

Abstract
Introduction
CDKN2A(p16) inactivation is common in lung cancer and occurs via homozygous deletions (HD), methylation of promoter region, or point mutations. While p16 promoter methylation has been linked to KRAS mutation and smoking, the associations between p16 inactivation mechanisms and other common genetic mutations and smoking status are still controversial or unknown.
Methods
We determined all three p16 inactivation mechanisms using multiple methodologies for genomic status, methylation, RNA and protein expression, and correlated them with EGFR, KRAS, STK11 mutations and smoking status in 40 cell lines and 45 tumor samples of primary NSCLC. We also performed meta-analyses to investigate the impact of smoke exposure on p16 inactivation.
Results
p16 inactivation was the major mechanism of RB pathway perturbation in NSCLC, with HD being the most frequent method, followed by methylation and the rarer point mutations. Inactivating mechanisms were tightly correlated with loss of mRNA and protein expression. p16 inactivation occurred at comparable frequencies regardless of mutational status of EGFR, KRAS and STK11, however, the major inactivation mechanism of p16 varied. p16 methylation was linked to KRAS mutation but was mutually exclusive with EGFR mutation. Cell lines and tumor samples demonstrated similar results. Our meta-analyses confirmed a modest positive association between p16 promoter methylation and smoking.
Conclusions
Our results confirm that all of the inactivation mechanisms are truly associated with loss of gene product and identify specific associations between p16 inactivation mechanisms and other genetic changes and smoking status.
Keywords: p16, CDKN2A, inactivation, homozygous deletion, methylation, lung cancer, adenocarcinoma, meta-analysis
INTRODUCTION
Lung cancer is the second most common type of cancer and the leading cause of cancer-related deaths in both men and women in the United States. According to the cancer statistics review from the Surveillance, Epidemiology, and End Results (SEER) program of the U.S. National Cancer Institute (NCI), the age-adjusted death rate was 51.6 per 100,000 in both men and women per year during 2004-2008.1 A total of 226,160 new cases of lung cancer and 160,340 deaths from lung cancer were projected to occur in the U.S. in 2012.2
There are two major types of lung cancer, non-small cell lung carcinoma (NSCLC) and small cell lung carcinoma (SCLC). NSCLC can be further divided into three main subtypes: large cell lung carcinoma, squamous cell lung carcinoma, and adenocarcinoma. Adenocarcinoma accounts for approximately 40% of lung cancers and is the most common type of lung cancer in the United States.3,4
Most cases of lung cancer, including adenocarcinoma, are due to tobacco smoking. However, approximately 25% of lung cancer cases worldwide and about 15% of the cases in the USA are not attributable to smoking.5,6 The most common type of lung cancer among never smokers is adenocarcinoma, and it is found to be more prevalent among East Asians and females. Despite advances in oncology, the five-year survival rate of patients with lung adenocarcinoma has not changed significantly over the past three decades. The NCI SEER program showed that about 84% of lung adenocarcinoma patients die within five years after diagnosis.1 We believe lung cancer prognosis can be improved by a combination of early detection7 with advances in biology and the subsequent implementation of effective targeted therapies such as those directed against EGFR mutations and ALK translocations.8,9
Studies have shown that patients with lung adenocarcinoma often have genetic mutations in EGFR, KRAS, STK11 (also known as LKB1), TP53 (also known as p53), and CDKN2A (also known as p16 or INK4a).10-14 Several new potential targets for therapeutic approaches have also been identified recently.15,16 A study of these mutations may eventually lead to novel therapeutic applications.
One of the most common genetic alterations in many forms of cancer including lung adenocarcinoma is inactivation of p16. p16 is located at chromosome region 9p21 and is encoded by the CDKN2A gene. In addition to p16, CDKN2A encodes a completely unrelated tumor suppressor protein, ARF, which interacts with TP53. The simple tandem arrangement is complicated by the presence of an additional exon 1β which is transcribed from its own promoter. The resulting RNA incorporates exons 2 and 3 but specifies a distinct protein because the exons are translated by an alternative reading frame. Thus, while exons 2 and 3 are shared by the two mRNAs, they encode different protein products, p16 and ARF. 17
p16 is a tumor suppressor, functions as an inhibitor of CDK4 and CDK6, the D-type cyclin-dependent kinases that initiate the phosphorylation of the retinoblastoma tumor suppressor protein RB, and induces cell cycle arrest.18 Both alleles must be inactivated before its function is eliminated. Three mechanisms have been implicated in its inactivation: homozygous deletion (HD), hypermethylation in the promoter CpG island (methylation), and point mutation. It has been reported that p16 is frequently inactivated by homozygous deletion or promoter hypermethylation, and rarely by point mutation in primary NSCLC.12,19 Studies demonstrated that the frequency of p16 methylation is significantly higher in lung adenocarcinoma with KRAS mutation, however, the associations between p16 inactivation mechanisms and other common genetic mutations in lung adenocarcinoma such as EGFR and STK11 remain controversial or have never been explored.20 Smoking has been reported to be associated with p16 methylation and p16 HD was found to be associated with never smokers in some studies, but these findings are inconsistent.12,19-21
Despite the large number of reports, many of these associations remain controversial, in part because the majority of the reports a) do not examine all the mechanisms of inactivation, and b) do not demonstrate that the inactivating mechanism(s) studied were truly associated with inactivation of the gene. Therefore, the aims of the present study were to: 1) examine all the three described inactivation mechanisms of p16 in lung cancer cell lines and tumor samples; 2) demonstrate that our methods of detecting inactivation are truly associated with inactivation; 3) correlate the data with tobacco exposure; and 4) correlate inactivation mechanisms with molecular features. Since one of our major interests was correlation with tobacco exposure, we wished to study sufficient numbers of cancers arising in never smokers. Thus, with a few exceptions, we limited our study to adenocarcinoma as they are by far the most common form of lung cancer arising in never smokers.
Materials and Methods
Cell Culture
Forty NSCLC cell lines were used in the study. Thirty-six of them were lung adenocarcinomas, one was large cell carcinoma, one was adenosquamous carcinoma, and two were unspecified NSCLC cell lines. Data for the cell lines have been reported in multiple previous studies.22-24 In this study, light smokers were defined as patients with less than 15 pack-year history. Heavy smokers were defined as those who had a smoking history of 15 pack-year or more. Refer to Supplementary Table 1 for further information.
Tumor Samples
Forty-five tumor samples were included in this study and all of them were obtained from patients with primary lung adenocarcinoma. IRB approval and informed consents were obtained from the patients for molecular analysis of the samples. Among the forty-five tumor samples, twenty-nine of them were obtained from smokers, either current or former smokers, and sixteen of them were from never smokers. Further details about smoking histories were not available. Refer to Supplementary Table 2 for further information.
DNA, RNA extraction and cDNA synthesis
Genomic DNA was obtained from cell lines and tumor samples by standard phenol-chloroform extraction or by using the DNeasy Tissue Kit (QIAGEN, Alameda, CA). Total RNA was extracted from cell lines using the RNeasy Plus Mini Kit (QIAGEN). The cDNA was prepared by reverse transcription of RNA using High-Capacity cDNA Reverse Transcription Kits (Applied Biosystems, Foster City, CA) according to the manufacturer's protocol.
p16 inactivation methodology
Because of the multiple methods used to determine and confirm inactivation, and their applications to tumors and cell lines, these methods are summarized in Supplementary Table 3 and detailed below.
Homozygous Deletions (HD)
HDs of p16 in cell lines were detected using SYBR green real-time PCR (qPCR) and SNP which will be discussed later. The sequences of the primers used were forward: 5′-GTGAAGCCATTGCGAGAACT-3′ and reverse: 5′-TTCTTTCAATCGGGGATGTC-3′, and both primers recognize sequences located in intron 2. The reactions were performed in a Bio-Rad DNA Engine Thermal Cycler. The thermal cycling conditions were set to 2 min at 50°C, 10 min at 95°C, followed by 40 cycles of 15 sec at 95°C alternating with 1 min at 60°C. Standard curves for the copy numbers were generated using human diploid genomic DNA as a reference. DNA copy number ratios were calculated as the average copy number of the target locus divided by the average copy number of line 1, which is used as the reference locus, and then normalized against the human genomic DNA. Homozygous deletion was defined as copy number equal to or less than 0.001 and the copy number of human genomic DNA was set at 2.
HDs of p16 in tumor samples were identified using SNP arrays. The Genome-Wide Human SNP Array 6.0 platform (Affymetrix Inc, Santa Clara, CA) was used to profile each tumor. Copy number profiles were generated with Partek Genomics Suite (PGS) software, using the Paired Copy Number Analysis workflow. In this analysis, matched nonmalignant lung tissue is used as a copy number baseline for defining copy number alterations in each tumor. Regions of copy number gain and loss were statistically detected using PGS's genomic segmentation algorithm with the following parameters: signal to noise ratio > 0.3, minimum number of 50 probes per segment, and p-value thresholds of 10-7 for the statistical differences between a) signal intensities of adjacent segments, and b) tumor and nonmalignant lung DNA. Copy numbers were estimated by the PGS genomic segmentation algorithm, and homozygous deletion was defined as a copy number less than 1. p16 was mapped to the genomic regions detected using the March 2006, hg18 genome coordinates from the UCSC Genome Browser.
Methylation analysis
The methylation status of CpG islands in the p16 gene promoter was determined by methylation-specific qPCR (MSP) using quantitative real-time PCR and fluorescent Taqman chemistry. The probes used to assess methylation were located in exon 1 and their selection was based upon the results of qPCR in our previous study.25 Bisulfite conversion of DNA was performed using the EZ DNA Methylation Gold kit by Zymo Research (Irvine CA). All other materials and methods for MSP were as described in prior studies.21,25
The methylation status of p16 in cell lines was confirmed using the Infinium HumanMethylation 450K BeadChip by Illumina (San Diego, CA). Because Illumina Human Methylation 450 beadchip data was not available for our tumor samples, we downloaded the data for squamous cell (LUSC) and adenocarcinoma lung cancer (LUAD) from The Cancer Genome Atlas (TCGA) data portal (https://tcga-data.nci.nih.gov/tcga). The beta values of all samples were merged into a data matrix. Tumor samples and non malignant samples were identified from the sample barcode according to the TCGA documentation. T test was then applied to each methylation probe to search for statistically significant differential methylation between tumor samples and non malignant samples. Methlyation was defined as tumor samples with beta scores of 0.3 or higher than the mean values for non malignant tissues.
Point mutation analysis
Sequencing of p16 was performed in all cell lines and tumor samples to detect point mutations. Sequencing of both genomic DNA and cDNA were performed dependent on material availability, and all of the reactions were performed using the Applied Biosystem GeneAmp PCR System 9700 instrument. For DNA sequencing, exon 1 was amplified by PCR using the following primers: forward: 5′-GAAGAAAGAGGAGGGGCT-3′ and reverse: 5′-GCGCTACCTGATTCCAATTC-3′. PCR was performed for 42 cycles consisting of denaturation at 94°C for 1 min, annealing at 57°C for 1 min, and extension at 72°C for 2 min. Exon 2 was amplified using the following primers: forward: 5′-AGCTTCCTTTCCGTCATGC-3′ and reverse: 5′-GGAAGCTCTCAGGGTACAAATTC-3′. PCR was performed for 42 cycles consisting of denaturation at 94°C for 1 min, annealing at 48°C for 1 min, and extension at 72°C for 2 min. For amplification of exon 3, the following primers were used: forward: 5′-CCTGGCATTGTGAGCAACC-3′ and reverse: 5′-GGTTCTGGCATTTGCTAGCAG-3′. PCR was performed for 42 cycles consisting of denaturation at 94°C for 1 min, annealing at 55°C for 1 min, and extension at 72°C for 2 min. For cDNA sequencing, the following primers were used: forward: 5′-CGGAGGAAGAAAGAGGAG-3′ and reverse: 5′-TTCTCAGAGCCTCTCTGGT. PCR was performed for 44 cycles consisting of denaturation at 94°C for 1 min, annealing at 50°C for 90 sec, and extension at 72°C for 2 min.
Exome sequencing was performed on an Illumina GAIIx platform using standard procedures. Post sequence data was processed with Illumina Pipeline software v 1.7.
Expression analysis
Taqman gene expression assay for p16 was used to quantitatively detect p16 mRNA levels according to the manufacturer's protocol (Applied Biosystems).
RNA-seq was also used to measure p16 transcript expression levels. One μg of total RNA per sample was poly-A tail purified, fragmented and adapter ligated using Illumina TruSeq RNA sample prep kit version 2 following the instructions written by manufacturers. Samples were multiplexed 4 to a lane on the Illumina HiSeq 2000 using the Single Read (SR) version 3 flowcell and underwent a 100 base pair single end run using TruSeq version 3 SBS kits. Sequencing was performed at the Southern California Genotyping Consortium, University of California Los Angeles. Sequence data was read using Illumina OBC (off-line base caller) software, was trimmed to remove adapter reads, and aligned to the NCBI GrCh37 (hg19) transcriptome using CLCbio Genomics Workbench version 5.1. Reads with more than 3 mismatches to the reference genome (poor alignment) or over 10 identical copies (PCR bias) were discarded. Coverage was determined by read density over the gene body and used to estimate expression levels.
Protein expression of p16 was assessed by mass spectrometric (MS) analysis. The collection of whole cell extracts was performed as described in a previous study.26 Protein digestion and identification by liquid chromatography (LC)-MS/MS were performed as described previously.26 Three International Protein Indexes (IPI) were identified for CDKN2A in which IPI00001560 and IPI00651662 correspond to p16 while IPI00478390 corresponds to ARF.
Mutation status of EGFR, KRAS, STK11, TP53 and RB
The forty cell lines and forty-five tumor samples in our study were examined for mutations in exons 18 to 21 of the EGFR gene and exons 1 and 2 of the KRAS gene by genomic PCR and direct sequencing as described in our previous studies.27-29 Mutations in exons 1 to 9 of the STK11 gene were detected by cDNA sequencing in both cell lines and tumors. The following primers were used: forward: 5′- GAAGGGAAGTCGGAACACAA -3′ and reverse: 5′- CCCTGGCTATGCAGGTACTC -3′; forward: 5′- ATGGAGTACTGCGTGTGTGG -3′ and reverse: 5′- CAGCCGGAGGATGTTTCTT -3′; forward: 5′- GTTTGAGAACATCGGG AAGG-3′ and reverse: 5′- AACCGGCAGGAAGACTGAG -3′. Mutations in exons 1 to 11 of the p53 gene were detected by cDNA sequencing only in cell lines. The following primers were used: forward: 5′- GCTTTCCACGACGGTGAC-3′ and reverse: 5′- TGACTGCTTGTAGAT GGCCA -3′; forward: 5′- GTCTGGGCTTCTTGCATTCT -3′ and reverse: 5′- TCTTGCGGA GATTCTCTTCC -3′; forward: 5′- CCTCACCATCATCACACTGG -3′ and reverse: 5′- TTCT GACGCACACCTTGC -3′. The mutation data for RB in 40 NSCLC cell lines were obtained from Sanger Institute Catalog Of Somatic Mutations In Cancer (COSMIC).
Copy number analysis of CCND1, CDK4 and CDK6:
The copy numbers of CCND1, CDK4 and CDK6 in forty cell lines were detected using SYBR green real-time PCR (qPCR). The sequences of the primers used for CCND1 were forward: 5′- GCGGAGGAGAACAAACAGAT-3′ and reverse: 5′- ACCCAGGTGGAGAGCAAGA-3′; for CDK4, forward: 5′- TTGTTGCTGCAGGCTCATAC-3′ and reverse: 5′- ATAGGCACCGACACCAATTT-3′; for CDK6, forward: 5′-ATTCAAAATCTGCCCAACCA-3′ and reverse: 5′- GCAGACGAGCTTGACATCAG-3′. The detailed method was described in the section on HD.
Statistical analysis
Frequencies of all three p16 inactivation mechanisms in each mutational group (EGFR, KRAS, and STK11 mutation), and smoking group were compared using Fisher exact test. Some of these genetic mutations are known to be associated with each other, smoking status, gender, and ethnicity. In order to prevent the potential confounding effect of multiple variables, a multivariate logistic regression model was used to analyze the association of specific genetic mutations or smoking status with the various mechanisms of p16 inactivation. Results with p-value < 0.05 were regarded as statistically significant.
Meta-analysis
Literatures published from 2001 to May 2012 in PubMed database were screened by using keywords “p16 or CDKN2A or INK4A” and “smoking or tobacco” and “promoter methylation or homozygous deletion or mutation or inactivation” to determine the impact of smoking. Keywords “never smoker or never smoke or never smoking” were used instead of “smoking or tobacco” in the meta-analysis for never smokers. Only articles providing raw data and published in English were selected. The meta-analyses were performed using R (www.r-project.org) with the Rmeta package. A Woolf's test was performed to identify heterogeneity among publications. The random effects model was used for heterogeneous studies while the fixed effects model was applied if the studies were homogeneous.30 To prevent one of the studies from dominating the results by contributing a larger number of samples or a big effect size, we adopted the leave-one-out strategy by removing data from each study one at a time and detected how the overall odds ratio (OR) had changed. The overall OR was found to remain significantly greater than 1 no matter which study was removed in both meta-analyses and hence none of the selected studies dominated the result.
Results
Multiple methodologies for the detection of p16 inactivation
p16 can be inactivated by HD, hypermethylation of promoter CpG islands, or point mutations. In this study, we investigated the inactivation mechanisms of p16 in forty NSCLC cell lines and forty-five NSCLC tumor samples. As shown in Table 1, the frequency of p16 inactivation was higher in cell lines (75%) compared to tumor samples (38%), and this has been observed in previous studies.14 Despite the higher frequency of inactivation in cell lines, no evidence of difference in the proportions of p16 inactivation mechanisms was found between cell lines and tumor samples. Hence, the results for tumors and cell lines are presented individually and were also combined to increase the sample size for our analyses (Table 1).
Table 1. Frequency of p16 inactivation and mechanisms of p16 inactivation.
| Frequency of p16 inactivation | Mechanisms of p16 inactivation | |||
|---|---|---|---|---|
|
| ||||
| HD | Methylation | Mutation | ||
|
| ||||
| Cell lines | 30/40 (75%) | 16/30 (53%) | 10/30 (33%) | 4/30 (13%) |
|
| ||||
| Tumors | 17/45 (38%) | 10/17 (59%) | 4/17 (24%) | 3/17 (18%) |
|
| ||||
| Combined | 47/85 (55%) | 26/47 (55%) | 14/47 (30%) | 7/47 (15%) |
HD was the most frequent mechanism of inactivation in both cell lines (53%) and tumors (59%), as detected by copy number changes (SNP arrays). Representative examples of HDs are shown in Figure 1. For cell lines, qPCR was used to confirm the results of the SNP arrays.
Figure 1.
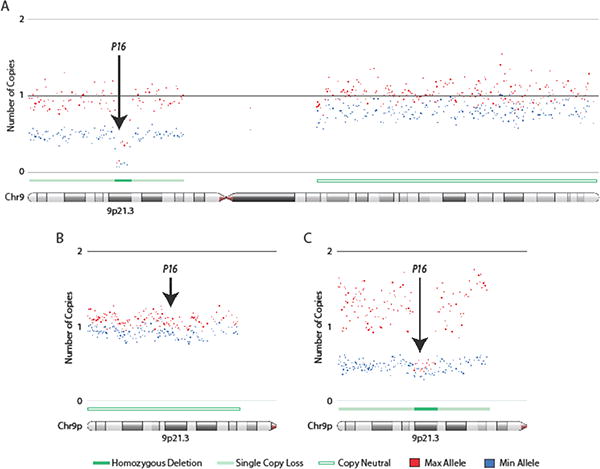
Homozygous deletions of p16 in lung adenocarcinoma tumors and cell lines. Affymetrix SNP 6 arrays were used to generate copy number profiles for lung adenocarcinoma cell lines and clinical lung tumors. Allele specific copy numbers were derived using the allele specific copy number workflow in Partek Genomics Suite (PGS) software. Copy number profiles for lung cancer cell lines were generated using a panel of 72 non-diseased HapMap individuals as a baseline while matched non-malignant lung tissue was used as a reference for defining copy number changes in tumors. Arrows indicate region of the p16 gene. (A) Focal homozygous deletion (HD) of p16 in the lung adenocarcinoma cell line, NCI-H1944. (B) Lung adenocarcinoma tumor illustrating neutral p16 copy number state. (C) Focal HD of p16 in a lung adenocarcinoma tumor. Both HDs in (A) and (C) occur in a background of single copy loss on chromosome 9p. Copy number states in regions of deletion do not drop right to zero due to cancer cell heterogeneity, aneuploidy, and/or the presence of non-malignant cells that contribute to array signals. Maximum (red) and minimum (blue) alleles are defined by copy number state. Each dot represents the smoothed copy number for 30 adjacent array probes. Copy number states (HD, single copy loss, or copy neutral) are indicated. Images were generated in PGS. Arrows indicate location of p16 gene at chromosome 9p21.3.
The methylation status of p16 was assessed by quantitative MSP analysis. p16 methylation was present in 30% (33% for cell lines and 24% for tumors) of samples with p16 inactivation. We also confirmed these results of 38 cell lines (data were not available for cell lines PC-9 and DFCI032) by Illumina infinium human methylation 450k BeadChip. Probe cg13601799 for detecting exon one of p16 (E1α) and probe cg03079681 located in exon one of p14ARF (E1β) were selected (Figure 2A). Only cell lines with methylated p16 detected by MSP analysis showed high levels of methylation using probe cg13601799, and these cell lines are not methylated in p14ARF exon 1 (probe cg03079681) (Figure 2B). Among them, NCI-H969 showed partial methylation of p16 as indicated by its beta value. Of interest, there are some values for beta scores in the cell lines carrying a homozygous deletion of the CDKN2A locus. These probably result from deletions in the region which are artifactually affecting the beta value resulting from a comparison of the unmethylated and methylated probes. Thus the results do not reflect the correct methylation status of the cell lines. We also examined the methylation status of NSCLC cancers in the TCGA data base (Figure 2C). We found that p16 methylation was more frequent in squamous cell carcinomas (36%) than in adenocarcinomas (17%), which is consistent with published reports.15
Figure 2.
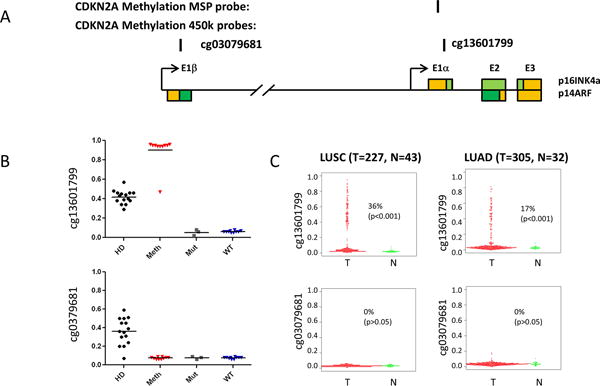
Methylation of p16 promoter in lung cancers and cell lines. (A) Schematic diagram of CDKN2A (p14ARF-p16INK4a) genomic locus. Colored boxes represent exons of p16INK4a or p14ARF. The coding regions are shown as green, while the non-coding regions shown as yellow. The locations of the CDKN2A methylation-specific PCR (MSP) probe and corresponding illumina methylation450 beadchip probes are indicated. (B) The methylation status of p16 in the NSCLC cell lines by illumina methylation450 beadchip probes. The methylation beta values from cell lines carrying a homozygous deletion of the CDKN2A locus (HD), DNA methylation of the CpG island in exon 1a (p16INK4a) (Meth), a point mutation in p16INK4a (Mut), or no detectable alteration of the locus (WT) are plotted. A value of 0 indicates non-methylation of the locus; a value of 1 means complete methylation. (C) The methylation status of p16 in lung cancers from TCGA data portal by illumina methylation450 beadchip probes. The methylation beta values from squamous cell lung cancer (LUSC) or adenocarcinoma lung cancer(LUAD) and non-malignant lung tissues are plotted. Methlyation was defined as tumor samples with beta scores of 0.3 or higher than the mean values for non malignant tissues.
The p16 mutational status was determined by direct sequencing for cell lines and tumors. P16 mutations were present in 15% (13% for cell lines and 18% for tumors) of p16 inactivation. Additionally, the results were confirmed by next-generation sequencing in the cell lines.
In summary, the results obtained by different methods designed to measure the same type of inactivation (HD, methylation, or sequence mutation) were concordant with each other in every case.
The association between p16 inactivation and its mRNA and protein expression
As shown in Fig. 3A (with details presented in Supplementary Table 4), the concordance between all of the three inactivation mechanisms (HD, methylation, point mutations) and RNA and protein expression were excellent. With the exception of one partially methylated cell line (NCI-H969), all HD and methylated cell lines showed absent RNA or protein expression. With the exception of PC9, cell lines with point mutations expressed low or absent levels of mRNA and protein. Thus 28 of 30 (93%) cell lines demonstrating any method of inactivation lacked p16 expression. By contrast, nine of ten (90%) of wild type lines expressed varying levels of mRNA expression and eight of nine (89%) had protein expression.
Figure 3.

Expression analysis of the CDKN2A locus in NSCLC cell lines. (A) Heat map shows RNA and protein expression levels in cell line carrying no detectable alteration of the CDKN2A locus (WT), DNA methylation of the CpG island in exon 1a (p16INK4a) (Meth), a point mutation in p16INK4a (Mut), or a homozygous deletion of the locus (HD). RNA expression was determined by QPCR. The relative mRNA levels of p16 were compared to non-malignant immortalized bronchial epithelial cells (HBECs). Protein expression was determined by mass spectrometry. * indicates Rb mutants. (B) RNA-Seq data for the CDKN2A locus for representative cell lines. The different splice variants arising from the locus are indicated. Genome-wide coverage was similar for all 4 lines. Peaks detected are shown to scale. No signal was detected in the HD line (NCI-H2228), p14ARF mRNA, but not p16INK4a was detected in the Mut (NCI-H1975) or Meth (NCI-H23) lines; mRNA containing exon 1a (p16INK4a) was only detected in WT line (NCI-H522).
For representative cell lines we confirmed our results using RNA-Seq data. We selected four NSCLC cell lines (NCI-H2228, NCI-H1975, NCI-H23 and NCI-H522) to represent the different statuses of p16 (HD, mutation, methylated and WT, respectively), as illustrated in Figure 3B. Unlike ARF (E1β) which was detected in the other 3 cell lines except for the p16 HD cell line, p16 expression (E1α) was only detected in the p16 wild-type cell line. These data indicate that all three p16 inactivation mechanisms (HD, methylation, point mutation) affect p16 expression levels.
P16 inactivation in lung adenocarcinoma with other genetic mutation
Because EGFR, KRAS, and STK11 are frequently mutated in lung adenocarcinomas9,11, we investigated the association between mutational status of these genes and p16 inactivation. Among the forty cell lines, ten were EGFR mutants, fifteen were KRAS mutants, and fifteen were STK11 mutants. As previously reported, EGFR and KRAS mutations were mutually exclusive.28 Mutations of EGFR and STK11 were also found to be mutually exclusive in the forty cell lines. KRAS and STK11 mutations coexisted in seven cell lines. Of the forty-five tumor samples, fourteen had EGFR mutations, twenty-one had KRAS mutations, and twenty had STK11 mutations. Again, the EGFR and KRAS mutations were mutually exclusive. Six of the tumor samples had coexisting STK11 and EGFR mutations, while seven tumor samples had mutations in both STK11 and KRAS. The detailed information of mutational status of each sample is provided in Supplementary Tables 1 and 2
Overall p16 inactivation was detected with comparable frequencies regardless of EGFR, KRAS, and STK11 mutational status. Figure 4 and Supplementary Table 5 summarizes the data. Among all EGFR mutants, p16 was mostly inactivated by HD (42% combined from cell lines and tumor samples, 50% in cell lines and 36% in tumor samples), and the remainder demonstrated p16 inactivation by point mutation (13% combined, 20% in cell lines and 7% in tumor samples). None of the EGFR mutated cases demonstrated p16 promoter methylation, which has been identified as a common mechanism of p16 inactivation in lung adenocarcinomas in this as well as in a number of other studies.9,12,25 Our analysis revealed that EGFR mutations and p16 methylation were mutually exclusive in all cases, although our sample size was modest. By contrast KRAS mutations were positively correlated with p16 methylation (for cell lines, p=0.025; for tumors, p=0.025; for combined, p=0.003). We did not note any significant relationships between STK11 or p53 (data not shown) mutations and mechanisms of p16 inactivation.
Figure 4.

The association between p16 inactivation and EGFR, KRAS, STK11 mutations and smoke exposure. The frequency of p16 WT, HD, methylation and point mutation cases in each mutation type or smoking exposure group are presented in the bar graphs for cell lines, tumors or combined. * indicates statistically significant value. (for cell lines, p=0.025; for tumors, p=0.025; for combined, p=0.003)
Other variables including smoke exposure, gender, and ethnicity, have been reported to be associated with specific mutations. For instance, EGFR mutation is more prevalent in females, Asians, and never smokers while KRAS and STK11 mutations were more often found in smokers. Given these known relationships, it is plausible that p16 inactivation mechanisms may also be associated with specific clinical features in addition to the molecular features described above. In order to decipher the effects of multiple clinical and genetic variables, a multivariate logistic regression model was used to determine the associations between particular genetic mutation and p16 inactivation mechanisms. The results were consistent with our initial univariate analyses described above.
We also investigated genetic alterations of other components of the pathway. Although 75% NSCLC cell lines contained p16 inactivation, only 2/40 (5%) had RB mutations (Figure 5 and Supplementary Table 6). We also analyzed the copy number changes of cyclin D1, CDK4 and CDK6 in these cell lines. Low level gains were present in 20-25% of the lines (Fig. 5, Supplementary Table 6), while high level amplifications were rare.
Figure 5.
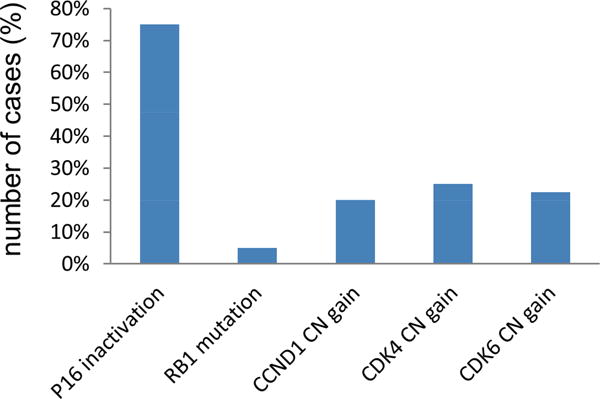
Other genetic alterations in p16-RB pathway in NSCLC cell lines. The percentages of cell lines containing RB mutations, CCND1, CDK4 and CDK6 copy number gains are plotted. Copy numbers >3 are considered as copy number gains.
P16 inactivation in smokers and never smokers
We failed to find a relationship between smoking status and frequency or method of inactivation of the p16 gene (Fig. 4, Supplementary Table 5). Because there are contradictory reports on the relationship of smoking status and p16 methylation, we performed meta-analyses to investigate the relationship of smoking status and p16 methylation as well as with any mechanism of inactivation.11,12,19-21,30,32,34-51
a) Meta-analysis of smoke exposure and p16 methylation
Promoter methylation of p16 is reportedly associated with smoking in some studies but not in others. In order to address these variable findings, we performed a meta-analysis in addition to analyzing our own data. A total of twenty-three studies containing 1903 smokers and 982 nonsmokers, were selected as described in the method section. A Woolf's test was performed to test for heterogeneity among the studies selected, and the results showed that the studies were heterogeneous (chi square = 39.29, df = 23, p = 0.018). A random effects model was therefore applied and the overall OR from the meta-analysis was 1.99, which was significant (p<0.05) with 95% CI between 1.48 and 2.72. Therefore, our meta-analysis based on the published literature confirmed a positive association between p16 promoter methylation and smoking (Figure 6).
Figure 6.

Meta-analysis of smoke exposure and p16 promoter methylation. A random effects model was applied due to the heterogeneity of the studies selected. The square and the connecting lines indicate the Odds Ratio (OR) and its confidence intervals (CI). The area of each square is proportional to the sample size of the study in the meta-analysis. The overall OR was 1.99 (p<0.05) with 95% CI between 1.48 and 2.72.
b) Meta-analysis Smoke exposure and other p16 inactivation mechanisms
While the association between promoter methylation and smoking has been extensively studied, little has been done to explore the potential association between smoke exposure and other p16 inactivation mechanisms. We performed a second meta-analysis to evaluate the association between smoke exposure and other methods of p16 inactivation. Five studies that contained data on homozygous deletions and/or point mutations with smoking history, were selected. There were total 305 smokers and 99 nonsmokers included in these studies. The Woolf's test showed that the studies were homogeneous (df = 4, p = 0.2680), and hence a fixed effects model rather than random effects model was applied. The meta-analysis did not identify any significant association between never smoking and any of the p16 inactivation mechanisms (Figure 7), although it did reveal a positive trend of p16 HD among never smokers. The overall OR was 0.61 (p=0.1068) with 95% CI between 0.338 and 1.11. These results corroborated the findings of our study.
Figure 7.
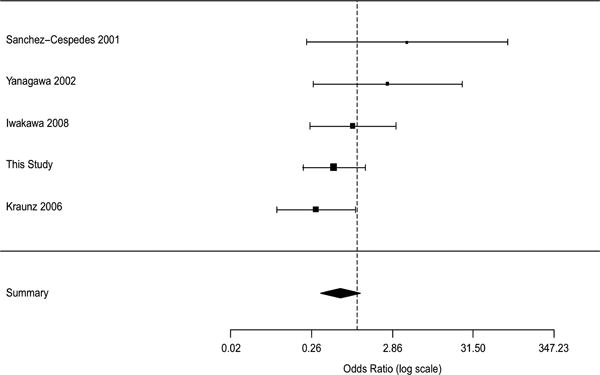
Meta-analysis of smoke exposure and p16 homozygous deletion. A fixed effects model was used. The square and the connecting lines indicate the OR and its CIs. The area of each square is proportional to the sample size of the study in the meta-analysis. The overall OR was 0.61 (p=0.1068) with 95% CI between 0.338 and 1.11.
Discussion
In order to achieve the goals of our study with confidence, we utilized multiple methodologies to study p16 inactivation and to confirm inactivation by RNA and protein expression. Reassuringly, the data from each of these methodologies gave concordant results, confirming the accuracy of our data and conclusions of this comprehensive study.
We found p16 inactivation in 75% of cell lines, mainly via HD (53%) or methylation (33%), and occasionally by point mutations (13%). Because the p16 gene forms a crucial component of the RB growth regulatory pathway, we investigated genetic alterations of other components of the pathway. Only 5% of cell lines had RB mutations. We also analyzed the copy number changes in these cell lines. Low level gains of cyclin D1, CDK4 and CDK6 were present in 20-25% of the cell lines while high level amplifications were rare. Thus, p16 inactivation was the major perturbation of the RB pathway noted in NSCLC cell lines.
Recent studies have shown the association between p16 promoter methylation and KRAS mutation in lung adenocarcinoma. However, the association between p16 inactivation mechanisms and other common genetic alterations in lung adenocarcinoma is much less understood or has never explored. In this study, we demonstrated that p16 inactivation occurred at similar frequencies regardless of the mutational status of EGFR, KRAS, and STK11 in lung adenocarcinoma; however, the patterns of p16 inactivation were significantly different depending on the mutational status of these genes. We also confirmed the link between KRAS mutation and p16 promoter methylation, and showed that EGFR mutation and p16 methylation were mutually exclusive. The negative and positive associations of p16 promoter methylation with EGFR and KRAS mutation respectively and the similar frequencies of p16 inactivation in both groups indicated the differences in the evolvement of epigenetic p16 alterations in these groups; despite the differences, it is clear that inactivation of the p16 gene is involved in the pathogenesis of both EGFR and KRAS mediated lung cancer.
More recently, STK11 has been identified as being frequently mutated in lung adenocarcinoma and could play an important role in lung cancer differentiation and metastasis.11,31 However, the relationship between STK11 mutation and p16 inactivation has never been investigated. Our results showed no significant correlations between STK11 and p16 inactivation. Because STK11 is usually inactivated by HDs, its true incidence may be underestimated. As with HD of p16, we examined STK11 by multiple means (sequencing, RNA expression, copy number) and are confident that are figures are accurate or near accurate.
Multiple studies have examined the relationship between smoking and mechanisms of p16 inactivation, however, the results have been variable. A number of studies reported that p16 promoter methylation was significantly associated with smoking but this finding was not reproduced by others.21,32 The effect of smoke exposure on p16 inactivation can be clarified by performing a meta-analysis. A recent study from China reported that smoking is positively correlated with p16 hypermethylation in NSCLC.33 However, their meta-analysis was limited to promoter methylation, which is not the most frequent mechanism of inactivation for p16. Our meta-analysis confirmed that smoking had a modest effect on p16 methylation. However when we performed a second meta-analysis to include all three mechanisms of p16 inactivation in smokers, we did not identify any significant association between never smoking and any specific p16 inactivation mechanism.
In conclusion, our results demonstrate that p16 inactivation occurs at similar frequencies regardless of the mutation status of EGFR, KRAS, and STK11 in lung adenocarcinoma. However, the patterns of inactivation mechanism differ significantly depending upon the genetic mutation present. We also confirm that p16 methylation is linked to KRAS mutation and is mutually exclusive with EGFR mutation. Our results indicate that these genes are involved in different pathways and play different roles in development of lung adenocarcinoma. Furthermore, our meta-analyses confirm the modest correlation between p16 methylation and smoking, and the trend of higher frequencies of p16 HD among never smokers. These findings support the concept that tumors arising in never smokers are driven by distinct molecular mechanisms in lung tumorigenesis.6 Future studies are needed to explore the associations between specific p16 inactivation mechanisms and other environmental and genetic alterations in order to further elucidate the different molecular mechanisms involved in the development of lung adenocarcinoma.
Supplementary Material
Acknowledgments
Support was provided by generous grants from the Canary Foundation, the Early Detection Research Network U01CA086402, National Cancer Institute, Bethesda, MD and the University of Texas SPORE in Lung Cancer PO # P50CA70907, National Cancer Institute, Bethesda, MD. Drs. Ma and Zhang were supported by HG001696 and ES017166 (to MQZ). We thank Dr. Guanghua Xiao and Dr. Ward Wakeland from UT Southwestern Medical Center for their contribution to the work.
References
- 1.Howlader N, Noone AM, Krapcho M, et al. SEER Cancer Statistics Review 1975-2009. National Cancer Institute; Bethesda MD: [Accessed August 20, 2012]. Available at: http://seer.cancer.gov/csr/1975_2009. [Google Scholar]
- 2.Siegel R, Naishadham D, Jemal A. Cancer statistics, 2012. CA: a cancer journal for clinicians. 2012;62:10–29. doi: 10.3322/caac.20138. [DOI] [PubMed] [Google Scholar]
- 3.Borczuk AC, Kim HK, Yegen HA, et al. Lung adenocarcinoma global profiling identifies type II transforming growth factor-beta receptor as a repressor of invasiveness. Am J Respir Crit Care Med. 2005;172(6):729–737. doi: 10.1164/rccm.200504-615OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shimosato Y. Histological Typing of Lung and Pleural Tumors (3rd edition, 1999): Malignant epithelial tumors. Nihon Rinsho. 2002 May;60(Suppl 5):123–31. [PubMed] [Google Scholar]
- 5.Subramanian J, Govindan R. Lung cancer in never smokers: A Review. J Clin Oncol. 2007;25(5):561–70. doi: 10.1200/JCO.2006.06.8015. [DOI] [PubMed] [Google Scholar]
- 6.Sun F, Schiller J, Gazdar AF. Lung cancer in never smokers – a different disease. Nature Reviews Cancer. 2007;7:778–790. doi: 10.1038/nrc2190. [DOI] [PubMed] [Google Scholar]
- 7.National Lung Screening Trial Research Team. Aberle DR, Adams AM, et al. Reduced lung-cancer mortality with low-dose computed tomographic screening. NEJM. 2011;365:395–409. doi: 10.1056/NEJMoa1102873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ladanyi M, Pao W. Lung adenocarcinoma: guiding EGFR-targetedtherapy and beyond. Modern Pathology. 2008;21:16–22. doi: 10.1038/modpathol.3801018. [DOI] [PubMed] [Google Scholar]
- 9.Ding L, Getz G, Wheeler D, et al. Somatic mutations affect key pathways in lung adenocarcinoma. Nature. 2008;455:1069–1075. doi: 10.1038/nature07423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jang TW, Oak CH, Chang HK, et al. EGFR and KRAS mutations in patients with adenocarcinoma of the lung. Korean J Intern Med. 2009;24(1):48–54. doi: 10.3904/kjim.2009.24.1.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sanchez-Cespedes M, Parrella P, Esteller M, et al. Inactivation of STK11/STK11 is a common event in adenocarcinoma of the lung. Cancer Research. 2002;62:3659–3662. [PubMed] [Google Scholar]
- 12.Iwakawa R, Kohno T, Anami Y, et al. Association of p16 homozygous deletions with clinicopathologic characteristics and EGFR/KRAS/p53 mutations in lung adenocarcinoma. Clin Cancer Res. 2008;14(12):374–353. doi: 10.1158/1078-0432.CCR-07-4552. [DOI] [PubMed] [Google Scholar]
- 13.Sanchez-Cespedes M, Reed A, Buta M, et al. Inactivation of the INK4A/ARF locus frequently coexists with TP53 mutations in non-small cell lung cancer. Oncogene. 1999;18:5843–5849. doi: 10.1038/sj.onc.1203003. [DOI] [PubMed] [Google Scholar]
- 14.Seike M, Gemma A, Hosoya Y, et al. Increase in frequency of p16INK4A gene inactivation by hypermethylation in lung cancer during the process of metastasis and its relation to the status of p53. Clin Cancer Res. 2000;6:4307–4313. [PubMed] [Google Scholar]
- 15.The Cancer Genome Atlas Research Network. Comprehensive genomic characterization of squamous cell lung cancers. Nature. 2012;489:519–525. doi: 10.1038/nature11404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Imielinski M, Berger AH, Hammerman PS, et al. Mapping the hallmarks of lung adenocarcinoma with massively parallel sequencing. Cell. 2012;150:1107–1120. doi: 10.1016/j.cell.2012.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gil J, Peters G. Regulation of the INK4b-ARF-INK4a tumour suppressor locus: all for one or one for all. Nat Rev Mol Cell Biol. 2006;7(9):667–677. doi: 10.1038/nrm1987. [DOI] [PubMed] [Google Scholar]
- 18.Ohtani N, Yamakoshi K, Takahashi A, et al. The p16INK4a-RB pathway: molecular link between cellular senescence and tumor suppression. J Med Invest. 2004;51:146–153. doi: 10.2152/jmi.51.146. [DOI] [PubMed] [Google Scholar]
- 19.Kraunz K, Nelson H, Lemos M, et al. Homozygous deletion of p16INK4a and tobacco carcinogen exposure in nonsmall cell lung cancer. Int J Cancer. 2006;118:1364–1369. doi: 10.1002/ijc.21522. [DOI] [PubMed] [Google Scholar]
- 20.Toyooka S, Suzuki M, Tsuda T, et al. Dose effect of smoking on aberrant methylation in non-small cell lung cancers. Int J Cancer. 2004;110:462–464. doi: 10.1002/ijc.20125. [DOI] [PubMed] [Google Scholar]
- 21.Kim D, Nelson H, Wiencke J, et al. p16INK4a and Histology-specific Methylation of CpG Islands by Exposure to Tobacco Smoke in Non-Small Cell Lung Cancer. Cancer Research. 2001;61:3419–3424. [PubMed] [Google Scholar]
- 22.Phelps R, Johnson B, Ihde D, et al. NCI-navy medical oncology branch cell line data base. Journal of Cellular Biochemistry. 1996;24:32–91. doi: 10.1002/jcb.240630505. [DOI] [PubMed] [Google Scholar]
- 23.Koivunen JP, Mermel C, Zejnullahu K, et al. EML4-ALK Fusion Gene and Efficacy of an ALK Kinase Inhibitor in Lung Cancer. Clin Cancer Res. 2008;14(13):4275. doi: 10.1158/1078-0432.CCR-08-0168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gandhi J, Zhang J, Xie Y, et al. Alterations in genes of the EGFR signaling pathway and their relationship to EGFR tyrosine kinase inhibitor sensitivity in lung cancer cell lines. PLoS One. 2009;4(2):e4576. doi: 10.1371/journal.pone.0004576. Published online February 24, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shivapurkar N, Stastny V, Suzuki M, et al. Application of a methylation gene panel by quantitative PCR for lung cancers. Cancer Letters. 2007;247(1):56–71. doi: 10.1016/j.canlet.2006.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Faca V, Krasnoselsky A, Hanash S. Innovative proteomic approaches for cancer biomarker discovery. Biotechniques. 2007;43(3):279–283. doi: 10.2144/000112541. [DOI] [PubMed] [Google Scholar]
- 27.Soh J, Okumura N, Lockwood WW, et al. Oncogene mutations, copy number gains and mutant allele specific imbalance (MASI) frequently occur together in tumor cells. PLoS One. 2009;4(10):e7464. doi: 10.1371/journal.pone.0007464. Published online October 14, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shigematsu H, Gazdar AF. Somatic mutations of epidermal growth factor receptor signaling pathway in lung cancers. Int J Cancer. 2006;118:25–262. doi: 10.1002/ijc.21496. [DOI] [PubMed] [Google Scholar]
- 29.Shigematsu H, Lin L, Takahashi T, et al. Clinical and biological features associated with epidermal growth factor receptor gene mutations in lung cancer. J Natl Cancer Inst. 2005;97(5):346. doi: 10.1093/jnci/dji055. [DOI] [PubMed] [Google Scholar]
- 30.DerSimonian R, Laird N. Meta-Analysis in Clinical Trials. Control Clin Trials. 1986;7(3):177–188. doi: 10.1016/0197-2456(86)90046-2. [DOI] [PubMed] [Google Scholar]
- 31.Ji H, Ramsey M, Hayes DN, et al. STK11 modulates lung cancer differentiation and metastasis. Nature. 2007;448:807–811. doi: 10.1038/nature06030. [DOI] [PubMed] [Google Scholar]
- 32.Vaissiere T, Hung R, Zaridze D, et al. Quantitative analysis of DNA methylation profiles in lung cancer identifies aberrant DNA methylation of specific genes and its association with gender and cancer risk factors. Cancer Research. 2009;69:243–252. doi: 10.1158/0008-5472.CAN-08-2489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang B, Zhu W, Yang P, et al. Cigarette smoking and p16INK4α gene promoter hypermethylation in non-small cell lung carcinoma patients: a meta-analysis. PLoS One. 2011;6(12):e28882. doi: 10.1371/journal.pone.0028882. Published online December 13, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Glass GV. Primary, Secondary, and Meta-Analysis of Research. Educational Researcher; 1976. [Google Scholar]
- 35.Borenstein M, Hedges LV, Higgins JP, et al. Introduction to meta-analysis. Chichester UK: John Wiley & Sons; 2009. [Google Scholar]
- 36.Zöchbauer-Müller S, Fong KM, Virmani AK, et al. Aberrant promoter methylation of multiple genes in non-small cell lung cancers. Cancer Res. 2001;61(1):249–255. [PubMed] [Google Scholar]
- 37.Toyooka S, Maruyama R, Toyooka KO, et al. Smoke exposure, histologic type and geography-related differences in the methylation profiles of non-small cell lung cancer. Int J Cancer. 2003;103(2):153–160. doi: 10.1002/ijc.10787. [DOI] [PubMed] [Google Scholar]
- 38.Yanagawa N, Tamura G, Oizumi H, et al. Promoter hypermethylation of tumor suppressor and tumor-related genes in non-small cell lung cancers. Cancer Sci. 2003;94(7):589–592. doi: 10.1111/j.1349-7006.2003.tb01487.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yanagawa N, Tamura G, Oizumi H, et al. Inverse correlation between EGFR mutation and FHIT, RASSF1A and RUNX3 methylation in lung adenocarcinoma: relation with smoking status. Anticancer Res. 2011;31(4):1211–1214. [PubMed] [Google Scholar]
- 40.Furonaka O, Takeshima Y, Awaya H, et al. Aberrant methylation of p14(ARF), p15(INK4b) and p16(INK4a) genes and location of the primary site in pulmonary squamous cell carcinoma. Pathol Int. 2004;54(8):549–555. doi: 10.1111/j.1440-1827.2004.01663.x. [DOI] [PubMed] [Google Scholar]
- 41.Liu Y, Lan Q, Siegfried JM, et al. Aberrant Promoter Methylation of p16 and MGMT Genes in Lung Tumors from Smoking and Never-Smoking Lung Cancer Patients. Neoplasia. 2006;8(1):46–51. doi: 10.1593/neo.05586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Georgiou E, Valeri R, Tzimagiorgis G, et al. Aberrant p16 promoter methylation among Greek lung cancer patients and smokers: correlation with smoking. Eur J Cancer Prev. 2007;16(5):396–402. doi: 10.1097/01.cej.0000236260.26265.d6. [DOI] [PubMed] [Google Scholar]
- 43.Jarmalaite S, Kannio A, Anttila S, et al. Aberrant p16 promoter methylation in smokers and former smokers with nonsmall cell lung cancer. Int J Cancer. 2003;106(6):913–918. doi: 10.1002/ijc.11322. [DOI] [PubMed] [Google Scholar]
- 44.Suga Y, Miyajima K, Oikawa T, et al. Quantitative p16 and ESR1 methylation in the peripheral blood of patients with non-small cell lung cancer. Oncol Rep. 2008;20(5):1137–1142. [PubMed] [Google Scholar]
- 45.Hong YS, Kim NY, Kim HK, et al. Hypermethylation of p16 in Korean non-small cell lung cancer patients. J Korean Med Science. 2007:S32–37. doi: 10.3346/jkms.2007.22.S.S32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Divine KK, Pulling LC, Marron-Terada PG, et al. Multiplicity of abnormal promoter methylation in lung adenocarcinomas from smokers and never smokers. Int J Cancer. 2005;114:400–405. doi: 10.1002/ijc.20761. [DOI] [PubMed] [Google Scholar]
- 47.Wu MF, Cheng YW, Lai JC, et al. Frequent p16INK4a promoter hypermethylation in human papillomavirus-infected female lung cancer in Taiwan. Int J Cancer. 2005;113(3):440–445. doi: 10.1002/ijc.20597. [DOI] [PubMed] [Google Scholar]
- 48.Kim DS, Cha SI, Lee JH, et al. Aberrant DNA methylation profiles of non-small cell lung cancers in a Korean population. Lung Cancer. 2007;58(1):1–6. doi: 10.1016/j.lungcan.2007.04.008. [DOI] [PubMed] [Google Scholar]
- 49.Tessema M, Yu YY, Stidley CA, et al. Concomitant promoter methylation of multiple genes in lung adenocarcinomas from current, former and never smokers. Carcinogenesis. 2009;30(7):1132–1138. doi: 10.1093/carcin/bgp114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kim JS, Kim H, Shim YM, et al. Aberrant methylation of the FHIT gene in chronic smokers with early stage squamous cell carcinoma of the lung. Carcinogenesis. 2004;25(11):2165–2171. doi: 10.1093/carcin/bgh217. [DOI] [PubMed] [Google Scholar]
- 51.Toyooka S, Tokumo M, Shigematsu H, et al. Mutational and epigenetic evidence for independent pathways for lung adenocarcinomas arising in smokers and never smokers. Cancer Res. 2006;66:1371–1375. doi: 10.1158/0008-5472.CAN-05-2625. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.