

Abstract
X-ROS signaling is a novel redox signaling pathway that links mechanical stress to changes in [Ca2+]i. This pathway is activated rapidly and locally within a muscle cell under physiological conditions, but can also contribute to Ca2+-dependent arrhythmia in heart and to the dystrophic phenotype in heart and skeletal muscle. Upon physiologic cellular stretch, microtubules serve as mechanotransducers to activate NADPH oxidase 2 in the transverse tubules and sarcolemmal membranes to produce reactive oxygen species (ROS). In heart, the ROS acts locally to activate ryanodine receptor Ca2+ release channels in the junctional sarcoplasmic reticulum, increasing the Ca2+ spark rate and “tuning” excitation-contraction coupling. In skeletal muscle, where Ca2+ sparks are not normally observed, the X-ROS signaling process is muted. However in muscular dystrophies, such as Duchenne Muscular Dystrophy and dysferlinopathy, X-ROS signaling operates at a high level and contributes to myopathy. Importantly, Ca2+ permeable stretch-activated channels are activated by X-ROS and contribute to skeletal muscle pathology. Here we review X-ROS signaling and mechanotransduction in striated muscle, and highlight important questions to drive future work on stretch-dependent signaling. We conclude that X-ROS provides an exciting mechanism for the mechanical control of redox and Ca2+ signaling, but much work is needed to establish its contribution to physiologic and pathophysiologic processes in diverse cell systems.
Introduction
Reactive oxygen species (ROS) have long been implicated in cellular pathology, but more recently have emerged as important physiologic signaling agents [1-6]. Much like subcellular Ca2+ signaling in the heart, redox signaling can be tightly controlled, spatially compartmentalized (“local”), and source specific [7]. Here we review a newly characterized ROS-dependent signaling cascade that exemplifies these properties and regulates Ca2+ signaling in cardiac and skeletal muscle [1, 6].
X-ROS signaling
Using new methods to stretch heart cells (Fig. 1, see below), a mechano-chemo transduction pathway was recently found to underlie stretch-dependent Ca2+ signaling in cardiac ventricular myocytes. Ca2+ sparks, the elementary unit of excitation-contraction coupling in heart [8-10], occur at a low rate during diastole. If a single myocyte is stretched within the physiologic sarcomere range, the rate of Ca2+ spark occurrence increases rapidly and reversibly [1, 11]. Specific experiments have revealed that the underlying subcellular process involves three necessary components: 1. a stabilized microtubule network, 2. NADPH oxidase 2 (Nox2) derived ROS, and 3. Ca2+ release channels in the sarcoplasmic reticulum (SR), ryanodine receptors type 2 (RyR2). In heart, cellular stretch activates local ROS production by Nox2 in a process requiring an intact microtubule network (Fig. 2). Local ROS directly or indirectly leads to post-translational modification of RyR2s, increasing the sensitivity of RyR2s to [Ca2+]i and promoting the fidelity of excitation-contraction (EC) coupling. We term this mechano-chemo signaling “X-ROS,” from the NoX2 dependence of the ROS signaling [1, 12].
Figure 1. Improved methods to study mechanotransduction in heart and skeletal muscle.
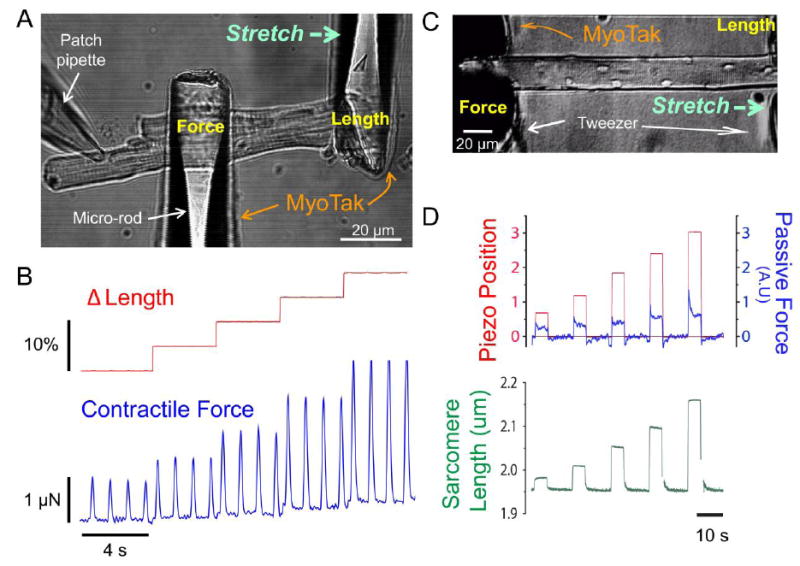
A, A ventricular myocyte is attached and stretched via MyoTak-coated micro-rods (Modified from [1]). B, This method is used to record isometric force (blue) in a ventricular myocyte stimulated at 1Hz and subjected to step changes in cell length (red, stretch from 5 – 20% of cell length). C, A skeletal FDB muscle fiber is attached and stretched via MyoTak-coated tweezers (Modified from [6]). D, Coated-tweezers are used to impose step changes in length (red) of an adult FDB muscle fiber while sarcomere length (green) and passive tension (blue) are recorded.
Figure 2. Summary of X-ROS signaling in heart.
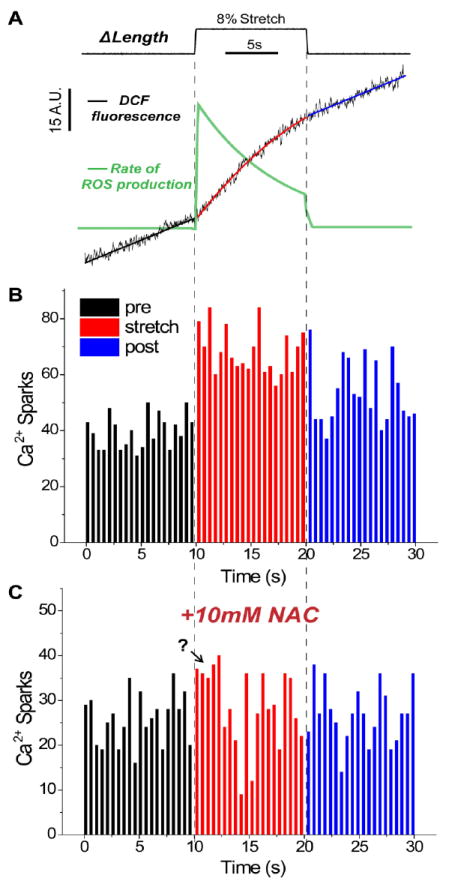
A, Average DCF fluorescence signal (black) from ventricular myocytes stretched 8% of cell length (n = 36 rat cells). The pre and post stretch intervals are well fit by a linear function, while the stretched interval is better fit by a polynomial function. Taking the derivative of these DCF fits gives the rate of ROS production (green). There is a rapid and transient increase in the rate of ROS production upon stretch. B, Histogram of Ca2+ sparks (n = 3,233 sparks) from 52 myocytes stretched as above. There is a rapid increase in the frequency of Ca2+ sparks (500ms bins) upon stretch. C, Histogram of Ca2+ sparks (n = 1,715 sparks) from 29 myocytes pre-treated with 10mM of the general anti-oxidant N-acetylcysteine and stretched as above. NAC blocks any statistically significant change in Ca2+ spark frequency; the question mark denotes a possible initial stretch-dependent increase in spark rate that is not blocked by NAC (Modified from [1]).
Many of the features of X-ROS signaling in heart are also found in skeletal muscle, but the signaling involves additional molecular components [6]. One prominent component in skeletal muscle is a mechanosensitive sarcolemmal channel whose opening is enhanced by Nox2-derived ROS. This signaling system is an important pathological component in Duchenne muscular dystrophy (DMD), where an increase in microtubule network density leads to a detrimental enhancement of X-ROS signaling.
Overview
In both cardiac and skeletal muscle Nox2-derived ROS is a central component in stretch-dependent signaling. Under physiological conditions it underlies fine adjustment of Ca2+ signaling in cardiac EC coupling. In pathological conditions, X-ROS signaling is increased and contributes to Ca2+-dependent arrhythmogenesis in heart and to ROS-linked pathology in dystrophic skeletal and cardiac muscle. While X-ROS is a provocative mechanical signaling pathway, much work is still needed to establish its role in the heart and other cell systems. Important questions will guide future work and will be addressed throughout this review. We will place our findings in context with the current state of the field, as well as take a critical look at the limitations of work to date and the future challenges ahead to unravel the physiologic and pathologic roles of stretch-dependent X-ROS signaling.
Results and Discussion
X-ROS signaling in heart
A new method to explore mechanical signaling in muscle cells
Mechanotransduction is the conversion of mechanical stimuli, such as cell stress or strain, into cellular responses. In the heart, stretching of heart cells during diastole and shortening during systole triggers diverse mechanotransduction signaling pathways that have broad impacts on cardiac patho/physiology [13-15]. While much impactful work has been done, investigations into mechanotransduction in single heart cells have been limited by the techniques available. Previously carbon filaments were used to attach and stretch intact cells [11, 16, 17], but the difficulty of this technique is great, and the strength of attachment is limited to around 2.5μN [18], which falls below the upper limit of cardiomyocyte force generation.
Fig. 1a shows a new method which has simplified our investigation into mechanotransduction [1]. Single, enzymatically isolated cells are attached to glass micro-rods coated with MyoTak™, a biological adhesive created from components of the extracellular matrix that provides robust attachment of muscle cells to experimental apparatus. The coated micro-rods are connected to a high sensitivity force transducer and piezo-electric length controller, permitting evaluation of length-tension relationships in single cells (Fig. 1B). Varying the length (and diameter) of the glass rods can render them stiff or compliant so that isometric to isotonic contractions can be measured. The greatest advantages of this system are the ease of use and high throughput – we have had multiple visitors to the lab “stick” and stretch a cell on their first attempt with MyoTak, and can conduct stretch experiments on 20 or more cells a day. However, perfecting the attachment certainly requires practice, and while we now routinely record contractile forces in excess of 3μN (Fig. 1B), cells can detach from MyoTak coated rods at high forces.
The microtubule network underlies mechanotransduction in heart
Rather than a static structure, the microtubule (MT) network is in dynamic equilibrium between stabilized cylindrical polymers of α and β tubulin and un-polymerized tubulin monomers. Our initial hypothesis that the MT network played a role in stretch-dependent mechanotransduction came from Ingber’s provocative concept of cellular “tensegrity,” in which he proposed that the stabilized MT network resists mechanical perturbations in cells and in doing so acts as a mechanotransducer [19-22].
Initial evidence for the MT network mechanically influencing cardiac Ca2+ signaling came from our group[1, 11]. where we demonstrated that MT destabilization with colchicine was solely sufficient to abrogate the stretch-dependent activation of Ca2+ sparks. As we had evidence from electron microscopy that microtubule filaments interdigitate with t-tubule/SR junctions, our initial hypothesis speculated on a direct interaction of MTs with clusters of RyR2s that somehow altered RyR2 sensitivity in a stretch-dependent fashion.
Stretch-dependent ROS production triggers Ca2+ sparks
Following these initial investigations we considered alternative hypotheses to a direct mechano-activation of RyR2. Here our studies focused on the possibility that a stretch-dependent diffusible messenger acted to modulate RyR2 sensitivity. Much attention had focused on ROS as potent diffusible messengers capable of modifying RyR2 sensitivity through redox modification.
ROS generation occurs at multiple sites in striated muscle including the mitochondria, membrane bound Nox2, xanthine oxidase (XO) lipoxygenase (LOX), and phospholipase A2 (PLA2). Recent work by our group and others implicated Nox2 as the major source of ROS during repetitive contraction [23] and osmotic stress [24-27] in striated muscle. Two sources of ROS production have been linked with mechanical stress. First, membrane stress may induce ROS production within the t-tubules [28] by activation of Nox2 [29, 30]. Second, Nox2-derived ROS has also been linked to stretch-dependent Ca2+ entry into the mitochondria, which may drive further mitochondrial ROS production [31, 32].
In cardiomyocytes loaded with the ROS indicator dichlorofluorescein (DCF), we showed that there was a rapid increase in local ROS production following physiologic mechanical stretch (Fig. 2A). As DCF is a non-specific ROS indicator, we used specific inhibitors and genetic approaches with stretch to reveal that the microtubule network acted as a mechanotransducer to activate NoX2 dependent ROS generation, a pathway we termed X-ROS signaling.
A note on measuring ROS production
For real time measurements of ROS production in living cells, there are limited options. DCF and its derivatives are commonly used due to cell permeability, a decent signal, and high temporal resolution, all of which are real advantages for studies such as our own. Yet there are significant limitations to DCF, including irreversible oxidation, time-dependent mitochondrial compartmentalization [33], light-induced oxidation of the dye [34], unknown selectivity for ROS species, and a tricky management of the signal-to-noise ratio. Each of these limitations can compromise interpretation of results. A significant number of test measures must be taken to ensure that one is working within the linear range of the dye and detecting cytosolic changes in ROS. We have not found a suitable alternative to measure rapid changes in subcellular cytosolic ROS. This represents a limitation of our studies, and there is a great need for an improved indicator in the field.
Rapid stretch-dependent activation of Nox2
One of the more remarkable aspects of X-ROS is the speed of signaling – in the heart both ROS production and Ca2+ spark rate are elevated within seconds of stretch (Fig. 2A). This speed is undoubtedly favored by the tight spatial organization of signaling components. Nox2 and its regulatory subunits are localized to the t-tubule membranes in heart [1, 35] and skeletal muscle [29], closely apposed (~15 nm) to RyR2 clusters in the junctional SR membrane. As ROS are short lived species confined to interaction within their immediate vicinity, this tight spatial organization is paramount for rapid, controlled and spatially resolved signaling.
Even given the co-localization of signaling components, the onset of X-ROS signaling is still exceptionally fast, as a stretch-dependent process must activate Nox2 within seconds. Nox2 activation typically requires phosphorylation and recruitment of multiple cytosolic regulatory subunits to its transmembrane catalytic subunit (gp91phox), a process which is likely slow by comparison. The rapid nature of X-ROS signaling implies some pre-assembly of the Nox2 complex which minimizes steps to activate ROS production. In support of this, Nox2 subunits can tether to the membrane in hetero-multimeric complexes primed for activation [36]. But the question remains: how is the cell stretch linked to the activation of Nox2?
Based on our results and the work of others, we propose a model where the small GTPase Rac1, an activating component of Nox2, is a critical part of a pre-assembled Nox2 complex that is ready to be activated by stetch. Several lines of evidence support this theory: 1) Rac is a required subunit for Nox2 activation and inhibition of Rac1 blocks X-ROS signaling [1]; 2) Rac1 binds microtubules [37] and translocates to gp91phox separately from other regulatory subunits [36]; 3) Microtubules have been suggested to facilitate the recruitment of activating subunits to gp91phox in the membrane [38]; 4) destabilization of the microtubule network inhibits Nox2 activity [38] and blocks X-ROS signaling [1, 6]. Still, a clear demonstration of stretch-dependent Rac1 recruitment to gp91phox in a microtubule-dependent fashion is needed to validate this model. Experiments that could reveal the spatial organization of Nox2 components at high temporal resolution before, during, and after stretch would be illuminating.
How does ROS affect RyR2 activity?
Once Nox2 is activated, there is strong evidence to support a rapid effect of ROS on RyR2s to increase Ca2+ spark frequency. ROS generation likely occurs in the t-tubule lumen, and while the immediate product of Nox2 is superoxide (O2·-), H2O2 is also rapidly generated due to spontaneous and enzymatic dismutation [39]. Negatively charged O2·- may pass through select membrane anion channels to access RyR2s, but more likely uncharged H2O2 freely diffuses across the t-tubule membrane or through water channels [40] to affect channel activity. Multiple groups have confirmed that extracellular application of H2O2 leads to an immediate increase in intracellular Ca2+ spark rate ([1, 41], suggesting that the time required from extracellular H2O2 generation to modulation of RyR2 function is only diffusion-limited.
But what are the specific ROS-dependent modifications that affect RyR2 activity? It is well documented that oxidation and nitrosylation of RyR2 reversibly stimulates channel activity in vitro and in vivo (for review, see [42]). Of the ~90 cysteine residues per RyR2 subunit, approximately 20 are in a reduced state. These reactive cysteine residues are likely targets for X-ROS signaling, as they exhibit rapid and reversible redox modifications under physiologic conditions. However, unlike for RyR1 [43], the specific hyper-reactive cysteine residues on RyR2 have not been identified. An alternative (or additional) possibility to consider is that X-ROS may indirectly modify RyR2 activity through an intermediate target(s) of oxidative modification. For example, oxidation of Ca2+/Calmodulin dependent protein Kinase II (CaMKII) can increase its activity and stimulate RyR2 through phosphorylation [41, 44], and oxidation of calmodulin itself can remove its tonic inhibitory action on RyR2 [45]. To identify the specific stretch-dependent modifications that affect RyR2 activity will require a detailed and unprecedented combination of physiology, proteomics, and molecular biology.
Multiple effects of stretch on RyR2 activity?
Examination of the spark frequency histogram in Fig. 2B suggests that spark rate is rapidly increased within 500ms of stretch. Inspection of the average DCF data (Fig. 2A) suggests that the DCF signal slope (and thus ROS production) is increased within 1 – 2 seconds of stretch, but it should be noted that due to the small signal change of DCF recordings (see above), we can only confidently conclude that there is a significant increase in DCF slope when measured over 10s of applied stretch. Thus the kinetic details linking the rapid increase in ROS and Ca2+ spark rate need further examination. When cells are treated with the ROS scavenger N-acetylcysteine (NAC), the stretch-dependent change in spark rate is significantly blocked (Fig. 2C, [1]). However, close inspection of the spark frequency histogram in NAC treated cells does not rule out a minor increase in spark rate within the first seconds of stretch (Fig. 2C). Thus the possibility of multiple stretch effects – e.g. an initial mechanical effect that slightly increases spark rate and a secondary oxidative effect that produces the larger and longer lasting increase – cannot be ruled out at this time.
The rapid off-rate of X-ROS signaling
The rapid off-rate of X-ROS signaling is also a provocative topic. In our recent work we concluded that the increase in ROS production with stretch returns to the pre-stretch level upon return to resting length [1]. In retrospect, while ROS production and spark rate have returned towards the pre-stretch level by the post stretch interval (Fig. 2A and B), it is unclear if this is because X-ROS “shuts off” upon release of stretch, or if ROS production simply declined sufficiently over the prolonged stretch duration. Recent experiments using shorter-duration high-frequency stretches suggest that if ROS production is still high upon release of stretch, there is some delay (on the order of seconds to 10s of seconds) between the release of stretch and the return of ROS production and Ca2+ spark rate to pre-stretch levels (BLP unpublished observations). Thus, while X-ROS certainly demonstrates a rapid off-rate, this may occur on a scale of seconds to 10s of seconds, and we must consider the above limitations when discussing the immediacy of X-ROS signaling.
Regardless, based on the reversibility and reproducibility of the X-ROS effect, it is clear that stretch does not lead to irreversible oxidative modifications of RyR2s (such as the formation of sulfonic acids). As far as reversing the oxidative modification, a strong reducing cytosolic environment, caused by the high concentration of reduced glutathione (GSH) and activity of thioredoxin and glutaredoxin systems [46, 47], drives cysteine residues towards a reduced state. Preferential local signaling of these enzymes around the CRU should be explored to explain such rapid changes in RyR2 activity. In support of such speed in redox reversibility, if spark rate is elevated by an acute application of low dose H2O2, spark rate can be returned to normal within seconds of wash out of H2O2[1]. Interestingly, this reversibility is compromised in mdx muscle [1], which is deficient in endogenous reducing systems [48].
Modulators and downstream targets of X-ROS signaling
A seemingly overwhelming number of cardiac enzymes, kinases, phosphatases, channels, exchangers, pumps, and transcription factors have been shown to be redox-sensitive in vitro (Reviewed in [7, 49], blurring the potential downstream targets of X-ROS signaling. However, a growing body of evidence suggests redox signaling is both source and compartment specific. This, coupled with the short life span of ROS and heavily reducing cytosolic environment suggest that the targets of X-ROS are likely limited in vivo.
EC coupling
In addition to RyR2, key EC coupling proteins including Na+/Ca2+ exchanger (NCX), the L-type Ca2+ channel (LCC), and SERCA pump are all sensitive to oxidation [49]. In general, a small controlled amount of ROS production such as that derived from Nox enzymes ramps up the activity of exchangers, channels and transporters. NCX and LCC co-localize with Nox2 at the t-tubule/SR junction, and therefore spatially form potential targets of X-ROS signaling. However it is difficult to predict (and will be equally difficult to experimentally tease out) which of the many redox-sensitive signaling cascades and channels may be influenced by X-ROS, particularly given the complex time and concentration dependent effects of ROS.
Mechanosensitive channels
One intriguing target is the “slow force response” (or SFR), an increase in Ca2+ transient amplitude and contractile force generation that occurs after minutes of sustained stretch (also known as the “Anrep” effect) [50]. The activation of mechanosensitive channels (MSC) that raise intracellular [Ca2+] and [Na+] has been postulated to underlie the SFR [51], along with other potentially convergent or independent signaling pathways [52, 53]. Interestingly, MSCs may co-localize with Nox2 in caveolae and are modulated by ROS [54], and X-ROS has recently been demonstrated to promote Ca2+ influx through MSCs in skeletal muscle [6]. Additionally, studies in patch-clamped ventricular myocytes suggest that mechanosensitive currents are modulated by the microtubule network and Nox2 activity[30, 55, 56]. Thus the possibility of X-ROS regulation of MSCs and the SFR warrants investigation.
CaMKIIδ
In an exciting report from the Anderson lab, it was shown that Nox-derived ROS can oxidize and activate CaMKIIδ both in vivo and in vitro [44]. ROS-dependent oxidation of CaMKIIδ (ox-CaMKIIδ) is a key contributor to Ca2+-dependent arrhythmogenesis and contractile dysfunction [41]. Ox-CaMKIIδ enhances the late, slowly inactivating Na+ current (late INa), which causes Na+ to accumulate in the cell; this in turn drives Ca2+ entry via Na+/Ca2+ exchange, leading to cellular Ca2+ “overload” and subsequent Ca2+ dependent arrhythmia [41, 57, 58]. Importantly, Wagner et al. (2011) showed that both ROS and Ca2+ release from the SR were critical co-factors required to activate ox-CaMKIIδ. This identifies X-ROS signaling (where stretch generates these co-factors) as a strong physiologic candidate to activate ox-CaMKIIδ dynamically.
Angiotensin signaling
Several studies have shown that passive stretch increases tubulin mRNA [59, 60] in cardiac myocytes and that pressure overload increases MT stability in the heart [61, 62]. Angiotensin signaling may be involved in this response to stretch, as Angiotensin II (AngII) is produced and released from both neonatal cardiac myocytes [63] and skeletal muscle myoblasts [64] following stretch, and angiotensin signaling has been shown to induce microtubule reorganization in various cell systems (see below, Fig. 3). These results suggest a stretch-dependent autocrine regulatory pathway that increases MT content and stability, a key component of X-ROS signaling. However to our knowledge these findings have not been verified in acutely isolated adult cardiomyocytes subjected to physiologic stretch. It is well established that blockade of the AngII type 1 receptor (AT1R) (by drugs such as losartan) provides protection from congestive heart disease, hypertension and acute cardiac injury, and it is tempting to speculate that a portion of this protection is due to an inhibition of X-ROS components as outlined above.
Figure 3. Angiotensin II increases microtubule content and network stability.
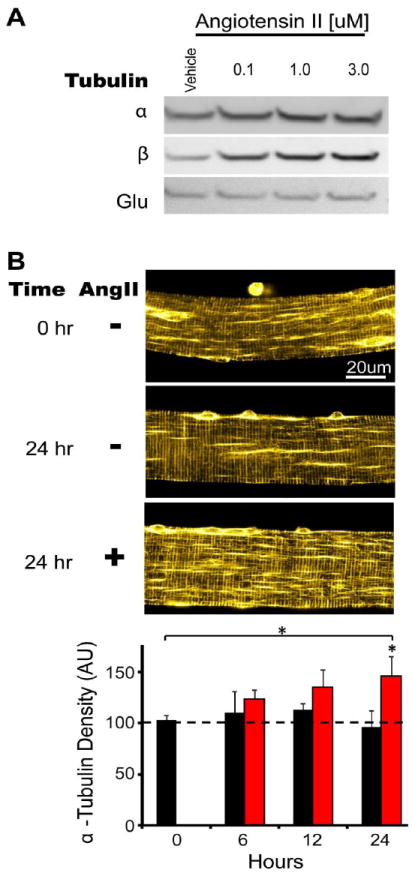
A, Differentiated C2C12 cell cultures were challenged with Angiotensin II (Ang II) or vehicle for 24 hours. Lanes were loaded with equal amounts of total protein and Western blot was conducted for α and β tubulin isoforms and Glu tubulin. B, Enzymatically isolated FDB myofibers from WT mice were cultured with vehicle (black bars) or 3uM Angiotensin II (red bars) for 24 hours. Fixed and permeablized myofibers were stained for α tubulin. Confocal flat plane projections were taken and analyzed for microtubule network density as previously described [6]. A significant increase in MT network density was seen after 24 hours of incubation with Ang II.
Additionally several lines of evidence have been put forth that reveals AT1R signaling can be activated by stretch independent of AngII ligand binding [65]. β-arrestin biased AT1R ligands were also shown to be cardioprotective [66, 67], and stretch or mechanical stress activates AT1R dependent ERK signaling via β-arrestin, but not through the traditional G protein signaling cascade. While stress-dependent ERK signaling does not require G proteins, it also does not need AngII ligand release. Despite the overlapping activation by stretch, it is not clear if X-ROS signaling may be linked to this process.
X-ROS signaling in cardiomyopathy
RyR2 hypersensitivity to [Ca2+]i
The impetus for investigating X-ROS in cardiomyopathy was driven by reports of increases in MTs and Nox2 activity in failing hearts [68-73]. Our initial studies focused on myocytes from the murine model of Duchenne Muscular Dystrophy (DMD, the mdx mouse), where both increased Nox2 expression and MT stability are associated with the pathogenic phenotype. In mdx cells or in cells under Ca2+ overload, the same stretch that normally triggers a burst of Ca2+ sparks instead often triggers a propagating “wave” of SR Ca2+ release. In both cases there is a background elevation of RyR2 Ca2+ sensitivity [74], so that when the cells are stretched the additional redox element creates hyperactive RyR2s that sustain the Ca2+ wave activity. The elevation in [Ca2+]i during a wave activates a depolarizing Na+/Ca2+ exchange current that can disrupt the electrical rhythm of the heart [75]. However, in an intact heart other factors contribute to this process and so the linkage between Ca2+ waves and arrhythmogenesis is an area of active investigation.
While the precise contribution of X-ROS signaling to the onset and progression of cardiomyopathy remains to be established, mounting evidence suggests that redox modification of RyR2 contributes to abnormal Ca2+ handling in disease. RyR2 may be redox modulated by various sources of ROS, including nitric oxide (NO) [76], XO [77], and Nox [1, 78], as well as indirectly modulated through phosphorylation by redox-sensitive kinases such as PKA [79] and CaMKII [41]. Nox-derived ROS can influence NO signaling and nitrosative stress through the conversion of NO to peroxynitrite and the reduction of bioavailable NO. Peroxynitrite nitrosylates RyR2s, causing “leaky” (or hypersensitive) RyR2s which have been linked to arrhythmia and dysfunction in diseased myocardium [80].
Oxidative stress
In addition to acute arrhythmogenic potential via redox modification of RyR2s, chronically elevated X-ROS signaling may also contribute to more generalized oxidative stress. Oxidative stress results from an imbalance between ROS and endogenous antioxidant systems. ROS-induced oxidative stress contributes to impaired Ca2+ handling and the progression of cardiomyopathy (including DMD) and correlates with the severity of heart failure (HF) [81-83]. A consensus has recently emerged that chronic oxidative stress leads to abnormal Ca2+ homeostasis and arrhythmia in the mdx heart, which exhibits a progressive dilated cardiomyopathy [80, 84-86]. X-ROS is a likely contributor to this oxidative stress, as we found that mdx myocytes generate excessive X-ROS with each diastolic stretch [1]. This mechanical sensitivity is consistent with the findings of Jung et al [87], who showed that severe mechanical stress (as osmotic shock) triggered excess Nox-derived ROS in mdx myocytes. As mentioned above, increased microtubule stability [1, 88] and Nox2 activity [86] in DMD may underlie the exacerbated X-ROS signaling. Extending these findings to broader cardiac pathology, X-ROS components are similarly upregulated in a more generalized model of HF induced by pressure-overload (transaortic constriction, TAC) – Nox2 expression and activity are increased [89, 90], and microtubule density shows a similar increase and correlation with disease progression [61, 62]. Thus it is tempting to speculate that HF myocytes may similarly generate excessive ROS upon stretch. Furthermore, Nox2-/- mice (which lack X-ROS [1]) subjected to TAC are protected against systolic and diastolic dysfunction at the myocyte and whole heart level [91], and Nox2-dependent oxidation of CaMKIIδ also contributes to impaired cardiac function in disease [44, 92]. Taken together, a growing body of evidence suggests that Nox2-derived ROS plays an important role in the pathophysiology of heart disease [93]. We propose that in diseased heart cells, upregulation of X-ROS signaling components may cause a normal physiologic stretch to produce excessive X-ROS, thus contributing to oxidative stress and abnormal Ca2+ signaling.
Ischemia Reperfusion
While excessive X-ROS may contribute to pathology, Nox-derived ROS may also protect the heart from hypoxia and ischemia-reperfusion injury such that occurs during myocardial infarction and ischemic heart disease. Numerous studies suggest that mitochondrial and Nox-derived ROS are involved in ischemic preconditioning, a phenomenon that provides powerful protection against subsequent ischemia-reperfusion injury [94, 95]. Sanchez et al. (2005 and 2008) demonstrated that preconditioning exercise and tachycardia (which increase the mechanical activity of the heart) increase Nox activity, RyR2 S-glutathionylation and SR calcium release. X-ROS provides a mechanism that may link these effects, which significantly reduced infarct size following coronary artery occlusion in dogs.
X-ROS in the whole heart
To better understand the patho/physiological ramifications of X-ROS signaling, an important future challenge will be to determine how the cellular signaling occurs in the beating heart, as recently suggested by Hidalgo and Donoso [12] in their perspective on our work. These investigations will be complicated by the presence of vascular and endothelial Nox enzymes, which may also produce ROS through X-ROS signaling in response to mechanical stress (see below). Developing methods to isolate X-ROS components in specific cell types will be critical in our attempt to unravel how X-ROS signaling contributes to function and disease in the whole heart.
The role of microtubules as mechanotransducers of stretch-dependent signaling must also be explored in the intact heart. MTs have previously been linked to stretch-induced arrythmias[96] consistent with our findings in single cells [1]. However, this link is controversial[97].and further work is needed[98].
X-ROS signaling in skeletal muscle
Muscle Stretch Tool: MuST
Progress in our understanding of stretch signaling in adult skeletal muscle fibers has also been limited by the assays available. Recent work by Allen and colleagues in dissected single muscle fiber preparations used stretched (i.e. eccentric) contractions and gained valuable information on the role of ROS and Ca2+ in stretch-induced injury [32, 99-101]. These experiments however are technically difficult and have low throughput. Furthermore, the combination of a mechanical stretch during a tetanic contraction (i.e., eccentric contraction) increases the number of parameters that are uncontrolled (mechanical damage, [Ca2+]i elevation due to EC coupling, etc.), such that the central question of “what effect does stretch have on ROS and Ca2+ signaling?” remains unclear.
Due to the dramatic difference in size between enzymatically isolated cardiomyocytes and single skeletal muscle cells (flexor digitorum brevis; FDB) we needed to develop new methods for cell attachment. We developed MuST (Muscle Stretch Tool), to make mechanical measurements and control length in enzymatically isolated skeletal myofibers. Fig. 1C demonstrates the components of this technology. MyoTak™ biological adhesive provides the necessary attachment interface between the intact muscle cell and tweezer; the tweezer is simply used to bracket the myofibers (without compression), thus providing a large surface area for attachment to handle the larger forces of muscle fibers. As seen in Fig. 1D, MuST enables the precise manipulation of cell length (i.e., stretch) and monitoring of sarcomere length and passive tension. Through cell electroporation we are able to incorporate new pharmacologic and genetic techniques to over express or silence targeted protein expression in FDB fibers [23], and we combine this with confocal imaging and MuST to address the mechanistic basis for stretch-dependent signaling in skeletal muscle.
Acute stretch does not reveal X-ROS signaling in wild-type muscle fibers
The key components of X-ROS signaling, (i.e., MT network and Nox2) are conserved in many mammalian cells including skeletal muscle, and Nox2 is a major source of ROS in response to repetitive contraction [23]. Despite these findings, we detected only a small non-significant X-ROS signal with a small physiologic stretch (i.e., acute single stretch to 10% of resting sarcomere length; ~1.9μm to ~2.1μm for 5 sec.). When the minimal stretch assay is contextualized with the use of a low signal-to-noise ROS indicator, we must consider that our mechanical stress was too brief to elicit a significant signal. This hypothesis is consistent with a report from Palomero et al. [102], who used 10s stretch-release cycles over 10 minutes to reveal a small, but statistically significant increase in ROS production in skeletal muscle. Our current experiments will address whether we reveal X-ROS in wild-type (WT) fibers with repetitive stretches as in exercising muscle.
X-ROS is prominent in mdx muscle
In considering a growing body of evidence, we hypothesized that MT-dependent X-ROS may be prominent in the skeletal muscle myopathy associated with DMD. First, the mechanical stressing of mdx muscle fibers triggers excessive sarcolemmal Ca2+ influx and Nox2-derived ROS [99]. Similarly, osmotic shock resulted in aberrant Ca2+ homeostasis and Ca2+ sparks, downstream of increased ROS production by Nox2 [25, 26, 99]. Second, in adult mdx muscle fibers α, β, and the stabilized detyrosinated tubulin (Glu-tubulin) are all increased, and the MT network is more stabilized and disorganized [88, 103]. Furthermore, there is a significant increase in Nox2 subunit expression (gp91phox, p47phox, p67phox) in mdx muscle [99]. Having confirmed the increase in Nox2 and MT expression/disorganization [6], we assayed the contribution of X-ROS in mdx skeletal muscle.
Mechanical stretch activates ROS production and Ca2+ influx in mdx skeletal muscle
Using a small physiologic stretch, DCF-loaded mdx muscle cells revealed a large and nearly instantaneous burst of ROS production. This ROS was produced by Nox2, as specific inhibition of the enzyme completely abrogated the effect. Given that skeletal muscle does not exhibit Ca2+ sparks under physiological conditions, we could not use spark occurrence as a downstream readout of X-ROS. As several reports indicated that mechanosensitive channels were up regulated in mdx and co-localize with Nox2 [54], we assayed calcium influx through MSCs as a potential downstream target for X-ROS. Passive stretch led to amplified Ca2+ influx through MSCs in mdx fibers, and this influx could be prevented by selective inhibition of either the MSC or Nox2.
Role of microtubule network stability in skeletal muscle X-ROS
Similar to X-ROS activation in cardiomyocytes (see above), X-ROS in adult mdx skeletal muscle was abrogated by de-stabilizing the MT network with colchicine. Furthermore, cells with a less stabilized microtubule cytoskeleton (i.e. WT or young mdx myocytes), did not display X-ROS. A critical supportive experiment revealed that the pharmacological stabilization (paclitaxel) of the MT cytoskeleton in WT or young mdx fibers revealed a robust ROS burst following stretch that was not present in the naïve cell [6]. Thus, the protein components necessary for robust X-ROS signaling (MT and Nox2) are present in WT muscle but are in a muted configuration. These results suggest that an increase in MT network stability is sufficient to enhance X-ROS signaling independent of increases in Nox2 content. The effect of an increase in Nox2 content independent of altered MT stability has not yet been explored.
Given the important role of MT stability in regulating the magnitude of X-ROS, further studies should examine the origin of the increased MT stability in dystrophic muscle. In mammalian cells, endogenous modulators regulate MT stability [70, 73, 104-106], however little is known about the expression or post-translational modification of these in dystrophic skeletal muscle. We are currently exploring these questions. As discussed above, passive stretch increases tubulin mRNA [104, 107] and MT stability in the heart. Recent evidence demonstrated stretch also triggers an angiotensin signaling system in skeletal muscle myoblasts [64]. AngII has been shown to induce microtubule reorganization in endothelial [108] and neuronal cells [109] as well as the model cell line PC12 and NG108-15 [110, 111]. Our preliminary experiments have shown that AngII increases tubulin protein content in C2C12 cells and increases MT network density in FDB fibers after a 24h incubation (Fig. 3). Of note, AngII stimulation in vascular smooth muscle cells has been shown to activate Nox2 in a Rac1 dependent manner [112], and the intramuscular Angiotensin system is activated in DMD [64]. Interestingly, we have previously reported that AngII receptor inhibition with Losartan reduced the muscle damage in the mdx mouse [113], and we now speculate that a portion of this protection may have come from reducing X-ROS signaling. Thus angiotensin signaling represents a prime candidate for the increased MT stability and X-ROS signaling in diseased muscle.
X-ROS and microtubules as therapeutic targets in the dystrophies
Our novel method and new insights in X-ROS prompted a preliminary exploration of X-ROS in models of muscular dystrophy disparate from dystrophin deficiency. Dysferlin-/- skeletal muscle fibers, a model of Limb girdle muscular dystrophy type 2b/Miyoshi myopathy, has a similar, albeit milder, in vivo contraction induced injury phenotype [114, 115] as the mdx mouse. Using identical methods to those used during X-ROS experiments in mdx fibers, we show that in adult (6+ month old) muscle fibers, X-ROS is significantly elevated in dysferlin-/- (A/J) FDB myofibers when compared to strain controls (A/WySnJ; n=3 fibers) (Fig. 4A and 4B). Furthermore, assays of stretch-dependent Ca2+ influx reveal an exaggerated mechano-sensitive influx in the dysferlin-/- muscle fibers (Fig. 4C). Finally, the dysferlin-/- muscle presents with increased MT and Nox2 protein content, which we hypothesize may mechanistically account for the presence of X-ROS in these mice (Fig. 4D). Ongoing studies are evaluating the stability of the MT network in fibers from each strain. These studies warrant the investigation of X-ROS as an underlying contributor to myopathy in the muscular dystrophies.
Figure 4. Dysferlin null myofibers exhibit enhanced X-ROS and stretch induced Ca2+ influx.
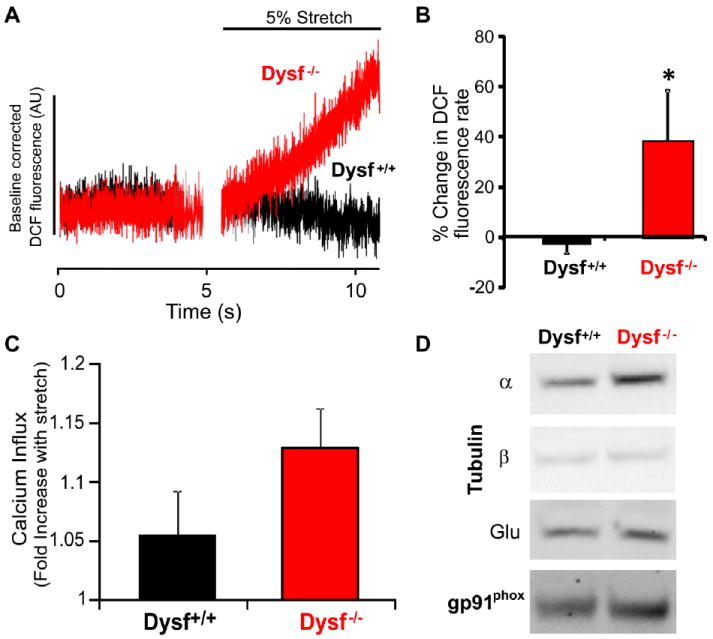
A-B, An acute 5% stretch in DCF loaded FDB muscle fibers from control (A/WySnJ) and dysferlin-/- (A/J) mice activated X-ROS in dysferlin null fibers but not WT fibers (n = 3 per genotype). C, Acute stretch increased Ca2+ influx in dysferlin-/- myofibers to a greater extent than in WT. D, Western analysis of tubulin and NOX2 in dysferlin-/- and WT muscle.
The importance of excessive X-ROS signaling in pathology is revealed with repetitive contraction in the injury prone mdx muscle. Whereas normal healthy muscle is capable of withstanding repeated and prolonged isometric and eccentric contractions, mdx muscle displays a rapid decrease in nerve evoked muscle force or muscle injury. Acute inhibition of X-ROS (~3 to 24h prior) by microtubule destabilization or Nox2 inhibition greatly diminishes the contraction-induced injury [6]. Thus, pharmacological interventions targeting X-ROS components may hold therapeutic promise in the treatment of DMD.
Is X-ROS a conserved mechanism in different cell types?
Many cellular processes may utilize X-ROS signaling, and our findings have been foreshadowed in recent investigations. In endothelial cells, mechanical strain stimulates Nox-dependent ROS production, a finding with critical links to atherosclerosis [116]. Cyclic stretch also increases ROS production and Rac1 activity in neonatal ventricular cells, findings relevant to hypertrophic gene regulation [117]. In fact, Nox activity is enhanced by various signaling cascades linked to hypertrophy and heart failure [118]. Several of these pathways share overlapping links to mechanical stretch and strain, including the angiotensin II-ROS signaling cascade [67, 119].
A role for microtubules in regulating Nox2 has also been suggested in the literature. For example in neurons paclitaxol (a microtubule stabilizing drug used in cancer treatment) enhances Nox activity and induces cell death [120]. A treatment related side effect for patients on these drugs is myalgia (i.e., muscle pain and weakness) and cardiomyopathy [121]; an effect that may be due to excessive X-ROS signaling. Devillard et al (2006) showed that destabilizing the microtubule network reduced Nox activity in the vasculature following ischemia-reperfusion injury and suggested that microtubules may recruit activating subunits of Nox to the membrane, which resonates strongly with our findings in heart and skeletal muscle. It is clear that components of X-ROS signaling are implicated and intertwined in numerous cell systems and disease models, but the degree of signaling conservation remains to be established. Time will tell if X-ROS signaling emerges as a conserved mechanism providing mechanical control of redox and Ca2+ signaling in diverse physiological and pathophysiological settings.
Highlights.
Review of the recently characterized stretch-dependent X-ROS signaling
New tools developed to stretch single cells
Stretch triggers the generation of reactive oxygen species (ROS)
ROS regulate subcellular calcium signaling in heart and skeletal muscle
X-ROS contributes to pathology in muscular dystrophy
Acknowledgments
The authors thank World Precision Instruments, Ionoptix, and Aurora Scientific for helping in the development of instrumentation critical to these works. We also thank Drs. Ajay Shah, Mark Anderson, and Stuart Martin for stimulating discussion and ongoing collaboration, and Dr. Bjorn Knollman for discussion and critical observations. We also extend recognition to the late George Cooper IV for important insights and reagents for our work. Finally we also thank Dr. Robert Bloch for kindly providing dysferlin-/- mice.
Sources of Funding: BLP is supported by a National Institutes of Health (NIH) training grant (T32 HL072751-07) in Cardiovascular Cell Biology and by 5K99HL114879-02. Additional support from NIH R01 HL106059, R01 HL36974; Leducq North American-European Atrial Fibrillation Research Alliance; European Union Seventh Framework Program (FP7), Georg August University, “Identification and therapeutic targeting of common arrhythmia trigger mechanisms.”
Footnotes
Disclosures: BLP, CWW, and WJL have filed a university sponsored US patent for MyoTak.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Prosser BL, Ward CW, Lederer WJ. X-ROS signaling: rapid mechano-chemo transduction in heart. Science. 2011;333:1440–5. doi: 10.1126/science.1202768. [DOI] [PubMed] [Google Scholar]
- 2.Shao D, Oka S, Brady CD, Haendeler J, Eaton P, Sadoshima J. Redox modification of cell signaling in the cardiovascular system. J Mol Cell Cardiol. 2012;52:550–8. doi: 10.1016/j.yjmcc.2011.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Choi WG, Swanson SJ, Gilroy S. High-resolution imaging of Ca2+, redox status, ROS and pH using GFP biosensors. Plant J. 2012;70:118–28. doi: 10.1111/j.1365-313X.2012.04917.x. [DOI] [PubMed] [Google Scholar]
- 4.Perjes A, Kubin AM, Konyi A, Szabados S, Cziraki A, Skoumal R, et al. Physiological regulation of cardiac contractility by endogenous reactive oxygen species. Acta Physiol (Oxf) 2011 doi: 10.1111/j.1748-1716.2012.02391.x. [DOI] [PubMed] [Google Scholar]
- 5.Dedkova EN, Blatter LA. Measuring mitochondrial function in intact cardiac myocytes. J Mol Cell Cardiol. 2012;52:48–61. doi: 10.1016/j.yjmcc.2011.08.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Khairallah RJ, Shi G, Sbrana F, Prosser BL, Borroto C, Mazaitis MJ, et al. Microtubules underlie dysfunction in duchenne muscular dystrophy. Sci Signal. 2012;5:ra56. doi: 10.1126/scisignal.2002829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Santos CX, Anilkumar N, Zhang M, Brewer AC, Shah AM. Redox signaling in cardiac myocytes. Free Radic Biol Med. 2011;50:777–93. doi: 10.1016/j.freeradbiomed.2011.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Williams GS, Chikando AC, Tuan HT, Sobie EA, Lederer WJ, Jafri MS. Dynamics of calcium sparks and calcium leak in the heart. Biophys J. 2011;101:1287–96. doi: 10.1016/j.bpj.2011.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cheng H, Lederer WJ. Calcium sparks. Physiol Rev. 2008;88:1491–545. doi: 10.1152/physrev.00030.2007. [DOI] [PubMed] [Google Scholar]
- 10.Cheng H, Lederer WJ, Cannell MB. Calcium sparks: elementary events underlying excitation-contraction coupling in heart muscle. Science. 1993;262:740–4. doi: 10.1126/science.8235594. [DOI] [PubMed] [Google Scholar]
- 11.Iribe G, Ward CW, Camelliti P, Bollensdorff C, Mason F, Burton RA, et al. Axial stretch of rat single ventricular cardiomyocytes causes an acute and transient increase in Ca2+ spark rate. Circ Res. 2009;104:787–95. doi: 10.1161/CIRCRESAHA.108.193334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hidalgo C, Donoso P. Cell signaling. Getting to the heart of mechanotransduction. Science. 2011;333:1388–90. doi: 10.1126/science.1212183. [DOI] [PubMed] [Google Scholar]
- 13.Knoll R, Hoshijima M, Chien K. Cardiac mechanotransduction and implications for heart disease. Journal of Molecular Medicine. 2003;81:750–6. doi: 10.1007/s00109-003-0488-x. [DOI] [PubMed] [Google Scholar]
- 14.Lammerding J, Kamm RD, Lee RT. Mechanotransduction in cardiac myocytes. Ann N Y Acad Sci. 2004;1015:53–70. doi: 10.1196/annals.1302.005. [DOI] [PubMed] [Google Scholar]
- 15.Cannell MB. Pulling on the heart strings: a new mechanism within Starling’s law of the heart? Circ Res. 2009;104:715–6. doi: 10.1161/CIRCRESAHA.109.195511. [DOI] [PubMed] [Google Scholar]
- 16.Calaghan S, White E. Activation of Na+-H+ exchange and stretch-activated channels underlies the slow inotropic response to stretch in myocytes and muscle from the rat heart. J Physiol. 2004;559:205–14. doi: 10.1113/jphysiol.2004.069021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Le Guennec JY, Peineau N, Argibay JA, Mongo KG, Garnier D. A new method of attachment of isolated mammalian ventricular myocytes for tension recording: length dependence of passive and active tension. J Mol Cell Cardiol. 1990;22:1083–93. doi: 10.1016/0022-2828(90)90072-a. [DOI] [PubMed] [Google Scholar]
- 18.Yasuda SI, Sugiura S, Kobayakawa N, Fujita H, Yamashita H, Katoh K, et al. A novel method to study contraction characteristics of a single cardiac myocyte using carbon fibers. Am J Physiol Heart Circ Physiol. 2001;281:H1442–6. doi: 10.1152/ajpheart.2001.281.3.H1442. [DOI] [PubMed] [Google Scholar]
- 19.Ingber DE. Tensegrity and mechanotransduction. J Bodyw Mov Ther. 2008;12:198–200. doi: 10.1016/j.jbmt.2008.04.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stamenovic D, Mijailovich SM, Tolic-Norrelykke IM, Chen J, Wang N. Cell prestress. II. Contribution of microtubules. Am J Physiol Cell Physiol. 2002;282:C617–24. doi: 10.1152/ajpcell.00271.2001. [DOI] [PubMed] [Google Scholar]
- 21.Wang N, Naruse K, Stamenovic D, Fredberg JJ, Mijailovich SM, Tolic-Norrelykke IM, et al. Mechanical behavior in living cells consistent with the tensegrity model. Proc Natl Acad Sci U S A. 2001;98:7765–70. doi: 10.1073/pnas.141199598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang N, Butler JP, Ingber DE. Mechanotransduction across the cell surface and through the cytoskeleton. Science. 1993;260:1124–7. doi: 10.1126/science.7684161. [DOI] [PubMed] [Google Scholar]
- 23.Michaelson LP, Shi G, Ward CW, Rodney GG. Mitochondrial redox potential during contraction in single intact muscle fibers. Muscle Nerve. 2010;42:522–9. doi: 10.1002/mus.21724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ullrich ND, Fanchaouy M, Gusev K, Shirokova N, Niggli E. Hypersensitivity of excitation-contraction coupling in dystrophic cardiomyocytes. Am J Physiol Heart Circ Physiol. 2009;297:H1992–2003. doi: 10.1152/ajpheart.00602.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shkryl VM, Martins AS, Ullrich ND, Nowycky MC, Niggli E, Shirokova N. Reciprocal amplification of ROS and Ca(2+) signals in stressed mdx dystrophic skeletal muscle fibers. Pflugers Arch. 2009;458:915–28. doi: 10.1007/s00424-009-0670-2. [DOI] [PubMed] [Google Scholar]
- 26.Martins AS, Shkryl VM, Nowycky MC, Shirokova N. Reactive oxygen species contribute to Ca2+ signals produced by osmotic stress in mouse skeletal muscle fibres. J Physiol. 2008;586:197–210. doi: 10.1113/jphysiol.2007.146571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Isaeva EV, Shkryl VM, Shirokova N. Mitochondrial redox state and Ca2+ sparks in permeabilized mammalian skeletal muscle. J Physiol. 2005;565:855–72. doi: 10.1113/jphysiol.2005.086280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Espinosa A, Leiva A, Pena M, Muller M, Debandi A, Hidalgo C, et al. Myotube depolarization generates reactive oxygen species through NAD(P)H oxidase; ROS-elicited Ca2+ stimulates ERK, CREB, early genes. J Cell Physiol. 2006;209:379–88. doi: 10.1002/jcp.20745. [DOI] [PubMed] [Google Scholar]
- 29.Hidalgo C, Sanchez G, Barrientos G, Aracena-Parks P. A transverse tubule NADPH oxidase activity stimulates calcium release from isolated triads via ryanodine receptor type 1 S - glutathionylation. J Biol Chem. 2006;281:26473–82. doi: 10.1074/jbc.M600451200. [DOI] [PubMed] [Google Scholar]
- 30.Dyachenko V, Rueckschloss U, Isenberg G. Modulation of cardiac mechanosensitive ion channels involves superoxide, nitric oxide and peroxynitrite. Cell Calcium. 2009;45:55–64. doi: 10.1016/j.ceca.2008.06.002. [DOI] [PubMed] [Google Scholar]
- 31.Whitehead NP, Streamer M, Lusambili LI, Sachs F, Allen DG. Streptomycin reduces stretch-induced membrane permeability in muscles from mdx mice. Neuromuscul Disord. 2006;16:845–54. doi: 10.1016/j.nmd.2006.07.024. [DOI] [PubMed] [Google Scholar]
- 32.Whitehead NP, Yeung EW, Allen DG. Muscle damage in mdx (dystrophic) mice: role of calcium and reactive oxygen species. Clin Exp Pharmacol Physiol. 2006;33:657–62. doi: 10.1111/j.1440-1681.2006.04394.x. [DOI] [PubMed] [Google Scholar]
- 33.Swift LM, Sarvazyan N. Localization of dichlorofluorescin in cardiac myocytes: implications for assessment of oxidative stress. Am J Physiol Heart Circ Physiol. 2000;278:H982–H90. doi: 10.1152/ajpheart.2000.278.3.H982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bilski P, Belanger AG, Chignell CF. Photosensitized oxidation of 2’,7’-dichlorofluorescin: singlet oxygen does not contribute to the formation of fluorescent oxidation product 2’,7’-dichlorofluorescein. Free Radic Biol Med. 2002;33:938–46. doi: 10.1016/s0891-5849(02)00982-6. [DOI] [PubMed] [Google Scholar]
- 35.Sanchez G, Escobar M, Pedrozo Z, Macho P, Domenech R, Hartel S, et al. Exercise and tachycardia increase NADPH oxidase and ryanodine receptor-2 activity: possible role in cardioprotection. Cardiovasc Res. 2008;77:380–6. doi: 10.1093/cvr/cvm011. [DOI] [PubMed] [Google Scholar]
- 36.Nauseef WM. Assembly of the phagocyte NADPH oxidase. Histochem Cell Biol. 2004;122:277–91. doi: 10.1007/s00418-004-0679-8. [DOI] [PubMed] [Google Scholar]
- 37.Best A, Ahmed S, Kozma R, Lim L. The Ras-related GTPase Rac1 binds tubulin. J Biol Chem. 1996;271:3756–62. doi: 10.1074/jbc.271.7.3756. [DOI] [PubMed] [Google Scholar]
- 38.Devillard L, Vandroux D, Tissier C, Brochot A, Voisin S, Rochette L, et al. Tubulin ligands suggest a microtubule-NADPH oxidase relationship in postischemic cardiomyocytes. Eur J Pharmacol. 2006;548:64–73. doi: 10.1016/j.ejphar.2006.08.004. [DOI] [PubMed] [Google Scholar]
- 39.Bedard K, Krause KH. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev. 2007;87:245–313. doi: 10.1152/physrev.00044.2005. [DOI] [PubMed] [Google Scholar]
- 40.Bienert GP, Moller AL, Kristiansen KA, Schulz A, Moller IM, Schjoerring JK, et al. Specific aquaporins facilitate the diffusion of hydrogen peroxide across membranes. J Biol Chem. 2007;282:1183–92. doi: 10.1074/jbc.M603761200. [DOI] [PubMed] [Google Scholar]
- 41.Wagner S, Ruff HM, Weber SL, Bellmann S, Sowa T, Schulte T, et al. Reactive oxygen species-activated Ca/calmodulin kinase IIdelta is required for late I(Na) augmentation leading to cellular Na and Ca overload. Circ Res. 2011;108:555–65. doi: 10.1161/CIRCRESAHA.110.221911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Donoso P, Sanchez G, Bull R, Hidalgo C. Modulation of cardiac ryanodine receptor activity by ROS and RNS. Front Biosci. 2011;16:553–67. doi: 10.2741/3705. [DOI] [PubMed] [Google Scholar]
- 43.Voss AA, Lango J, Ernst-Russell M, Morin D, Pessah IN. Identification of hyperreactive cysteines within ryanodine receptor type 1 by mass spectrometry. J Biol Chem. 2004;279:34514–20. doi: 10.1074/jbc.M404290200. [DOI] [PubMed] [Google Scholar]
- 44.Erickson JR, Joiner ML, Guan X, Kutschke W, Yang J, Oddis CV, et al. A dynamic pathway for calcium-independent activation of CaMKII by methionine oxidation. Cell. 2008;133:462–74. doi: 10.1016/j.cell.2008.02.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yamaguchi N, Xu L, Pasek DA, Evans KE, Meissner G. Molecular basis of calmodulin binding to cardiac muscle Ca(2+) release channel (ryanodine receptor) Journal of Biological Chemistry. 2003;278:23480–6. doi: 10.1074/jbc.M301125200. [DOI] [PubMed] [Google Scholar]
- 46.Hayes JD, McLellan LI. Glutathione and glutathione-dependent enzymes represent a coordinately regulated defence against oxidative stress. Free Radic Res. 1999;31:273–300. doi: 10.1080/10715769900300851. [DOI] [PubMed] [Google Scholar]
- 47.Berndt C, Lillig CH, Holmgren A. Thiol-based mechanisms of the thioredoxin and glutaredoxin systems: implications for diseases in the cardiovascular system. Am J Physiol Heart Circ Physiol. 2007;292:H1227–36. doi: 10.1152/ajpheart.01162.2006. [DOI] [PubMed] [Google Scholar]
- 48.Ragusa RJ, Chow CK, St Clair DK, Porter JD. Extraocular, limb and diaphragm muscle group-specific antioxidant enzyme activity patterns in control and mdx mice. J Neurol Sci. 1996;139:180–6. [PubMed] [Google Scholar]
- 49.Zima AV, Blatter LA. Redox regulation of cardiac calcium channels and transporters. Cardiovasc Res. 2006;71:310–21. doi: 10.1016/j.cardiores.2006.02.019. [DOI] [PubMed] [Google Scholar]
- 50.Allen DG, Kurihara S. The effects of muscle length on intracellular calcium transients in mammalian cardiac muscle. J Physiol. 1982;327:79–94. doi: 10.1113/jphysiol.1982.sp014221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ward ML, Williams IA, Chu Y, Cooper PJ, Ju YK, Allen DG. Stretch-activated channels in the heart: contributions to length-dependence and to cardiomyopathy. Prog Biophys Mol Biol. 2008;97:232–49. doi: 10.1016/j.pbiomolbio.2008.02.009. [DOI] [PubMed] [Google Scholar]
- 52.Ennis IL, Garciarena CD, Perez NG, Dulce RA, Camilion de Hurtado MC, Cingolani HE. Endothelin isoforms and the response to myocardial stretch. Am J Physiol Heart Circ Physiol. 2005;288:H2925–30. doi: 10.1152/ajpheart.01202.2004. [DOI] [PubMed] [Google Scholar]
- 53.Cingolani OH, Kirk JA, Seo K, Koitabashi N, Lee DI, Ramirez-Correa G, et al. Thrombospondin-4 is required for stretch-mediated contractility augmentation in cardiac muscle. Circ Res. 2011;109:1410–4. doi: 10.1161/CIRCRESAHA.111.256743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gervasio OL, Whitehead NP, Yeung EW, Phillips WD, Allen DG. TRPC1 binds to caveolin-3 and is regulated by Src kinase - role in Duchenne muscular dystrophy. J Cell Sci. 2008;121:2246–55. doi: 10.1242/jcs.032003. [DOI] [PubMed] [Google Scholar]
- 55.Isenberg G, Kazanski V, Kondratev D, Gallitelli MF, Kiseleva I, Kamkin A. Differential effects of stretch and compression on membrane currents and [Na+]c in ventricular myocytes. Prog Biophys Mol Biol. 2003;82:43–56. doi: 10.1016/s0079-6107(03)00004-x. [DOI] [PubMed] [Google Scholar]
- 56.Dyachenko V, Husse B, Rueckschloss U, Isenberg G. Mechanical deformation of ventricular myocytes modulates both TRPC6 and Kir2.3 channels. Cell Calcium. 2009;45:38–54. doi: 10.1016/j.ceca.2008.06.003. [DOI] [PubMed] [Google Scholar]
- 57.Zeitz O, Maass AE, Van Nguyen P, Hensmann G, Kogler H, Moller K, et al. Hydroxyl radical-induced acute diastolic dysfunction is due to calcium overload via reverse-mode Na(+)-Ca(2+) exchange. Circ Res. 2002;90:988–95. doi: 10.1161/01.res.0000018625.25212.1e. [DOI] [PubMed] [Google Scholar]
- 58.Lindegger N, Hagen BM, Marks AR, Lederer WJ, Kass RS. Diastolic transient inward current in long QT syndrome type 3 is caused by Ca2+ overload and inhibited by ranolazine. J Mol Cell Cardiol. 2009;47:326–34. doi: 10.1016/j.yjmcc.2009.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Watson PA, Hannan R, Carl LL, Giger KE. Contractile activity and passive stretch regulate tubulin mRNA and protein content in cardiac myocytes. Am J Physiol. 1996;271:C684–C9. doi: 10.1152/ajpcell.1996.271.2.C684. [DOI] [PubMed] [Google Scholar]
- 60.Yutao X, Geru W, Xiaojun B, Tao G, Aiqun M. Mechanical stretch-induced hypertrophy of neonatal rat ventricular myocytes is mediated by beta(1)-integrin-microtubule signaling pathways. Eur J Heart Fail. 2006;8:16–22. doi: 10.1016/j.ejheart.2005.05.014. [DOI] [PubMed] [Google Scholar]
- 61.Hein S, Kostin S, Heling A, Maeno Y, Schaper J. The role of the cytoskeleton in heart failure. Cardiovasc Res. 2000;45:273–8. doi: 10.1016/s0008-6363(99)00268-0. [DOI] [PubMed] [Google Scholar]
- 62.Tagawa H, Koide M, Sato H, Zile MR, Carabello BA, Cooper Gt. Cytoskeletal role in the transition from compensated to decompensated hypertrophy during adult canine left ventricular pressure overloading. Circ Res. 1998;82:751–61. doi: 10.1161/01.res.82.7.751. [DOI] [PubMed] [Google Scholar]
- 63.Sadoshima J, Xu Y, Slayter HS, Izumo S. Autocrine release of angiotensin II mediates stretch-induced hypertrophy of cardiac myocytes in vitro. Cell. 1993;75:977–84. doi: 10.1016/0092-8674(93)90541-w. [DOI] [PubMed] [Google Scholar]
- 64.Sun G, Haginoya K, Dai H, Chiba Y, Uematsu M, Hino-Fukuyo N, et al. Intramuscular renin-angiotensin system is activated in human muscular dystrophy. J Neurol Sci. 2009;280:40–8. doi: 10.1016/j.jns.2009.01.020. [DOI] [PubMed] [Google Scholar]
- 65.Zou Y, Akazawa H, Qin Y, Sano M, Takano H, Minamino T, et al. Mechanical stress activates angiotensin II type 1 receptor without the involvement of angiotensin II. Nat Cell Biol. 2004;6:499–506. doi: 10.1038/ncb1137. [DOI] [PubMed] [Google Scholar]
- 66.Kim KS, Abraham DM, Williams B, Violin JD, Mao L, Rockman HA. beta-Arrestin-biased AT1R stimulation promotes cell survival during acute cardiac injury. Am J Physiol Heart Circ Physiol. 2012 doi: 10.1152/ajpheart.00475.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Rakesh K, Yoo B, Kim IM, Salazar N, Kim KS, Rockman HA. beta-Arrestin-biased agonism of the angiotensin receptor induced by mechanical stress. Sci Signal. 2010;3:ra46. doi: 10.1126/scisignal.2000769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Iribe G, Ward CW, Camelliti P, Bollensdorff C, Mason F, Burton RA, et al. Axial stretch of rat single ventricular cardiomyocytes causes an acute and transient increase in Ca2+ spark rate. Circ Res. 2009;104:787–95. doi: 10.1161/CIRCRESAHA.108.193334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Cheng G, Kasiganesan H, Baicu CF, Wallenborn JG, Kuppuswamy D, Cooper G. Cytoskeletal Role in Protection of the Failing Heart by beta-Adrenergic Blockade. Am J Physiol Heart Circ Physiol. 2011 doi: 10.1152/ajpheart.00867.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Chinnakkannu P, Samanna V, Cheng G, Ablonczy Z, Baicu CF, Bethard JR, et al. Site-specific microtubule-associated protein 4 dephosphorylation causes microtubule network densification in pressure overload cardiac hypertrophy. J Biol Chem. 2010;285:21837–48. doi: 10.1074/jbc.M110.120709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Cheng G, Takahashi M, Shunmugavel A, Wallenborn JG, DePaoli-Roach AA, Gergs U, et al. Basis for MAP4 dephosphorylation-related microtubule network densification in pressure overload cardiac hypertrophy. J Biol Chem. 2010;285:38125–40. doi: 10.1074/jbc.M110.148650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Koide M, Hamawaki M, Narishige T, Sato H, Nemoto S, DeFreyte G, et al. Microtubule depolymerization normalizes in vivo myocardial contractile function in dogs with pressure-overload left ventricular hypertrophy. Circulation. 2000;102:1045–52. doi: 10.1161/01.cir.102.9.1045. [DOI] [PubMed] [Google Scholar]
- 73.Sato H, Nagai T, Kuppuswamy D, Narishige T, Koide M, Menick DR, et al. Microtubule stabilization in pressure overload cardiac hypertrophy. J Cell Biol. 1997;139:963–73. doi: 10.1083/jcb.139.4.963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Prosser BL, Ward CW, Lederer WJ. Subcellular Ca2+ signaling in the heart: the role of ryanodine receptor sensitivity. J Gen Physiol. 2010;136:135–42. doi: 10.1085/jgp.201010406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Schlotthauer K, Bers DM. Sarcoplasmic reticulum Ca(2+) release causes myocyte depolarization Underlying mechanism and threshold for triggered action potentials. Circulation Research. 2000;87:774–80. doi: 10.1161/01.res.87.9.774. [DOI] [PubMed] [Google Scholar]
- 76.Petroff MG, Kim SH, Pepe S, Dessy C, Marban E, Balligand JL, et al. Endogenous nitric oxide mechanisms mediate the stretch dependence of Ca2+ release in cardiomyocytes. Nat Cell Biol. 2001;3:867–73. doi: 10.1038/ncb1001-867. [DOI] [PubMed] [Google Scholar]
- 77.Khan SA, Lee K, Minhas KM, Gonzalez DR, Raju SV, Tejani AD, et al. Neuronal nitric oxide synthase negatively regulates xanthine oxidoreductase inhibition of cardiac excitation-contraction coupling. Proc Natl Acad Sci U S A. 2004;101:15944–8. doi: 10.1073/pnas.0404136101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Sanchez G, Pedrozo Z, Domenech RJ, Hidalgo C, Donoso P. Tachycardia increases NADPH oxidase activity and RyR2 S-glutathionylation in ventricular muscle. J Mol Cell Cardiol. 2005;39:982–91. doi: 10.1016/j.yjmcc.2005.08.010. [DOI] [PubMed] [Google Scholar]
- 79.Marx SO, Reiken S, Hisamatsu Y, Jayaraman T, Burkhoff D, Rosemblit N, et al. PKA phosphorylation dissociates FKBP12.6 from the calcium release channel (ryanodine receptor): defective regulation in failing hearts. Cell. 2000;101:365–76. doi: 10.1016/s0092-8674(00)80847-8. [DOI] [PubMed] [Google Scholar]
- 80.Fauconnier J, Thireau J, Reiken S, Cassan C, Richard S, Matecki S, et al. Leaky RyR2 trigger ventricular arrhythmias in Duchenne muscular dystrophy. Proc Natl Acad Sci U S A. 2010;107:1559–64. doi: 10.1073/pnas.0908540107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.McMurray J, Chopra M, Abdullah I, Smith WE, Dargie HJ. Evidence of oxidative stress in chronic heart failure in humans. Eur Heart J. 1993;14:1493–8. doi: 10.1093/eurheartj/14.11.1493. [DOI] [PubMed] [Google Scholar]
- 82.Dhalla AK, Hill MF, Singal PK. Role of oxidative stress in transition of hypertrophy to heart failure. J Am Coll Cardiol. 1996;28:506–14. doi: 10.1016/0735-1097(96)00140-4. [DOI] [PubMed] [Google Scholar]
- 83.Sia YT, Lapointe N, Parker TG, Tsoporis JN, Deschepper CF, Calderone A, et al. Beneficial effects of long-term use of the antioxidant probucol in heart failure in the rat. Circulation. 2002;105:2549–55. doi: 10.1161/01.cir.0000016721.84535.00. [DOI] [PubMed] [Google Scholar]
- 84.Ullrich ND, Fanchaouy M, Gusev K, Shirokova N, Niggli E. Hypersensitivity of excitation-contraction coupling in dystrophic cardiomyocytes. Am J Physiol Heart Circ Physiol. 2009 doi: 10.1152/ajpheart.00602.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Fanchaouy M, Polakova E, Jung C, Ogrodnik J, Shirokova N, Niggli E. Pathways of abnormal stress-induced Ca2+ influx into dystrophic mdx cardiomyocytes. Cell Calcium. 2009;46:114–21. doi: 10.1016/j.ceca.2009.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Williams IA, Allen DG. The role of reactive oxygen species in the hearts of dystrophin-deficient mdx mice. Am J Physiol Heart Circ Physiol. 2007;293:H1969–H77. doi: 10.1152/ajpheart.00489.2007. [DOI] [PubMed] [Google Scholar]
- 87.Jung C, Martins AS, Niggli E, Shirokova N. Dystrophic cardiomyopathy: amplification of cellular damage by Ca2+ signalling and reactive oxygen species-generating pathways. Cardiovasc Res. 2008 doi: 10.1093/cvr/cvm089. [DOI] [PubMed] [Google Scholar]
- 88.Prins KW, Humston JL, Mehta A, Tate V, Ralston E, Ervasti JM. Dystrophin is a microtubule-associated protein. J Cell Biol. 2009;186:363–9. doi: 10.1083/jcb.200905048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Heymes C, Bendall JK, Ratajczak P, Cave AC, Samuel JL, Hasenfuss G, et al. Increased myocardial NADPH oxidase activity in human heart failure. J Am Coll Cardiol. 2003;41:2164–71. doi: 10.1016/s0735-1097(03)00471-6. [DOI] [PubMed] [Google Scholar]
- 90.Li JM, Gall NP, Grieve DJ, Chen M, Shah AM. Activation of NADPH oxidase during progression of cardiac hypertrophy to failure. Hypertension. 2002;40:477–84. doi: 10.1161/01.hyp.0000032031.30374.32. [DOI] [PubMed] [Google Scholar]
- 91.Grieve DJ, Byrne JA, Siva A, Layland J, Johar S, Cave AC, et al. Involvement of the nicotinamide adenosine dinucleotide phosphate oxidase isoform Nox2 in cardiac contractile dysfunction occurring in response to pressure overload. J Am Coll Cardiol. 2006;47:817–26. doi: 10.1016/j.jacc.2005.09.051. [DOI] [PubMed] [Google Scholar]
- 92.Zhang R, Khoo MS, Wu Y, Yang Y, Grueter CE, Ni G, et al. Calmodulin kinase II inhibition protects against structural heart disease. Nat Med. 2005;11:409–17. doi: 10.1038/nm1215. [DOI] [PubMed] [Google Scholar]
- 93.Murdoch CE, Zhang M, Cave AC, Shah AM. NADPH oxidase-dependent redox signalling in cardiac hypertrophy, remodelling and failure. Cardiovasc Res. 2006;71:208–15. doi: 10.1016/j.cardiores.2006.03.016. [DOI] [PubMed] [Google Scholar]
- 94.Bell RM, Cave AC, Johar S, Hearse DJ, Shah AM, Shattock MJ. Pivotal role of NOX-2-containing NADPH oxidase in early ischemic preconditioning. FASEB J. 2005;19:2037–9. doi: 10.1096/fj.04-2774fje. [DOI] [PubMed] [Google Scholar]
- 95.Kimura S, Zhang GX, Nishiyama A, Shokoji T, Yao L, Fan YY, et al. Role of NAD(P)H oxidase- and mitochondria-derived reactive oxygen species in cardioprotection of ischemic reperfusion injury by angiotensin II. Hypertension. 2005;45:860–6. doi: 10.1161/01.HYP.0000163462.98381.7f. [DOI] [PubMed] [Google Scholar]
- 96.Parker KK, Taylor LK, Atkinson JB, Hansen DE, Wikswo JP. The effects of tubulin-binding agents on stretch-induced ventricular arrhythmias. Eur J Pharmacol. 2001;417:131–40. doi: 10.1016/s0014-2999(01)00856-1. [DOI] [PubMed] [Google Scholar]
- 97.Dick DJ, Lab MJ. Mechanical modulation of stretch-induced premature ventricular beats: induction of mechanoelectric adaptation period. Cardiovasc Res. 1998;38:181–91. doi: 10.1016/s0008-6363(97)00314-3. [DOI] [PubMed] [Google Scholar]
- 98.White E. Mechanical modulation of cardiac microtubules. Pflugers Arch. 2011;462:177–84. doi: 10.1007/s00424-011-0963-0. [DOI] [PubMed] [Google Scholar]
- 99.Whitehead NP, Yeung EW, Froehner SC, Allen DG. Skeletal muscle NADPH oxidase is increased and triggers stretch-induced damage in the mdx mouse. PLoS One. 2010;5:e15354. doi: 10.1371/journal.pone.0015354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Allen DG, Zhang BT, Whitehead NP. Stretch-induced membrane damage in muscle: comparison of wild-type and mdx mice. Adv Exp Med Biol. 2010;682:297–313. doi: 10.1007/978-1-4419-6366-6_17. [DOI] [PubMed] [Google Scholar]
- 101.Allen DG, Gervasio OL, Yeung EW, Whitehead NP. Calcium and the damage pathways in muscular dystrophy. Can J Physiol Pharmacol. 2010;88:83–91. doi: 10.1139/Y09-058. [DOI] [PubMed] [Google Scholar]
- 102.Palomero J, Pye D, Kabayo T, Jackson MJ. Effect of passive stretch on intracellular nitric oxide and superoxide activities in single skeletal muscle fibres: influence of ageing. Free Radic Res. 2012;46:30–40. doi: 10.3109/10715762.2011.637203. [DOI] [PubMed] [Google Scholar]
- 103.Percival JM, Gregorevic P, Odom GL, Banks GB, Chamberlain JS, Froehner SC. rAAV6-microdystrophin rescues aberrant Golgi complex organization in mdx skeletal muscles. Traffic. 2007;8:1424–39. doi: 10.1111/j.1600-0854.2007.00622.x. [DOI] [PubMed] [Google Scholar]
- 104.Watson PA, Hannan R, Carl LL, Giger KE. Contractile activity and passive stretch regulate tubulin mRNA and protein content in cardiac myocytes. Am J Physiol. 1996;271:C684–9. doi: 10.1152/ajpcell.1996.271.2.C684. [DOI] [PubMed] [Google Scholar]
- 105.Poulain FE, Sobel A. The microtubule network and neuronal morphogenesis: Dynamic and coordinated orchestration through multiple players. Mol Cell Neurosci. 2010;43:15–32. doi: 10.1016/j.mcn.2009.07.012. [DOI] [PubMed] [Google Scholar]
- 106.Akhmanova A, Steinmetz MO. Tracking the ends: a dynamic protein network controls the fate of microtubule tips. Nat Rev Mol Cell Biol. 2008;9:309–22. doi: 10.1038/nrm2369. [DOI] [PubMed] [Google Scholar]
- 107.Yutao X, Geru W, Xiaojun B, Tao G, Aiqun M. Mechanical stretch-induced hypertrophy of neonatal rat ventricular myocytes is mediated by beta(1)-integrin-microtubule signaling pathways. Eur J Heart Fail. 2006;8:16–22. doi: 10.1016/j.ejheart.2005.05.014. [DOI] [PubMed] [Google Scholar]
- 108.Hashimoto-Komatsu A, Hirase T, Asaka M, Node K. Angiotensin II induces microtubule reorganization mediated by a deacetylase SIRT2 in endothelial cells. Hypertens Res. 2011;34:949–56. doi: 10.1038/hr.2011.64. [DOI] [PubMed] [Google Scholar]
- 109.Cote F, Do TH, Laflamme L, Gallo JM, Gallo-Payet N. Activation of the AT(2) receptor of angiotensin II induces neurite outgrowth and cell migration in microexplant cultures of the cerebellum. J Biol Chem. 1999;274:31686–92. doi: 10.1074/jbc.274.44.31686. [DOI] [PubMed] [Google Scholar]
- 110.Laflamme L, Gasparo M, Gallo JM, Payet MD, Gallo-Payet N. Angiotensin II induction of neurite outgrowth by AT2 receptors in NG108-15 cells. Effect counteracted by the AT1 receptors. J Biol Chem. 1996;271:22729–35. doi: 10.1074/jbc.271.37.22729. [DOI] [PubMed] [Google Scholar]
- 111.Stroth U, Meffert S, Gallinat S, Unger T. Angiotensin II and NGF differentially influence microtubule proteins in PC12W cells: role of the AT2 receptor. Brain Res Mol Brain Res. 1998;53:187–95. doi: 10.1016/s0169-328x(97)00298-2. [DOI] [PubMed] [Google Scholar]
- 112.Seshiah PN, Weber DS, Rocic P, Valppu L, Taniyama Y, Griendling KK. Angiotensin II stimulation of NAD(P)H oxidase activity: upstream mediators. Circ Res. 2002;91:406–13. doi: 10.1161/01.res.0000033523.08033.16. [DOI] [PubMed] [Google Scholar]
- 113.Cohn RD, van Erp C, Habashi JP, Soleimani AA, Klein EC, Lisi MT, et al. Angiotensin II type 1 receptor blockade attenuates TGF-beta-induced failure of muscle regeneration in multiple myopathic states. Nat Med. 2007;13:204–10. doi: 10.1038/nm1536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Lovering RM, O’Neill A, Muriel JM, Prosser BL, Strong J, Bloch RJ. Physiology, structure, and susceptibility to injury of skeletal muscle in mice lacking keratin 19-based and desmin-based intermediate filaments. Am J Physiol Cell Physiol. 2011;300:C803–13. doi: 10.1152/ajpcell.00394.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Roche JA, Lovering RM, Bloch RJ. Impaired recovery of dysferlin-null skeletal muscle after contraction-induced injury in vivo. Neuroreport. 2008;19:1579–84. doi: 10.1097/WNR.0b013e328311ca35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Hwang J, Saha A, Boo YC, Sorescu GP, McNally JS, Holland SM, et al. Oscillatory shear stress stimulates endothelial production of O2- from p47phox-dependent NAD(P)H oxidases, leading to monocyte adhesion. J Biol Chem. 2003;278:47291–8. doi: 10.1074/jbc.M305150200. [DOI] [PubMed] [Google Scholar]
- 117.Pimentel DR, Amin JK, Xiao L, Miller T, Viereck J, Oliver-Krasinski J, et al. Reactive oxygen species mediate amplitude-dependent hypertrophic and apoptotic responses to mechanical stretch in cardiac myocytes. Circ Res. 2001;89:453–60. doi: 10.1161/hh1701.096615. [DOI] [PubMed] [Google Scholar]
- 118.Akki A, Zhang M, Murdoch C, Brewer A, Shah AM. NADPH oxidase signaling and cardiac myocyte function. J Mol Cell Cardiol. 2009;47:15–22. doi: 10.1016/j.yjmcc.2009.04.004. [DOI] [PubMed] [Google Scholar]
- 119.Ren Z, Raucci FJ, Jr, Browe DM, Baumgarten CM. Regulation of swelling-activated Cl(-) current by angiotensin II signalling and NADPH oxidase in rabbit ventricle. Cardiovasc Res. 2008;77:73–80. doi: 10.1093/cvr/cvm031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Jang HJ, Hwang S, Cho KY, Kim do K, Chay KO, Kim JK. Taxol induces oxidative neuronal cell death by enhancing the activity of NADPH oxidase in mouse cortical cultures. Neuro sci Lett. 2008;443:17–22. doi: 10.1016/j.neulet.2008.07.049. [DOI] [PubMed] [Google Scholar]
- 121.Chen X, Green PG, Levine JD. Abnormal muscle afferent function in a model of Taxol chemotherapy-induced painful neuropathy. J Neuro physiol. 2011;106:274–9. doi: 10.1152/jn.00141.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]