 4). Notably, the reductive coupling of TMS alkyne 107 delivers 108 – an isomer not typically observed in related coupling reactions.
Table 4.

| entry | imine | unsaturated alkoxide |
yield[a] [%] |
major regioisomer |
|---|---|---|---|---|
| 1 |
|
|
65 |
|
| 2 | 100 | 103;R=Et | 60 | 104;R=Et |
| 3 | 100 | 105;R=iPr | 53 | 106;R=iPr |
| 4 | 100 | 107; R = TMS | 55 | 108; R = TMS |
Reaction conditions: Ti(OiPr)4, cC5H9MgCl, Et2O, −78 to −40 °C, then add unsaturated alkoxide (−40 to 0 °C), quench with sat. NH4Cl(aq).
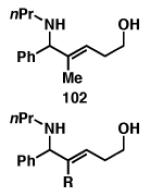
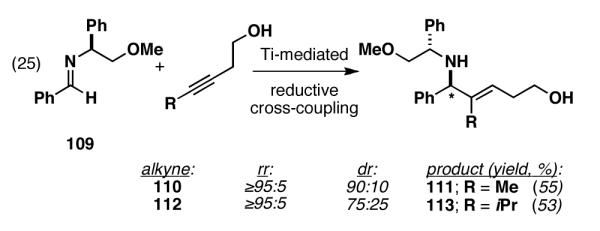
The problem of stereoselection in this reductive coupling process is one that can be addressed by the use of a chiral imine. Here, use of a phenylglycine-based imine 109 in reductive coupling reactions with simple alkynes delivered allylic amines with good levels of regioselection and modest levels of stereoinduction (Figure 20).[53] Interestingly, diastereoselection was affected by the size of the alkyne substituent, with a drop in stereoselection observed in the case of the i-Pr-substituted alkyne 113.
Figure 20.
Asymmetric Class II alkoxide-directed reductive cross-coupling reaction.
Finally, in this subset of reaction, alkoxide-directed C–C bond formation can be used to override the electronic effect that dictates site-selectivity in a myriad of related coupling reactions. As depicted in Figure 21, conjugated alkynes do little to affect the dominant control imparted by the presence of the tethered free hydroxy substituent. Here, reductive coupling with aromatic imines delivers products where C–C bond formation occurs α-to the π-conjugation (this is an opposite sense of regioselection as that seen with related metal-catalyzed coupling processes).[12, 13, 54]
Figure 21.

Regioselective Class II alkoxide-directed reductive cross-coupling reactions of conjugated alkynes with aromatic imines. [a] General reaction conditions: To a preformed Ti–imine complex (1eq) at −40 °C was dropwise added a preformed solution of the lithium alkoxide of the homopropargylic alcohol (1.5-3 eq). This solution was warmed slowly from −40 to 0 °C, then treated with saturated aqueous solution of NaHCO3.
The unique regioselectivity offered from this emerging class of reductive cross-coupling can be employed for novel complexity-generating processes in organic synthesis. In the present case, the site-selective reductive cross-coupling depicted in Figure 21 forms the foundation of a two-step three component coupling process for heterocycle assembly.[54] As illustrated in Figure 22, cationic annulation via acid mediated reaction with an aldehyde (117 → 125; eq 26), or simple intramolecular cyclization (123 → 126; eq 27) defines a simple process for the stereoselective synthesis of polycyclic heterocycles.
Figure 22.
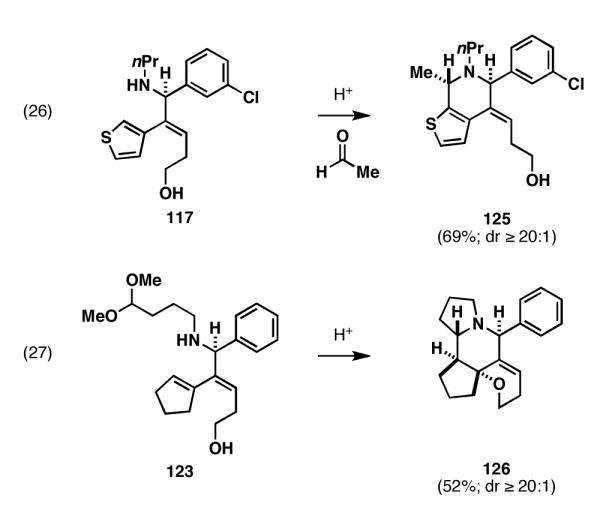
Cationic annulation processes for the conversion of reductive cross-coupling products to stereodefined heterocycles.
5. Summary and Outlook
Over the past fifty years, C–C bond formation reactions that proceed through metallacyclopentane-like intermediates have grown from observations made in the trimerization of acetylenes to the development of a wide range of useful synthetic methods. Early advances were made in defining intramolecular C–C bond forming processes, with recent contributions defining catalytic asymmetric variants for the union of alkynes with carbonyl-based electrophiles. Despite this significant growth, the utility of metallacycle-mediated C–C bond formation in synthesis has been largely limited to either intramolecular processes, or the bimolecular union of only a small subset of unsaturated coupling partners. Independent of the metal employed, or whether or not that metal is used in a substoichiometric fashion, basic issues associated with reactivity and control (homo- vs. heterocoupling, regio- and stereoselectivity), have remained as central barriers for reaction discovery and development in this broad area of chemical reactivity.
This Microreview has presented a brief historical background to modern reductive cross-coupling chemistry, described the state-of-the-art in regioselective coupling of internal alkynes, and defined the unique potential of alkoxide-directed titanium-mediated reductive cross-coupling for accomplishing novel bond construction. Overall, titanium-mediated reductive cross-coupling has emerged as a subset of metallacycle-mediated bimolecular C–C bond forming reactions that can be controlled in a unique and highly selective manner. The ability to generate C–C bonds in the presence of unprotected hydroxy groups, while rendering these processes “directed” by association of a pendant alkoxide to the Ti-center, has led to the development of a range of unique fragment coupling processes. The survey presented here has focused on regioselective reductive coupling chemistry of unsymmetrical internal alkynes, as these processes are the most well-established of all reductive cross-coupling methods. We hope to convey that alkoxide-directed titanium-mediated reductive cross-coupling has the potential to define unique bond constructions not yet available with other methods. From a more broad perspective, we are confident that the conceptual pathways presented here have the potential to support the discovery and development of a large class of novel fragment coupling reactions for chemical synthesis.[55, 56] We hope that this discussion serves to clarify the major challenges to reaction development that exist in the field, and provide a conceptual framework for the development of countless new reactions in organic chemistry.
Acknowledgments
We thank the National Institutes of Health, the American Cancer Society, the American Chemical Society, the Arnold and Mabel Beckman Foundation, Boehringer Ingelheim, Eli Lilly, Novartis, Bristol-Myers Squibb and Pfizer for financial support of our research program (in the form of grants and graduate student fellowships).
Author Portrait
Holly A. Reichard received a B.S. in Chemistry from The Pennsylvania State University in 2004. Currently, she is enrolled in the graduate program in chemistry of Yale University and finishing up at her studies in the Department of Chemistry at The Scripps Research Institute. Her graduate research in the laboratories of Professor Glenn C. Micalizio focuses on the design of new reductive cross-coupling methods and their application to the total synthesis of biologically important natural products.
Martin McLaughlin received his B. Sc. in chemistry from Boston College in 2002. After working for two years for Array Biopharma Inc., he moved to New Haven, CT where he joined the group of Professor Glenn Micalizio at Yale University. He is currently finishing his Ph.D. with Prof. Micalizio in The Department of Chemistry at The Scripps Research Institute, where he studies titanium-mediated convergent coupling reactions for use in heterocycle synthesis.
Ming Z. Chen received her B.A. in chemistry from Middlebury College in 2007 working with Prof. Jeffrey H. Byers and M.S. in organic chemistry from Yale University in 2008 working with Prof. Glenn C. Micalizio. Currently, she is enrolled in the chemistry graduate program at The Scripps Research Institute. Her graduate research in the laboratory of Prof. Glenn C. Micalizio focuses on developing new methodology for convergent C-C bond formation via titanium-mediated reductive cross coupling.
Glenn C. Micalizio obtained a Ph.D. at the University of Michigan in 2001 under the supervision of Professor William R. Roush. After postdoctoral study as a Fellow of the Helen Hay Whitney Foundation at Harvard University in the laboratory of Professor Stuart L. Schreiber, he moved to Yale University as an Assistant Professor in the Department of Chemistry (2003). In 2008, he moved to the Department of Chemistry at The Scripps Research Institute as an Associate Professor. His research is focused on the development of new synthetic methods and application of these methods to complex molecule synthesis.
References
- [1].For an interesting discussion in the context of process chemistry, see: Zhang TY. Chem. Rev. 2006;106:2583–2595. doi: 10.1021/cr040677v.
- [2] a).For discussions regarding the “maturity” of organic chemistry as a scientific discipline, see: Seebach D. Angew. Chem. Int. Ed. Engl. 1990;29:1320–1367. Heathcock CH. Angew. Chem. Int. Ed. Engl. 1992;31:665–681. (Eplogue and Editorial section) Sierra MA, de la Torre MC. Angew. Chem. Int. Ed. 2000;39:1538–1559. doi: 10.1002/(sici)1521-3773(20000502)39:9<1538::aid-anie1538>3.0.co;2-o.
- [3] a).Reppe W, Schweckendick WJ. Liebigs Ann. Chem. 1948;560:104. For a recent review of metal-mediated [2+2+2], see: Agenet N, Buisine O, Slowinski F, Gandon V, Aubert C, Malacria M. Org. React. 2007;68:1–301.
- [4].Vol’pin ME, Dubovitskii VA, Nogina OV, Kursanov DN. Doklady Akademii Nauk Sssr. 1963;151:1100. [Google Scholar]
- [5] a).For recent reviews of Pauson-Khand chemistry, see: Gibson SE, Stevanazzi A. Angew. Chem. Int. Ed. 2003;42:1800–1810. doi: 10.1002/anie.200200547. Gibson SE, Mainolfi N. Angew. Chem. Int. Ed. 2005;44:3022–3037. doi: 10.1002/anie.200462235. For a review of higher order annulations metal-catalyzed annulations, see: Wender PA, Bi FC, Gamber GG, Gosselin F, Hubbard RD, Scanio MJC, Sun R, Williams TJ, Zhang L. Pure Appl. Chem. 2002;74:25–31. For reviews of Ru-mediated processes, see: Trost BM, Toste FD, Pinkerton AB. Chem. Rev. 2001;101:2067–2096. doi: 10.1021/cr000666b. Trost BM, Frederiksen MU, Rudd MT. Angew. Chem. Int. Ed. 2005;44:6630–6666. doi: 10.1002/anie.200500136. For a review of metal catalyzed cycloisomerization, see: Michelet V, Toullec PY, Genet J-P. Angew. Chem. Int. Ed. 2008;47:4268–4315. doi: 10.1002/anie.200701589. For reviews of Zr-mediated processes, see: Buchwald SL, Nielsen RB. Chem. Rev. 1988;88:1047–1058. Negishi E-I. J. Chem. Soc. Dalton Trans. 2005:827–848. doi: 10.1039/b417134a. For reviews of Ti-mediated processes, see: Kulinkovich OG, de Meijere A. Chem. Rev. 2000;100:2789–2834. doi: 10.1021/cr980046z. Sato F, Urabe H, Okamoto S. Chem. Rev. 2000;100:2835–2886. doi: 10.1021/cr990277l. For a review of Ni-catalyzed processes, see: Montgomery J, Sormunen GJ. Top. Curr. Chem. 2007;279:1–23. For a review of a collection of Ru-, Ir- and Rh-catalyzed processes, see: Patman RL, Bower JF, Kim IS, Krische MJ. Aldrich. Acta. 2008;41:95–104. For a recent review of Co- and Ni-catalyzed reductive coupling of alkyns, allenes and alkenes with alkenes, see: Jeganmohan M, Cheng C-H. Chem. Eur. J. 2008;14:10876–10886. doi: 10.1002/chem.200800904.
- [6] a).Khand IU, Knox GR, Pauson PL, Watts WE. Chem. Commun. 1971;36 [Google Scholar]; b) Khand IU, Knox GR, Pauson PL, Watts WE, Foreman MI. J. Chem. Soc. Perkin. 1973;1:977–981. [Google Scholar]; c) Cohen SA, Auburn PR, Bercaw JE. J. Am. Chem. Soc. 1983;105:1136–1143. [Google Scholar]; d) Cohen SA, Bercaw JE. Organometallics. 1985;4:1006–1014. [Google Scholar]; e) Buchwald SL, Watson BT. J. Am. Chem. Soc. 1987;109:2544–2546. [Google Scholar]
- [7] a).For early discussions concerning the reversibility of metallacyclopentane formation, see: McDermott JX, Whitesides GM. J. Am. Chem. Soc. 1974;96:947–948. McDermott JX, Wilson ME, Whitesides GM. J. Am. Chem. Soc. 1976;98:6529–6536. Grubbs RH, Miyashita A. J. Chem. Soc. Chem. Comm. 1977:864–865. For an example of regioselective reductive coupling where thermodynamic equilibration of a mixture of metallacyclopentadienes leads to selective formation of a regiodefined 1,3-diene, see: Buchwald SL, Nielsen RB. J. Am. Chem. Soc. 1989;111:2870–2874.
- [8] a).For an example of a silyl-alkyne in regioselective Alder-Ene chemistry, see: Trost BM, Machacek MR, Ball ZT. Org. Lett. 2003;5:1895–1898. doi: 10.1021/ol034463w. For an example of regioselective Alder-Ene chemistry with alkynylboronates, see: Hansen EC, Lee D. J. Am. Chem. Soc. 2005;127:3252–3253. doi: 10.1021/ja0424629.
- [9] a).Buchwald SL, Watson BT, Wannamaker MW, Dewan JC. J. Am. Chem. Soc. 1989;111:4486–4494. [Google Scholar]; b) Grossman RB, Davis WM, Buchwald SL. J. Am. Chem. Soc. 1991;113:2321–2322. [Google Scholar]
- [10] a).Harada K, Urabe H, Sato F. Tetrahedron Lett. 1995;36:3203–3206. For an example of a Ta-mediated regioselective reductive coupling of alkynes with aldehydes where regioselectivity is derived from C–C bond formation distal to a TMS- or t-Bu substituent, see: Takai K, Kataoka Y, Utimoto K. J. Org. Chem. 1990;55:1707–1708. For a regioselective reductive cross-coupling of a stannylalkyne with an aldehyde, see: Launay V, Beaudet I, Quintard J-P. Synlett. 1997:821–823.
- [11] a).Huang W-S, Chan J, Jamison TF. Org. Lett. 2000;2:4221–4223. doi: 10.1021/ol006781q. [DOI] [PubMed] [Google Scholar]; b) Sa-ei K, Montgomery J. Org. Lett. 2006;8:4441–4443. doi: 10.1021/ol061579u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].For regioselective coupling of phenylacetylene derivatives, see Ref 10b, and: Patel SJ, Jamison TF. Angew. Chem. Int. Ed. 2003;42:1364–1367. doi: 10.1002/anie.200390349. Mahandru GM, Liu G, Montgomery J. J. Am. Chem. Soc. 2004;126:3698–3699. doi: 10.1021/ja049644n. Miller KM, Jamison TF. Org. Lett. 2005;7:3077–3080. doi: 10.1021/ol051075g. Ngai M-Y, Barchuk A, Krische MJ. J. Am. Chem. Soc. 2007;129:280–281. doi: 10.1021/ja0670815. Patman RL, Chaulagain MR, Williams VM, Krische MJ. J. Am. Chem. Soc. 2009;131:2066–2067. doi: 10.1021/ja809456u. For a study of regioselective Ta-mediated reductive coupling of acetylenic esters and amides with carbonyl compounds, see: Takai K, Tezuka M, Utimoto K. J. Org. Chem. 1991;56:5980–5982.
- [13] a).For regioselective coupling of 1,3-enynes and 1,3-diynes, see: Huddleston RR, Jang H-Y, Krische MJ. J. Am. Chem. Soc. 2003;125:11488–11489. doi: 10.1021/ja030415v. Miller KM, Luanphaisarnnont T, Molinaro C, Jamison TF. J. Am. Chem. Soc. 2004;126:4130–4131. doi: 10.1021/ja0491735. Jang H-Y, Huddleston RR, Krische MJ. J. Am. Chem. Soc. 2004;126:4664–4668. doi: 10.1021/ja0316566. Kong J-R, Cho C-W, Krische MJ. J. Am. Chem. Soc. 2005;125:11269–11276. doi: 10.1021/ja051104i. Komanduri V, Krische MJ. J. Am. Chem. Soc. 2006;128:16448–16449. doi: 10.1021/ja0673027. Cho C-W, Krische MJ. Org. Lett. 2006;8:3873–3876. doi: 10.1021/ol061485k. Cho C-W, Skucas E, Krische MJ. Organometallics. 2007;26:3860–3867. Hong Y-T, Cho C-W, Skucas E, Krische MJ. Org. Lett. 2007;9:3745–3748. doi: 10.1021/ol7015548.
- [14].For an example in silacyclopropane-mediated coupling of alkynes with nitriles, see: Anderson LL, Woerpel KA. Org. Lett. 2009;11:425–428. doi: 10.1021/ol802412b.
- [15] a).For an example in Co-catalyzed Alder-Ene chemistry, see: Hilt G, Treutwein J. Angew. Chem. Int. Ed. 2007;46:8500–8502. doi: 10.1002/anie.200703180. For a range of selectivities observed in Ru-catalyzed Alder-Ene chemistry, see: Trost BM, Shen HC, Pinkerton AB. Chem. Eur. J. 2002;8:2341–2349. doi: 10.1002/1521-3765(20020517)8:10<2341::AID-CHEM2341>3.0.CO;2-A.
- [16] a).An interesting exception to this trend was reported in the Ti-alkoxide promoted reductive cross-coupling of an phenylpropyne with cyclohexanecarboxaldehyde: Harada K, Urabe H, Sato F. Tetrahedron Lett. 1995;36:3203–3206. Surprisingly, this selectivity was reported to be reversed in these authors study of a Ti-alkoxide-mediated reductive cross-coupling of the same alkyne with an imine, see: Gao Y, Harada K, Sato F. Tetrahedron Lett. 1995;36:5913–5916.
- [17].Chaulagain MR, Sormunen GJ, Montgomery J. J. Am. Chem. Soc. 2007;129:9568–9569. doi: 10.1021/ja072992f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Barchuk A, Ngai M-Y, Krische MJ. J. Am. Chem. Soc. 2007;129:8432–8433. doi: 10.1021/ja073018j. [DOI] [PubMed] [Google Scholar]
- [19].Miller KM, Jamison TF. J. Am. Chem. Soc. 2004;126:15342–15343. doi: 10.1021/ja0446799. [DOI] [PubMed] [Google Scholar]
- [20] a).While not included in the discussion due to the highly focused nature of this review, regioselective coupling reactions of other heteroatom-substituted alkynes have been reported. For RS-substitution, see: Van Wagenen BC, Livinghouse T. Tetrahedron Lett. 1989;30:3495–3498. Takai K, Miyai J, Kataoka Y, Utimoto K. Organometallics. 1990;9:3030–3031. Kataoka Y, Miyai J, Tezuka M, Takai K, Utimoto K. J. Org. Chem. 1992;57:6796–6802. For RN-based substitution, see: Tanaka R, Hirano S, Urabe H, Sato F. Org. Lett. 2003;5:67–70. doi: 10.1021/ol027209x. For RP-based substitution, see: Quntar A, Srebnik M. Org. Lett. 2003;5:357–359. doi: 10.1021/ol027429a. For an example of regioselective coupling of an R3SiO-substituted alkyne in a silycyclopropene-mediated coupling reaction, see: Ref 14.
- [21] a).Hamada T, Suzuki D, Urabe H, Sato F. J. Am. Chem. Soc. 1999;121:7342–7344. [Google Scholar]; b) Urabe H, Narita M, Sato F. Angew. Chem. Int. Ed. 1999;38:3516–3518. doi: 10.1002/(sici)1521-3773(19991203)38:23<3516::aid-anie3516>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- [22].Yamashita K, Urabe H, Sato F. Tetrahedron Lett. 1997:4619–4622. [Google Scholar]
- [23] a).Luanphaisarnnont T, Ndubaku CO, Jamison TF. Org. Lett. 2005;7:2937–2940. doi: 10.1021/ol050881k. Sa-ei K, Montgomery J. Org. Lett. 2006;8:4441–4443. doi: 10.1021/ol061579u.
- [24] c).For recent reviews that describe catalytic asymmetric reductive cross-coupling chemistry, see: a) Ref. 5k, b) Ref 5l, and Moslin RM, Miller-Moslin K, Jamison TF. Chem. Commun. 2007:4441–4449. doi: 10.1039/b707737h.
- [25].For a review of substrate-directable reactions in organic chemistry, see: Hoveyda AH, Evans DA, Fu GC. Chem. Rev. 1993;93:1307–1370.
- [26] a).For hydroxyl-directed carbometalation reactions based on titanium, see: Coleman RA, O’Doherty CM, Tweedy HE, Harris TV, Thompson DW. J. Organomet. Chem. 1976;107:C15–C17. Brown DC, Nichols SA, Gilpin AB, Thompson DW. J. Org. Chem. 1979;44:3457–3461. Ewing JC, Ferguson GS, Moore DW, Schultz FW, Thompson DW. Org. Chem. 1985;50:2124–2128. For hydroxyl-directed carbometalation based on magnesium, see: Eisch JJ, Merkley JH. J. Organomet. Chem. 1969;20:P27–P31. Eisch JJ. J. Organomet. Chem. 1980;200:101–117. Richey HG, Von Rein FW. J. Organomet. Chem. 1969;20:P32–P35. Hoveyda AH, Xu Z. J. Am. Chem. Soc. 1991;113:5079–5080. Hoveyda AH, Xu Z, Morken JP, Houri AF. J. Am. Chem. Soc. 1991;113:8950–8952. Itami K, Mitsudo K, Yoshida J-I. Angew. Chem. Int. Ed. 2001;40:2337–2339. For directed carbometalation based on tantalum, see: Takai K, Yamada M, Odaka H, Utimoto K. J. Org. Chem. 1994;59:5852–5853. For directed carbometalation based on aluminum/zirconium, see: Ma S, Negishi E-I. J. Org. Chem. 1997;62:784–785. doi: 10.1021/jo970009s.
- [27] a).For examples of S- and P- directed C–C bond formation, see: Burke SD, Cobb JE. Tetrahedron Lett. 1986;27:4237–4240. Krafft ME, Juliano CA, Scott IL, Wright C, McEachin MD. J. Am. Chem. Soc. 1991;113:1693–1703. Krauss IJ, Wang CC-Y, Leighton JL. J. Am. Chem. Soc. 2001;123:11514–11515. doi: 10.1021/ja016978t. Breit B, Demel P. Tetrahderon. 2000;56:2833–2846. Breit B, Breuninger D. J. Am. Chem. Soc. 2004;126:10244–10245. doi: 10.1021/ja0467364. Bruch A, Gebert A, Breit B. Synthesis. 2008:2169–2176. For an interesting approach to relay transient formation of a C–O bond to P-directed hydroformylation, see: Lightburn TE, Dombrowski MT, Tan KL. J. Am. Chem. Soc. 2008;130:9210–9211. doi: 10.1021/ja803011d. For an example of a pyiridyl-directed Pauson-Khand reaction, see: Itami K, Mitsudo K, Fujita K, Ohashi Y, Yoshida J-I. J. Am. Chem. Soc. 2004;126:11058–11066. doi: 10.1021/ja047484+.
- [28] a).For selected examples of heteroatom-directed C–H activation followed by C–C bond formation, see: Murai S, Kakiuchi F, Sekine S, Tanaka Y, Kamatani A, Sonoda M, Chatani N. Nature. 1993;366:529–532. Thalji RK, Ahrendt KA, Bergman RG, Ellman JA. J. Am. Chem. Soc. 2001;123:9692–9693. doi: 10.1021/ja016642j. Kalyani D, Deprez NR, Desai LV, Sanford MS. J. Am. Chem. Soc. 2005;127:7330–7331. doi: 10.1021/ja051402f. Hull KL, Sanford MS. J. Am. Chem. Soc. 2007;129:11904–11905. doi: 10.1021/ja074395z. Deprez NR, Sanford MS. J. Am. Chem. Soc. 2009;131:11234–11241. doi: 10.1021/ja904116k. For a recent review of Rh-catalyzed C–C bond formation via heteroatom-directed C–H bond activation, see: Colby DA, Bergman RG, Ellman JA. Chem. Rev. 2009 doi: 10.1021/cr900005n. DOI: 10.1021/cr900005n.
- [29].Katsuki T, Martin VS. Org. React. 1996;48:1–299. [Google Scholar]
- [30] a).Nugent WA, Calabrese JC. J. Am. Chem. Soc. 1984;106:6422–6424. Mashima K, Takaya H. Organometallics. 1985;4:1464–1466. Nugent WA, Thorn DL, Harlow RL. J. Am. Chem. Soc. 1987;109:2788–2796. RajanBabu TV, Nugent WA, Taber DF, Fagan PJ. J. Am. Chem. Soc. 1988;110:7128–7135. Mashima K, Sakai N, Takaya H. Bull. Chem. Soc. Jpn. 1991;64:2475–2483. Berk SC, Bergman RB, Buchwald SL. J. Am. Chem. Soc. 1993;115:4912–4913. Kablaoui NM, Buchwald SL. J. Am. Chem. Soc. 1995;117:6785–6786. Crowe WE, Rachita MJ. J. Am. Chem. Soc. 1995;117:6787–6788. Kablaoui NM, Hicks FA, Buchwald SL. J. Am. Chem. Soc. 1996;118:5818–5819. Kablaoui NM, Buchwald SL. J. Am. Chem. Soc. 1996;118:3182–3191. Hicks FA, Kablaoui NM, Buchwald SL. J. Am. Chem. Soc. 1996;118:9450–9451. Kablaoui NM, Hicks FA, Buchwald SL. J. Am. Chem. Soc. 1997;119:4424–4431. Aoki K, Peat AJ, Buchwald SL. J. Am. Chem. Soc. 1998;120:3068–3073. Sturla SJ, Kablaoui NM, Buchwald SL. J. Am. Chem. Soc. 1999;121:1976–1977. Hicks FA, Kablaoui NM, Buchwald SL. J. Am. Chem. Soc. 1999;121:5881–5898. Hicks FA, Buchwald SL. J. Am. Chem. Soc. 1999;121:7026–7033. Mandal SK, Amin R, Crowe WE. J. Am. Chem. Soc. 2001;123:6457–6458. doi: 10.1021/ja005568m.
- [31] a).For chemistry related to Ti-aryloxides, see: Chamberlain LR, Durfee LD, Fanwick PE, Kobriger L, Latersky SL, McMullen AK, Rothwell IP, Folting K, Huffman JC, Streib WE, Wang R. J. Am. Chem. Soc. 1987;109:390–402. doi: 10.1021/ja00236a017. Chamberlain LR, Durfee LD, Fanwick PE, Kobriger LM, Latesky SL, McMullen AK, Steffey BD, Rothwell IP, Folting K, Huffman JC. J. Am. Chem. Soc. 1987;109:6068–6076. doi: 10.1021/ja00236a017. Thorn MG, Hill JE, Waratuke SA, Johnson ES, Fanwick PE, Rothwell IP. J. Am. Chem. Soc. 1997;119:8630–8641. For early examples documenting the reactivity of Ti-alkoxides, see: Kulinkovich OG, Sviridov SV, Vasilevski DA, Pritytskaya TS. J. Org. Chem. USSR (Eng. Transl.) 1989;25:2027–2028.
- [32].For recent reviews, see: Kulinkovich OG. Eur. J. Org. Chem. 2004:4517–4529.
- [33].Marek I, editor. Titanium and Zirconium in Organic Synthesis. Vol. 512. Weinheim; Wiley-VCH: 2002. [Google Scholar]
- [34].The calculation presented in Figure 12 is based on the use of 5-10 mol% of each catalyst (as reported in the literature for typical catalytic processes). The cost associated with additional reagents for these processes has not been considered in calculations presented. That said, the current cost of 2 moles of i-PrMgCl is approximately $184 whereas the additional cost associated with use of 5 mol% of (+/−)-BIPHEP is $1,359 (Strem-2008).
- [35].Although not related to the reductive cross-coupling chemistry discussed, the cost of 1 mol% Pd(PPh3)4 for a hypothetical reaction run on 1 mol scale would be approximately $220 (Strem 2008).
- [36].Ti(Oi-Pr)4 is a free flowing liquid that can be purified by distillation and transferred by syringe. No glove box is needed to conduct any operation required for setting up a reductive cross-coupling reaction with stoichiometric Ti(Oi-Pr)4, and no significant precautions need to be taken to exclude trace quantities of air or water from the reaction mixture.
- [37].For an early report of a directed Kulinkovich reaction that embraced a potential bicyclic metallacyclopropane as an intermediate; see: Quan LG, Kim S-H, Lee JC, Cha JK. Angew. Chem. Int. Ed. 2002;41:2160–2162.
- [38].For an early report of a related design in Ta-mediated reductive coupling that was limited to the reaction of internal alkynes with terminal alkenes, see: Takai K, Yamada M, Odaka H, Utimoto K. J. Org. Chem. 1994;59:5852–5853. Regiochemical control in alkyne functionalization was not achieved as a result of the preorganization envoked. Rather, reactivity of a preformed monocyclic Ta-alkyne complex was achieved with a terminal alkene containing a tethered hydroxy substituent. Also, the position of the hydroxy substituent played a central role in the site-selective reaction of dienes with a preformed Ta-alkyne complex.
- [39].While the use of substituted alkenes, allenes and alkynes are well known in intramolecular metallacycle-mediated bond construction, barriers associated with reactivity broadly impact the viability of such bond constructions in a bimolecular manifold.
- [40] (a).For a review of carbonyl olefination methods, see: Takeda T, editor. Modern Carbonyl Olefination: Methods and Applications. Vol. 365. Wiley-VCH; 2004. Maryanoff BE, Reitz AB. Chem. Rev. 1989;89:863–927. For selected examples of carbonyl olefination for the stereoselective generation of 1,3-dienes in the context of natural product total synthesis, see: Bonazzi S, Güttinger S, Zemp I, Kutay U, Gademann K. Angew. Chem. Int. Ed. 2007;46:8707–8710. doi: 10.1002/anie.200703134. Arai N, Chikaraishi N, Omura S, Kuwajima I. Org. Lett. 2004;6:2845–2848. doi: 10.1021/ol049219z. Marshall JA, Bourbeau MP. J. Org. Chem. 2002;67:2751–2754. doi: 10.1021/jo016025d. Lautens M, Stammers TA. Synthesis. 2002:1993–2012. Smith AB, Brandt BM. Org. Lett. 2001;3:1685–1688. doi: 10.1021/ol0158922. Kobayashi M, Wang W, Tsutsui Y, Sugimoto M, Murakami N. Tetrahedron Lett. 1998;39:8291–8294. Schnermann MJ, Romero FA, Hwang I, Nakamaru-Ogiso E, Yagi T, Boger DL. J. Am. Chem. Soc. 2006;128:11799–11807. doi: 10.1021/ja0632862. Evans DA, Rajapakse HA, Chiu A, Stenkamp D. Angew. Chem. Int. Ed. 2002;41:4573–4576. doi: 10.1002/1521-3773(20021202)41:23<4573::AID-ANIE4573>3.0.CO;2-S.
- [41] (a).For reviews of palladium-catalyzed cross-coupling, see: Miyaura N, Suzuki A. Chem. Rev. 1995;95:2457–2483. Vinicius M, De Souza N. Curr. Org. Synth. 2006;3:313–326. Negishi E-I, Huang Z, Wang G, Mohan S, Wang C, Hattori H. Acc. Chem. Res. 2008;41:1474–1485. doi: 10.1021/ar800038e. For selected examples of metal-mediated cross-coupling for the assembly of dienes in the context of natural product synthesis, see: Langille NF, Panek JS. Org. Lett. 2004;6:3203–3206. doi: 10.1021/ol048664r. Marshall JA, Bourbeau MP. Org. Lett. 2002;4:3931–3934. doi: 10.1021/ol026791m. Tortosa M, Yakelis NA, Roush WR. J. Am. Chem. Soc. 2008;130:2722–2723. doi: 10.1021/ja710238h. Nicolaou KC, Chakraborty TK, Piscopio AD, Minowa N, Bertinato P. J. Am. Chem. Soc. 1993;115:4419–4420.
- [42].For examples of regioselective reductive cross-coupling reactions of internal alkynes with terminal alkynes, see Refs 7d and 21. Also, for a summary of previously required substitution patterns of the internal alkyne to render this type of reductive cross-coupling reaction regioselective, see Figure 10 and the accompanying discussion.
- [43].Shimp HL, Micalizio GC. Org. Lett. 2005;7:5111–5114. doi: 10.1021/ol052241n. [DOI] [PubMed] [Google Scholar]
- [44].Perez LJ, Shimp HL, Micalizio GC. J. Org. Chem. 2009;74 doi: 10.1021/jo901451c. accepted. [DOI] [PubMed] [Google Scholar]
- [45] a).Bahadoor AB, Flyer A, Micalizio GC. J. Am. Chem. Soc. 2005;127:3694–3695. doi: 10.1021/ja050039+. [DOI] [PubMed] [Google Scholar]; b) Bahadoor AB, Micalizio GC. Org. Lett. 2006;8:1181–1184. doi: 10.1021/ol0600786. [DOI] [PubMed] [Google Scholar]
- [46].Otera J, editor. Modern Carbonyl Chemistry. Weinheim; Wiley-VCH: 2000. [Google Scholar]
- [47].Belardi JK, Micalizio GC. Org. Lett. 2006;8:2409–2412. doi: 10.1021/ol0607995. [DOI] [PubMed] [Google Scholar]
- [48] a).Ryan J, Micalizio GC. J. Am. Chem. Soc. 2006;128:2764–2765. doi: 10.1021/ja057352w. For related examples, see: Takai K, Kataoka Y, Utimoto K. J. Org. Chem. 1990;55:1707–1708. Kataoka Y, Oguchi Y, Yoshizumi K, Miwatashi S, Takai K, Utimoto K. Bull. Chem. Soc. Jpn. 1992:1543–1549. Takai K, Miwatashi S, Kataoka Y, Utimoto K. Chem. Lett. 1992:99–102. Colby EA, Jamison TF. J. Org. Chem. 2003;68:156–166. doi: 10.1021/jo0264123.
- [49] a).In the reaction of a Ti–alkyne complex with a carbonyl electrophile, conversion of the metallacyclopropene species to the metallacyclopentene intermediate is coupled to the formation of a strong Ti–O bond. In the related reaction with an alkyne, formation of the metallacyclopentadiene is coupled to the formation of a relatively weak Ti–C bond. For a discussion of the relative position of transition states as a function of the relative exothermicity of the reaction, see: Leffler JE. Science. 1953;117:340–341. doi: 10.1126/science.117.3039.340. Hammond GS. J. Am. Chem. Soc. 1955;77:334–338.
- [50].Hoffmann RW. Chem. Rev. 1989;89:1841–1860. [Google Scholar]
- [51].Gao Y, Yoshida Y, Sato F. Synlett. 1997:1353–1354. [Google Scholar]
- [52].McLaughlin M, Takahashi M, Micalizio GC. Angew. Chem. Int. Ed. 2007;46:3912–3914. doi: 10.1002/anie.200605060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Fukuhara K, Okamoto S, Sato F. Org. Lett. 2003;5:2145–2148. doi: 10.1021/ol034599u. [DOI] [PubMed] [Google Scholar]
- [54].Chen MZ, Micalizio GC. Org. Lett. 2009;11 doi: 10.1021/ol902169k. accepted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55] a).For an alkene–alkyne reductive cross-coupling, see: Reichard HA, Micalizio GC. Angew. Chem. Int. Ed. 2007;46:1440–1443. doi: 10.1002/anie.200603515. For an alkene–imine coupling process, see: Takahashi M, Micalizio GC. J. Am. Chem. Soc. 2007;129:7514–7516. doi: 10.1021/ja071974v. For an allylic alcohol–alkyne coupling, see: Kolundzic F, Micalizio GC. J. Am. Chem. Soc. 2007;129:1511–15113. doi: 10.1021/ja075678u. For an allene–imine coupling, see: McLaughlin M, Shimp HL, Navarro R, Micalizio GC. Synlett. 2008:735–738. doi: 10.1055/s-2008-1042808. For an allylic alcohol–vinylsilane coupling, see: Belardi JK, Micalizio GC. J. Am. Chem. Soc. 2008;130:16870–16872. doi: 10.1021/ja8074242. For an allylic alcohol–imine coupling, see: Takahashi M, McLaughlin M, Micalizio GC. Angew. Chem. Int. Ed. 2009;48:3648–3652. doi: 10.1002/anie.200900236.
- [56] a).For examples of complex Ti-mediated reductive cross-coupling reactions in the synthesis of natural products, see: Macklin TK, Micalizio GC. J. Am. Chem. Soc. 2009;131:1392–1393. doi: 10.1021/ja809491b. Belardi JK, Micalizio GC. Angew. Chem. Int. Ed. 2008;47:4005–4008. doi: 10.1002/anie.200800400. Reichard HA, Rieger JC, Micalizio GC. Angew. Chem. Int. Ed. 2008;47:7837–7840. doi: 10.1002/anie.200803031.